

ABSTRACT
CX3CR1 has been identified as a highly attractive target for several therapeutic interventions. Despite this potential, no potent antagonists, either small molecule or monoclonal antibody, have been identified. Here we describe the lead finding and engineering approach that lead to the identification of BI 655088, a potent biotherapeutic antagonist to CX3CR1. BI 655088 is a potent CX3CR1 antagonist that, upon therapeutic dosing, significantly inhibits plaque progression in the standard mouse model of atherosclerosis. BI 655088 represents a novel and highly selective biotherapeutic that could reduce inflammation in the atherosclerotic plaque when added to standard of care treatment including statins, which could result in a significant decrease in atherothrombotic events in patients with existing cardiovascular disease.
KEYWORDS: VHH Antibodies, GPCR, atherosclerosis, BI 655088, biophysical assessment, pharmacokinetic profile, humanization
Introduction
CX3CR1, a chemokine receptor expressed on monocytes/macrophages, dendritic cells, and memory effector T cells, is associated with atherosclerosis and proposed to mediate cell migration and survival within plaques.1,2 CX3CR1 expression has been shown to be increased in human atherosclerotic plaques where, in addition to inflammatory cells, the receptor is up-regulated on smooth muscle cells.3 CX3CR1 has a unique ligand fractalkine (CX3CL1), which modulates leukocyte adhesion functions and acts as a chemotactic agent. Fractalkine is released into the circulation where it can be used as a systemic biomarker for CX3CR1 target engagement.4 In humans, a CX3CR1 variant with reduced activity has been associated with a lower risk of cardiovascular disease.5,6 In addition, several studies using CX3CR1-deficient mice have shown a beneficial effect on atherosclerosis.7,8
Based on the known biology of CX3CR1, the development of an antagonist has the potential to provide a novel approach to treat inflammatory diseases. To date, the only examples of CX3CR1 antagonists include the small molecule inhibitor AZD8797 and a CX3CL1 variant.9,10 AZD8797 was shown to selectivity inhibit CX3CR1, but in vivo efficacy in a rat experimental autoimmune encephalomyelitis model required subcutaneous (s.c.) administration and high plasma concentrations (2 µM) to produce efficacy. The fractalkine variant, F1, was shown to be a potent inhibitor (IC50 values of 5–50 nM) of CX3CR1, but again the dosing regimen (~2000 mg/kg, 3x/wk) was not supportive for use as a therapeutic agent.
To date, antagonistic monoclonal antibodies to CX3CR1 have not been identified and our attempt to identify one was also unsuccessful. We hypothesized that Variable VHH antibodies (variable domains of camelid heavy chain-only) may provide an advantage to conventional monoclonal antibodies due to their small size and long complementarity-determining region (CDR) loops in targeting a G-protein-coupled receptor (GPCR) such as CX3CR1. Here, we present llama-derived VHHs that effectively block the fractalkine-induced signaling mediated by CX3CR1. We demonstrate the multivalent VHHs exhibit desirable potency and selectivity. Furthermore, through the inclusion of an anti-albumin VHH, we constructed an antagonist that is predicted to have desirable pharmacokinetics (PK) in humans. Finally, we show that an optimized VHH, BI 655088, can block the CX3CR1 receptor in vivo and concomitantly reduce the progression of atherosclerosis in an apoE KO mouse model.
Results
Immunization strategy and immunogen designs
To identify VHH antagonists of CX3CR1, llamas were immunized using a variety of strategies and immunogens. One group of llamas was immunized first with CX3CR1 DNA followed by four boosts with Caki cells, a camelid cell line, over-expressing human CX3CR1.11 The second group of llamas was immunized first with the Caki cell line over-expressing human CX3CR1 followed by four boosts of peptides derived from the extracellular loops of CX3CR1 adjuvanted with bovine serum albumin (BSA). Serology was performed using a Chinese hamster ovary (CHO) cell line over-expressing human CX3CR1 by flow cytometry (Table 1).
Table 1.
Serum titer analysis for immunized animals by FACS.
| Llama | Immunogen | Titer After Prime | Titer After Boost |
|---|---|---|---|
| 1 | Caki huCX3CR1 + NT/EC3 peptide | - | - |
| 2 | Caki huCX3CR1 + NT/EC3 peptide | + | + |
| 3 | Caki huCX3CR1 + NT/EC3 peptide | - | + |
| 4 | DNA + Caki huCX3CR1 | ++ | ++ |
| 5 | DNA + Caki huCX3CR1 | - | - |
| 6 | DNA + Caki huCX3CR1 | - | - |
Library generation
Peripheral blood mononuclear cells (PBMCs) and lymph nodes were isolated from the animals and used as a source of mRNA for phage heavy chain variable domain library construction. VHHs binding to CX3CR1 were identified through two different panning strategies, with alternative panning on the Ba/F3 and CHO-K1 human CX3CR1 over-expressing cell lines or multiple rounds of panning with CX3CR1-expressing virus-like particles (VLP). Binders were then selected based on their ability to compete with the binding of AF647-labeled fractalkine to human CX3CR1-expressing CHO cells using flow cytometry in a screening mode (n = 1, Supplemental Figure 1). Due to the necessity of using a non-human primate species to evaluate the PK and safety of the VHHs, molecules were selected that were able to compete with AF647-fractalkine binding to cynomolgus monkey CX3CR1 with an EC50 that was within 10-fold of the value for human CX3CR1.
Lead identification
VHHs that were screened through the process above were then sequenced and consolidated sequences were grouped into four families (9, 13, 14 and 101) based on phylogenetic tree (Supplemental Figure 2). Family 9 showed very potent binding to human CX3CR1 and fractalkine blocking, but no cross-reactivity to cynomolgus monkey CX3CR1. Family 13 showed good binding to human and cynomolgus CX3CR1 but were only partial blockers of fractalkine. Family 14 showed complete blocking of fractalkine with only very weak binding affinity (triple digit nM, data not shown) to both human and cynomolgus CX3CR1. Finally, panning with CX3CR1-expressing VLP led to the identification of Family 101 showing all the desired functionalities with potent binding and blocking to both human and cynomolgus CX3CR1.
Family 101 was further pursued for lead identification. Monovalent VHHs were generated in E. coli and the titer was measured as an evaluation of their manufacturability (Table 2). Specificity of the VHHs for the human CX3CR1 receptor was evaluated by performing binding experiments on CHO-K1 parental cells or CHO cells expressing human CCR2 or human CCR5. No binding to either CCR2 or CCR5 was observed when VHHs were tested up to 1 µM. The VHHs were profiled in a fluorescence-activated cell sorting (FACS) competition assay with AF647-labeled human fractalkine to generate IC50s against human and cynomolgus CX3CR1 (Table 2, Supplemental Figure 1). The competition assay was performed at the EC30 of AF647-labeled fractalkine, and IC50s were calculated based on the VHH dose response (Supplemental Figure 3). Percent block was calculated as the ability to block fractalkine completely from the cell surface.
Table 2.
