

ABSTRACT
Nonsense mutations are relatively frequent in the rare X-linked lysosomal α-galactosidase A (α-Gal) deficiency (Fabry disease; FD), but have been poorly investigated. Here, we evaluated the responsiveness of a wide panel (n = 14) of GLA premature termination codons (PTCs) to the RNA-based approach of drug-induced readthrough through expression of recombinant α-Gal (rGal) nonsense and missense variants.
We identified four high-responders to the readthrough-inducing aminoglycoside G418 in terms of full-length protein (C56X/W209X, ≥10% of wild-type rGal) and/or activity (Q119X/W209X/Q321X, ~5-7%), resulting in normal (Q119X/Q321X) or reduced (C56X, 0.27 ± 0.11; W209X, 0.35 ± 0.1) specific activity.
To provide mechanistic insights we investigated the predicted amino acid substitutions mediated by readthrough (W209C/R, C56W/R), which resulted in correct lysosomal localization and appreciable protein/activity levels for the W209C/R variants. Differently, the C56W/R variants, albeit appreciably produced and localized into lysosomes, were inactive, thus indicating detrimental effects of substitutions at this position.
Noticeably, when co-expressed with the functional W209C or W209R variants, the wild-type rGal displayed a reduced specific activity (0.5 ± 0.2 and 0.6 ± 0.2, respectively) that, considering the dimeric features of the α-Gal enzyme, suggested dominant-negative effects of missense variants through their interaction with the wild-type.
Overall, we provide a novel mechanism through which amino acids inserted during readthrough might impact on the functional protein output. Our findings may also have implications for the interpretation of pathological phenotypes in heterozygous FD females, and for other human disorders involving dimeric or oligomeric proteins.
KEYWORDS: Nonsense mutations, translational readthrough, lysosomal disorders, fabry disease
Introduction
Lysosomal storage disorders are a group of inherited metabolic diseases associated with the accumulation of undegraded substrates within lysosomes due to mutations in genes coding for lysosomal proteins [1].
Fabry disease (FD, OMIM #301500) is a rare (1:40,000–117,000 worldwide) [2,3] X-linked glycolipid storage disorder caused by mutations in the GLA gene (Xq22; MIM #300644), encoding the lysosomal α-galactosidase A (α-Gal) hydrolase.
The α-Gal enzyme is synthesized as a ~50 kDa precursor of 429 amino acids that undergoes maturation in endoplasmic reticulum (ER) and Golgi apparatus and is finally delivered to lysosomes through the mannose-6-phosphate (M6P) pathway [4]. In the ER, the signal sequence (residues 1–31) is removed [5], resulting in the mature 46 kDa form, and three N-linked carbohydrates are attached to residues N139, N192 and N215, which are then modified in the Golgi by addition of phosphate groups to form the M6P moieties essential for transport to lysosomes [6]. Once in lysosomes, the functional α-Gal enzyme is formed as a homodimer [4,7], in which an extensive interface consisting of 30 residues from each monomer maintains the correct packing [8]. Each monomer, composed of two domains, contains the catalytic residues (D170 and D231) enclosed by the side-chains of 13 residues forming the active site, which acts through a double displacement reaction mechanism [9].
The deficiency of α-Gal activity leads to a failure in the metabolism of glycosphingolipids, mainly globotriaosylceramide (Gb3), resulting in the progressive deposition of Gb3 in plasma and lysosomes of all tissues [10], with severe clinical consequences for kidneys, heart and brain [11]. The disease is mainly classified into classic or late-onset FD subtypes. Very low activity levels (<1% of normal) or the complete loss-of-function are associated with classic FD, while residual activity might lead to a slow progression of the disease and a milder late-onset phenotype [11,12].
To date, more than 500 FD mutations have been identified (human gene mutation database, HGMD, http://www.hgmd.cf.ac.uk/ac/index.php; http://fabry-database.org/) [13,14]. Among these, nonsense mutations, relatively frequent in FD, show the highest association with the classic form.
Nonsense mutations, accounting for ~11% of all described disease-causing gene lesions [15], are caused by premature termination codons (PTCs) that may lead to the so-called null phenotypes. However, misrecognition of PTCs and incorporation of an amino acid at the nonsense position results in the synthesis of the full-length protein through translational readthrough [16]. The efficiency of this recoding process is influenced by PTCs sequence contexts [17,18] and by the type of amino acid inserted at the PTC [19–21], with the latter dictating the protein output in terms of biosynthesis and function [22–24].
Since translational readthrough can be potentiated by drugs, it has been proposed as therapy for several disorders [25–28]. The proof-of-principle of this RNA-based correction approach in FD would make it particularly relevant in the view of new readthrough-inducing compounds that, due to their small size, have been shown the potential ability to cross the blood–brain barrier [29], which still represents an unmet need in FD therapy [12,30]. However, despite these premises, the approach based on translational readthrough has never been extensively investigated for FD.
Here, by exploring drug-induced readthrough on a wide panel of GLA nonsense mutations, we observed three variants with appreciably rescued enzyme activity (≥5%) and a discrepancy between protein and functional levels that was not compatible with amino acid insertions as the unique determinant. Indeed, for the first time, we suggest a novel readthrough-mediated output resulting in potential dominant-negative effects on α-Gal enzyme activity due to the interaction of wild-type and missense variants.
Results
A total of 14 GLA nonsense mutations were rationally selected according to the features of PTCs sequence contexts (Fig. 1A), which represent a first hallmark of responsiveness to drug induction [17]. In particular, we included a panel of PTCs (TGA-C/A/G) predicted to result in a high degree of readthrough, and TAG PTCs with similar sequence contexts as comparators. In addition, all these contexts predict the insertion of the original residue [19]. The selected PTCs were challenged with the readthrough-inducing agent G418, the most potent drug among aminoglycosides [17].
Figure 1.
