

ABSTRACT
In addition to its role in translation termination, eRF3A has been implicated in the nonsense-mediated mRNA decay (NMD) pathway through its interaction with UPF1. NMD is a RNA quality control mechanism, which detects and degrades aberrant mRNAs as well as some normal transcripts including those that harbour upstream open reading frames in their 5ʹ leader sequence. In this study, we used RNA-sequencing and ribosome profiling to perform a genome wide analysis of the effect of either eRF3A or UPF1 depletion in human cells. Our bioinformatics analyses allow to delineate the features of the transcripts controlled by eRF3A and UPF1 and to compare the effect of each of these factors on gene expression. We find that eRF3A and UPF1 have very different impacts on the human transcriptome, less than 250 transcripts being targeted by both factors. We show that eRF3A depletion globally derepresses the expression of mRNAs containing translated uORFs while UPF1 knockdown derepresses only the mRNAs harbouring uORFs with an AUG codon in an optimal context for translation initiation. Finally, we also find that eRF3A and UPF1 have opposite effects on ribosome protein gene expression. Together, our results provide important elements for understanding the impact of translation termination and NMD on the human transcriptome and reveal novel determinants of ribosome biogenesis regulation.
KEYWORDS: eRF3, GSPT1, translation termination, UPF1, nonsense-mediated mRNA decay, uORF, ribosome protein genes
Introduction
At the final step of mRNA translation, the stop codon is recognized by the translation termination complex which induces the release of the nascent polypeptide [1]. In some cases, translation termination events are recognized by the nonsense-mediated mRNA decay (NMD) machinery which triggers mRNA degradation [2–4]. mRNA degradation by the NMD pathway requires translation termination to proceed and these processes are intimately related at the molecular level. It also requires the recognition of a termination event as ‘premature’ by the NMD machinery. NMD triggered by premature termination codons (PTCs) was first discovered for aberrant mRNAs harbouring a mutation in the open reading frame (ORF) or resulting from incorrect splicing [5–7]. Later, PTCs were detected in non-faulty mRNAs having long 3ʹ untranslated region (3ʹUTR) or carrying uORFs in their 5ʹ leader sequence or with splicing in the 3ʹUTR or in regular alternative splice products [8–12]. However, the definition of a PTC remains imprecise and the extent of stop codon recognition as NMD substrate is still a matter of intensive research. Recent studies on the molecular mechanisms governing the relationship between NMD and translation termination shed new light on the definition of a PTC which seems to be strongly correlated to the efficiency of translation termination [3,4].
In mammals, the eRF1-eRF3A translation termination complex binds to the terminating ribosome with a stop codon positioned in the A site and triggers polypeptide release. The efficiency of translation termination is enhanced by the interaction of eRF3A with the cytoplasmic poly(A)-binding protein PABPC1 [13,14]. The translation termination complex also mediates NMD through its interactions with SMG1 and UPF1 in the SURF complex that assembles at PTCs [15]. However, this view of NMD and translation termination interplay based on direct interaction between UPF1 and eRF3A was recently challenged by the finding that this interaction is actually mediated by UPF3B, a factor which could promote NMD through its interaction with UPF1 and controls translation termination efficiency through its interaction with eRF3A [4,16]. Studies on NMD factors have established that the key determinants to trigger NMD are those that delay translation termination together with those that prevent eRF3A-PABPC1 interaction. These NMD determinants are the presence of an exon-junction complex (EJC) downstream of the stop codon (> 50 nucleotides), the phosphorylation of UPF1 by SMG1, and the distance between the terminating ribosome and the poly(A) tail-associated PABPC1 [11,17,18]. The current view is therefore that of a tight functional association between NMD and translation termination.
Genome wide studies have shown that the NMD pathway is not only implicated in mRNA surveillance of aberrant transcripts but also regulates the steady state level of many physiological transcripts, contributing to the fine-tuning of their expression [10,12,19–26]. Some of these studies revealed that transcripts coding for NMD factors are among the NMD targets and thus participate to a feedback regulatory mechanism that protects cells from the deleterious effect of NMD perturbation [12,23,27]. Together the numerous reports on the NMD pathway uncover its substantial role in many cellular processes, from development and differentiation to stress response and immunity [28].
Conversely, the impact of translation termination efficiency on gene expression is poorly documented. The release factor eRF3A is a key actor of translation termination and NMD through its binding to eRF1 and UPF3B/UPF1 complex. By controlling translation termination efficiency, eRF3A may also influence the expression of uORF-carrying mRNAs as previously shown for the particular case of ATF4 [29]. Apart from its role in translation termination, eRF3A was shown to act in the control of cell cycle [30,31], in mTOR signalling [32], in apoptosis [33,34] and in mRNA deadenylation through its interaction with the poly(A)-binding protein PABPC1 [35,36]. Thus, eRF3A may potentially regulate the expression of many mRNAs including a number of NMD targets.
To address this issue, we evaluated translation by ribosome profiling and mRNA level by RNA-seq in human cells subjected to either eRF3A- or UPF1 depletion. Our bioinformatics analyses allow to delineate the features of the transcripts controlled by eRF3A and UPF1 and to compare the impact of each of these factors on gene expression. We also drew up a transcriptome-wide map of translated uORFs that yields new insights on the importance of translation termination and NMD in uORF-driven mRNAs expression. Lastly, we found that eRF3A and UPF1 have opposite effects on the expression of ribosome protein genes.
Results
Genome-wide analysis of the transcriptome and translatome of eRF3A- and UPF1-depleted human HCT 116 cells
To explore the effects of translation termination factor eRF3A and NMD factor UPF1 on gene expression regulation, we performed a transcriptome-wide map of ribosome-protected mRNA sequences in human HCT 116 cell line depleted in either eRF3A or UPF1. Parallel cell cultures were electroporated with plasmids expressing either a non-silencing shRNA sequence or shRNAs targeting eRF3A or UPF1 mRNAs [29]. The non-silencing shRNA sequence (sh-Ctrl) served to generate reference experiments and control data set. Three days after electroporation, cells were blocked in translation elongation by cycloheximide treatment and cell extracts were prepared. For the two biological replicates, the Western blot analyses showed that eRF3A and UPF1 protein levels decreased by approximately five-fold under knockdown conditions (Fig. 1A) and the RNA-seq data showed that eRF3A and UPF1 mRNA levels were reduced to 22% and 37% of control levels, respectively (Fig. 1B).
Figure 1.
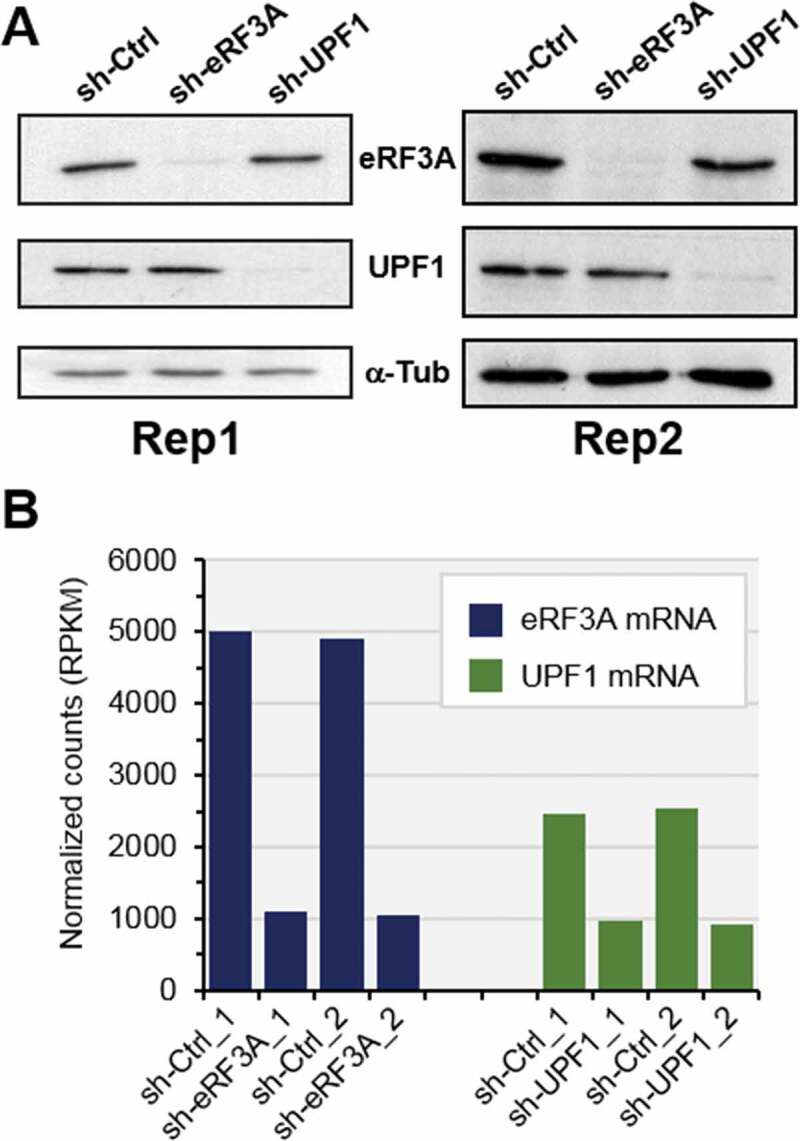
Monitoring of eRF3A and UPF1 knockdown in HCT 116 cells. (A) Western blot analysis of eRF3A and UPF1 in the two biological replicates (Rep1 and Rep2) of eRF3A-depleted cells (sh-eRF3A), UPF1-depleted cells (sh-UPF1) and control cells (sh-Ctrl); α-Tubulin (α-Tub) served as a loading control. (B) eRF3A and UPF1 mRNA levels in eRF3A-depleted (sh-eRF3A) and UPF1-depleted (sh-UPF1) and control (sh-Ctrl) cells for the two biological replicates of RNAseq experiments; normalized counts are expressed in RPKM (reads per kilobase million).