VHH competition with fractalkine.
| Mono-valent VHH | Titer(mg/L) | Percent Block (human) | hFractalkine Competition IC50 (nM) | Percent Block (cyno) | cyFractalkine Competition IC50 (nM) | Cyno Ratio(IC50) Cyno/Human) |
|---|---|---|---|---|---|---|
| 54A12 | 17.4 | 96 | 6.8 | 94 | 67 | 9.9 |
| 54A04 | 0.4 | 100 | 49 | 91 | 31 | 0.6 |
| 54A07 | 8.5 | 52 | 23 | 69 | 280 | 12.2 |
| 54B03 | 19.3 | 92 | 67 | 95 | 470 | 7.0 |
| 54D05 | 6.3 | 94 | 5.3 | 91 | 64 | 12.1 |
| 54G03 | 26.1 | 89 | 270 | 89 | 470 | 1.7 |
| 54H01 | 2.8 | 64 | 10 | 92 | 68 | 6.8 |
| 61B02 | 36.4 | 94 | 10 | 92 | 45 | 4.5 |
| 61B04 | 22.5 | 97 | 57 | 93 | 87 | 1.5 |
| 61E02 | 0.7 | 98 | 82 | 96 | 46 | 0.6 |
| 61F11 | 9.7 | 96 | 46 | 95 | 160 | 3.5 |
| 61G03 | 6.4 | 96 | 60 | 89 | 300 | 5.0 |
| 61G04 | 11 | 96 | 42 | 100 | 200 | 4.8 |
| 66B02 | 13.2 | 102 | 2.5 | 97 | 19 | 7.6 |
| 66G01 | 9.2 | 99 | 14 | 100 | 120 | 8.6 |
Four VHHs were selected based on potency in ligand competition for further functional profiling in a human fractalkine-induced chemotaxis of BAF/3 cells over-expressing human CX3CR1 (Table 3). While several of these VHHs showed the desired functional profile and specificity and selectivity profiles, none of the monovalent VHHs were able to meet the IC50 criteria of <1 nM.
Table 3.
Percent block of fractalkine-induced chemotaxis by VHHs.
| VHH | Percent Block | Fractalkine-Induced Chemotaxis IC50 (nM) |
|---|---|---|
| 54A12 | 84 | 80 |
| 54D05 | 81 | 70 |
| 66B02 | 87 | 20 |
| 66G01 | 54 | 400 |
VHH formatting and half-life extension
Bivalent constructs were made to determine whether avidity could improve the binding potency of the VHHs. The bivalent constructs were formatted with 35 GLY-SER linkers between the VHHs. The four bivalent VHHs were expressed in E. coli and demonstrated saturable (100 percent block), dose-dependent binding with IC50 values <1 nM against human CX3CR1-expressing Ba/F3 cells (Figure 1, Table 4) or cynomolgus monkey CX3CR1-expressing HEK293 cells. The ability of candidate VHHs to bind to endogenously expressed human CX3CR1 was explored using the Alexa Fluor 647-labeled VHHs. Labeled VHHs were incubated with human PBMCs from healthy donors and flow cytometry was used to evaluate binding affinity for selected VHHs. The binding affinities are comparable to those observed with the Baf3-hCX3CR1 cell line (data not shown). The selection of the best candidate for further optimization was based on binding to primary cells in addition to performance in expression and purification as a predictor of manufacturability. From these data monovalent 66B02 was selected as the best lead candidate and named BI 18 as a bivalent VHH.
Figure 1.
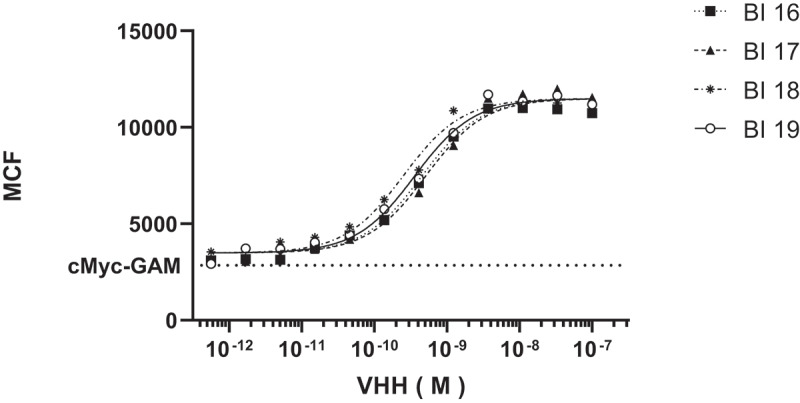
Binding of bivalent family101 VHHs to Ba/F3/hCX3CR1 cells. Mean fluorescence per cell was measured by flow cytometry of various concentrations of Alexa Fluor labeled VHHs. IC50s were calculated to be <1 nM for all four constructs tested.
Table 4.
Functional profiling of Bi-valent VHHs.
| hFractalkine Competition |
cyFractalkine Competition |
Chemotaxis |
Inhibition of Fractalkine Internalization |
|||||
|---|---|---|---|---|---|---|---|---|
| Bi-valent VHH | Percent Block | IC50 (nM) | Percent Block | IC50 (nM) | Percent Block | IC50 (nM) | Percent Block | IC50 (nM) |
| 54A12-35GS-54A12 (BI 16) | 102 | 0.19 | 98 | 0.69 | 88 | 2.0 | 90 | 0.8 |
| 54D05-35GS-54D05 (BI 17) | 99 | 0.31 | 95 | 1.6 | 89 | 3.0 | 91 | 0.6 |
| 66B02-35GS-66B02 (BI 18) | 102 | 0.03 | 97 | 0.10 | 98 | 0.6 | 92 | 0.33 |
| 66G01-35GS-66G01 (BI 19) | 100 | 0.29 | 96 | 0.82 | 85 | 2.0 | 89 | 0.90 |
Sequence optimization
Parental wildtype llama VHH sequences were mutated to yield VHH sequences that are more identical to human VH3-JH germline consensus sequences. An alignment of the wildtype llama VHH sequence with humanVH3-JH is shown in supplemental Figure 4. The following amino acids: E1D, S11L, A14P, E16G, R44Q, D46E, A74S, K83R, and Q108L were investigated and evaluated to confirm that protein structure, activity, and stability were kept intact. In addition, a consensus sequence for a predicted glycosylation site (NS) was identified at residues 52 and 53. Two additional libraries were constructed, one for position 52 and one for position 53, which were designed to include all possible amino acids at the respective positions. All sequence optimization variants were compared with the parental VHH in three different assays. The final optimized sequence was based on (1) determination of the melting temperature (Tm) in a Thermal Shift Assay (TSA), (2) analysis of in vitro potency in fractalkine competition FACS, and (3) analysis of in vitro potency in the fractalkine-induced chemotaxis assay. A comparison between the wildtype and sequence optimized clone is shown in Table 5.
Table 5.
Comparison between wildtype and sequence optimized VHH.
| hFractalkine Competition |
||||
|---|---|---|---|---|
| Clone | Percent Block | IC50 (nM) | Tm at pH 7 | Chemotaxis IC50 (nM) |
| 66B02 | 101 | 2.5 | 65.7 | 36 |
| 66B02-SO* | 97 | 1.9 | 68.1 | 63 |
*66B02-SO: Sequence optimized 66B02 VHH
VHHs formatted as single variable domains are known to have a very short half-life (<1 day).12 To achieve the new therapeutic concept, which requires chronic dosing, two half-life extension technologies were evaluated. To achieve the required potency and PK properties, the sequence-optimized VHH was formatted to be bivalent and fused to either an albumin-binding VHH (BI 655088) or to a human lgG1 Fc domain (BI 655089). For the Fc-fusion, two mutations were incorporated in the CH2 domain to abrogate potential Fc-mediated effector function in this construct.11
BI 655088 and BI 655089 retained the binding potency and functional activity in both the chemotaxis and internalization assays that we observed in the bivalent VHH molecules (Table 6 and Figure 2). To extend the characterization of the VHH, we evaluated the effect on fractalkine-stimulated calcium mobilization in CHO cells that express both Gα16 and human CX3CR1. In order to confirm that BI 655088 and BI 655089 do not have agonist activity, BI 655088 and BI 655089 were also evaluated for induction of calcium influx in the CHO-K1 huCX3CR1 cells. While pre-incubation with BI 655088 and BI 655089 inhibited fractalkine-mediated calcium influx with an IC50 of 1.3 nM and 3.2 nM, respectively, no increase in cytosolic calcium levels were observed when the polypeptide alone was added at concentrations up to 1 μΜ.
