

ABSTRACT
This 2020 installment of the annual ‘Antibodies to Watch’ series documents the antibody therapeutics approved in 2019 and in regulatory review in the United States or European Union, as well as those in late-stage clinical studies, as of November 2019*. At this time, a total of 5 novel antibody therapeutics (romosozumab, risankizumab, polatuzumab vedotin, brolucizumab, and crizanlizumab) had been granted a first approval in either the US or EU, and marketing applications for 13 novel antibody therapeutics (eptinezumab, teprotumumab, enfortumab vedotin, isatuximab, [fam-]trastuzumab deruxtecan, inebilizumab, leronlimab, sacituzumab govitecan, satralizumab, narsoplimab, tafasitamab, REGNEB3 and naxituximab) were undergoing review in these regions, which represent the major markets for antibody therapeutics. Also as of November 2019, 79 novel antibodies were undergoing evaluation in late-stage clinical studies. Of the 79 antibodies, 39 were undergoing evaluation in late-stage studies for non-cancer indications, with 2 of these (ublituximab, pamrevlumab) also in late-stage studies for cancer indications. Companies developing 7 (tanezumab, aducanumab, evinacumab, etrolizumab, sutimlimab, anifrolumab, and teplizumab) of the 39 drugs have indicated that they may submit a marketing application in either the US or EU in 2020. Of the 79 antibodies in late-stage studies, 40 were undergoing evaluation as treatments for cancer, and potentially 9 of these (belantamab mafodotin, oportuzumab monatox, margetuximab, dostarlimab, spartalizumab, 131I-omburtamab, loncastuximab tesirine, balstilimab, and zalifrelimab) may enter regulatory review in late 2019 or in 2020. Overall, the biopharmaceutical industry’s clinical pipeline of antibody therapeutics is robust, and should provide a continuous supply of innovative products for patients in the future. *Note on key updates through December 18, 2019: 1) the US Food and Drug Administration granted accelerated approval to enfortumab vedotin-ejfv (Padcev) on December 18, 2019, bringing the total number of novel antibody therapeutics granted a first approval in either the US or EU during 2019 to 6; 2) the European Commission approved romosozumab on December 9, 2019; 3) the European Medicines Agency issued a positive opinion for brolucizumab; 4) Sesen Bio initiated a rolling biologics license application (BLA) on December 6, 2019; 5) GlaxoSmithKline submitted a BLA for belantamab mafodotin; and 6) the status of the Phase 3 study (NCT04128696) of GSK3359609, a humanized IgG4 anti-ICOS antibody, in patients with head and neck squamous cell carcinoma was updated to recruiting from not yet recruiting.
KEYWORDS: Antibody therapeutics, Food and Drug Administration, European Medicines Agency, cancer, immune-mediated disorders, monoclonal antibodies
Introduction
The development of prophylactic and therapeutic agents for infections or disease is a slow, costly process that requires substantial knowledge and expertise in a wide variety of areas, including relevant biological pathways, creation and characterization of drug molecules, manufacturing, clinical studies, and regulatory affairs. It is critical, however, that this knowledge and expertise expands and improves over time, incorporating new techniques and approaches as they become available, as new challenges are presented.
Over the past decade, the ‘Antibodies to Watch’ article series has documented the results of the global biopharmaceutical industry’s efforts to bring innovative antibody therapeutics to patients in need.1–12 Each annual paper has reported the number of unique antibodies in late-stage clinical studies, in regulatory review and approved for marketing as of November or early December of the year in which it was written. In particular, we have focused on first transitions of novel monoclonal antibodies (mAbs), (e.g., first approval in either the United States (US) or European Union (EU)) as a measure of innovation and success. This approach is stringent, as it necessarily excludes any subsequent development from the quantitative assessment. For example, if a product is first approved in the US, any subsequent approval in the EU or supplemental approval is not included in the product totals. By this measure, the number of antibody therapeutic products approved in 2019, as of November, (5 products) is the lowest since 2013, when only 2 products were approved. However, when looking at all US or EU first approvals, the data shows the biopharmaceutical industry has in fact made substantial advances in developing antibody therapeutics during 2010–19, nearly tripling the number of antibody therapeutics on the market (Figure 1).
Figure 1.