Then, each cell lysate was used to prepare both ribosomal footprint and mRNA libraries and thus obtain matched data. Two biological replicates of ribosomal footprint and mRNA libraries were generated and subjected to deep sequencing to obtain the Ribo-seq and the RNA-seq data sets, respectively. Ribo-seq and RNA-seq reads were aligned against the human genome assembly hg38 (Genome Reference Consortium Human Build 38).
After discarding rRNA and multiple aligned reads, we obtained ~1.8x107 and ~1.2x108 unique mapped reads per sample for RNA-seq and Ribo-seq data, respectively (see Supplemental Figure S1 for details). Our Ribo-seq and RNA-seq data sets were highly reproducible across deep-sequencing replicates as well as across biological replicates, with Pearson correlation coefficient (R) values > 0.97 (Supplemental Figure S2, A and B). Reads data were therefore pooled over replicates for further analysis. For Ribo-seq data, ribosome-protected fragment-length assays generated a peak of 29–30 nucleotides for all samples, as expected for mammalian ribosomal footprints (Supplemental Figure S2C). Moreover, providing additional evidence for footprint of ribosomes decoding the message, a 3-bp periodicity at the 5ʹ end of the read coverage was apparent as a peak at 0.33 Hertz on the periodogram (Periodicity = 1/Frequency) for the coding sequences (Supplemental Figure S2D, CDS panel), and not for the 5ʹUTRs (Supplemental Figure S2D, 5ʹUTR panel).
Then, we used the RNA-seq and Ribo-seq data sets to establish a transcriptome map of HCT 116 cells (see Material and Methods section for details). This allowed us to eliminate the poorly-supported transcripts and to obtain homogeneous sets of mRNAs that were used for further analyses.
Differential expression analysis of eRF3A and UPF1-depleted cells
A differential expression analysis of mRNA (RNA-seq) and translational (Ribo-seq) level changes was conducted using DESeq2 [37] to compute log2 fold changes (log2FC) between two conditions, either eRF3-depleted (sh-eRF3A) versus control (sh-Ctrl) cells or UPF1-depleted (sh-UPF1) versus control cells. The adjusted p-value (p adj) threshold for significant changes was set to 0.05 (Supplemental file S1).
For eRF3A-depleted cells (eRF3A knockdown or eRF3A KD), we identified 2,688 genes that had significant changes in mRNA level (1,574 up-regulated and 1,114 down-regulated) and 2,645 genes that had significant changes in translation (1,450 up-regulated and 1,195 down-regulated; see Supplemental file S1). The vast majority of these genes (2,144) exhibited significant changes at mRNA level as well as translational level (Supplemental Figure S3A). For UPF1-depleted cells (UPF1 KD), we identified 784 genes with significant changes in mRNA level (533 up-regulated and 251 down-regulated) and 815 genes that had significant changes in translation (502 up-regulated and 313 down-regulated). Again, the majority of these genes (563) were significantly changed, at both, mRNA and translational levels (Supplemental Figure S3B).
Interestingly, when comparing the modified sets of genes between both conditions, i.e., eRF3A and UPF1 knockdowns, we found a low overlap of only ~250 targets when analysing either the mRNA or the translational levels (Fig. 2A,B). The fact that the target sets of these two factors were poorly overlapping suggested that eRF3A and UPF1 impacted very differently cellular processes. These differences were confirmed by Gene Ontology analysis [38] (Supplemental Figure S4). eRF3A knockdown targets showed highly significant enrichment for genes associated with cellular response to stress, regulation of transcription and cell cycle progression. This is in agreement with the reported literature showing that eRF3 is involved in the control of cell cycle progression [30–32] and in the cellular response to stress through its effect on the translational control of the transcriptional activator ATF4 [29].
Figure 2.
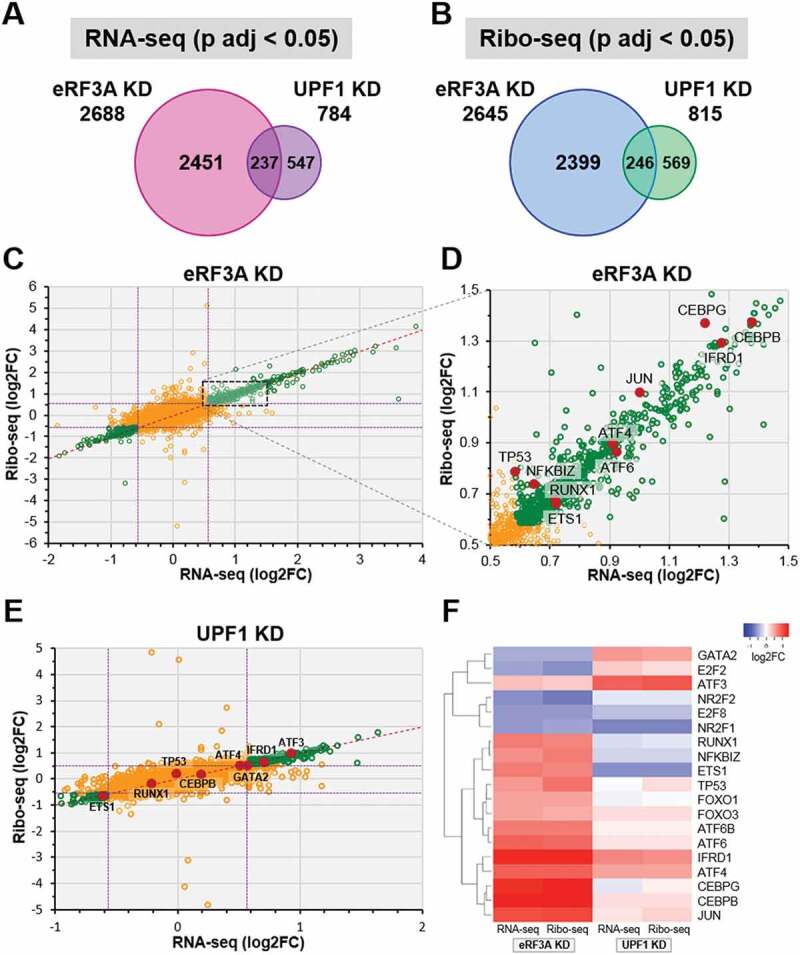
Comparison of differentially expressed genes in eRF3A and UPF1 knockdown cells. (A and B) Proportional Venn diagrams showing the overlap of differentially expressed genes (adjusted p‐value, p adj < 0.05, DESeq2) between eRF3A knockdown and UPF1 knockdown targets for RNA-seq (A) and Ribo-seq (B) data. For each Venn diagram, the number of differentially expressed genes are indicated. (C) Scatter plot comparing Ribo-seq (y axis) and RNA-seq (x axis) log2 Fold Change (log2FC) for eRF3A-depleted versus control cells (eRF3A KD). Green circles indicate genes with p adj < 0.05, dotted purple lines indicate 1.5 fold change (log2FC = ± 0.585). (D) Enlargement of the dotted rectangle in C. Some transcriptional regulator genes are indicated by red dots. (E) Scatter plot comparing Ribo-seq (y axis) and RNA-seq (x axis) log2 Fold Change (log2FC) for UPF1-depleted versus control cells (UPF1 KD). Green circles indicate genes with p adj < 0.05, dotted purple lines indicate 1.5 fold change. Some transcriptional regulator genes are indicated by red dots. (F) Expression heatmap of a selection of transcriptional regulators for eRF3A and UPF1 knockdown cells. Heatmap was performed using Heatmapper website http://www2.heatmapper.ca/expression/[85] and Complete Linkage clustering method.
Although UPF1 knockdown targets were also enriched in genes related to cell cycle and stress, this enrichment is much lower than that observed for eRF3A knockdown (Supplemental Figure S4). The moderate overlap between eRF3A and UPF1 targets could be explained by UPF1 functions that are not linked to mRNA translation. Indeed, UPF1 is partly located in the nucleus where it acts in telomere maintenance [39] and in the early events of mRNA biogenesis including transcription elongation [40] and nonsense-associated alternative splicing [41].
The differences in gene expression described above suggested that eRF3A and UPF1 most likely had divergent effects on translation and mRNA level regulations. Thus, we plotted the fold change in translation (Ribo-seq) against the fold change in mRNA levels (RNA-seq). For the vast majority of both eRF3A and UPF1 target genes, that showed significantly changed expression (Fig. 2C,E, green circles), the fold change of the Ribo-seq was proportional to the fold change of the RNA-seq (i.e., the majority of the points fall on the diagonal) indicating that the regulation is mainly at the mRNA level, and impacts either mRNA stability or transcription. This prompted us to examine the status of transcriptional modulators involved in a broad range of cellular processes. As shown on Fig. 2 (panels D and E), the expression level of a number of transcriptional regulators (red dots) was significantly affected. Some were altered either by eRF3A or by UPF1 depletion while others were affected by both. This profound rearrangement of the transcriptional landscape was particularly clear for eRF3A knockdown. Interestingly, for some transcriptional regulators such as GATA2, ETS1, E2F2, RUNX1 and NFKBIZ, eRF3A- and UPF1-depleted cells exhibited opposite changes in expression (Fig. 2F). These differences in the mRNA level of transcription factors may explain the poor overlapping of eRF3A and UPF1 targets. At the same time, some other transcriptional modulators such as ATF4 and IFRD1 were impacted in the same way by both knockdowns. Interestingly, these two factors contain regulatory uORFs that control the main ORF expression via mechanisms involving translation as well as mRNA stability such as NMD [29,42,43].