Table 6.
Functional profile of BI 655088 and BI 655089.
| Assay | BI 655088 | BI 655089 |
|---|---|---|
| hFractalkine Competition IC50 (nM) | 0.4 | 0.7 |
| Chemotaxis IC50 (nM) | 2.7 | 3.0 |
| Ligand Internalization IC50 (nM) | 0.33 | 0.37 |
| Calcium Influx IC50 (nM) | 1.3 | 3.2 |
Figure 2.
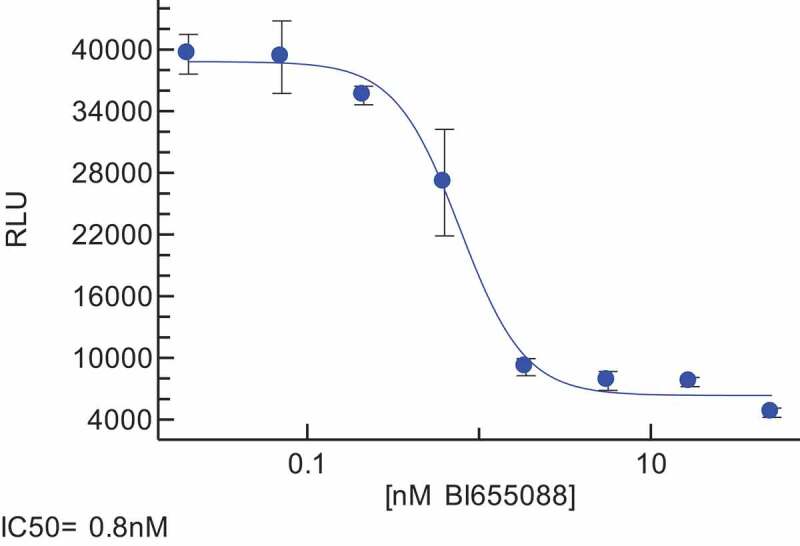
Exemplar Chemotaxis Assay data showing the functional activity of BI 655088.
To assess whether the binding of human serum albumin (HSA) to BI 655088, which contains an albumin-binding VHH, could impact the affinity for human CX3CR1, ligand competition was performed in the presence of HSA. Full blocking efficacy was reached in the presence of HSA, although the potency was reduced four-fold (0.43 nM vs. 1.6 nM). As expected, there is no impact on binding potency for BI 655089 in the presence of HSA. Using a surface plasmon resonance technique, the affinity (KD) of the albumin-binding domain of BI 655088 was previously determined to be 31 nM against HSA or 180 nM against mouse albumin.12
Cynomolgus monkey PK/PD
Non-compartmental analysis (Phoenix, WinNonlin v 5.3) of the PK for BI 655088 and BI 655089 were compared in cynomolgus monkey after intravenous (i.v.) (0.2, 2.0 mg/kg) and s.c. (2.0 mg/kg) administration. In additional an equimolar i.v. dose (5 and 10 mg/kg for BI 655088 and BI 655089, respectively) was added to compare the linear PK of both molecules. Mean systemic clearance for both molecules was dose-dependent, consistent with saturable target-mediated drug disposition (TMDD) frequently observed for protein therapeutics targeting cell-associated antigens. Unexpectedly, clearance at all doses was substantially higher for BI 655089 than for BI 655088 (Table 7). For BI 655088, clearance was comparable between the 2 and 5 mg/kg doses, suggesting that TMDD saturation was substantially achieved at the 2 mg/kg dose, whereas for BI 655089, the dose nonlinearity persisted between the mid and high doses. Mean bioavailability of BI 655088 following s.c. administration was 55.2%, with exposure being maintained at >70 nM for 14 days. Plasma concentration versus time profile for BI 655089 following subcutaneous administration suggested possible “flip-flop” pharmacokinetics, with the absorption rate being significantly slower than the rate of elimination, potentially resulting in an overestimation of bioavailability. Plasma concentration versus time profile for BI 655089 following subcutaneous administration suggested possible “flip-flop” pharmacokinetics, with absorption rate being significantly slower than the rate of elimination, potentially resulting in an overestimation of bioavailability. Due to its superior PK profile, BI 655088 was selected as the top candidate for further profiling.
Table 7.
Pharmacokinetic profile of BI 655088 and BI 655089.
| Parameter | BI 655088 Mean (SD) |
BI 655089 Mean (SD) |
|---|---|---|
| 0.2 mg/kg i.v. | ||
| CL (mL/d/kg) | 112.6 (24.9) | 145.6 (28.9) |
| AUCinf (nM * h) | 1126 (249) | 438 (91) |
| T1/2 (d) | 1.3 (0.2) | 0.4 (0.1) |
| 2 mg/kg i.v. | ||
| CL (mL/d/kg) | 9.4 (1.2) | 92 (29.8) |
| AUCinf (nM * h) | 155841 (9732) | 7040 (1957) |
| T1/2 (d) | 9.2 (2.1) | 1.8 (0.5) |
| 2 mg/kg s.c. | ||
| AUCinf (nM * h) | 73530 (10583) | 10713 (1770) |
| F% | 55.2 (7.9) | >100% |
hCX3CR1 KI mouse model
Consistent with the sequence divergence in the extracellular domains of human and mouse CX3CR1, we observed that none of the anti-human CX3CR1 VHHs were able to compete with fractalkine binding to mouse CX3CR1. To enable pharmacological profiling of the VHHs, human CX3CR1 knock-in (hCX3CR1 KI) mice were generated and back-crossed with apoE-/- mice to create an atherosclerosis model for evaluation of the molecules. These mice express similar levels of CX3CR1 in PBMCs compared to wildtype mice (Supplemental Figure 5). Mouse fractalkine is a functional ligand for human CX3CR1, with an EC50 of 4.8 nM in the calcium flux assay, and thus we expected that this knock-in mouse would retain productive fractalkine-CX3CR1 signaling.
To evaluate the pharmacodynamics of the anti-CX3CR1 VHH, BI 655088 was dosed to hCX3CR1 KI mice, and serum samples were evaluated for fractalkine concentration and for free-receptor concentration. As shown in Figure 3, serum levels of BI 655088 were indirectly correlated with levels of free receptor and directly correlated with the concentration of fractalkine. The increase in serum fractalkine is consistent with a decrease in the clearance of the ligand caused by the blockade of the receptor.
Figure 3.