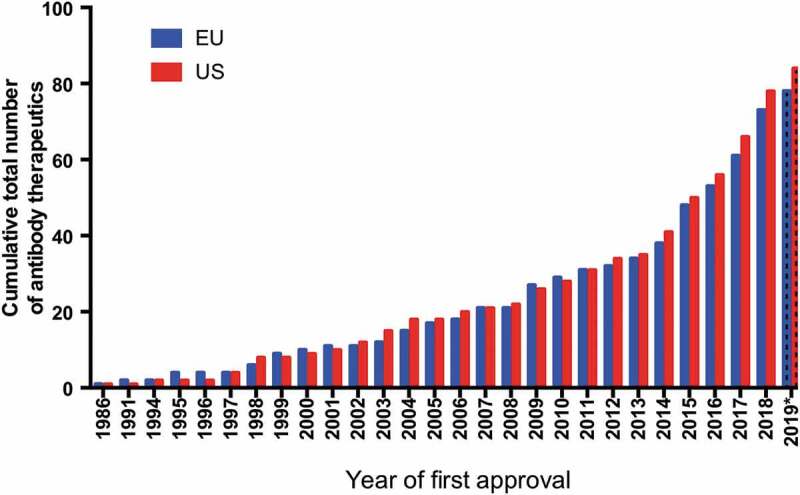
Cumulative number of antibody therapeutics first approved in the US or EU, 1986–2019.
*Data available as of November 28, 2019. Biosimilar antibody and Fc fusion protein products were excluded. A table of US- and EU-approved antibody therapeutics is available at https://www.antibodysociety.org/resources/approved-antibodies/.
Looking toward the future, our data suggest that the relatively low number of 2019 first approvals was anomalous, and likely due to a transient decrease in the rate of application submissions during late 2018 and early 2019. Overall, our projections suggest that the number of first approvals granted in the 2020s may exceed that of the 2010s. Based on the number of antibody therapeutics currently in regulatory review in either the US or EU, we anticipate that at least 13 products will be granted first approvals in 2020. The scale of approvals later in the decade depends entirely on the clinical pipeline of these products. We report here that the late-stage clinical pipeline is robust, and we anticipate that more antibody therapeutics will be in late-stage studies in 2020 than any year we previously documented (Figure 2). Remarkably, compared to 2010, the number of antibody therapeutics currently in late-stage studies has more than tripled (to 79). The early-stage commercial clinical pipeline is also robust, with at least 550 novel antibody therapeutics at Phase 1, 1/2 or Phase 2 as their most advanced stage of clinical studies (data not shown).
Figure 2.
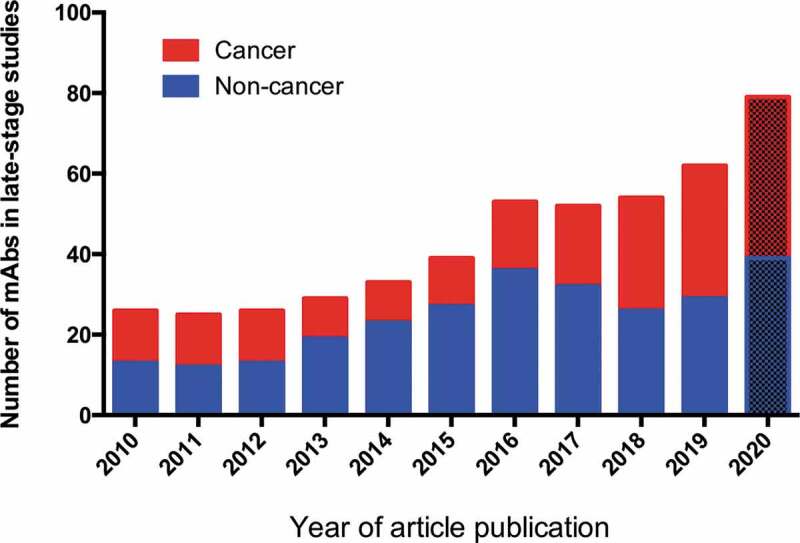
Antibody therapeutics in late-stage clinical studies.
2020 values based on data publicly available as of December 5, 2019; other data from ‘Antibodies to watch’ articles published in mAbs starting in 2010. “Late-stage” clinical studies include pivotal or registrational Phase 2, Phase 2/3 or Phase 3 studies. Biosimilar antibody and Fc fusion protein products were excluded. A table of antibodies in late-stage studies sponsored by commercial firms is maintained by The Antibody Society.
In keeping with our previous reports, ‘Antibodies to watch in 2020ʹ includes updates on recent and anticipated events relevant to antibody therapeutics in clinical development. Data for antibody therapeutics that were first approved in either the US or EU during 2019, as well as several products first approved in Russia or India, are provided. Antibody therapeutics undergoing regulatory review by the Food and Drug Administration (FDA) or the European Medicines Agency (EMA) as of November 2019 are also discussed. Finally, we provide brief summaries of antibody therapeutics in late-stage clinical study that may progress to regulatory review in late 2019 or 2020, based on public disclosures by the sponsoring companies. Due to the substantial amount of data available for these antibody therapeutics, we have focused on the indications of late-stage clinical studies and include references for recent information only.