Then, RT-qPCR assays were used to verify the results of the gene expression changes in RNA-seq data, with particular focus on the transcription modulators mentioned above. The mRNA for β2-microglobulin (B2M) was used as the endogenous reference mRNA in the qRT-PCR assays. eRF3A and UPF1 mRNAs were used as controls to assess knockdown efficiencies. As shown in Fig. 3A, RT-qPCR data correlated well with the RNA-seq results (Fig. 2 and Supplemental File S1). To validate our differential expression analysis of Ribo-seq data, we also examined the protein level of ATF4 and IFRD1 together with the transcriptional regulator C/EBPβ whose translation is controlled by an uORF located within the long activating form (LAP*) coding sequence, downstream of the main CDS start codon (Fig. 3B). The expression of the short inhibitory form (LIP) of C/EBPβ is regulated at the translation level by this internal uORF [44,45]. The results of Western blot analysis of ATF4, IFRD1 and C/EBPβ proteins in eRF3A- and UPF1-depleted cells (Fig. 3C) confirmed our differential analysis, showing an increase in ATF4 and IFRD1 translation for both knockdown conditions. For C/EBPβ translation, we observed an increase in the LIP form in eRF3A-depleted cells only (Fig. 3C). This last result was expected as C/EBPB gene encoding C/EBPβ lacks introns and is therefore probably not sensitive to NMD.
Figure 3.
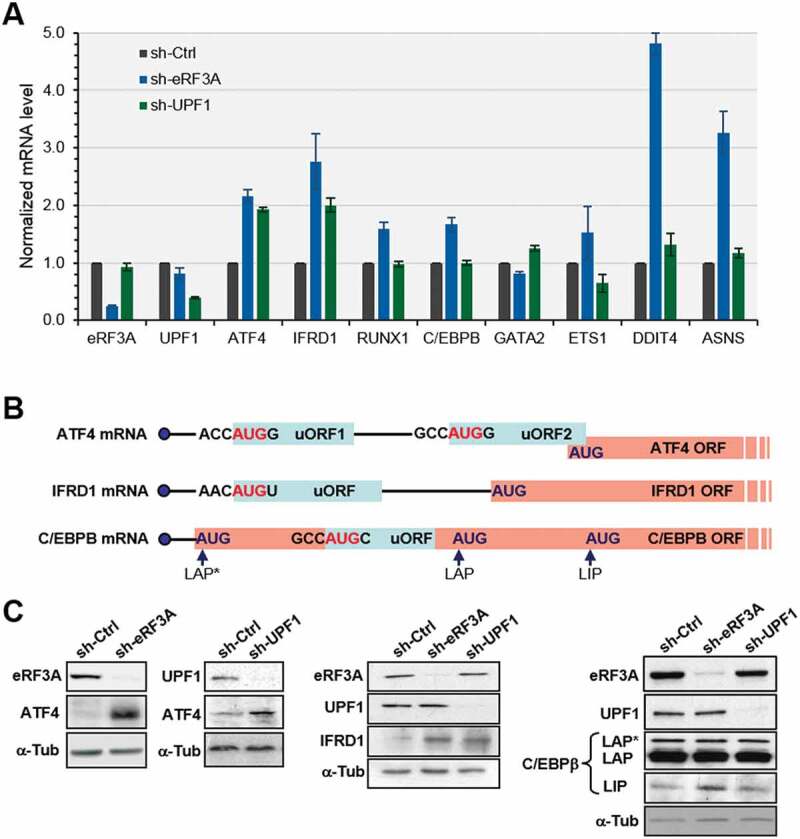
Validation of differentially expressed genes in eRF3A and UPF1 knockdown cells. (A) RT-qPCR performed on total RNA of HCT 116 cells 3 days after electroporation with shRNAs targeting eRF3A mRNA (sh-eRF3A) or UPF1 mRNA (sh-UPF1) or control shRNA (sh-Ctrl). mRNA levels of eRF3A, UPF1 and selected eRF3A and UPF1 targets are shown. The ratio of mRNA levels in either eRF3A knockdown (sh-eRF3A) or UPF1 knockdown (sh-UPF1) versus control cells (sh-Ctrl) was calculated, mRNA levels in the control cells was set to 1.0. Bars and error bars correspond to mean values and standard deviations from two independent experiments. (B) Schematic illustration of the organization of uORFs in the transcripts of ATF4, IFRD1 and C/EBPβ. The start codon context of the uORFs is indicated. ATF4 mRNA carries two uORFs with uORF2 overlapping ATF4 main ORF, IFRD1 mRNA presents only one uORF and C/EBPβ mRNA present a single uORF located within its main ORF. The different isoforms of C/EBPβ termed LAP* for liver-activating protein*, LAP and LIP for liver inhibitory protein are translated from three consecutive in-frame AUG codons[86]. (C) Western blot analysis of eRF3A, UPF1 and ATF4 (left panels), IFRD1 (central panel), or C/EBPβ proteins (right panel) in control cells (sh-Ctrl), eRF3A-depleted cells (sh-eRF3A) and UPF1-depleted cells (sh-UPF1); α-Tubulin (α-Tub) served as a loading control.
Regulation of mRNAs carrying uoRFs in eRF3A- and UPF1-depleted cells
We then decided to draw up a comprehensive map of the expression of uORF-carrying genes in HCT 116 cells with the aim to identify those that are regulated by translation termination and those regulated by NMD. To compute the detection of uORFs with translation signals in HCT 116 cells, we used a two-step strategy. We first inferred all potential uORFs in the human transcriptome. For this purpose, an uORF was predicted as a sequence in mRNA 5ʹUTRs starting either by a canonical AUG or by a non-AUG codon in the RCCNNNG (R being a purine nucleotide and NNN the initiation codon) optimal context for translation initiation [46] and ending with an in frame stop codon. This led to a list of 8,827 transcripts with predicted uORFs. In parallel, for each transcript, we processed the Ribo-seq data to detect translation signals (ribosome protected fragments) in the 5ʹUTRs, regardless of the sequence information. We then compared the Ribo-seq detected signals with the ‘predicted uORFs’ to determine truly translated uORFs, i.e., a predicted uORF containing Ribo-seq signals, within HCT 116 transcriptome. This task was achieved using the algorithm presented in Supplemental Figure S4 and homemade Python 3 scripts. However, assigning a codon as an initiation site is also dependent on the ribosome footprint coverage threshold that is used to distinguish starts from non-starts. Thus, we set the threshold of read coverage per nucleotide to 10. The overall results mapped 2,726 mRNAs carrying ~7,000 translated uORFs. One can note that a high number (4,179) of transcripts with predicted uORFs were not selected through our algorithm (Supplemental Figure S5). This was due predominantly to the fact that these transcripts were not part of our HCT 116 cells transcriptome, and, for the remaining ones, to the absence of ribosome footprint coverage matching the predicted uORF. In addition, among the Ribo-seq signals detected in 5ʹUTRs, 1,922 signals (noted ‘unverified’ in Supplemental Figure S5) could not be attributed to uORFs either through our algorithm or by visual inspection. Because we were unable to decide whether these signals corresponded to truly translated sequences, they were excluded from our analysis.
We next used our generated data of translated uORFs (tuORFs), to analyse the status of mRNAs carrying uORFs in eRF3A- and UPF1-depleted HCT 116 cells. Thus, we examined the differentially expressed mRNAs (p adj < 0.05) in the Ribo-seq data sets of eRF3A (2,645 mRNAs) and UPF1 (815 mRNAs) knockdowns, and found 596 uORF-carrying transcripts for eRF3A depletion and 186 for UPF1 depletion (Fig. 4A). Interestingly, among these transcripts, only a small fraction (63 mRNAs) was common to both knockdowns (Figure 4A) including ATF4 and IFRD1 mRNAs. This result not only confirmed that different sets of transcripts are regulated at the translational level by either eRF3A or UPF1 (Fig. 2B) but also suggested that there are at least two different classes of uORFs, the one being involved in the control of mechanisms related to eRF3A function including NMD, and the second acting on the regulation of UPF1-driven processes beyond NMD[3].
Figure 4.