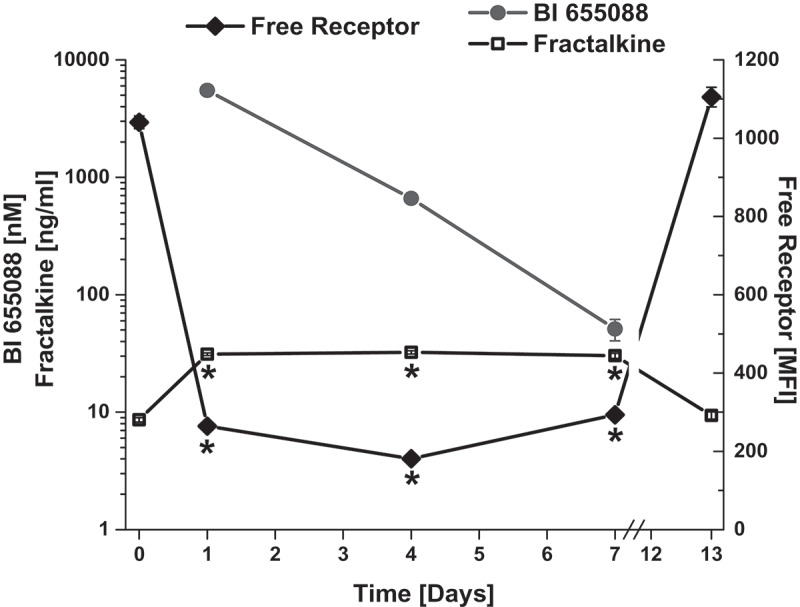
Characterization of Target Engagement Biomarkers in hCX3CR1 KI Mouse Model. Free receptor levels, serum concentrations of BI 655088 and serum fractalkine levels in hCX3CR1 KI mice after receiving two doses at 30 mg/kg of BI 655088 or vehicle. Day 0 represents baseline concentrations for free receptor and serum fractalkine of hCX3CR1 KI mice dosed with vehicle control (Mean ± SD, *p < .0001 vs. Day 0 and Day 13). BI 655088 and Fractalkine levels were determined by immunoassay and free receptor levels assessed by FACS.
CX3CR1 VHH reduces atherosclerosis in mice
CX3CR1 staining has been reported in atherosclerotic lesions of both mice and humans.13 We confirmed that CX3CR1 was present in plaques of hCX3CR1 KI/apoE-/- mice, as well as in atherosclerotic plaque from cross-sections of human coronary artery samples (Figure 4). To determine the effect of CX3CR1 antagonism on the progression of atherosclerosis, mice were placed on a high-fat diet for 16 weeks and treatment was performed during the last 6 weeks to simulate a therapeutic treatment protocol. A 30 mg/kg (twice a week) dose of BI 655088 produced a significant, three-fold increase in serum fractalkine and a concomitant 62% decrease in plaque area (Figure 5). A lower dose of BI 655088 (10 mg/kg once a week) did not significantly modulate serum fractalkine levels or plaque area compared to vehicle-treated mice. The trough plasma concentration of BI 655088 was 88 nM for the 30 mg/kg dose (samples collected 3.5 days after the last dose) and below quantitative limits (<0.5 nM) for the 10 mg/kg dose (samples collected 7 days after the last dose). Treatment with BI 655088 did not affect serum cholesterol, serum triglycerides, or body weight (Supplemental Table 1). A comparable extent of atherosclerotic plaque reduction was observed in CX3CR1-/-/apoE-/- mice compared to hCX3CR1 KI apoE-/- mice (Supplemental Figure 6).
Figure 4.
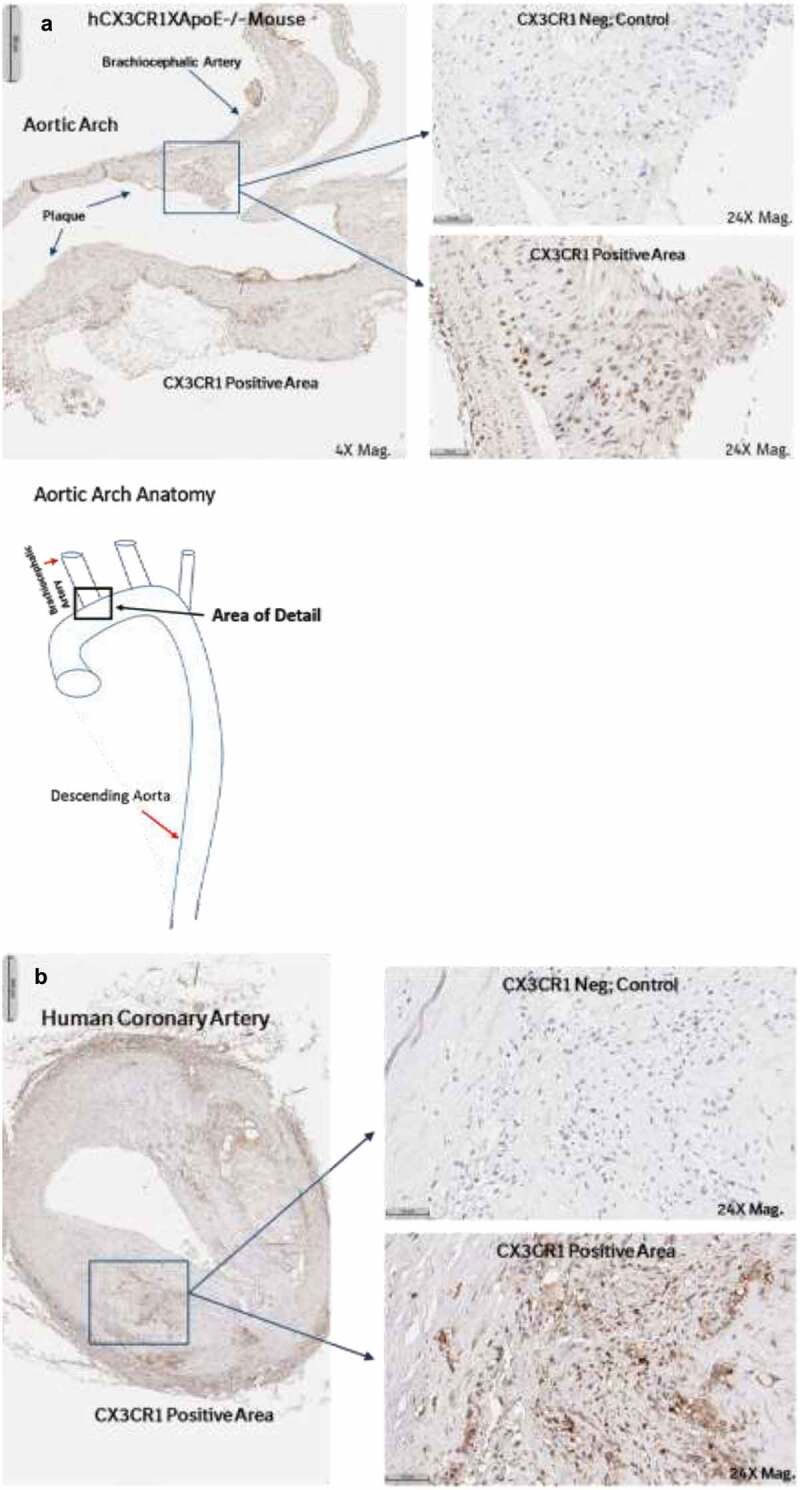
Human CX3CR1 is expressed in the atherosclerotic plaques of hCX3CR1/ApoE KO mice (brachiocephalic artery) after feeding with an HF/HC diet (a). IHC was done using a human anti-CX3CR1 antibody and compared to a no-antibody negative control. Similar expression of CX3CR1 was also seen in the plaque from a cross-section of human coronary artery from an atherosclerotic patient (b).
Figure 5.
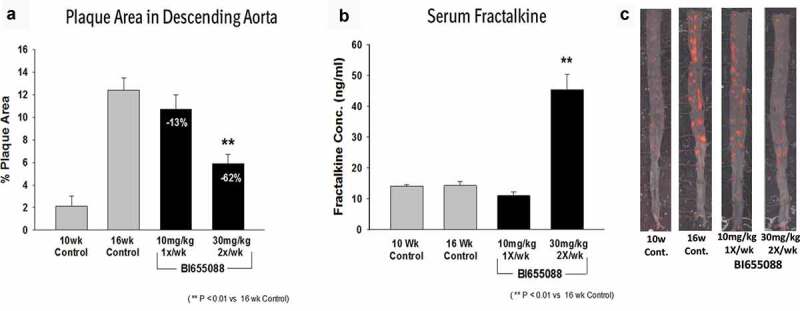
BI 665088 treatment significantly reduced the descending aorta plaque area (a) with a concomitant increase in serum fractalkine (b). Representative images of descending aortas after Oil Red O staining for plaque area (c).