Antibody therapeutics granted a first approval in the US or EU in 2019
As of November 2019, a total of 5 novel antibody therapeutics (romosozumab, risankizumab, polatuzumab vedotin, brolucizumab, crizanlizumab) had been granted a first approval in either the US or EU (Table 1). On a per year basis, this is the lowest number of approvals since 2013, when only 2 antibody therapeutics were approved in these two regions. In particular, it is substantially lower than the number of first US or EU approvals granted in 2018 (13 products; 12 first approved in the US, and 1 first approved (caplacizumab) in the EU).1 All 5 products first approved in 2019 (as of November) were granted approvals by FDA; risankizumab was also approved in the EU. Documents relating to FDA review and approval of these products can be found by searching drugs@fda using the international nonproprietary name of the mAb. As of November 2019, FDA had approved a total of 6 mAb therapeutics, namely the 5 noted above as well as caplacizumab-yhdp (Cablivi),13 which was approved by FDA on February 6, 2019 after being granted a first approval in the EU on August 31, 2018.14
Table 1.
Antibody therapeutics granted first approvals in the European Union or the United States during 2019*.
| International non- proprietary name | Brand name | Target; Format | Indication first approved | Date of first EU approval | Date of first US approval |
|---|---|---|---|---|---|
| Romosozumab | Evenity | Sclerostin; Humanized IgG2 | Osteoporosis in postmenopausal women at increased risk of fracture | EC decision pending | 4/9/2019 |
| Risankizumab | Skyrizi | IL-23 p19; Humanized IgG1 | Plaque psoriasis | 4/26/2019 | 4/23/2019 |
| Polatuzumab vedotin | Polivy | CD79b; Humanized IgG1 ADC | Diffuse large B-cell lymphoma | EC decision pending | 6/10/2019 |
| Brolucizumab | BEOVU | VEGF; Humanized scFv | Neovascular age-related macular degeneration | In review | 10/7/2019 |
| Crizanlizumab | Adakveo | CD62 (aka P-selectin); Humanized IgG2 | Sickle cell disease | In review | 11/15/2019 |
*Data available as of November 28, 2019.
Table notes: Romosozumab first approved in Japan on January 8, 2019; risankizumab was first approved in Japan on March 26, 2019.
International nonproprietary names for products approved in the US are: brolucizumab-dbll, crizanlizumab-tmca, polatuzumab vedotin-pllq, risankizumab-rzaa, romosozumab-aqqg.
Romosozumab (Amgen, Inc., UCB, Amgen Astellas Biopharma K.K.)
Romosozumab (AMG785, EVENITY™) is an IgG2 humanized monoclonal antibody that binds and inhibits sclerostin, which is secreted by osteocytes, leading to transient stimulation of bone formation and inhibition of bone resorption. Romosozumab was first approved in Japan on January 8, 2019 for the treatment of osteoporosis in patients at high risk of fracture.15 Amgen and UCB are co-developing EVENITY worldwide, with development in Japan being led by Amgen Astellas BioPharma K.K., a joint venture between Amgen and Astellas Pharma Inc.
On April 9, 2019, the FDA approved romosozumab-aqqg for treatment of osteoporosis in postmenopausal women at high risk for fracture, defined as a history of osteoporotic fracture, or multiple risk factors for fracture; or patients who have failed or are intolerant to other available osteoporosis therapy. The safety and efficacy of Evenity were demonstrated in two Phase 3 studies, FRAME (NCT01575834) and ARCH (NCT01631214).16 In the FRAME study, 1 year of treatment with Evenity (210 mg) lowered the risk of a new vertebral fracture by 73% compared to placebo. This benefit was maintained over the second year of the trial when Evenity was followed by 1 year of denosumab compared to placebo followed by denosumab. In the ARCH study, 1 year of treatment with Evenity followed by 1 year of alendronate reduced the risk of a new vertebral fracture by 50% compared to two years of alendronate alone. More than 11,000 women with postmenopausal osteoporosis participated in the 2 studies.
Showing a difference of opinion with FDA, the EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended the refusal of the marketing authorization for Evenity in the EU because their evaluation indicated that the benefits of Evenity did not outweigh the risk of serious effects on the heart or circulatory system. EMA issued its opinion on June 27, 2019, but the applicant, UCB Pharma S.A., requested a reexamination of the opinion. On October 18, 2019, UCB and Amgen announced that, after reexamination, CHMP adopted a positive opinion recommending marketing authorization for EVENITY® for the treatment of severe osteoporosis in postmenopausal women at high risk of fracture and with no history of myocardial infarction or stroke.17 The European Commission’s decision, which will be based on CHMP’s recommendation, is expected by the end of 2019.