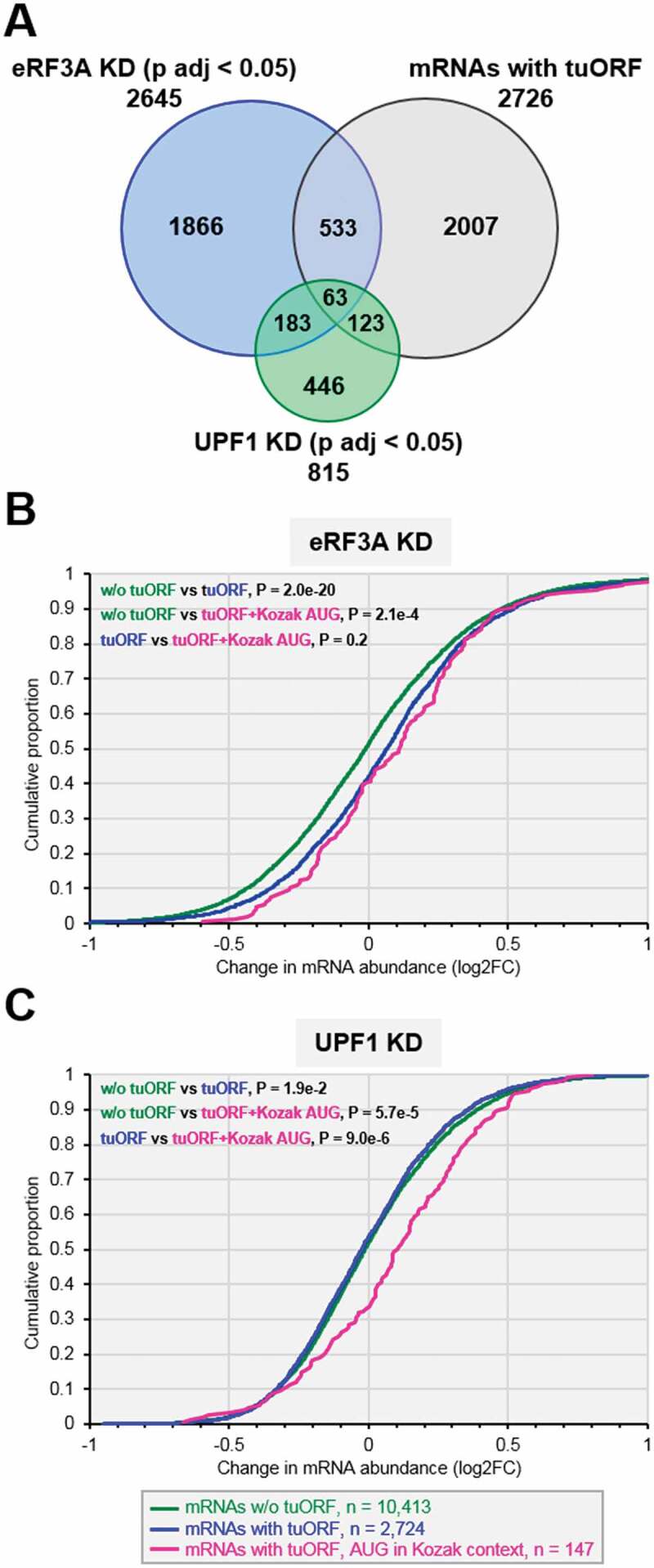
Expression of uORF carrying mRNAs in eRF3A and UPF1 knockdown cells. A. Proportional Venn diagram showing the overlap between three sets of transcripts: differentially expressed genes (main coding sequence changes in Ribo-seq data; p adj < 0.05) in eRF3A knockdown cells (blue circle, eRF3A KD), UPF1 knockdown cells (green circle, UPF1 KD) and mRNAs carrying translated uORFs – tuORF (purple circle). The number of genes is indicated for each class of mRNA. (B and C) Cumulative distribution functions of changes in mRNA abundance (plotted as log2FC) following eRF3A depletion (B, eRF3A KD) or UPF1 depletion (C, UPF1 KD) for mRNAs without translated uORF corresponding to mRNA devoid of uORF and to mRNAs with non-translated uORFs (w/o tuORF, green line), mRNAs with translated uORFs (tuORF, blue line) and mRNAs carrying a translated uORF with an AUG initiation codon surrounded by a Kozak context (pink line). In B and C, P-values were determined by Wilcoxon rank sum test for the two sided hypothesis with a 95% confidence interval. The number of genes in each category is indicated below the graphs.
Several studies reported that uORFs drive broad repression of downstream translation and correlate with lower steady-state mRNA levels [47,48]. We thus sought to characterize the overall effects of eRF3A and UPF1 knockdown on uORF regulatory capacity. To this end, we compared the changes in mRNA abundance and main ORF translation for mRNAs containing at least one translated uORF with that of all mRNAs devoid of translated uORFs, i.e., those without uORFs and those with non-translated uORFs (noted ‘w/o tuORFs’ in Fig. 4). For mRNAs with a translated uORF, this analysis revealed a broad up-regulation of mRNA abundance (p = 2.0 x 10−20, p-values determined by Wilcoxon rank sum test) following eRF3A depletion (Fig. 4B) and a global and significant (p = 6.5 x 10−15) increase in main ORF translation (Supplemental Figure S6). These results indicate that eRF3A depletion globally derepressed the expression of mRNAs containing translated uORFs and suggest that translation termination plays a key role in the modulation of translation driven by uORFs. In contrast, the level of mRNAs with translated uORFs was not more sensitive to UPF1 depletion than was that of mRNAs without translated uORFs (w/o tuORF versus tuORF in Fig. 4C). These findings suggest that most of the translated uORFs do not affect the mRNA level and hence do not trigger NMD. Interestingly, when we selected only mRNAs with uORFs starting at an AUG initiation codon surrounded by the RCCaugG optimal nucleotide context for translation initiation, also called the ‘Kozak context’[46], we found a clear increase of mRNA levels in UPF1 knockdown cells (Fig. 4C). This suggests that solely uORFs with high translation initiation efficiency are bona fide NMD activators. Thus, the context surrounding the initiation codon of uORFs appears to be one of the key determinants for triggering NMD. It is to be mentioned that the presence of an AUG in the Kozak context does not significantly increase the level of mRNAs carrying tuORFs in eRF3A depleted cells (Fig. 4B).
Regulation of ribosomal protein genes in eRF3A- and UPF1-depleted cells
Previous genome-wide studies showed that mRNAs coding for NMD factors are themselves targeted by NMD resulting in a feedback regulation of NMD [12,23,27]. To assess this feedback regulation in our samples, we verified whether the knockdown of UPF1 had an effect on the expression of NMD genes. As shown in Fig. 5 (panels A and B), most of the constituents of the NMD pathway were indeed overexpressed in UPF1-depleted cells, conversely to eRF3A-depleted cells. We next extended our analysis to the entire translation process. Thus, we sought to examine the effects of eRF3A and UPF1 depletions on the expression of factors involved in the initiation, elongation and termination steps of translation. We did not observe any clear effect of either eRF3A or UPF1 depletion on the expression level of translation factors (Supplemental Figure S7).
Figure 5.
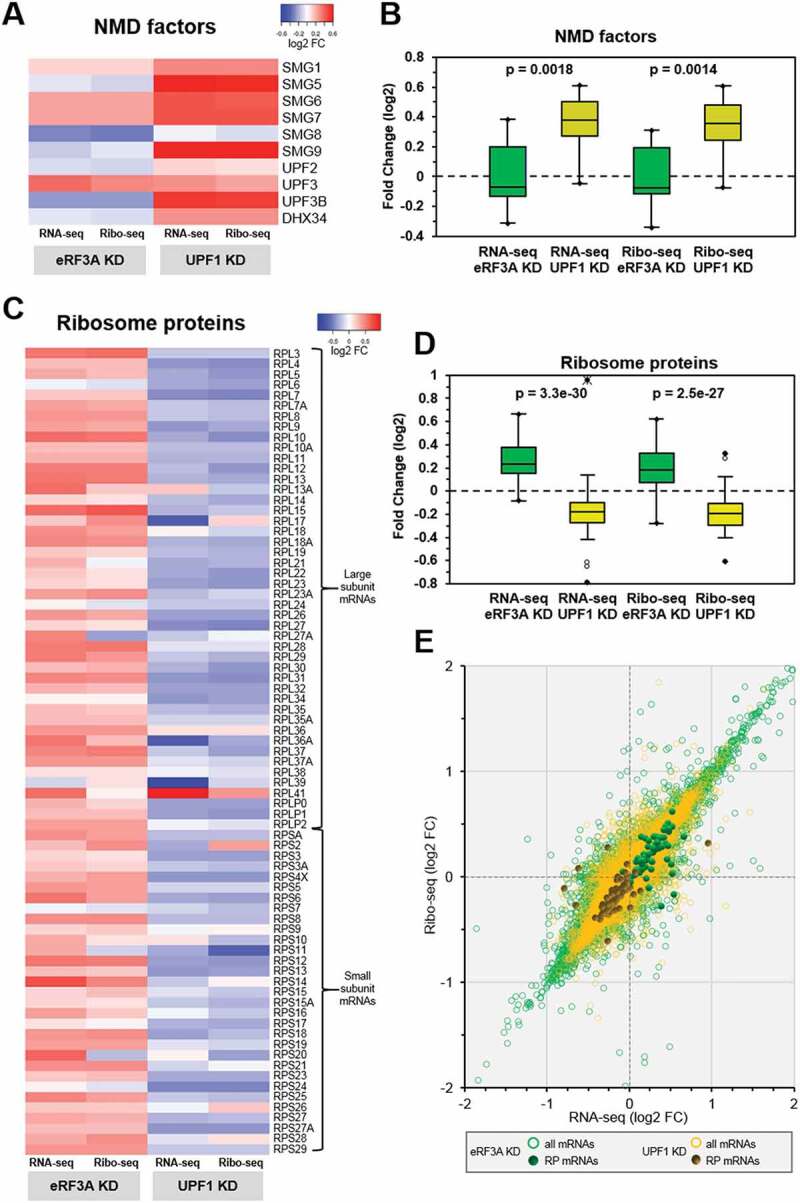
Feedback control loops in eRF3A and UPF1 knockdown cells. (A) Heatmap representation of the differential expression levels (log2 FC scale) of the NMD factor mRNAs in the transcriptome (RNA-seq) and translatome (Ribo-seq) following eRF3A knockdown (eRF3A KD) or UPF1 knockdown (UPF1 KD). Heatmap was performed using Heatmapper website http://www2.heatmapper.ca/expression/[85] without linkage clustering method. (B) Corresponding box plot of log2FC values for NMD factor mRNAs in RNA-seq and Ribo-seq experiments. The central lines show the medians; the box limits indicate the 25th and 75th percentiles. Two-tailed t-test was used to determine p-values. (C) Heatmap representation of the expression levels of ribosomal protein mRNAs in the transcriptome (RNA-seq) and translatome (Ribo-seq) following eRF3A knockdown (eRF3A KD) or UPF1 knockdown (UPF1 KD). Heatmap was performed as in A. (D) Corresponding box plot of log2FC values for ribosomal protein mRNAs in RNA-seq and Ribo-seq experiments. Box plot was performed as in B. (E) Scatter plot of the Ribo-seq versus RNA-seq differential expression for eRF3A depletion (all mRNAs: green circles and ribosome protein (RP) mRNAs: green dots) and UPF1 depletion (all mRNAs: yellow circles and ribosome protein (RP) mRNAs: dark yellow dots).