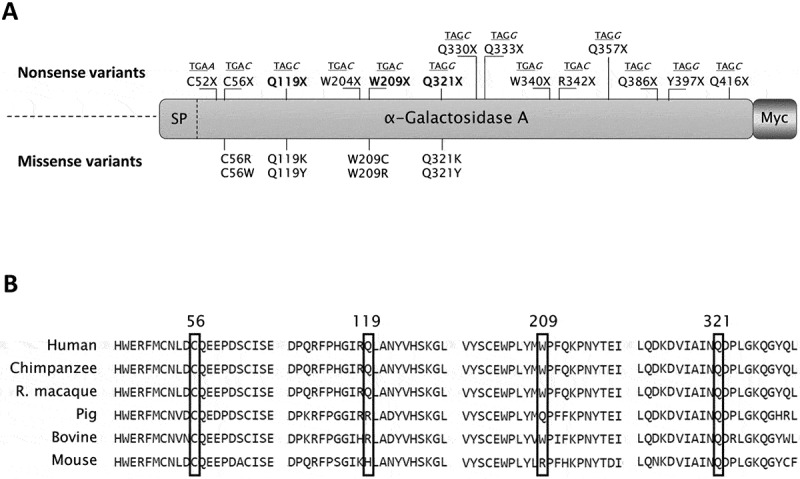
Sequence features and positions of GLA nonsense mutations selected for the study.
(A) Schematic representation of α-Gal and relative position of nonsense mutations (top) and of predicted missense changes arising from translational readthrough (bottom). The sequence contexts of nonsense mutations, namely the PTC (underlined) and the following nucleotide (italic), are also indicated above each nonsense mutation. SP, signal peptide; Myc, carboxyl-terminal Myc tag. (B) Sequence alignment among human (NP_000160.1), chimpanzee (XP_003954083.2), rhesus macaque (XP_001093625.1), pig (NP_001171396.1), bovine (NP_001179665.1) and mouse (NP_038491.2) α-Gal proteins. Positions 56, 119, 209 and 321 are marked by black rectangles.
Readthrough induction resulted in detectable full-length rGal proteins
As a first attempt to assess the responsiveness of the whole panel of GLA nonsense mutations to translational readthrough, we performed Western blotting analysis on lysates from cells expressing Myc-tagged recombinant α-Gal (rGal) nonsense variants in the absence or presence of G418. In this setting, we did not observe a band compatible with the full-length rGal form in basal conditions (data not shown), but only upon drug induction. The differential intensity of bands suggested a different degree of PTCs suppression (Fig. 2A).
Figure 2.
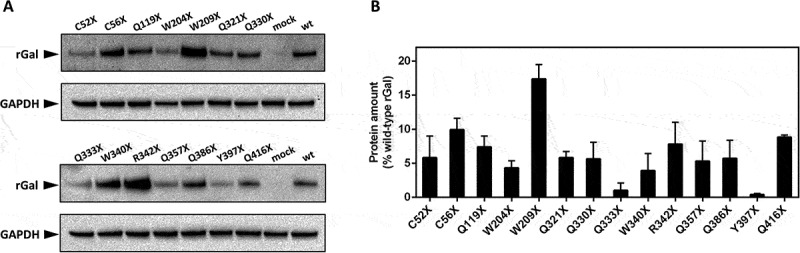
Evaluation of drug-induced readthrough over GLA nonsense mutations.
(A) Representative Western blotting analysis on full-length rGal variants resulting from treatment of transiently transfected HEK293 cells with G418. For rGal detection, wild-type lysate was further diluted (1:20) and used as reference. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as control. Mock, cells transfected with the gutted vector; wt, wild-type rGal. (B) ELISA-based analysis of full-length rGal variants transiently expressed in HEK293 cells treated with G418. Results, indicated as % of wild-type rGal, are reported as mean ± standard deviation from three independent experiments. In both analyses, the full-length rGal arising from nonsense variants was not observed in untreated conditions.
To quantify the amount of full-length rGal produced, we carried out a double ELISA assay that takes advantage of a capture antibody recognizing the Myc epitope tag, whose presence ensures the immobilization of the full-length protein produced as a result of readthrough. Two variants (C56X, 10.0 ± 1.7% of wild-type rGal; W209X, 17.4 ± 2.1%) revealed full-length protein levels ≥10% of wild-type rGal, while the majority (n = 10) showed protein levels ranging from ~4% to ~9% and two (Q333X and Q397X) displayed a very low response (<1%) (Fig. 2B). Full-length proteins were undetectable in lysates from untreated cells.
These first data indicate that all GLA nonsense mutations tested in our experimental system were differentially susceptible to induction of translational readthrough and resulted in the synthesis of detectable full-length rGal proteins.
Activity of rGal variants was partially restored upon drug-induced readthrough
The major goal of a correction approach such as drug-induced readthrough, together with responsiveness at the protein level, is represented by the rescue of enzyme activity. Since no protein levels were detectable in the absence of induction, the functional impact of readthrough was assessed only on cell lysates collected after treatment of cells with the readthrough-inducing agent G418.
The Q119X, W209X and Q321X variants revealed the highest range of activity rescue (~5–7% of wild-type rGal) upon treatment, whereas the majority of rGal nonsense variants (n = 7) showed functional levels ranging from 1% to 3%. Activity of the remaining variants (C52X, Q333X, W340X and Y397X) was undetectable (Fig. 3A). As a consequence, only two variants (Q119X and Q321X) showed a specific activity (0.94 ± 0.03 and 0.84 ± 0.01, respectively), referred as the ratio between activity and protein levels, similar to that of wild-type rGal. The majority of variants, as the C56X (0.27 ± 0.11) and W209X (0.35 ± 0.1), high responders in terms of full-length protein levels, showed a specific activity ranging from 0.24 to 0.42 (Fig. 3B). No specific activity was calculable for those nonsense variants with undetectable enzyme activity.
Figure 3.