Risankizumab (Boehringer Ingelheim, AbbVie)
Risankizumab (risankizumab-rzaa, SKYRIZI™) is a humanized IgG1 monoclonal antibody that inhibits interleukin (IL)-23, a cytokine involved in inflammatory processes, by binding to its p19 subunit. Risankizumab was first approved in Japan on March 26, 2019 for the treatment of plaque psoriasis, generalized pustular psoriasis, erythrodermic psoriasis and psoriatic arthritis in adult patients who have an inadequate response to conventional therapies. By the end of April 2019, the product had been granted approvals in Canada, the US and the EU. On April 23, 2019, FDA approved SKYRIZI™ for the treatment of moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.18 SKYRIZI is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization of SKYRIZI globally.
The product’s approval is supported by data from 4 randomized, placebo and/or active-controlled (ustekinumab or adalimumab) pivotal studies, IMMvent (NCT02694523), IMMhance (NCT02672852), ultIMMA-1 (NCT02684370), and ultIMMa-2 (NCT02684357), that evaluated the safety and efficacy of risankizumab in more than 2,000 patients with moderate-to-severe chronic plaque psoriasis. The co-primary endpoints of the studies were Psoriasis Area and Severity Index and static Physician Global Assessment (sPGA) score of clear or almost clear (sPGA 0/1) at 16 weeks versus placebo. In these 4 studies, all co-primary and ranked secondary outcome measures were met and no new safety signals were observed. Risankizumab showed superior efficacy to placebo, ustekinumab, or adalimumab in the treatment of moderate-to-severe plaque psoriasis in the IMMvent,19 IMMhance,20 UltIMMa-1 and UltIMMa-221 studies.
Polatuzumab vedotin (Genentech, Inc.)
On June 10, 2019, FDA granted accelerated approval to Polivy (polatuzumab vedotin-piiq), in combination with the chemotherapy bendamustine and a rituximab product (BR), to treat adult patients with diffuse large B-cell lymphoma (DLBCL) that has progressed or returned after at least 2 prior therapies.22 Polivy is composed of a humanized anti-CD79b IgG1 antibody conjugated to the antimitotic agent monomethyl auristatin E (MMAE). The antibody’s target is highly expressed on B cells of patients with lymphoma. The biologics license application (BLA) for Polivy was granted FDA’s Breakthrough Therapy and priority review designations. Polivy was designated an Orphan Drug for the approved indication.
The drug also has EMA’s Priority Medicines (PRIME) designation, and EU Orphan Drug designations for DLBCL. On November 14, 2019, EMA’s CHMP adopted a positive opinion, and recommended granting a conditional marketing authorization for Polivy (polatuzumab vedotin) for the treatment of relapsed/refractory diffuse large B-cell lymphoma, a rare type of cancer of the white blood cells. The European Commission’s decision, which will be based on CHMP’s recommendation, will normally be issued 67 days from adoption of the opinion.
The drug’s efficacy was evaluated in a study of 80 patients with relapsed or refractory DLBCL who were randomized to receive Polivy with BR or BR alone. Efficacy was based on complete response rate and duration of response (DOR), defined as the time the disease stays in remission. At the end of treatment, the complete response rate was 40% with Polivy plus BR compared to 18% with BR alone. Of the 25 patients who achieved a partial or complete response to Polivy plus BR, 16 (64%) had a DOR of at least 6 months and 12 (48%) had a DOR of at least 12 months.22
Brolucizumab (Novartis)
On October 7, 2019, FDA approved brolucizumab-dbll (BEOVU®) for the treatment of neovascular age-related macular degeneration (nAMD).23 Brolucizumab is a humanized antibody single-chain variable fragment (scFv) that binds to the 3 major isoforms of human vascular endothelial growth factor (VEGF), thereby interfering with their interaction with receptors VEGFR-1 and VEGFR-2 and suppressing endothelial cell proliferation, neovascularization and vascular permeability. BEOVU is administered by intravitreal injection, and the recommended dose is 6 mg monthly for the first 3 doses, followed by 1 dose of 6 mg every 8–12 weeks. A marketing application for brolucizumab is undergoing review by EMA.