Because the ribosome is a key component of the translation process, we next compared the effect of eRF3A or UPF1 knockdown on the expression of ribosomal protein (RP) genes. Unexpectedly, the heatmap and scatter-plot representations revealed a broad up-regulation of ribosomal protein mRNAs in eRF3A-depleted cells whereas the large majority of these mRNAs were globally down-regulated in UPF1-depleted cells (Fig. 5C–E). The statistical two-tailed t-test analysis confirmed that these differences in RP expression were highly significant (p = 3.3x10−30 for RNA-seq and p = 2.5x10−27 for Ribo-seq, Fig. 5D). Therefore, the important question was the nature of the mechanism leading to these opposite effects. A correlation of RP gene expression between both conditions would be a strong indication for a common control mechanism. However, correlation coefficient measurement for RP mRNAs showed that there is likely no correlation in RP gene expression between the two conditions (Spearman coefficient ρ = 0.272 for RNA-seq and 0.192 for Ribo-seq data, Supplemental Figure S8). This result is consistent with the possibility that two independent regulatory processes are responsible for the reverse effects of eRF3A and UPF1 on RP gene expression.
Discussion
Whereas several genome-wide RNA-seq and RNA half-life analyses have previously identified the transcripts that are dysregulated in response to UPF1 knockdown [20,21,23], the effect of eRF3A depletion on mammalian transcriptome has not been documented previously. In the present study, we established the genome-wide landscape of gene expression in human cells depleted either in the translation termination factor eRF3A or in UPF1, a key actor of the NMD pathway, using RNA sequencing and ribosome profiling [49,50]. The RNA-seq and Ribo-seq data sets allowed us to compare for both, eRF3A and UPF1 knockdowns, the modifications of mRNA and translation levels for the ~13,000 transcripts expressed in HCT 116 cells. Our comparison highlights a number of interesting features on the role of translation termination and NMD in physiological gene expression.
Our analyses show that both factors participate in the fine-tuning of gene expression however with highly divergent effects. The differential analysis of eRF3A-depleted versus control cells reveals that most of the genes that are regulated at the mRNA level, are also affected at the translational level (Fig. 2D). Interestingly, among the up-regulated genes, we found a number of positive regulators of transcription such as ATF4, ATF6, IFRD1, RUNX1, TP53, JUN, C/EBPβ, C/EBPγ (Fig. 2). The translational changes of these transcription factors might consequently cause important changes in the rate of transcription of their target genes, which could then indirectly affect their translational level. The combined effects of changes in mRNA levels and translational control on transcriptional regulators provides for versatility in regulating gene expression program. A well-documented example of this versatility is given by ATF4 for which this combination of transcriptional regulation and translational control allows to selectively repress or activate key regulatory genes that are essential for maintaining the balance between stress remediation and apoptosis [51]. This may explain the particular enrichment of genes involved in stress response and apoptosis in eRF3A-depleted samples (Supplementary Figure S3). In addition, these transcriptional regulators are downstream targets of signalling pathways controlling the cell cycle progression such as the mTOR pathway which was previously shown to be inhibited by eRF3A depletion [32]. Interestingly, the analysis of the impact of UPF1-knockdown on gene expression revealed that eRF3A and UPF1 have largely non-overlapping sets of targets (less than ~250 targets are common to both factors, Fig. 2A). Nevertheless, we observed that, as eRF3A knockdown, UPF1 depletion also induced altered expression of the same set of transcription factors, though to a slightly lesser extent or with an opposite effect. This agrees with previous reports showing that a number of transcriptional regulators are direct or indirect targets of NMD [20,21,23]. As an example, the up-regulation of ATF4, ATF3 and IFRD1 transcripts following NMD inhibition has been well documented [10,23,43,52] and validates our observations. The important point revealed by our differential analyses, is that the efficiency of translation termination as well as that of NMD have considerable and unexpected influence on the mRNA landscape, although these actions might be largely indirect.
Sequence-based analyses have identified uORFs in the transcriptomes of a variety of organisms and have shown that genes with an uORF represent up to 50% of the human genome [53,54]. In addition, transcripts with uORFs have seemingly lower protein expression levels than transcripts without uORFs [47,55]. However, as noticed by Wethmar et al.[56], remarkably few uORFs have been functionally studied when compared to the number of predicted uORFs. Thus, it is still largely unclear what fraction of uORFs effectively undergoes translation, and, besides mRNA sequence elements, what are the factors that influence uORF function. eRF3A and UPF1 are good candidates to be part of such regulating factors. Indeed, by decreasing translation termination efficiency, depletion of release factors promotes translational readthrough of stop codons [57] and hence is potentially regulating the translation of mRNAs harbouring uORFs. In addition, by creating a premature stop codon, uORFs may render these mRNAs susceptible to NMD. In this study, we establish a comprehensive map of the functional uORFs of human HCT 116 cells to bring new insights on how translation termination and NMD may influence uORF function. Using a computational analysis based on Ribo-seq data, we obtained a list of 2,726 mRNAs carrying translated uORFs. Strikingly, the number of mRNAs with translated uORF appears rather low, constituting only ~40% of the predicted uORF-carrying mRNAs (6,896) in our reference HCT 116 transcriptome (see Supplemental file S1). The regulatory potential of a uORF is mainly controlled by the uORF structural properties such as its length, its position in the mRNA leader sequence, the distance from the main ORF, and importantly, the context of the initiation codon [56]. The influence of translation termination efficiency could therefore be limited to the small portion of uORFs with favourable structural properties, such as those of ATF4 and IFRD1 mRNAs. However, we observed that eRF3A knockdown globally increased mRNA levels and main ORF translation for mRNAs carrying translated uORFs (Fig. 4B and Supplemental Figure S6A). This suggests that eRF3A depletion relieves the inhibitory effect of uORFs regardless of their structural properties. This finding strengthens the idea that the accuracy of translation termination is one of the determinants of the regulatory function of uORFs either by controlling the efficiency of stop codon readthrough or by influencing the reinitiation capacity of post-termination ribosomes [56].
An overall up-regulation of transcripts harbouring translated uORFs was not observed for UPF1-depleted cells. Nevertheless, a clear up-regulation of mRNA levels was observed when the list of uORFs was restricted to those having an initiator AUG codon in the optimal Kozak context (Fig. 4C). These findings lead to the conclusion that one should be cautious with the suggestion that translated uORFs commonly trigger NMD. Several reports have questioned the mechanisms that allow mRNA to escape NMD in the case of either AUG-proximal nonsense mutation [58–60] or uORFs [61]. These studies clearly show that NMD resistance mainly depends on the ability of post-termination ribosomes to reinitiate translation at a downstream start codon, the favourable features for an efficient reinitiation being a short uORF with unstructured sequence and a sufficiently long distance to the downstream initiation site [58–60]. The set-up of the reinitiation mechanisms prevents the association of NMD components with the termination complex, and thus, the reinitiation machinery, i.e., a 40S ribosomal sub-unit associated with initiation factors, resumes scanning and removes downstream EJC. It is interesting to note that the major difference between the 40S pre-initiation complex scanning the mRNA 5ʹUTR and by-passing an AUG in weak initiation context, the so-called ‘leaky scanning’[62], and the post-termination 40S ribosomal complex scanning downstream a stop codon towards the next initiation codon is the presence of the ternary complex eIF2-GTP-Met-tRNAi on the 40S pre-initiation complex. In contrast, the post-termination 40S ribosomal complex must be reloaded with the ternary complex to become initiation competent. However, both of these initiation complexes may remove EJC while scanning the mRNA, and hence, may prevent the NMD activation. Thus, the leaky scanning mechanism could explain the absence of NMD activation in the case of mRNAs harbouring uORFs with weak initiation context. As strongly suggested by our results, among mRNAs having an uORF, solely those with the initiator AUG in a strong context may be direct targets of NMD. After careful review of the literature, we found only one experiment showing the influence of the AUG context on NMD activation [63]. This important point is now being investigated.