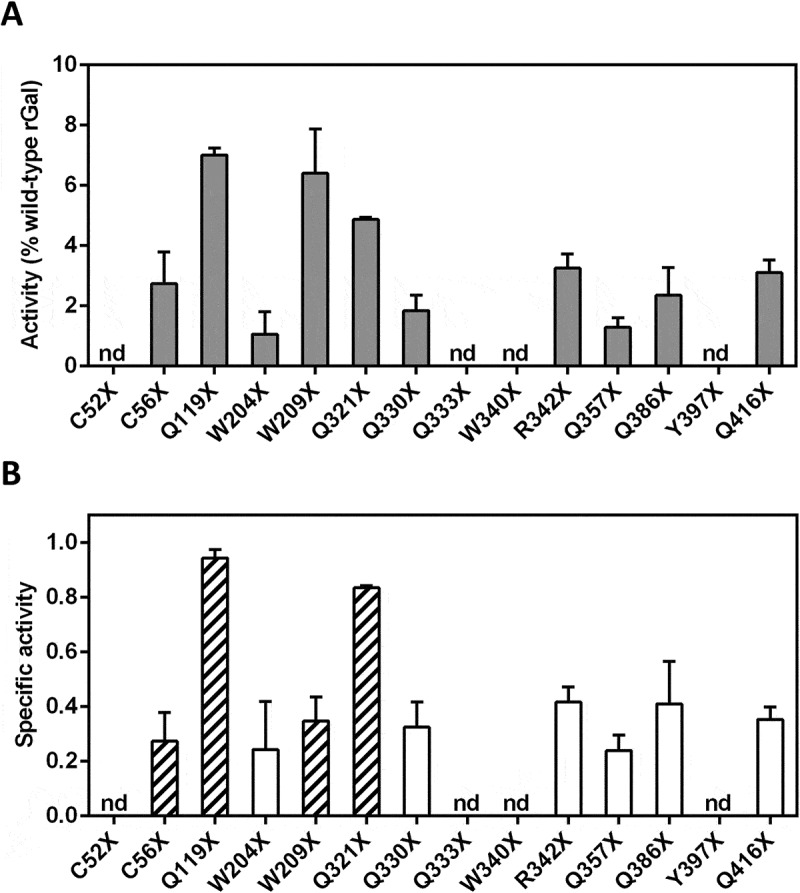
Functional evaluation of rGal variants after drug-induced translational readthrough.
(A) Activity of full-length rGal variants, produced by readthrough induction, towards the 4-MU-α-D-galactopyranoside fluorogenic substrate. Results, indicated as % of wild-type rGal, are reported as mean ± standard deviation from three independent experiments. (B) Specific activity, referred as the ratio between enzyme activity (panel A) and protein amount (Fig. 2B), of rGal nonsense variants after induction of translational readthrough. The most responsive rGal variants in terms of specific activity (Q119X and Q321X) or protein levels (C56X and W209X) were selected (striped bars) for further characterization. nd, not detectable.
These observations indicate that only three variants showed a relevant functional rescue after drug-induced translational readthrough (≥5% of wild-type activity), and provide the evidence for a low response in comparison with measured full-length protein levels.
Amino acid substitutions predicted from readthrough showed a differential impact on rGal protein and activity
To provide insights into the effects of amino acid changes produced by readthrough over the GLA nonsense variants displaying the highest full-length protein levels (C56X and W209X) or specific activity (Q119X and Q321X), we characterized the predicted missense variants. Indeed, prediction of readthrough-deriving amino acid insertions, based on the type of PTC [19], prompted us to produce the Q119K/Y, Q321K/Y, C56W/R and W209C/R missense variants.
Expression studies with rGal variants bearing amino acid substitutions at positions 119 and 321 showed protein levels of 49.4 ± 8.9% (Q119K), 66.1 ± 12.6% (Q119Y), 37.3 ± 2.5% (Q321K) and 3.7 ± 0.8% (Q321Y). Functional studies showed activity levels similar to those of wild-type rGal for protein variants at position 119 (Q119K, 90.6 ± 1.1%; Q119Y, 101.7 ± 4.3%), while slightly lower than protein levels (Q321K, 24.4 ± 3.2%) or undetectable (Q321Y) for variants at position 321 (Fig. 4, left). Protein levels were well-detectable for all missense variants at positions 56 (C56W, 32.6 ± 12.4%; C56R, 40.7 ± 8.1%) and 209 (W209C, 45.1 ± 6.4; W209R, 64.1 ± 24%), while activity levels were clearly detectable only for the W209C (40.8 ± 8.6%) and W209R (70.8 ± 16%) variants (Fig. 4, right).
Figure 4.
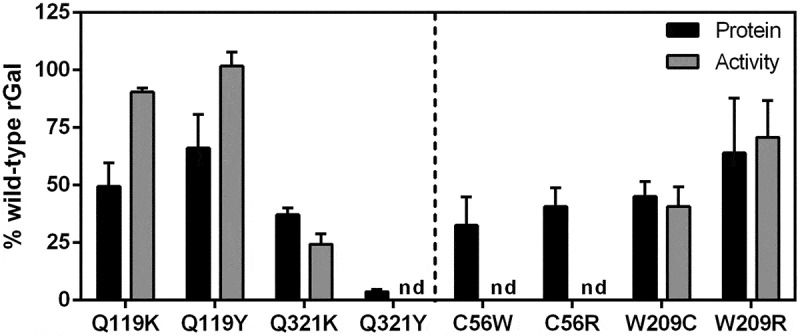
Expression studies with readthrough-deriving missense variants.
Protein amount (black bars) and activity (grey bars) of rGal missense variants (see striped bars in Figure 3B) predicted to arise from translational readthrough of GLA PTCs. Results, indicated as % of wild-type rGal, are reported as mean ± standard deviation from three independent experiments. nd, not detectable.
These data on rGal missense variants indicate that amino acid substitutions were compatible with detectable protein levels but exerted a differential impact on enzyme activity, particularly evident for those at positions 56 and 209.