FDA’s approval was supported by data from two Phase 3 studies, HAWK (NCT02307682) and HARRIER (NCT02434328),24 comparing the efficacy and safety of intravitreal injections of brolucizumab versus aflibercept in nAMD. Brolucizumab met the primary efficacy endpoint of non-inferiority to aflibercept (EYLEA®) in mean change in best-corrected visual acuity (BCVA) at Week 48 in both trials, with a mean change in BCVA of 6.6 letters for brolucizumab 6 mg versus 6.8 letters for aflibercept in the HAWK trial and 6.9 letters versus 7.6 letters, respectively, in the HARRIER study. Additionally, at Week 48, brolucizumab was superior to aflibercept in secondary endpoints considered key parameters of the disease, such as central subfield retinal thickness and retinal fluid (intraretinal fluid or subretinal fluid). Results at 96 weeks reaffirmed the superiority of brolucizumab 6 mg in reduction of retinal fluid, and patients who received this dose continued to demonstrate reductions in central subfield thickness.25
Crizanlizumab (Novartis)
FDA approved crizanlizumab (Adakveo, crizanlizumab-tmca) on November 15, 2019 as a treatment to reduce the frequency of vaso-occlusive crisis (VOC), which occurs when blood circulation is obstructed by sickled red blood cells, for patients that are 16 years and older.26 Also known as SEG101, crizanlizumab is a humanized antibody directed against P-selectin, which contributes to the pathogenesis of sickle cell disease, including vaso-occlusive events and hemolytic anemia. Crizanlizumab was granted Orphan Drug designation in the US and EU for the treatment of VOC in patients with sickle cell disease, as well as FDA’s Breakthrough Therapy designation for prevention of VOCs in patients of all genotypes with sickle cell disease. A marketing application for crizanlizumab is undergoing review by EMA.
FDA’s approval was based on Phase 2 results from the SUSTAIN study (NCT01895361), which demonstrated that crizanlizumab provided significant benefit over placebo, such as: 1) the percentage of crizanlizumab-treated patients (5 mg/kg) who did not experience any vaso-occlusive crisis (VOC) was higher compared to those treated with placebo (36% vs 17%, P = .010); 2) 45% reduction in the median annual rate of VOCs leading to health care visits in patients with or without hydroxyurea therapy compared to placebo (1.63 vs 2.98, P = .010); 3) 42% reduction in median annual rate of days hospitalized versus placebo (4.00 vs 6.87 P = .45), and 4) a 3-fold longer median time to first VOC vs placebo (4.07 vs 1.38 months, P < .001).27,28
Antibody therapeutics approved outside the US or EU in 2019
Most antibody therapeutics developed by major biopharmaceutical firms are first approved in either the US or EU. However, smaller firms may seek first approvals elsewhere, especially if the firm’s headquarters is located in a region other than the US or EU. In 2019, 1 antibody therapeutic was granted a first approval in Russia (netakimab) and 1 (Rabimab) was granted a first approval in India.
Netakimab (BIOCAD)
On May 7, 2019, BIOCAD announced the registration of netakimab (Efleira®, BCD-085) in Russia for the treatment of moderate-to-severe plaque psoriasis.29 Netakimab is a humanized IgG1 in which the VH domain is replaced by a llama VHH domain possessing a long complementarity-determining region (CDR-H3).30 The mAb targets IL-17, a pro-inflammatory cytokine that plays a critical role in the pathogenesis of psoriasis. The registration is the first for an innovative mAb developed in Russia. BIOCAD has indicated that they will seek approval for netakimab in the EU.
The efficacy and safety of Efleira® in psoriasis patients was confirmed in the Phase 3 BCD-085-7/PLANETA study (NCT03390101), which was conducted in 22 study sites in Russia and 2 study sites in the Republic of Belarus. After 12 weeks of the treatment, 83.3% of patients who received netakimab once a month after induction for the first 3 weeks achieved a 75% improvement in Psoriasis Area and Severity Index. The total duration of therapy and follow-up in this study is 3 years. BIOCAD, which is based in Moscow, is also evaluating netakimab in Phase 3 studies of patients with psoriatic arthritis (NCT03598751) and ankylosing spondylitis (NCT03447704).
Rabimab (Zydus Cadila)
On September 3, 2019, Zydus announced that it received marketing authorization for TwinrabTM (RabiMabs) from the Drug Controller General of India.31 The product, which is composed of an equipotent mixture of 2 murine monoclonal antibodies that bind to 2 different epitopes on the G protein expressed on the surface of rabies virus, is indicated in combination with rabies vaccine for rabies post-exposure prophylaxis. Antibodies M777-16-3 (IgG1) and 62-71-3 (IgG2b) bind to site II and site III, respectively, on the G protein of rabies virus envelope.32 The FDA granted Orphan
Drug designation to this candidate in May 2019.