In mammalian cells, intricate networks of regulatory loops allow to maintain the homeostasis
of normal gene expression, to adapt the cell response to changes in cellular environment and to protect cells from the deleterious consequences of various forms of stress. The negative regulation of NMD factors by NMD itself is a well-documented example of these feedback mechanisms [12,23,27]. Our results corroborate the feedback control of UPF1 on NMD factors and furthermore uncover new feedback regulatory loops that fine-tune the level of RP mRNAs (Fig. 5). In eukaryotes, the expression of RP genes is tightly regulated by multiple control mechanisms at transcriptional and translational levels [64,65]. These controls, that lead to the coordinate regulation of RP genes expression [66], allow maintaining ribosome biosynthesis at the appropriate level dictated by cell growth conditions and cell environment constraints. This coordinate regulation of RP genes is strikingly well illustrated by our results showing that the majority of the RP mRNAs are clustered either above (eRF3A knockdown) or below (UPF1 knockdown) the control cell level (log2 Fold Change = 0 in Fig. 5E). However, a few RP mRNAs escaped this clustering, and particularly RPL41 in UPF1 knockdown cells (Fig. 5C). A previous omic study on human ribosome has noted this striking difference for RPL41 mRNA and suggested that its very short ORF, encoding only 25 amino acids, was responsible for a lower translation efficiency and for the need of a compensatory increase in mRNA abundance [67]. It has also been proposed that some RPs, which exert extra-ribosomal functions, are driven by additional specific regulatory mechanisms [68,69]. This could be the case for RPL41 which was shown to be involved in a variety of processes such as degradation of transcription factor ATF4 and mitosis through its association with several cytoskeleton components [70,71].
Another unexpected observation is that eRF3A and UPF1 display opposite effects on RP gene expression. Although the transcriptional regulation of RP genes in eukaryotes is far from being completely elucidated, literature data argue that transcription of RP genes is controlled by the combination of a number of transcription factors including MYC, p53, RUNX1, GATA2, YY1, AP1 [64,72–74]. As noted above, in our study the expression of several of the transcription factors known to be involved in RP genes regulation is modified by eRF3A or UPF1 knockdowns. However, it is hard to determine whether, in our experiments, RP gene regulation is modified due to the altered expression of a single transcription factor or a combination of them. It is also possible that different combinations of transcription factors with modified expression are responsible for the divergent effects observed. Regardless of the precise mechanisms involved, our results suggest the existence of a feedback network that accurately adjust the expression level of RP genes to achieve the optimal ribosome synthesis required to maintain efficient translation together with cell homeostasis and low energy consumption. In normal cell growth conditions, the high efficiency of the translation process may negatively regulate RP gene expression, decreasing ribosome synthesis and thus lowering energy consumption. The outcome of a translation defect such as that induced by eRF3A depletion would be then a global up-regulation of RP gene expression.
Our results also indicate that, in conjunction with its role in mRNA quality control, NMD may promote translation accuracy and ribosome renewal through the global up-regulation of RP gene expression. Thus, NMD perturbation could not only up-regulate NMD factors, in order to restore its efficiency, but could also trigger the down-regulation of RP genes to slowdown the overall translation thus preventing the deleterious consequence of aberrant mRNA translation. Confirming our results, we also found a down-regulation of RP mRNAs in the data of previously published experiments on the effect of UPF1 knockdown in mammalian cells [21,23] (Supplemental Figure S9). Future studies will help to decipher the control mechanisms of these feedback responses and to assess their biological importance.
In summary, our comparative study emphasizes the divergent effects of eRF3A and UPF1 on mammalian transcriptome and identifies new regulatory loops controlling the expression of RP genes.
Material and methods
Plasmids and short interfering RNA
Plasmid expressing small interfering RNAs targeting eRF3A mRNA have been previously described [57]. Plasmids pSUPERpuro-hUPF1/I and pSUPERpuro-hUPF1/II targeting two different sequences in human UPF1 [75] and pSUPERcontrol with the non-silencing shRNA sequence (5ʹ-ATTCTCCGAACGTGTCACG-3ʹ) used as negative control shRNA (sh-Ctrl) were a generous gift from O. Mühlemann.
Cell culture, electroporation and transfection
The HCT 116 cell line (ATCC number: CCL-247) was maintained in McCoy medium (Invitrogen) supplemented with 10% foetal calf serum, 100 mg/ml streptomycin and 100 U/ml penicillin at 37°C under 5% CO2 atmosphere. Electroporation of cells was performed with a Gene pulser II electroporation system (Bio-Rad) using 4.8 × 106 cells and 20 µg of plasmid DNA. eRF3 depletion was performed with sh-3a1 [29]. Depletion of UPF1 was induced by co-transfection of an equimolar mix of pSUPERpuro-hUPF1/I and pSUPERpuro-hUPF1/II plasmids [75]. The cells were collected 72 h after electroporation.
Western blot analysis
Cell pellets were resuspended in 100 µl of a lysis buffer (50 mM TrisHCl pH 8, 120 mM NaCl, 1% NP40, 1 mM EDTA) containing, complete EDTA-free cocktail of protease inhibitors and phosphatase inhibitor cocktail (Roche) and 1 mg/ml pepstatine. Cells were lysed on ice, centrifuged for 20 min at 16,000 x g and supernatant retained as cell extracts. Protein concentrations of extracts were determined using MicroBCA Reagent (Sigma), bovine serum albumin being used as standard. For each sample, 30 µg of total protein were loaded on polyacrylamide gel and subjected to electrophoresis. Western blotting was then performed as described [32]. Antibodies directed against human eRF3 were previously described [57]. Mouse monoclonal antibodies against α-Tubulin were purchased from GE Healthcare. Antibodies directed against human IFRD1 and ATF4 were purchased from Proteintech. Antibodies directed against human C/EBPβ (C-19) recognizer both LAP and LIP forms and were purchased from Santa Cruz. Antibodies directed against human UPF1 were purchased from Cell Signalling. Proteins were detected by chemiluminescence and exposure to X-ray film.
Purification of ribosome-protected RNA fragments
Ribosome Profiling was performed as described previously [76]. Briefly, monosomes were prepared by digesting polysome extracts (see above) for 1 hour at room temperature with 15 units RNaseI (Ambion) per OD unit. To pellet the resulting 80S monosomes, the RNaseI-digested polysomes were purified on a 24% sucrose cushion in polysomes extraction buffer (50 mM Tris-Acetate pH 7.6, 50 mM NH4Cl, 12 mM MgCl2) centrifuged for 2 h 15 min at 4°C, at 100.000 rpm in a TLA110 rotor using OptimaMAX-XP Beckman centrifuge. Each pellet was washed and resuspended in 750 µl of polysomes extraction buffer by pipetting up and down. Ribosome protected fragments (RPFs) of mRNA were purified using acid phenol-chloroform solution and ethanol precipitation. Then the RNA fragments were recovered in 500 µl of TE (10 mM Tris-HCl pH 7, 1 mM EDTA) containing SUPERase-IN 0.1 U/µl (Ambion) and stored at −20°C. Subsequently, RNA fragments were loaded on 17% polyacrylamide gel containing 7 M urea and subjected to electrophoresis for about 6 h at 150V with heating at 65°C. After migration, the gel was stained with SYBER Gold (Invitrogen) for 30 min, RPFs were visualized with a UV lamp at 300 nm, and the gel pieces containing the 28 nt RNA fragments were excised. Then, RNA fragments were eluted from the excised gel pieces and precipitated with ethanol in the presence of glycogen (20 mg/ml) overnight at −20°C. Finally, RPFs were depleted of major ribosomal RNA (rRNA) contamination present within the 28 nt eluted RNA fragments by subtractive hybridization using biotinylated oligonucleotides and were captured by MageneShere Paramagnetic streptavidin particles (Ribo-Zero Gold rRNA Removal Kit Illumina). The supernatants containing the ribosome footprints were recovered and the RNA was precipitated in ethanol in the presence of glycogen overnight at −20°C.
Purification of total RNA for RNA-seq
Total RNA was purified from HCT 116 cell extracts by hot acid phenol extraction. An equal volume of acid phenol was added to the extract that was vortexed for 1 h at 65°C, then centrifuged 10 min at full speed. Then, the RNA was extracted by adding an equal volume of chloroform to the first supernatant, vortexed for 5 min and centrifuged at full speed. The RNA was then precipitated over night at −20°C with 0.3 M Sodium Acetate pH 5.2 and 3 volumes ethanol 100%. Centrifuged at full speed for 20 min at 4°C, RNA were solubilized in 500 µl TE (10 mM Tris-HCl pH 7, 1 mM EDTA) containing SUPERase-IN 0.1 U/µl (Ambion) and subjected to rRNA depletion, as described above.
Library construction and sequencing
cDNA library from 100 ng RNA was prepared by the Imagif platform (Institut de Biologie Intégrative de la Cellule, Gif sur Yvette, France), using the TruSeq Small RNA Sample Prep Kit with 3ʹ sRNA Adapter (Illumina) according to the manufacturer’s protocol. RNA integrity and quality was verified using Bioanalyzer Small RNA Analysis kit (Agilent). Sequencing was performed at the genomic platform of the Institut de Biologie de l’École Normale Supérieure (Paris, France) on an Illumina Nextseq 500 and with a single-read 75 cycle runs. Four sequencing libraries were prepared from purified RPFs and total RNA, for each of the two biological replicates of eRF3A-depleted, UPF1-depleted and control cells. A minimum of 7 × 108 reads were achieved for bioinformatics analysis.