Characterization of developability of BI 655088
After establishing in vivo proof of concept with BI 655088, a developability assessment was performed to determine whether this molecule might encounter setbacks in the manufacturing process. To assess the expression and purification potential, BI 655088 was expressed in P. pastoris. Expression levels were good with a titer of 1.1 g/L. BI 655088 was purified by Protein A affinity chromatography followed by cation exchange chromatography. The Protein A yield for BI 655088 was 70%. These results, coupled with a > 97% monomer content (Table 8, Supplemental Figure 7) after two-step purification, suggest this molecule should behave well in a standard platform expression and purification process.
Table 8.
Developability of BI 655088.
| Assessment | BI 655088 |
|---|---|
| Expression system | P. pastoris |
| Titer | 1.1 g/L (whole broth) |
| AUC (% monomer) | 97% |
| Valence | + 10.7 |
| Thermal Stability | 60.2°C |
One predictor of potential challenge in developing a high concentration liquid formulation (HCLF) is protein valence.14 Protein valence is a system property that determines how protein molecules interact with their neighbors in solution, and hence plays an important role in protein solubility. In a homogeneous solution, all protein molecules carrying the same average charge repel each other. The intermolecular charge-charge repulsion keeps the molecules apart and prevents the attractive short-range energies from being dominant, and hence helps proteins remain soluble. Low net charge (approaching 0) on macromolecules reduces the activation energy barrier to aggregation, resulting in poor solubility. Based on our internal valence data for human IgG1s, a measured valence of 15 indicates sufficient electrostatic repulsion to enable the achievement of HCLF. CAs compared to antibodies (stokes radius of ~6nm), the measured stokes radius of BI 655088 is approximately 3.5nm, and therefore calculations based on Equation 1 indicate that a measured valence criterion of 7.3 would indicate that HCLF was possible for VHHs. The measured valence for BI 655088 is +10.7 (Table 8).
The selection process used a thermal shift assay to select for stable immunoglobulin single variable domains to ensure that the final sequence-optimized molecules would have good thermal stability, a surrogate parameter often linked to shelf-life. Differential scanning calorimetry (DSC) is widely used to assess thermal stability and is a surrogate parameter to evaluate shelf-life. Using this technique, the measured melting temperature for BI 655088 is 60.2ºC. (Table 8, Supplemental Figure 7)
Discussion
Among the antigen classes used for antibody therapeutics discovery, GPCRs are generally considered to be one of the most challenging. Due to the unusual structure of VHHs, we hypothesized that the unique CDR loop structures may have the potential to engage binding pockets in GPCRs. To test this hypothesis, a llama immunization approach was taken. We explored multiple approaches to generate an immune response to the target, including the use of camelid cell lines expressing CX3CR1, peptides of the extracellular loops and DNA. Ultimately, DNA immunizations followed with multiple boosts of the overexpressing cell lines produced the highest titer to the target. Interestingly, for the three llamas immunized with this protocol, only one showed a strong response. This likely reflects the out-bred nature of the species versus a typical immunization in mice, where a more consistent response is observed.
GPCRs pose additional challenges in enabling panning as recombinant protein cannot easily be made. We explored both conventional panning on cells, but also included panning on VLPs because the surface presentation of receptors on VLPs is generally 10- to 100-fold greater.15 The higher density of CX3CR1 on the VLPs allowed the identification of a diverse set of VHHs with different profiles with respect to cross-reactivity and potency. We determined that single VHHs could fully block the fractalkine interaction, but not at the targeted concentration. Higher potency was later achieved by formatting the VHHs to be multi-valent either by linking two VHHs together or via an Fc domain. Since fractalkine is the only ligand demonstrated to activate CX3CR1, antagonizing the binding of this chemokine to the receptor was expected to block activation. A competition FACS assay quantitating antagonism of the binding of Alexa Fluor 647-labeled fractalkine to Ba/F3 cells over-expressing hCX3CR1 was used as the primary screening assay. BI 655088 was shown to potently inhibit fractalkine binding to CX3CR1. The VHH also blocked the internalization of Alexa Fluor 647-labeled fractalkine in hCX3CR1 CHO cells using an imaging platform. To confirm the ability of the VHHs to antagonize the CX3CR1 function, we used a chemotaxis assay monitoring the migration of hCX3CR1 Baf3 cells in response to fractalkine. BI 655088 was evaluated both in the presence and absence of HSA to determine the impact of albumin binding on in vitro activity. Up to a 3- to four-fold decrease in activity in the presence of albumin was observed. Calcium influx assays in CHO cells over-expressing various chemokine receptors were used for selectivity and species cross-reactivity analyses. This assay format was also used to confirm that the bivalent VHH functions solely as a receptor antagonist and not as an agonist (no activity was observed in the absence of fractalkine at concentrations up to 1 µM).
The bivalent lead wildtype VHH BI 18 was fluorescently labeled (AF647 018) to facilitate the characterization of VHH binding to both primary and recombinant cells and to enable the establishment of free receptor assays.
To achieve the desired PK profile for the new therapeutic concept, we tested both the Fc-fusion (BI 655089) and the addition of an albumin-binding domain (BI 655088) to the VHHs. In a cynomolgus PK study, the Fc fusion had significantly faster clearance compared to the HSA binding domain construct. For this reason, BI 655088 was chosen as the clinical candidate.
To demonstrate that BI 655088 blocked the CX3CR1 receptor in vivo, its effect on target engagement biomarkers was determined in mice. Two biomarker assays were established. The first was a whole blood-free receptor assay where fluorescently labeled bivalent VHH BI 18 (AF647 BI 18) was used in FACS and its binding to a gated monocyte (CD115+) population was monitored. AF647 BI 18 binding was blocked by the presence of dosed BI 655088. It has also been shown in CX3CR1-/- mice that serum fractalkine levels are elevated.16 Quantitation of serum fractalkine levels was used as the second biomarker assay. Given that the initially identified anti-CX3CR1 VHHs did not cross-react with mouse CX3CR1, a hCX3CR1 KI mouse model was developed and validated. Following administration of BI 655088 to hCX3CR1 KI mice, serum fractalkine levels were elevated up to 7 days. This correlated with low levels of free receptor on CD115+ monocytes seen during the same time period. By day 13, serum fractalkine concentrations decreased and free receptor increased to the levels seen in untreated mice.
The efficacy of BI 655088 was evaluated in the atherosclerosis disease relevant model. Therapeutic treatment over 6 weeks with BI 655088 at 30 mg/kg reduced lesion formation in the descending aorta by 62%. Dosing of BI 655088 with 10 mg/kg once weekly did not result in a significant reduction in plaque area. Undetectable VHH levels and baseline fractalkine concentrations at this dosing regimen are consistent with the lack of target coverage. This result demonstrates the efficacy of BI 655088 in the mouse atherosclerosis model. These studies show for the first time that the therapeutic administration of a CX3CR1 antagonist can inhibit plaque progression in an atherosclerosis disease model.