Antibody therapeutics undergoing first regulatory review in the US or EU
According to the information publicly available as of November 2019, marketing applications (BLA or Marketing Authorization Application (MAA)) for a total of 13 novel antibody therapeutics are undergoing review in either the US or EU (Table 2). The applications are the first submitted for: eptinezumab, teprotumumab, enfortumab vedotin, isatuximab, [fam-] trastuzumab deruxtecan, inebilizumab, leronlimab, sacituzumab govitecan, satralizumab, narsoplimab, tafasitamab, REGNEB3 and naxitamab. A brief summary of each antibody and the clinical studies relevant to the marketing applications are below. The summaries are listed in the chronological order in which the approvals may occur, as predicted by the Prescription Drug User Fee Act (PDUFA) date or date of application submission.
Table 2.
Investigational antibody therapeutics in regulatory review in the European Union or the United States*.
| International non- proprietary name | Target; Format | Indication under review | FDA or EMA designations for indication under review | Review status in EU | Review status in US; PDUFA date |
|---|---|---|---|---|---|
| Eptinezumab | CGRP; Humanized IgG1 | Migraine prevention | NA | NA | In review; 2/21/2020 |
| Teprotumumab | IGF-1R; Human IgG1 | Thyroid eye disease | BT, FT, US Orphan Drug | NA | Priority review; 3/8/2020 |
| Enfortumab vedotin | Nectin-4; Human IgG1 ADC | Urothelial cancer | BT | NA | Priority review; 3/15/2020 |
| Isatuximab | CD38; Humanized IgG1 | Multiple myeloma | EU/US Orphan Drug | In review | In review; 4/30/2020 |
| [fam-]trastuzumab deruxtecan | HER2; Humanized IgG1 ADC | HER2+ metastatic breast cancer | BT, FT | NA | Priority review; Q2-2020 |
| Inebilizumab | CD19; Humanized IgG1 | Neuromyelitis optica and neuromyelitis optica spectrum disorders | BT, EU/US Orphan Drug | NA | In review |
| Leronlimab | CCR5; Humanized IgG4 | HIV infection | FT | NA | In review |
| Sacituzumab govitecan | TROP-2; Humanized IgG1 ADC | Triple-neg. breast cancer | BT, FT | NA | Priority review |
| Satralizumab | IL-6R; Humanized IgG2 | Neuromyelitis optica spectrum disorders | BT, EU/US Orphan Drug | In review, Accelerated assessment | In review |
| Narsoplimab | MASP-2, Human IgG4 | Hematopoietic stem cell transplant-associated thrombotic microangiopathies | BT, EU/US Orphan Drug | NA | In review |
| Tafasitamab | CD19; Humanized IgG1 | Diffuse large B-cell lymphoma | BT, FT, EU/US Orphan Drug | NA | In review |
| REGNEB3 | Ebola virus; Human IgG1 mixture (3 mAbs) | Ebola virus infection | BT, EU/US Orphan Drug | NA | In review |
| Naxitamab | GD2; Humanized IgG1 | High-risk neuroblastoma and refractory osteomedullary disease | BT, Rare Pediatric Drug, EU/US Orphan Drug | NA | In review |
*Data available as of November 28, 2019. Abbreviations: ADC, antibody-drug conjugate; BT, FDA Breakthrough Therapy designation; EMA, European Medicines Agency; EU, European Union; FDA, Food and Drug Administration; FT, FDA Fast Track designation; NA, Not applicable; PDUFA, Prescription Drug User Fee Act; US, United States.
Eptinezumab (Alder BioPharmaceuticals, Inc.)
Like the marketed mAbs fremanezumab and galcanezumab, eptinezumab targets calcitonin gene-related peptide and has been developed for patients with migraine. FDA has accepted eptinezumab’s BLA, which has a PDUFA target action date of February 21, 2020.33 The BLA is supported by positive results from the PROMISE 1 (NCT02559895) and PROMISE 2 (NCT02974153) Phase 3 clinical trials, which investigated eptinezumab for episodic and chronic migraine prevention, respectively. Data from the 2 studies presented at the American Headache Society’s 61st Annual Scientific Meeting in Philadelphia, PA (July 11–14, 2019) show 18.1% of episodic migraine patients treated with 100 mg of eptinezumab experienced no migraine days for at least half of the study period (≥ 6 months), compared with 12.6% of placebo-treated patients, and 14.0% of chronic migraine patients treated with 100 mg of eptinezumab experienced no migraine days for at least half of the study period (≥ 3 months), compared with 4.9% of placebo-treated patients.33 Alder BioPharmaceuticals, Inc. anticipates a commercial launch by early 2020. The company initiated a Phase 3 clinical trial (NCT04152083) evaluating eptinezumab as a treatment for acute migraine in patients who are candidates for prevention therapy in November 2019.