Data analysis
Reads from transcriptome (RNA-seq) and ribosome profiling (Ribo-seq) libraries were trimmed using Cutadapt [77], to remove the 3ʹ adapter of the reads 5ʹTGGAATTCTCGGGT-GCCAAGGAACTCCAGTCA3ʹ. Reads derived from rRNA were removed using Bowtie [78] and by aligning to the rRNA reference downloaded from the Ensembl BIOMART database [79]. The remaining non-ribosomal reads were then aligned to the human reference genome hg38 using TopHat2 [80], allowing 2 mismatches and keeping only uniquely mapped reads. At the end, we obtained ~1.2x108 RPFs mapping principally to the CDS. These reads were used for subsequent analysis. In parallel to Ribo-seq data, we obtained transcriptomic data, generating ~8x107 reads from total RNA. As for ribosome profiling, we retained only uniquely mapped reads excluding rRNA sequences. After discarding all reads mapping to rRNA and all reads with multiple alignments, we obtained ~1.8x107 reads.
Differential gene expression
Raw counts were obtained using FeatureCounts [81] based on hg38 human annotations. The counts were realized on the exons (feature) and regrouped by transcripts (metafeature) keeping only stranded counts. Then, the counts were normalized using DESeq2 [37] through the SARTools package using R 3.3.1 [82]. RNA-seq and Ribo-seq counts and their normalizations were computed separately. P-value threshold for significant changes was set to 5 × 10−2. Samples correlations were computed using the SERE statistics [83]. Gene Ontology analysis [38] was carried out using the DAVID gene ontology functional annotation tool (https://david.ncifcrf.gov/) with default parameters. We ranked terms according to the p-value.
Detection of principal transcript isoform
For most of the genes, annotations of human genome hg38 provide more than one transcript derived either from multiple transcription start sites, or alternative splicing or alternative polyadenylation sites. In addition, some of these annotated transcripts are poorly supported by primary data. However, only a sub-set of these transcripts are expressed in a given cell type, and constitute its transcriptome. To select the most representative transcripts for each gene, we first used the APPRIS annotation [84] keeping only transcripts containing the ‘appris_principal’ tags. This allowed us to eliminate the poorly-supported transcripts. Then, for genes with more than one transcript left, we selected the most expressed transcript. This selection was done using the normalized count of transcripts in control cells (sh-Ctrl), dividing by their length to obtain a comparable number. Transcripts of a same gene were then compared between each other and the most covered (Ribo-seq counts) species was selected. Comparison of the resulting transcriptome of control cells with those of eRF3A- and UPF1-depleted cells showed only a very small number of changes in the sets of selected transcripts. Finally, using Ribo-seq data and visual inspection, these differences, mainly due to alternative splicing variations in the coding sequence, were standardized by retaining the transcript selected in the control cell transcriptome.
Real-time quantitative PCR analysis
Total RNA was extracted 72 h after electroporation using NucleoSpin RNAII Kit (Macherey-Nagel) and treated with DNase I, Amp Grade (Invitrogen) prior to cDNA synthesis. Total RNA (1 µg) was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer’s instructions. Real-time PCRs were performed on a CFX384 Touch Real-Time PCR detection system (Bio-Rad) in 384-well plates. A mastermix containing 0.5 µM of forward primer, 0.5 µM of reverse primer and 5µL SYBR Green Master mix (Applied Biosystems) was prepared for each target or reference gene mRNAs. Primer pairs are listed in Table 1. Reactions were carried out in 10 µL with 4 µL of cDNA from a 100X dilution of the RT reaction as PCR template and 6 µL of master mix. All reactions were performed in triplicate. Serial dilutions of a mixture of all cDNAs were used to generate a PCR standard curve. A negative control without cDNA template was run with every assay to assess the overall specificity. Absence of genomic DNA contamination was assessed using enzyme lacking reverse transcriptase reactions as template (no-RT). Data were analysed using Bio-Rad CFX Manager software v3.1. The amount of the target mRNA was normalized to the B2M (β2-microglobulin) reference gene mRNA.
Table 1.
Oligonucleotides used in qPCR.
| Name | Sequence 5ʹ>3’ |
|---|---|
| ATF4 fw | CCAACAACAGCAAGGAGGAT |
| ATF4 rev | GGGGCAAAGAGATCACAAGT |
| DDIT4 fw | GGTTTGACCGCTCCACGAG |
| DDIT4 rev | GGTAAGCCGTGTCTTCCTCC |
| ASNS fw | ATCACTGTCGGGATGTACCC |
| ASNS rev | CTTCAACAGAGTGGCAGCAA |
| B2M fw | TATCCAGCGTACTCCAAAGA |
| B2M rev | GACAAGTCTGAATGCTCCAC |
| GSPT1 fw | GAGGAGGAAGAGGAAATCCC |
| GSPT1 rev | TCCTTTTGTCAACCATTCCA |
| UPF1 fw | AGATCACGGCACAGCAGA |
| UPF1 rev | GTGGCAGAAGGGTTTTCCT |
| RUNX1 fw | ACAAACCCACCGCAAGTC |
| RUNX1 rev | CATCTAGTTTCTGCCGATGTCTT |
| C/EBPB fw | TTTGTCCAAACCAACCGCAC |
| C/EBPB rev | GCATCAACTTCGAAACCGGC |
| ACTB fw | TCCCTGGAGAAGAGCTACGA |
| ACTB rev | AGCACTGTGTTGGCGTACAG |
| IFRD1 fw | TGGCCAGAGGAATAGAGAGT |
| IFRD1 rev | TGGCCCGGTGTTT A TTTCCA |
| GATA2 fw | CAGCAAGGCTCGTTCCTGTT |
| GATA2 rev | GGCTTGATGAGTGGTCGGT |
| ETS1 fw | TACACAGGCAGTGGACCAATC |
| ETS1 rev | CCCCGCTGTCTTGTGGATG |
Funding Statement
A.A. was supported by a Doctoral Contract from the Université Pierre et Marie Curie. P.F. is supported by the ANR [Rescue Ribosome ANR-17-CE12-0024-01 to O.N.]) and P.B. is supported by INCa [RiboTEM 2014-092] to O.N.
Acknowledgments
This work benefited from the facilities and expertise of the Platform of I2BC for NGS library preparation. We thank Ammara Mohammad and Stéphane Le Crom from the Genomic Paris Centre for assistance with sequencing. This work was supported by the France Génomique national infrastructure, funded as part of the ‘Investissements d’Avenir’ program managed by the ANR (Contract ANR-10-INBS-0009)
Data availability
The data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE126385.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Jackson RJ, Hellen CUT, Pestova TV.. Termination and post-termination events in eukaryotic translation. Adv Protein Chem Struct Biol. 2012;86:45–93. [DOI] [PubMed] [Google Scholar]
- [2].Lejeune F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017;50:175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim YK, Maquat LE. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA. 2019;25:407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Raimondeau E, Bufton JC, Schaffitzel C. New insights into the interplay between the translation machinery and nonsense-mediated mRNA decay factors. Biochem Soc Trans. 2018;46:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Losson R, Lacroute F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc Natl Acad Sci USA. 1979;76:5134–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Maquat LE, Kinniburgh AJ, Rachmilewitz EA, et al. Unstable β-globin mRNA in mRNA-deficient β0 thalassemia. Cell. 1981;27:543–553. [DOI] [PubMed] [Google Scholar]
- [7].Leeds P, Peltz SW, Jacobson A, et al. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev. 1991;5:2303–2314. [DOI] [PubMed] [Google Scholar]
- [8].Bühler M, Steiner S, Mohn F, et al. EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3ʹ UTR length. Nat Struct Mol Biol. 2006;13:462–464. [DOI] [PubMed] [Google Scholar]
- [9].Hansen KD, Lareau LF, Blanchette M, et al. Genome-wide identification of alternative splice forms down-regulated by nonsense-mediated mRNA decay in Drosophila. PLoS Genet. 2009;5:e1000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mendell JT, Sharifi NA, Meyers JL, et al. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–1078. [DOI] [PubMed] [Google Scholar]
- [11].Singh G, Rebbapragada I, Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008;6:e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yepiskoposyan H, Aeschimann F, Nilsson D, et al. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA. 2011;17:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cosson B, Berkova N, Couturier A, et al. Poly(A)-binding protein and eRF3 are associated in vivo in human and Xenopus cells. Biol Cell. 2002;94:205–216. [DOI] [PubMed] [Google Scholar]
- [14].Hoshino S, Imai M, Kobayashi T, et al. The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3ʹ-Poly(A) tail of mRNA. Direct association of erf3/GSPT with polyadenylate-binding protein. J Biol Chem. 1999;274:16677–16680. [DOI] [PubMed] [Google Scholar]
- [15].Kashima I, Yamashita A, Izumi N, et al. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006;20:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Neu‐Yilik G, Raimondeau E, Eliseev B, et al. Dual function of UPF3B in early and late translation termination. EMBO J. 2017;36:2968–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Eberle AB, Stalder L, Mathys H, et al. Posttranscriptional gene regulation by spatial rearrangement of the 3ʹ untranslated region. PLoS Biol. 2008;6:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ivanov PV, Gehring NH, Kunz JB, et al. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 2008;27:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Celik A, Baker R, He F, et al. High-resolution profiling of NMD targets in yeast reveals translational fidelity as a basis for substrate selection. RNA. 2017;23:735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Colombo M, Karousis ED, Bourquin J, et al. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA. 2017;23:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hurt JA, Robertson AD, Burge CB. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013;23:1636–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pan Q, Saltzman AL, Kim YK, et al. Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev. 2006;20:153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tani H, Imamachi N, Salam KA, et al. Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA Biol. 2012;9:1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wittmann J, Hol EM, Jäck H-M. hUPF2 silencing identifies physiologic substrates of mammalian nonsense-mediated mRNA decay. Mol Cell Biol. 2006;26:1272–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Malabat C, Feuerbach F, Ma L, et al. Quality control of transcription start site selection by nonsense-mediated-mRNA decay. eLife. 2015;4:e06722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chapin A, Hu H, Rynearson SG, et al. In vivo determination of direct targets of the nonsense-mediated decay pathway in Drosophila. G3 Genes Genom Genet. 2014;4:485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang L, Lou C-H, Chan W, et al. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell. 2011;43:950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665–677. [DOI] [PubMed] [Google Scholar]