The developability considerations and assessments made for BI 655088 reflect our thinking and approach circa 2011. BI 655088 was characterized in a series of biophysical assays. The data show that BI 655088 can be purified to a high monomer content using a simple two-step process. Stability assessments have shown that the thermostability of the BI 655088 is in the lower range of acceptable (60°C), but within the range of marketed biotherapeutics. When subjected to thermal and chemical stress (37°C, pH 9.0), BI 655088 maintained monomer content above 90%. At the time, it had been demonstrated to maintain stability at a concentration of 50 mg/mL in 20 mM sodium citrate, 115 mM sodium chloride pH 6.0. Since then, BI 655088 progressed into the clinic (Phase 1 study: NCT02696616) and has been successfully formulated at a concentration of 100 mg/ml in 21.2 mM sodium acetate trihydrate, 3.80 mM acetic acid, 220 mM trehalose dihydrate, 0.489 mM polysorbate 20 pH 5.5. In conclusion, through the identification of potent binding and blocking VHHs to CX3CR1, we generated a promising biotherapeutic. BI 655088 has been shown to inhibit plaque progression in mice, making it an attractive clinical candidate for the treatment of atherosclerosis in humans. The favorable PK and biophysical profile also supports its potential for successful use as a therapeutic agent.
Materials and methods
Cell lines and virus-like particles
Ba/F3 cells stably expressing human CX3CR1 were generated using a retrovirus infection system. The cell line was produced by Boehringer Ingelheim, Ridgefield, CT, USA.17 CHO-K1 parental cells (ATCC) or CHO-K1 cells expressing human CCR2, human CCR5, and human CX3CR1 cell lines were produced by Boehringer Ingelheim, Ridgefield, CT.17 HEK293T (ATCC) cell line stably expressing cynomolgus CX3CR1 was produced by Boehringer Ingelheim, Ridgefield, CT. VLPs derived from the CHO human CX3CR1 overexpressing cell line were purchased from Integral Molecular (Philadelphia, PA).
VHH expression and purification immunizations and library generation
Llamas were immunized intramuscularly with 2 mg of pVAX1-huCX3CR1 plasmid vector (Invitrogen) weekly or bi-weekly. Some animals were given boosts consisting of 4 s.c. injections of huCX3CR1 overexpressing Caki cells (camel kidney cells provided by Ablynx).18 Cells were resuspended in 1X Dulbecco’s Phosphate-Buffered Saline (PBS) and resuspended on ice prior to injection. An additional group of llamas was immunized with the same Caki cells and then boosted with peptides from recombinant CX3CR1 (NeoMPS) coupled to BSA. These peptides are the N-terminal region MDQFPESVTENFEYDDLAEACYIGDIVVFGT and the extracellular loop 3 TKQKENECLGDYPEVLQEIWPVLRNVET and together are denoted CX3CR1 NT/EC3. Details of llama immunization procedures were followed as given by Ghahroudi et al.19 A phage display library was generated as described in Ghahroudi et al. Selections on the prepared libraries were performed: (1) in solution using multiple concentrations of Ba/F3 or CHO huCX3CR1 overexpressing cells, and (2) onto plates using VLP that display huCX3CR1. This procedure was completed following Maussang et al. and Veugelen et al.20,21 Ba/F3 cells stably expressing human CX3CR1 were maintained in the medium in a humidified, 37°C incubator with 5% CO2 and passaged twice weekly. A detailed description of materials used and immunization and library generation methods can be found in the associated patent.17
VHH expression and purification
VHHs derived from phage display were cloned into pAX100 expression vectors (kanamycin resistance) and expressed in TG1 E. coli cells. Overnight pre-cultures were diluted 1:100 in TB-0.1% glucose-50 µg/ml kanamycin, and incubated for 3 h at 37°C, 250 rpm. After inducing the cultures with 1 mM isopropyl β-D-1-thiogalactopyranoside for 4 h at 37°C, cultures were pelleted and stored at −20°C. Periplasmic extracts were prepared and his-tagged VHHs were purified through affinity chromatography (IMAC) using Histrap FF crude columns (GE Healthcare) and size exclusion chromatography. The purity and integrity of VHHs were verified by reducing SDS-PAGE.
Determination of selectivity
Binding to related chemokine receptors was evaluated by performing flow cytometry binding experiments on CHO-K1 parental cells or CHO cells expressing human CCR2 or human CCR5. The VHHs were incubated with the respective cell lines for 30 min at 4°C and subsequently incubated with the detection reagents. For detection, a mouse anti-c-myc antibody (Serotec, MCA2200) followed by a goat anti-mouse antibody coupled to PE (Jackson 115-116-071) was used. For each cell line, a quality control with receptor-specific antibodies was included. In addition, the highest concentration of each VHH was also incubated with CHO cells expressing human CX3CR1 as a positive control.
FACS competition assay with Alexa Fluor 647-labeled human or cynomolgus fractalkine
The VHHs were evaluated for their ability to block the binding of labeled fractalkine to human or cynomolgus CX3CR1 expressed in CHO cells. The recombinant fractalkine protein with both the chemokine and the mucin-rich stalk domains were purchased from R&D systems (365-FR/CF Lot# AF5051204A) and labeled with Alexa Fluor 647 according to manufacturer’s instructions (ThermoFisher Scientific, Catalog number A20173). Labeled material had a degree of labeling of 0.84. Cynomolgus fractalkine (ppt5-cyno CX3CL ECD-6HIS) was produced in HEK293 cells and purified via NI-NTA Fast Flow (Biorad) followed by size exclusion chromatography in to 50 mM HEPES, 100 mM NaCl and 5% glycerol. Cynomolgus fractalkine was labeled with Alexa Fluor 647 according to manufacturer’s instructions (ThermoFisher Scientific, Catalog number A20173). Labeled material had a degree of labeling of 1.0. Cells were transiently transfected with the receptor and binding of the labeled fractalkine was evaluated. A fixed concentration of labeled fractalkine, corresponding to the EC30 concentration as determined from a dose titration, was used in a competition setup to determine the IC50 values of the VHHs. The VHHs were serially diluted 1:4. The percent block was calculated based on the top and bottom value of the determined curve fit and the baseline value as determined from the competition with an excess of cold fractalkine.
Fractalkine-induced chemotaxis
Ba/F3-hCX3CR1 cells were washed in assay buffer (RPMI + 0.1% BSA) prior to use. Chemotaxis was performed using disposable chambers with a pore diameter of 5 µm (Neuroprobe) in a 96-well format. The bottom chamber was filled with 320 pM huFractalkine and 1.3 × 105 cells were placed on top of the filter after which chemotaxis was performed for 3 h at 37°C. Upon removal of the membrane, the number of migrated cells was determined by quantification of the ATP level using CellTiterGlo reagent (Promega). Readout was performed on an Envision instrument (Perkin Elmer) with standard settings for luminescence.
Production of bi-valent constructs
Bivalent constructs were generated by genetically linking two VHHs with a 35GS linker and expressed in TG1 E. coli cells with c-Myc and polyhistidine tags. Bivalent constructs were then purified by IMAC and size exclusion chromatography.
Cx3cr1-mediated fractalkine internalization assay
CHO-K1 cells stably expressing human CX3CR1 were seeded at 10,000 cells per well into a BD imaging plate and grown overnight. Cells were washed once with Hanks Buffer containing Ca+2/Mg+2, then 80 μl of assay buffer (Hanks Buffer with Ca+2/Mg+2, 10 mM HEPES and 0.1% BSA) was added to each well. For those cells assayed in the presence of HSA, HSA was added to all the buffers to a final concentration of 6.3 μM. After the addition of the assay buffer, ± 6.3 μM HSA, the cells were incubated for 30 min and then 10 μl VHH (single or formatted) to be tested was added at various concentrations. The cells were incubated for an additional 15 min and then 10 μl of 100 nM fractalkine-A647 was added to a final concentration of 10 nM. This represented the EC90 of the labeled ligand. After this addition, the cells were incubated for an additional 60 min and then fixed for 10 min with 3.7% formaldehyde diluted in PBS. The formaldehyde was removed, and after washing with PBS, 100 μl of 10 μM Hoechst diluted in PBS was added to each well. Plates were sealed and imaged on a BD Pathway 435 (Becton Dickinson).