The rGal C56R and W209C variants showed correct localization into lysosomes
Since amino acid substitutions may impact on protein function as well as on correct localization into lysosomes, which is essential for rGal to achieve full enzyme function [4,7], we performed immunofluorescence studies. The C56R and W209C variants were chosen as paradigmatic examples due to their overlapping protein levels. Both missense variants, clearly detectable by anti-rGal antibodies (Fig. 5, red signal), were appreciably visualized into lysosomes (Fig. 5, green signal), as indicated by the co-localization of the rGal and the lysosomal LAMP1 fluorescence signals, which were qualitatively comparable to that of wild-type rGal (Fig. 5).
Figure 5.
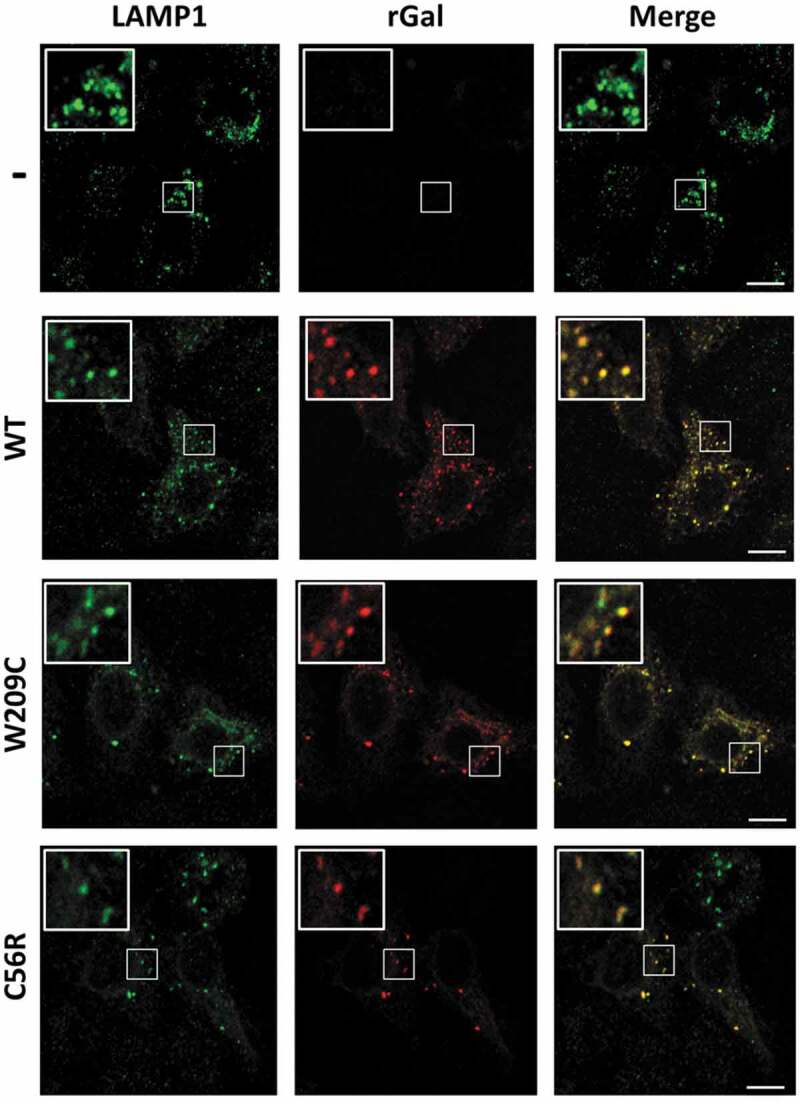
Co-localization studies with C56R and W209C variants.
Intracellular localization of rGal missense variants. Fluorescently labelled secondary antibodies were used to visualize LAMP-1 (lysosomes, green) or rGal (red) as well as their co-localization (yellow). Image magnifications (white squares) for each channel are also shown. Cells transfected with the gutted vector (-) or expressing wild-type rGal (WT) were used as controls.
These results indicate that the functional defect of the C56R variant is not due to altered lysosomal localization, and leaves unexplained the discrepancy between protein/functional levels of the nonsense variant at position 209 upon readthrough.
Readthrough-deriving missense variants exerted potential dominant-negative effects
The discrepancy between protein and functional levels observed for the W209X nonsense variant as well as the data obtained on predicted missense variants at the same position, prompted us to hypothesize that, by introducing amino acid changes, readthrough might result in missense monomeric variants endowed with virtually normal specific activity but exerting dominant-negative effects on the wild-type molecule.
To test our hypothesis, we performed co-expression studies with wild-type rGal and the readthrough-deriving missense variants at position 209 (W209C/R), as well as those at position 56 (C56W/R) as comparison (Fig. 6A). Co-expression of wild-type rGal with W209C/R or C56W/R resulted in protein levels higher than (W209C, 86.2 ± 17.3% of wild-type rGal; W209R, 110.7 ± 25.5%; C56W, 85.4 ± 5.7%) or similar to (C56R, 55.6 ± 8.5%) those obtained with wild-type rGal (50.3 ± 30.8%). Enzyme activity was not increased (W209C, 46.9 ± 22%; W209R, 63.0%±16%; C56W, 47.2 ± 4.6%) or was even decreased (C56R, 26.7 ± 1.3%) as compared with wild-type (55.6 ± 27.5%). As expected, the specific activity of wild-type rGal co-expressed with C56W (0.55 ± 0.02, p = 0.0085) or C56R (0.44 ± 0.04, p = 0.0045), both devoid-of-function variants, was significantly lower than that of control wild-type (1.2 ± 0.2). Noticeably, the specific activity of wild-type rGal co-expressed with W209C or W209R was also significantly lower (W209C 0.5 ± 0.2, p = 0.0027; W209R 0.6 ± 0.2, p = 0.0061) than that of wild-type rGal, despite both variants had shown relevant catalytic activity when expressed alone.
Figure 6.