- [29].Ait Ghezala H, Jolles B, Salhi S, et al. Translation termination efficiency modulates ATF4 response by regulating ATF4 mRNA translation at 5ʹ short ORFs. Nucleic Acids Res. 2012;40:9557–9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hoshino S, Miyazawa H, Enomoto T, et al. A human homologue of the yeast GST1 gene codes for a GTP-binding protein and is expressed in a proliferation-dependent manner in mammalian cells. EMBO J. 1989;8:3807–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kikuchi Y, Shimatake H, Kikuchi A. A yeast gene required for the G1-to-S transition encodes a protein containing an A-kinase target site and GTPase domain. EMBO J. 1988;7:1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chauvin C, Salhi S, Jean-Jean O. Human eukaryotic release factor 3a depletion causes cell cycle arrest at G1 phase through inhibition of the mTOR pathway. Mol Cell Biol. 2007;27:5619–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hashimoto Y, Inagaki H, Hoshino S. Calpain mediates processing of the translation termination factor eRF3 into the IAP-binding isoform p-eRF3. FEBS Lett. 2015;589:2241–2247. [DOI] [PubMed] [Google Scholar]
- [34].Hegde R, Srinivasula SM, Datta P, et al. The polypeptide chain-releasing factor GSPT1/eRF3 is proteolytically processed into an IAP-binding protein. J Biol Chem. 2003;278:38699–38706. [DOI] [PubMed] [Google Scholar]
- [35].Jolles B, Aliouat A, Stierlé V, et al. Translation termination-dependent deadenylation of MYC mRNA in human cells. Oncotarget. 2018;9:26171–26182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Osawa M, Hosoda N, Nakanishi T, et al. Biological role of the two overlapping poly(A)-binding protein interacting motifs 2 (PAM2) of eukaryotic releasing factor eRF3 in mRNA decay. RNA. 2012;18:1957–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Azzalin CM, Reichenbach P, Khoriauli L, et al. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007;318:798–801. [DOI] [PubMed] [Google Scholar]
- [40].Hong D, Park T, Jeong S. Nuclear UPF1 is associated with chromatin for transcription-coupled RNA surveillance. Mol Cells. 2019;42:523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mendell JT. ap Rhys CMJ, Dietz HC. Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science. 2002;298:419–422. [DOI] [PubMed] [Google Scholar]
- [42].Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhao C, Datta S, Mandal P, et al. Stress-sensitive regulation of IFRD1 mRNA decay is mediated by an upstream open reading frame. J Biol Chem. 2010;285:8552–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Calkhoven CF, Müller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000;14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- [45].Wethmar K, Bégay V, Smink JJ, et al. C/EBPbetaDeltauORF mice–a genetic model for uORF-mediated translational control in mammals. Genes Dev. 2010;24:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. [DOI] [PubMed] [Google Scholar]
- [47].Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci USA. 2009;106:7507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Johnstone TG, Bazzini AA, Giraldez AJ. Upstream ORFs are prevalent translational repressors in vertebrates. EMBO J. 2016;35:706–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Guo H, Ingolia NT, Weissman JS, et al. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ingolia NT, Ghaemmaghami S, Newman JRS, et al. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dey S, Baird TD, Zhou D, et al. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem. 2010;285:33165–33174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol. 2008;28:3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Iacono M, Mignone F, Pesole G. uAUG and uORFs in human and rodent 5′untranslated mRNAs. Gene. 2005;349:97–105. [DOI] [PubMed] [Google Scholar]
- [54].Brar GA, Yassour M, Friedman N, et al. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ye Y, Liang Y, Yu Q, et al. Analysis of human upstream open reading frames and impact on gene expression. Hum Genet. 2015;134:605–612. [DOI] [PubMed] [Google Scholar]
- [56].Wethmar K. The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdiscip Rev RNA. 2014;5:765–778. [DOI] [PubMed] [Google Scholar]
- [57].Chauvin C, Salhi S, Le Goff C, et al. Involvement of human release factors eRF3a and eRF3b in translation termination and regulation of the termination complex formation. Mol Cell Biol. 2005;25:5801–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Neu-Yilik G, Amthor B, Gehring NH, et al. Mechanism of escape from nonsense-mediated mRNA decay of human -globin transcripts with nonsense mutations in the first exon. RNA. 2011;17:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pereira FJC, Teixeira A, Kong J, et al. Resistance of mRNAs with AUG-proximal nonsense mutations to nonsense-mediated decay reflects variables of mRNA structure and translational activity. Nucleic Acids Res. 2015;43:6528–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang J, Maquat LE. Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J. 1997;16:826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Stockklausner C, Breit S, Neu-Yilik G, et al. The uORF-containing thrombopoietin mRNA escapes nonsense-mediated decay (NMD). Nucleic Acids Res. 2006;34:2355–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kozak M. The scanning model for translation: an update. J Cell Biol. 1989;108:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wang J, Vock VM, Li S, et al. A quality control pathway that down-regulates aberrant T-cell receptor (TCR) transcripts by a mechanism requiring UPF2 and translation. J Biol Chem. 2002;277:18489–18493. [DOI] [PubMed] [Google Scholar]
- [64].Hu H, Li X. Transcriptional regulation in eukaryotic ribosomal protein genes. Genomics. 2007;90:421–423. [DOI] [PubMed] [Google Scholar]
- [65].Russo A, Russo G. Ribosomal proteins control or bypass p53 during nucleolar stress. Int J Mol Sci. 2017;18:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Li BB, Qian C, Gameiro PA, et al. Targeted profiling of RNA translation reveals mTOR-4EBP1/2-independent translation regulation of mRNAs encoding ribosomal proteins. Proc Natl Acad Sci USA. 2018;115:E9325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gupta V, Warner JR. Ribosome-omics of the human ribosome. RNA. 2014;20:1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell. 2009;34:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhou X, Liao W-J, Liao J-M, et al. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol. 2015;7:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Geng W, Qin F, Ren J, et al. Mini-peptide RPL41 attenuated retinal neovascularization by inducing degradation of ATF4 in oxygen-induced retinopathy mice. Exp Cell Res. 2018;369:243–250. [DOI] [PubMed] [Google Scholar]
- [71].Wang A, Xu S, Zhang X, et al. Ribosomal protein RPL41 induces rapid degradation of ATF4, a transcription factor critical for tumour cell survival in stress. J Pathol. 2011;225:285–292. [DOI] [PubMed] [Google Scholar]
- [72].Cai X, Gao L, Teng L, et al. Runx1 deficiency decreases ribosome biogenesis and confers stress resistance to hematopoietic stem and progenitor cells. Cell Stem Cell. 2015;17:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ishii K, Washio T, Uechi T, et al. Characteristics and clustering of human ribosomal protein genes. BMC Genomics. 2006;7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Li X, Zheng Y, Hu H, et al. Integrative analyses shed new light on human ribosomal protein gene regulation. Sci Rep. 2016;6:28619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Paillusson A. A GFP-based reporter system to monitor nonsense-mediated mRNA decay. Nucleic Acids Res. 2005;33:e54–e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Baudin-Baillieu A, Hatin I, Legendre R, et al. Translation analysis at the genome scale by ribosome profiling. Methods Mol Biol. 2016;1361:105–124. [DOI] [PubMed] [Google Scholar]
- [77].Martin F, Barends S, Jaeger S, et al. Cap-assisted internal initiation of translation of histone H4. Mol Cell. 2011;41:197–209. [DOI] [PubMed] [Google Scholar]
- [78].Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kinsella RJ, Kähäri A, Haider S, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database. 2011;2011:bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kim D-Y, Kim W, Lee K-H, et al. hnRNP Q regulates translation of p53 in normal and stress conditions. Cell Death Differ. 2013;20:226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- [82].Varet H, Brillet-Guéguen L, Coppée J-Y, et al. SARTools: a DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-seq data. PloS One. 2016;11:e0157022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Schulze SK, Kanwar R, Gölzenleuchter M, et al. SERE: single-parameter quality control and sample comparison for RNA-Seq. BMC Genomics. 2012;13:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Rodriguez JM, Maietta P, Ezkurdia I, et al. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 2013;41:D110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Babicki S, Arndt D, Marcu A, et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016;44:W147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Descombes P, Schibler U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell. 1991;67:569–579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE126385.