Sequence optimization
During sequence optimization, parental wildtype VHH sequences were mutated to yield sequences more closely identical to human VH3-JH germline. Throughout the optimization process, variants were compared to parental VHH using thermal shift assay and in vitro potency using fractalkine competition FACS and fractalkine-induced chemotaxis assay.21,22 General principles of the VHH sequence optimization were followed as described in Vincke et al.23
Production of BI 655088 (VHH) and BI 655089 (FC-fusion)
BI 655088 was expressed in P. pastoris as described in Rahbarizadeh et al.24 For BI 655089, a stable pool was generated in NS0 cells for production. Cells were transfected with linearized NS0 expression vector, resuspended in NS0-10 media + 10% fetal bovine serum and allowed to recover for 48 h. Cells were then placed under selection with G418 and HXM for 2 weeks in 96-well plates. Cells were expanded and the highest titer pool was selected for scale up. Cells were passaged and expanded, and after 5 L of cells at 1.5 × 105 cell/ml were generated, a stir tank was filled with 20 L of media and inoculated with cells to a density of 2.5 × 105 cells/ml. The bioreactor was stirred at 70 rpm, pH 7.1, at 37ºC, and harvested by centrifugation after 168 h.
For both BI 655088 and BI 655089, harvested supernatant was captured by affinity chromatography using MabSelect SuRe resin (GE Healthcare) as previously described.25 Polishing was completed using cation exchange chromatography with BioRad S Support resin. Protein was loaded onto the column pre-equilibrated with 60 mM sodium acetate pH 5.0. Following loading, the column was washed with 20 mM sodium citrate pH 6.0. Protein was eluted on a gradient over 20 column volumes up to 20 mM sodium citrate, 1 M NaCl pH 6.0. Fractions were pooled based on purity. The purity and integrity of VHHs were verified by reducing SDS-PAGE.
Calcium influx assay
CHO cells stably expressing human CX3CR1 were washed with assay buffer Hank’s Balanced Salt Solution + 20 mM HEPES and seeded into 96-well assay plates (Costar #3904) at a density of 50,000 cells per well. After an overnight incubation (37°C, 5% CO2), cells were dye-loaded (Calcium 4 (Molecular Devices) and probenecid) for 1 h at 37°C. VHH was diluted into assay buffer and combined with cells during the last 15 min of dye-loading. Finally, fractalkine (10 nM final concentration) was added to stimulate cellular fluorescence as measured on a FLIPR Tetra (Molecular Devices) instrument.
Ligand competition in presence of human serum albumin
The constructs under evaluation and fractalkine were pre-incubated with HSA for 30 min before addition to the cells. The cells were then re-suspended in FACS buffer supplemented with HSA. The final concentration of HSA was 50-fold excess of the highest VHH concentration. Subsequently, competition was allowed for 2 h and further processing and assay set up was completed as described in section FACS competition assay with Alexa Fluor 647-labeled human or cynomolgus fractalkine.
Cynomolgus monkey pharmacokinetic analysis
The PK properties of BI 655088 and BI 655089 were evaluated in two independent single rising dose cynomolgus monkey studies. Both studies were approved by the Boehringer Ingelheim Pharmaceuticals, Inc.’s Institutional Animal Care and Use Committee and were conducted at Ricerca Biosciences in Concord, OH. Both molecules were administered either by s.c. injection or i.v. infusion. The serum concentrations of antibodies were determined using a validated, antigen-capture MSD assay. A detailed description of the study design and assay set-up is described by Banik et al.15
HuCX3CR1 KI mouse atherosclerosis model
To enable evaluation of the human-specific CX3CR1 antagonists, a human CX3CR1 KI mouse was created on a C57BL/6N background (Taconic) by inserting the full open reading frame of human CX3CR1 (exon 2) into the corresponding region of the mouse CX3CR1 gene to retain the mouse regulatory elements and signal peptide sequence. Mice were then back-crossed with B6.129P2-Apoetm1/unc/J mice (Jackson Laboratories) for 10 generations to generate hCX3CR1/apoE-/- mice. Female hCX3CR1/apoE-/- mice were fed a high fat/high cholesterol diet (Research Diets D12108C supplemented with 1.5% cholesterol, 0.025% soybean oil, and 0.15% cocoa butter) for 16 weeks beginning at 4 weeks of age.
After 10 weeks, the animals continued on the same diet for 6 weeks with either no treatment (control) or treatment with BI 655088 or BI 655089 at a dose of 30 mg/kg by i.p. injection two times per week or 10 mg/kg by i.p. injection once per week. Blood samples for compound exposure and fractalkine levels were taken 2 days after injection at 3 weeks and 3.5 and 7 days after injection after 6 weeks of treatment. On the day of sacrifice, mice were fasted for 4 h prior to surgery. Mice were anesthetized by gas anesthesia of isoflurane and a blood sample was taken (200–400 µL) by brachial artery bleed for plasma lipids, fractalkine levels and for plasma BI 655088 exposure. The mice were then perfused with 0.9% saline to remove blood. The descending aorta to the ileac bifurcation was removed, opened longitudinally, fixed in formalin, and stained with Sudan IV for 15 min, followed by 2 min with 70% methanol. The vessels were then washed under running water and covered with PBS to store at 4⁰C. The aortas were photographed with a digital camera using Spot Advance software. The percentage of lipid staining was determined with image analysis software (ImagePro Plus) and expressed as a percentage positive staining of the vessel. A one-way ANOVA was used to compare treatment groups to the control group using a Dunnett’s test.
Target engagement biomarkers
Analysis of free CX3CR1 receptor was conducted in situ using whole blood collected in EDTA-coated vials and processed immediately. Staining of mouse blood was performed by mixing 20 µl whole blood in FACS buffer (BD Biosciences) containing CD115-AF488 (1%, eBioscience) and BI18 (Bi-valent formatted VHH lead, 1 nM) in a prechilled plate. After incubation of the plate for 60 min in the dark on ice, 250 µl of 1-step Fix/Lyse Solution (1X, eBioscience) was added to each well and the samples were mixed. The plate was incubated for an additional 60 min in the dark at room temperature, and then centrifuged at 500 g for 5 min. The cells were washed twice with 250 µl/well FACS buffer and the samples were analyzed on an LSRII flow cytometer (BD Biosciences). Forward and side scatter voltages were set using unstained cells and an interval gate was drawn around the live cells. A second gate was then drawn around the CD115+ cells, which represented approximately 1.5 – 2% of the total cells. Instrument conditions were set to enable collection of a minimum of 50,000 events for each sample. Median Alexa Fluor fluorescence corresponding to free CX3CR1 receptors was measured in the gated CD115+ monocyte population. To determine the concentration of serum fractalkine, 10 µl of serum was tested in a Milliplex immune assay (Millipore) and analyzed using a Luminex 200 analyzer.
Histology
Formalin Fixed Paraffin Embedded tissue sections of Aortic Arch were de-paraffinized, hydrated, heat-treated to reveal the epitope (100ºC for 20 min), stained with a rabbit anti-CX3CR1 antibody (Abcam cat# ab8021, 1:200 dilution, 30 min) and then an anti-rabbit IgG secondary antibody (biotinylated anti-rabbit IgG-Vector, Cat # BA-100, 1:200 dilution, 30 min). Antibody-epitope complex was detected using Romulin AEC Chromogen (Biocare Medical Cat#RAEC810L). Negative controls consisted of either the antibody diluent (omitting the CX3CR1 antibody) or the inclusion of a rabbit IgG isotype control diluted at 1:200.