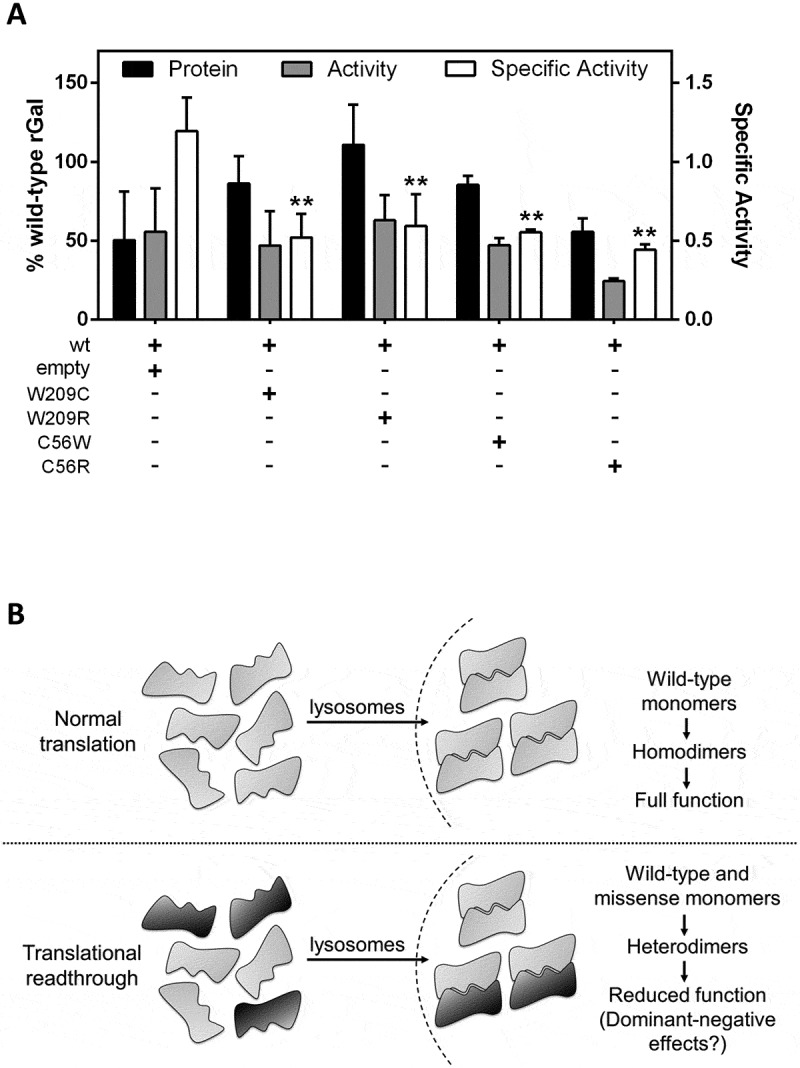
Potential dominant-negative effects of missense variants co-expressed with wild-type rGal.
(A) Co-expression studies of wild-type rGal with W209C/R or C56W/R variants. The empty plasmid was used as control. Results, indicated as % of wild-type rGal, are reported as mean ± standard deviation from three independent experiments. **, p < 0.05. (B) Schematic representation of the impact of readthrough-deriving amino acid insertions on full-length α-Gal biology. Drug-induced readthrough is predicted to result in a full-length α-Gal with wild-type features or bearing missense changes. Once transported into lysosomes, α-Gal monomers dimerize to form functional homodimers (upper panel) or heterodimers due to the interaction of monomers bearing both the original residue (grey monomer) or a missense change (black monomer) resulting in dominant-negative effects on enzyme function (lower panel).
These results suggest that amino acid substitutions introduced by readthrough might produce rGal monomers exerting potential dominant-negative effects on activity of the resulting enzymes due to interaction of wild-type and missense variants, thus lowering the functional rescue mediated by readthrough.
Discussion
In this study, we explored the approach based on drug-induced translational readthrough as a correction strategy for a wide panel of FD-associated nonsense mutations in the GLA gene. While several lysosomal disorders have been challenged with drugs inducing readthrough [31–33], a broad investigation of this approach has never been extensively attempted in FD.
The interplay between PTCs sequence contexts and amino acid(s) inserted upon readthrough induction resulted in a low degree of responsiveness for the majority of nonsense variants. This observation is partially explained by naturally-occurring missense mutations, mainly associated with classic FD (Supplementary Table 1), thus indicating that many positions do not tolerate amino acid substitutions. Indeed, unfavourable amino acid insertions help also explaining the absence of responsiveness of the GLA R227X (TGA, c.679C>T) mutation, the unique attempt of readthrough induction in FD [34]. This residue contributes to the formation of the active site of the enzyme [8], thus being unlikely that a missense change at this position would produce a functional α-Gal enzyme. On the other hand, in our system, three variants (Q119X, W209X and Q321X) revealed an enzyme activity ≥5%, an arbitrary threshold for appreciable functional readthrough. Interestingly, an activity range of 5–10% has been hypothesized to be sufficient to prevent clinically relevant Gb3 accumulation [35].
Evaluation of specific activity of rGal nonsense variants upon readthrough indicated that two (Q119X and Q321X) out of three high responders showed values similar to those of wild-type rGal. Conversely, we clearly observed a reduced specific activity for the C56X and W209X variants (Fig. 3B), despite the high response to readthrough induction in terms of full-length protein (both) or functional (W209X) levels (Figures 2B and 3A).
To provide mechanistic insights we expressed missense variants bearing amino acid substitutions predicted to arise from readthrough [19], namely tyrosine/lysine (Q119 and Q321) for TAG, and tryptophan/arginine (C56) or cysteine/arginine (W209) for TGA PTCs. Expression studies further highlighted a gradient of impact of amino acid changes on protein/activity. In particular, both residues inserted at position 119 produced variants (Q119K/Y) with unaffected function, while those at position 321 slightly (Q321K) or strongly (Q321Y) affected protein synthesis/activity, a finding compatible with the degree of conservation (119, low; 321, high) of these residues (Fig. 1B). Missense changes at position 56 (C56W/R) were tolerated in terms of protein biosynthesis but abolished rGal activity (see also Supplementary Table 1) due to disruption of a conserved disulphide bridge, thus explaining the low activity/protein ratio observed for the C56X nonsense variant after readthrough induction. Conversely, substitutions at position 209 (W209C/R) resulted in appreciable/normal protein and activity levels. This is consistent with the poor conservation of residues at this position (Fig. 1B), at which naturally-occurring mutations are not reported, but not with the protein/activity discrepancy observed for the W209X nonsense variant.