Teprotumumab (Horizon Pharma Plc)
Teprotumumab (HZN-001), a human mAb targeting insulin growth factor 1 receptor, was developed as a treatment for active thyroid eye disease (TED) leading to proptosis (eye bulging), strabismus and diplopia and blindness in some cases. Teprotumumab was granted Fast Track, Breakthrough Therapy and Orphan Drug designations for Graves’ orbitopathy by the FDA.34 Based on positive data from both Phase 2 (NCT01868997)35 and Phase 3 (NCT03298867) studies, Horizon Pharma submitted a BLA to the FDA for teprotumumab as a treatment for TED. The BLA was granted priority review, and FDA’s PDUFA target action date is March 8, 2020.36 FDA will hold a public advisory committee meeting of the Dermatologic and Ophthalmic Drugs Advisory Committee on December 13, 2019 to discuss the BLA for teprotumumab.
The efficacy, tolerability, and safety of teprotumumab were studied in the randomized, placebo-controlled, OPTIC Phase 3 study (NCT03298867). Teprotumumab met the study’s primary endpoint, which was a responder rate of ≥ 2 mm reduction of proptosis in the study eye (without deterioration in the fellow eye) at Week 24. Data from the OPTIC study showed that 82.9% of patients receiving teprotumumab were proptosis responders compared to 9.5% of patients receiving placebo at Week 24 (p < .001). All secondary endpoints were also met, including: 1) overall responder rate at Week 24 defined by the percent of patients with ≥2 point reduction in Clinical Activity Score (CAS) and ≥2 mm reduction in proptosis from baseline, provided there is no corresponding deterioration (≥2-point/mm increase) in CAS or proptosis in the fellow eye; 2) percent of patients with a CAS value of 0 or 1 at Week 24 in the study eye; 3) percent of patients with a change from baseline of at least one grade in diplopia; 4) mean change in proptosis measurement from baseline to Week 24 in the study eye; and 5) mean change in Graves’ Ophthalmopathy Quality of Life from baseline to Week 24.34
Enfortumab vedotin (Seattle Genetics, Inc., Astellas Pharma Inc.)
Enfortumab vedotin (ASG22ME) is an antibody-drug conjugate (ADC) composed of an antibody targeting Nectin-4, which is a target highly expressed in urothelial cancer, conjugated to monomethyl auristatin E (MMAE). Enfortumab vedotin was granted FDA’s Breakthrough Therapy designation for locally advanced or metastatic urothelial cancer in March 2018. Seattle Genetics and Astellas Pharma Inc. have submitted a BLA for enfortumab vedotin as monotherapy treatment for patients with locally advanced or metastatic urothelial cancer who have received a programmed cell death-1 (PD-1)/ligand (PD-L1) inhibitor and who have received a platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting.37 The BLA was granted a priority review and has a target action date of March 15, 2020.
In March 2019, Seattle Genetics and Astellas Pharma Inc. announced positive results from the pivotal, open-label, multi-center single-arm, 2 cohort Phase 2 EV-201 study (NCT03219333), which evaluated the efficacy and safety of enfortumab vedotin in patients previously treated with immune checkpoint therapy (ICP) and platinum chemotherapy (cohort 1) and in patients treated with ICP and no prior chemotherapy (cohort 2). Top-line results from cohort 1 demonstrated that the confirmed objective response rate (ORR) was 42% (95% CI: 33.6%–51.6%), with 9% complete response.38 Enfortumab vedotin is also undergoing evaluation in the Phase 3 confirmatory EV-301 trial (NCT03474107) in the same indication to support global US registration. This study has a primary completion date, i.e., the date that the final participant was examined or received an intervention for the purposes of final collection of data for the primary outcome, in September 2021.
Isatuximab (Sanofi)
Developed by Sanofi, isatuximab (SAR650984) is a chimeric IgG1 antibody directed against CD38 expressed on malignant plasma cells. The antibody acts through a combination of mechanisms, which may depend on the expression level of the target.39 EMA has accepted for review the MAA for isatuximab for the treatment of relapsed/refractory multiple myeloma (RRMM).40 FDA has accepted a BLA for isatuximab as a treatment for patients with RRMM; the target action date for the FDA’s decision is April 30, 2020.41 Both FDA and EMA granted isatuximab Orphan Drug designation for RRMM.