Analytical ultra centrifugation
All sedimentation velocity analytical ultracentrifugation experiments were conducted using ProteomeLab XL-1 analytical ultracentrifuges (Beckman Coulter) at 20ºC with absorbance detection at 280 nm. Samples were prepared at a concentration of 0.4 mg/ml in pH 6.0 20 mM citrate, 115 mM NaCl buffer. The rotor was run at 50,000 rpm for BI 655088. Data was fitted using the continuous c(S) distribution model in Sedfit software.
Valence
Valence measurement for BI 655088 was performed at 20ºC using ProteomeLab TM PA800 (Beckman coulter) capillary electrophoresis instrument using UV (ultraviolet detection) at 214 nm. Protein samples were prepared at a concentration of 1 mg/ml in 10 mM acetate 50 mM KCl pH 5.0 buffer. 0.005% dimethyl formamide in 10 mM acetate 50 mM KCl pH 5.0 buffer was used as the neutral marker to measure electroosmotic flow. The instrumentation, methodology, and data analysis were followed as discussed by Singh et al.25
Differential scanning calorimetry
DSC experiments were run with 1 mg/mL protein solutions in 20 mM sodium citrate, 115 mM sodium chloride pH 6.0 using automated capillary DSC (MicroCal LLC, North Hampton, MA). Experimental details and data analysis were completed following previously described protocols.24
Acknowledgments
The authors would like to acknowledge Alisa Waterman and Sanjaya Singh as the team leaders who lead this project during the Discovery Research phase.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takag IS, Nomiyama H, Schall TJ, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–12. doi: 10.1016/S0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 2.Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgame R, Shagdarsuren E, Lira SA, Weissman IL, Weber C, Jung S.. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113(4):963–72. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- 3.Stolla M, Pelisek J, von Brühl ML, Schäfer A, Barocke V, Heider P, Lorenz M, Tirniceriu A, Steinhart A, Bauersachs J, et al. Fractalkine is expressed in early and advanced atherosclerotic lesions and supports monocyte recruitment via CX3CR1. PLoS One. 2012;7(8):e43572. doi: 10.1371/journal.pone.0043572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ.. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385(6617):640–44. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 5.McDermott DH, Fong AM, Yang Q, Sechler JM, Cupples LA, Merrell MN, Wilson PW, D’Agostino RB, O’Donnell CJ, Patel DD, et al. Chemokine receptor mutant CX3CR1-M280 has impaired adhesive function and correlates with protection from cardiovascular disease in humans. J Clin Invest. 2003;111(8):1241–50. doi: 10.1172/JCI16790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghilardi G, Biondi ML, Turri O, Guagnellini E, Scorza R. Internal carotidartery occlusive disease and polymorphisms of fractalkine receptor CX3CR1: a genetic risk factor. Stroke. 2004;35(6):1276–79. doi: 10.1161/01.STR.0000128528.56009.d4. [DOI] [PubMed] [Google Scholar]
- 7.Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1-/-mice reveals a role for fractalkine in atherogenesis. J Clin Invest. 2003;111(3):333–40. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Combadière C, Potteaux S, Gao JL, Esposito B, Casanova S, Lee EJ, Debré P, Tedgui A, Murphy PM, Mallat Z. Decreased atherosclerotic lesion formationin CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107(7):1009–16. doi: 10.1161/01.CIR.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- 9.Karlström S, Nordvall G, Sohn D, Hettman A, Turek D, Åhlin K, Kers A, Claesson M, Slivo M, Lo-Alfredsson Y, et al. Substituted 7-Amino-5-thio-thiazolo[4,5-d]pyrimidines as potent and selective antagonists of the fractalkine receptor (CX3CR1). J Med Chem. 2013;56:3177–90. doi: 10.1021/jm3012273. [DOI] [PubMed] [Google Scholar]
- 10.Dorgham K, Ghadiri A, Hermand P, Rodero M, Poupel L, Iga M, Hartley O, Gorochov G, Combadière C, Deterre P. An engineered CX3CR1 antagonist endowed with anti- inflammatory activity. Leukoc Biol. 2009;86:903–11. [DOI] [PubMed] [Google Scholar]
- 11.Labrijn AF, Aalberse RC, Schuurman J. When binding is enough: nonactivating antibody formats. Curr Opin Immunol. 2008;20:479–585. doi: 10.1016/j.coi.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Hoefman S, Ottevaere I, Baumeister J, Sargentini-Maier ML. Pre-clinical intravenous serum pharmacokinetics of albumin binding and non-half-life extended VHHs. Antibodies. 2015;4:141–56. doi: 10.3390/antib4030141. [DOI] [Google Scholar]
- 13.Wong B, Wong D, McManus BM. Characterization of fractalkine (CX3CL1) and CX3CR1 in human coronary arteries with native atherosclerosis, diabetes mellitus, and transplant vascular disease. Cardio Path. 2002;11:332–38. doi: 10.1016/S1054-8807(02)00111-4. [DOI] [PubMed] [Google Scholar]
- 14.Yadav S, Laue TM, Kalonia DS, Singh SN, Shire SJ. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Mol Pharm. 2012;9:791–802. PMID:22352470. doi: 10.1021/mp200566k. [DOI] [PubMed] [Google Scholar]
- 15.Banik S, Laing C, Doranz BJ. Tools for studying membrane protein targets. Genetic Engineering & Biotechnology News; 2009. January 22–23.
- 16.Liu H, Jiang D. Fractalkine/CX3CR1 and atherosclerosis. Clinica Chimica Acta. 2011;412:1180–86. doi: 10.1016/j.cca.2011.03.036. [DOI] [PubMed] [Google Scholar]
- 17.Singh S, Waterman A, Depla E, Laeremans T, Van Hoorick D, Ververken D. WO2013130381A1. 2013.
- 18.Nguyen VK, Desmyter A, Muyldermans S. Functional heavy-chain antibodies in camelidae. Adv Immunol. 2001;79:261–96. [DOI] [PubMed] [Google Scholar]
- 19.Ghahroudi MA, Desmyter A, Wyns L, Hamers R, Muyldermans S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997;414:521–26. doi: 10.1016/S0014-5793(97)01062-4. [DOI] [PubMed] [Google Scholar]
- 20.Maussang D, Mujic-Delic A, Descamps F, Stortelers C, Vanlandschoot P, Walsum M, Vischer H, van Roy M, Vosjan M, Gonzalez-Pajuelo M, et al. Llama-derived single variable domains (nanobodies) directed aganist chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J Biol Chem. 2013;288:29562–72. doi: 10.1074/jbc.M113.498436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veugelen S, Dewilde M, De Strooper B, Chavez-Gutierrez L. Screening and characterization strategies for nanobodies targeting membrane proteins. Methods Enzymol. 2017;584:59–88. [DOI] [PubMed] [Google Scholar]
- 22.Lo M-C, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332:153–59. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 23.Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284:3273–84. doi: 10.1074/jbc.M806889200. [DOI] [PubMed] [Google Scholar]
- 24.Rahbarizadeh F, Rasaee MJ, Forouzandeh M, Allameh -A-A. Over expression of anti-MUC1 single-domain antibody fragments in the yeast Pichia pastoris. Mol Immunol. 2006;43:426–35. doi: 10.1016/j.molimm.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 25.Singh S, Kroe-Barrett R, Canada K, Zhu X, Sepulveda E, Wu H, He Y, Raymond E, Ahlberg J, Frego L, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7:778–91. doi: 10.1080/19420862.2015.1032491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.