The absence of α-Gal activity could be attributable to the impact of readthrough-mediated amino acid substitutions on enzyme activity itself [22,23] or on altered lysosomal localization, which is required for full enzyme function [4,7]. Immunofluorescence studies on cells expressing the W209C or the C56R variants demonstrated a correct targeting of the proteins to lysosomes, while leaving unexplained the low activity/protein ratio observed for the W209X after readthrough. These data prompted us to hypothesize that the interaction of wild-type molecules with missense monomeric variants arising from readthrough might result in potential dominant-negative effects on enzyme activity due to formation of heterodimers.
As a matter of fact, in co-expression experiments of wild-type rGal with the W209C/R or C56W/R variants, we observed a reduced specific activity for both mutants, which showed null (C56W/R) or virtually normal (W209C/R) specific activity when expressed alone. This observation could be explained by two different mechanisms. Missense variants at position 56 showed detectable, albeit reduced, rGal protein levels, but undetectable function. This indicates that the effects of C56 missense variants, when co-expressed with the wild-type, would be additive in terms of protein levels but not of catalytic activity, thus resulting in a decrease of specific activity. Differently, missense variants at position 209, with virtually normal catalytic activity when expressed alone, showed a reduction in specific activity only when co-expressed with wild-type rGal, thus suggesting their mutual interaction with the formation of dysfunctional heterodimers. This observation could also be interpreted in light of the symmetric nature of the dimer formed by α-Gal monomers (PDB 1R46), in which even a small distortion caused by an amino acid substitution in one of the two monomers might lead to a heterodimer with reduced catalytic efficiency. Overall, these findings depict a scenario in which, once transported into lysosomes, part of the amino acid changes potentially introduced by readthrough leads to the assembly of wild-type and missense monomeric variants resulting in a fraction of functional homodimers as well as heterodimers with lower or no function (Fig. 6B).
Our previous results on a wide panel of nonsense mutations associated with bleeding disorders such as factor VII deficiency [22] or haemophilia B [23,24] highlighted that favourable conditions such as i) re-insertion of the original residue, ii) tolerated missense changes, iii) rare gain-of-function effects, or iv) localization of nonsense mutations in regions that are removed during protein processing (i.e. signal peptides) may result in appreciable functional readthrough. Here, we suggest a fifth and novel mechanism dictating the functional protein output after translational readthrough. Noticeably, responsiveness of α-Gal might be further influenced by the formation of dysfunctional heterodimers stemming from missense monomeric variants combined with the wild-type monomer. To the best of our knowledge, this is the first evidence for translational readthrough on a dimeric enzyme that results in potentially dominant-negative effects related to the heterodimerization process.
Our findings prompt us to speculate that the potential dominant-negative effects, already hypothesized by others [36,37], might have implications for the large proportion (60–75%) of heterozygous females presenting with signs and symptoms of FD [38, 39]. Interestingly, these pathological phenotypes in heterozygous females are still not explained by skewed X-inactivation alone [40,41], and our hypothesis could provide an additional mechanism to interpret these observations. Our data foster additional studies on the effects of missense changes on altered α-Gal dimerization.
Conclusions
Taken together our results i) identify GLA nonsense mutations (i.e. Q119X, W209X and Q321X) that, due to favourable nucleotide and protein features, can be rescued by readthrough induction, ii) provide the first evidence for a novel mechanism acting through dominant-negative effects and influencing the functional response to drug-induced readthrough, and iii) might have implications for the understanding of pathological phenotypes in heterozygous FD female carriers, as well as for other human disorders affecting dimeric or oligomeric proteins.
Materials and methods
Nomenclature
All residues are reported according to the Human Genome Variation Society (HGVS) nomenclature [42], with numbering starting at the A (+1) nucleotide of the AUG (codon 1) translation initiation codon.
Creation of expression vectors for recombinant α-Gal variants
The human GLA cDNA (reference sequence NM_000169.2) was cloned into the pCMV6-XL4 expression plasmid through EcoRI-SacII restriction sites. The 5ʹCATTAAAAGACTTACTTGGATCCAAAAAAAAAAAAAAAAC3ʹ oligonucleotide was used to delete the natural GLA stop codon and to insert the BamHI restriction site (underlined). The Myc-tag sequence, obtained by annealing of the forward 5ʹgatccGAGCAGAAACTCATCTCAGAAGAGGATCTGTAAg3ʹ and reverse 5ʹgatccTTACAGATCCTCTTCTGAGATGAGTTTCTGCTCg3ʹ oligonucleotides (BamHI compatible ends in italic), was inserted downstream of the GLA coding sequence through cloning with BamHI to obtain the pCMV6-XL4-GLA-Myc plasmid expressing the Myc-tagged recombinant α-Gal protein (abbreviated as rGal).
Expression vectors for rGal nonsense and missense variants were created by site-directed mutagenesis (QuickChange® II XL Site-Directed Mutagenesis Kit, Agilent Technologies, Santa Clara, CA, USA) [43] of the human GLA cDNA with the oligonucleotides listed in Supplementary Table 2.
Cell culture and transient expression of rGal variants
Human embryonic kidney (HEK) 293 cells were maintained in DMEM medium (Gibco, Life Technologies, Carlsbad, CA, USA) supplemented with 10% foetal bovine serum (FBS; Gibco, Life Technologies) and 1% penicillin/streptomycin (Gibco, Life Technologies). Cells were incubated at 37°C under 5% CO2 controlled atmosphere.
Cells were seeded in 12-well culture plates (2.5x105 cells/well) 24 h before transfection and then transiently transfected in serum-free medium (Opti-MEM, Gibco, Life Technologies) with the Lipofectamine 2000 reagent (Life Technologies), according to manufacturer’s protocol. Briefly, a mixture of plasmid DNA (2 µg) and Lipofectamine 2000 (2 µL) in 150 µL Opti-MEM medium was incubated at room temperature for 20 min and then added to cells. For co-transfection studies, cells were transfected with a mixture of 1 μg of wild-type plasmid and 1 μg of plasmid either gutted or encoding missense variants. Wild-type alone (2 μg) was used as reference. Transfection medium was removed 4–6 h after transfection and 1 mL of fresh DMEM was added, with or without an optimized concentration (100 µg/ml) of geneticin (G418, SIGMA-Aldrich, St. Louis, MO, USA) [22–24], depending on the experimental set-up. Cell lysates were prepared 48 h post-transfection as previously described [44].
Evaluation of rGal protein levels
The total protein amount for each lysate was measured by BCA (Pierce BCA Protein Assay Kit; ThermoFisher Scientific, Waltham, MA, USA).
Cell lysates, normalized for total protein amount, were loaded on Bolt 4–12% Bis-Tris Plus gels (Invitrogen, ThermoFisher Scientific) and transferred onto nitrocellulose membranes. Western blotting analysis was conducted essentially as described [23]. Full-length rGal was detected by polyclonal goat anti-Myc and donkey anti-goat HRP-conjugated antibodies (Bethyl Laboratories, Montgomery, TX, USA). Blotting images were acquired through the Image Laboratory Software version 4.0 (Bio-Rad, Hercules, CA, USA).
A double ELISA-based assay was performed with coated anti-Myc antibodies followed by detection with polyclonal rabbit anti-α-Gal (SIGMA) and goat anti-rabbit HRP-conjugated (DAKO, Agilent) antibodies. Serial dilutions of cell lysates containing wild-type rGal were used as reference.
Evaluation of rGal activity with fluorogenic functional assays
Activity levels were evaluated according to a previously described protocol [44]. Briefly, rGal activity in cell lysates, normalized for the total protein, was revealed by the 4-methylumbelliferone-α-D-galactopyranoside fluorogenic substrate on a microplate fluorometer (TECAN, Salzburg, Austria). Cell lysates from HEK293 cells expressing wild-type rGal, whose activity was preliminarily evaluated and validated in comparison with untagged recombinant rGal (Supplementary Fig. 1), were used as reference. The endogenous α-Gal activity measured in lysates from cells transfected with the gutted vector was subtracted from the activity measured in lysates from cells expressing mutant or wild-type rGal [45]. Specific activity was calculated as the ratio between activity and protein levels.
Lysosomal localization studies by immunofluorescence
Immunofluorescence studies on cells expressing the rGal variants were essentially as described [46]. Briefly, transiently transfected cells, seeded on glass coverslips, were fixed (4% paraformaldehyde) and permeabilized (0.1% Triton X-100). Non-specific binding sites were blocked with PBS-5% milk (w/v)-0.1% Triton X-100 (Blocking buffer). Polyclonal goat anti-Myc (Bethyl Laboratories), monoclonal mouse anti-lysosomal-associated membrane protein 1 (LAMP-1) (clone eBioH4A3; ThermoFisher Scientific) antibodies were diluted in Blocking buffer and incubated overnight at 4°C. Detection was performed by Alexa Fluor-conjugated donkey anti-goat (594 nm; Myc tag) and anti-rabbit (488 nm; LAMP-1) antibodies (ThermoFisher Scientific) incubated for 1 h at room temperature. Images were acquired on the Zeiss LSM510 confocal microscope [47].
Statistical analysis
Data were analysed with GraphPad Prism 5 software (San Diego, CA, USA) and statistical differences were analysed by t-test.
Funding Statement
This work was supported by the ‘Fondo Ateneo Ricerca (FAR)’ from the University of Ferrara.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
S.L. performed expression as well as functional studies with nonsense and missense variants and analysed data, M.F. created all recombinant plasmids, S.M. performed immunofluorescence studies; P.P. analysed immunofluorescence images and revised the manuscript; A.B. conceived the study and designed research; A.B., F.B and M.P. analysed and interpreted data and wrote the manuscript. All authors revised and approved the final version of the manuscript.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Ballabio A, Gieselmann V.. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. [DOI] [PubMed] [Google Scholar]
- [2].Meikle PJ, Hopwood JJ, Clague AE, et al. Prevalence of lysosomal storage disorders. J Am Med Assoc. 1999;281:249–254. [DOI] [PubMed] [Google Scholar]
- [3].Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372:1427–1435. [DOI] [PubMed] [Google Scholar]
- [4].Lemansky P, Bishop DF, Desnick RJ, et al. Synthesis and processing of alpha-galactosidase A in human fibroblasts. Evidence for different mutations in Fabry disease. J Bio Chemi. 1987;262:2062–2065. [PubMed] [Google Scholar]
- [5].Bishop DF, Calhoun DH, Bernstein HS, et al. Human alpha-galactosidase A: nucleotide sequence of a cDNA clone encoding the mature enzyme. Proc Natl Acad Sci U S A. 1986;83:4859–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta. 2009;1793:605–614. [DOI] [PubMed] [Google Scholar]
- [7].Siekierska A, De Baets G, Reumers J, et al. α-Galactosidase aggregation is a determinant of pharmacological chaperone efficacy on Fabry disease mutants. J Biol Chem. 2012;287:28386–28397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol. 2004;337:319–335. [DOI] [PubMed] [Google Scholar]
- [9].Guce AI, Clark NE, Salgado EN, et al. Catalytic mechanism of human alpha-galactosidase. J Biol Chem. 2010;285:3625–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rombach SM, Dekker N, Bouwman MG, et al. Plasma globotriaosylsphingosine: diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta. 2010;1802:741–748. [DOI] [PubMed] [Google Scholar]
- [11].Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Desnick RJ, Ioannou YA. α-Galactosidase a deficiency. Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and molecular bases of inherited disease. 8th. New York: McGraw-Hill; 2001. p. 3733–3774. DOI: 10.1036/ommbid.181 [DOI] [Google Scholar]
- [13].Garman SC. Structure-function relationships in alpha-galactosidase A. Acta Paediatrica. 2007;96:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ranieri M, Bedini G, Parati EA, et al. Fabry Disease: recognition, Diagnosis, and Treatment of Neurological Features. Curr Treat Options Neurol. 2016;18:33. [DOI] [PubMed] [Google Scholar]
- [15].Mort M, Ivanov D, Cooper DN, et al. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29:1037–1047. [DOI] [PubMed] [Google Scholar]
- [16].Rospert S, Rakwalska M, Dubaquié Y. Polypeptide chain termination and stop codon readthrough on eukaryotic ribosomes. Rev Physiol Biochem Pharmacol. 2005;155:1–30. [DOI] [PubMed] [Google Scholar]
- [17].Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J Mol Med. 2002;80:367–376. [DOI] [PubMed] [Google Scholar]
- [19].Blanchet S, Cornu D, Argentini M, et al. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42:10061–10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pranke I, Bidou L, Martin N, et al. Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons. ERJ Open Res. 2018;4:00080–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Roy B, Leszyk JD, Mangus DA, et al. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc Natl Acad Sci U S A. 2015;112:3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Branchini A, Ferrarese M, Lombardi S, et al. Differential functional readthrough over homozygous nonsense mutations contributes to the bleeding phenotype in coagulation factor VII deficiency. J Thromb Haemost. 2016;14:1994–2000. DOI: 10.1111/jth.13443 [DOI] [PubMed] [Google Scholar]
- [23].Branchini A, Ferrarese M, Campioni M, et al. Specific factor IX mRNA and protein features favor drug-induced readthrough over recurrent nonsense mutations. Blood. 2017;129:2303–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ferrarese M, Testa MF, Balestra D, et al. Secretion of wild-type factor IX upon readthrough over F9 pre-peptide nonsense mutations causing hemophilia B. Hum Mutat. 2018;39:702–708. [DOI] [PubMed] [Google Scholar]
- [25].Fazzari M, Frasca A, Bifari F, et al. Aminoglycoside drugs induce efficient read-through of CDKL5 nonsense mutations, slightly restoring its kinase activity. RNA Biol. 2019:1–10 Epub ahead of print. DOI: 10.1080/15476286.2019.1632633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bukowy-Bieryllo Z, Dabrowski M, Witt M, et al. Aminoglycoside-stimulated readthrough of premature termination codons in selected genes involved in primary ciliary dyskinesia. RNA Biol. 2016;13:1041–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Keeling KM, Xue X, Gunn G, et al. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet. 2014;15:371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bidou L, Bugaud O, Belakhov V, et al. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol. 2017;14:378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang D, Belakhov V, Kandasamy J, et al. The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Mol Genet Metab. 2012;105:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471–486. [DOI] [PubMed] [Google Scholar]
- [31].Gómez-Grau M, Garrido E, Cozar M, et al. Evaluation of aminoglycoside and non-aminoglycoside compounds for stop-codon readthrough therapy in four lysosomal storage diseases. PLoS One. 2015;10:e0135873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Keeling KM. Nonsense suppression as an approach to treat lysosomal storage diseases. Diseases. 2016;4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Banning A, Schiff M, Tikkanen R. Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria. Biochimica Et Biophysica Acta Mol Basis Dis. 2018;1864:668–675. [DOI] [PubMed] [Google Scholar]
- [34].Matalonga L, Arias Á, Tort F, et al. Effect of readthrough treatment in fibroblasts of patients affected by lysosomal diseases caused by premature termination codons. Neurotherapeutics. 2015;12:874–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Clarke JT. Narrative review: fabry disease. Ann Intern Med. 2007;146:425–433. [DOI] [PubMed] [Google Scholar]
- [36].Miyamura N, Araki E, Matsuda K, et al. A carboxy-terminal truncation of human alpha-galactosidase A in a heterozygous female with Fabry disease and modification of the enzymatic activity by the carboxy-terminal domain. Increased, reduced, or absent enzyme activity depending on number of amino acid residues deleted. J Clin Investig. 1996;98:1809–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Keshav S. Gastrointestinal manifestations of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis; 2006. Chapter 28. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11570/. [Google Scholar]
- [38].Wang RY, Lelis A, Mirocha J, et al. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. 2007;9:34–45. [DOI] [PubMed] [Google Scholar]
- [39].Deegan PB, Baehner AF, Barba Romero MA, et al., European FOS Investigators . Natural history of fabry disease in females in the fabry outcome survey. J Med Genet. 2006;43:347–352. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Juchniewicz P, Kloska A, Tylki-Szymańska A, et al. Female fabry disease patients and X-chromosome inactivation. Gene. 2018;641:259–264. [DOI] [PubMed] [Google Scholar]
- [41].Maier EM, Osterrieder S, Whybra C, et al. Disease manifestations and X inactivation in heterozygous females with Fabry disease. Acta Paediatrica Suppl. 2006;95:30–38. [DOI] [PubMed] [Google Scholar]
- [42].Den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–569. [DOI] [PubMed] [Google Scholar]
- [43].Branchini A, Baroni M, Burini F, et al. The carboxyl-terminal region is NOT essential for secreted and functional levels of coagulation factor X. J Thromb Haemost. 2015;13:1468–1474. [DOI] [PubMed] [Google Scholar]
- [44].Lukas J, Giese AK, Markoff A, et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet. 2013;9:e1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Benjamin ER, Della Valle MC, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19:430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pignani S, Todaro A, Ferrarese M, et al. The chaperone-like sodium phenylbutyrate improves factor IX intracellular trafficking and activity impaired by the frequent p.R294Q mutation. J Thromb Haemost. 2018;16:2035–2043. [DOI] [PubMed] [Google Scholar]
- [47].Marchi S, Corricelli M, Branchini A, et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca(2+) levels and tumor growth. Embo J. 2019;38:e99435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.