The BLA is supported by data from the Phase 3 ICARIA-MM study (NCT02990338) demonstrating a statistically significant improvement in progression-free survival (PFS) (HR 0.596, 95% CI 0.44–0.81, p = .001); the median PFS was longer in patients treated with isatuximab in combination with pomalidomide/dexamethasone (11.53 months, 95% CI: 8.936 to 13.897) compared to those treated with pomalidomide/dexamethasone alone (6.47 months, 95% CI: 4.468 to 8.279).40
[fam-] Trastuzumab deruxtecan (AstraZeneca, Daiichi Sankyo Company, Limited)
[fam-] Trastuzumab deruxtecan (DS-8201a) is an ADC composed of an anti-human epidermal growth factor receptor 2 (HER2) humanized antibody fused to a potent topoisomerase I inhibitor evaluated as a treatment of cancer, including HER2-positive metastatic breast cancer. In October 2019, AstraZeneca and Daiichi Sankyo announced that the FDA accepted a BLA for trastuzumab deruxtecan for HER2-positive metastatic breast cancer and granted the application priority review. The companies have indicated that FDA’s first action date is set for the second quarter of 2020,42 which suggests an approval is possible as early as April 2020. The FDA previously granted this ADC Breakthrough Therapy and Fast Track Designations for HER2-positive patients in the advanced or refractory breast cancer setting.43 In September 2019, Daiichi Sankyo announced the submission of a New Drug Application (NDA) to Japan’s Ministry of Health, Labor and Welfare (MHLW) based on results of the pivotal Phase 2 DESTINY-Breast 01 study (NCT03248492).44
The safety and efficacy of trastuzumab deruxtecan is being evaluated in the 2-part pivotal Phase 2 DESTINY-Breast 01 trial in patients with HER2-positive unresectable and/or metastatic breast cancer previously treated with trastuzumab emtansine (KADCYLA®). The aim of the first part of the study was to identify the recommended dose of trastuzumab deruxtecan, which is 5.4 mg/kg.43 The second part enrolled patients into 2 cohorts: 1) patients resistant or refractory to trastuzumab emtansine (part 2a); and 2) patients who discontinued treatment with trastuzumab emtansine for reasons other than resistant or refractory disease (part 2b). ORR was the primary endpoint of the study. Secondary objectives included duration of response, disease control rate, clinical benefit rate, PFS and OS. In May 2019, AstraZeneca and Daiichi Sankyo Company announced the DESTINY-Breast01 trial met its primary endpoint and the response rate confirmed clinical activity observed in a Phase 1 trial (NCT02564900).43
The Phase 1 study included patients with HER2-positive metastatic breast cancer previously treated with trastuzumab emtansine received trastuzumab deruxtecan at a recommended expansion dose of 5.4 or 6.4 mg/kg in the dose escalation or dose expansion parts of the Phase 1 trial. In these patients, a confirmed ORR of 59.5% (95% CI: 49.7, 68.7) and a disease control rate of 93.7% (95% CI: 87.4, 97.4) were observed with trastuzumab deruxtecan. The median duration of response was 20.7 months (0.0 to 21.8), median PFS was 22.1 months (0.8 to 27.9), and median overall survival (OS) had not yet been reached.45
Trastuzumab deruxtecan is also being evaluated as a treatment for other types of cancers, including gastric cancer (Phase 2 NCT04014075 study) and non-small cell lung cancer (Phase 2 NCT03505710 study). The drug has received MHLW’s SAKIGAKE designation for the treatment of HER2-positive advanced gastric or gastro-esophageal junction cancer.45
Inebilizumab (Viela Bio)
Inebilizumab (MEDI-551) is a humanized anti-CD19 monoclonal antibody developed by Viela Bio for the treatment of neuromyelitis optica spectrum disorder (NMOSD). Most patients with this rare disorder have autoantibodies to aquaproin-4 (AQP4), which lead to recurrent attacks of optic neuritis or myelitis. By targeting CD19, inebilizumab depletes CD19 + B cells and plasmablasts responsible for the production of autoantibodies directed against AQP4. In August 2019, Viela Bio announced that the FDA accepted for review a BLA for inebilizumab for NMOSD.46 Inebilizumab was granted FDA’s Breakthrough Therapy designation for the treatment of NMOSD, as well as Orphan Drug designation by the FDA and the EMA.
The BLA includes safety and efficacy results from the Phase 2/3 N-MOmentum trial (NCT02200770). This study enrolled 230 patients with and without the AQP4-IgG antibody who were randomized 3:1 to receive either 2 doses of 300 mg of inebilizumab as a monotherapy or placebo at Day 1 and Day 15, and then followed for a total of 28 weeks. The primary endpoint (time to first attack) and a majority of the secondary endpoints of the study were met. Results demonstrated that inebilizumab reduced the risk of developing an NMOSD attack by 77% compared to placebo in AQP4-IgG seropositive patients after 28 weeks of treatment.46 Of 174 patients who received inebilizumab, 12% had an attack versus 39% of 56 patients who received placebo (hazard ratio 0 · 272 [95% CI 0 · 150–0 · 496]; p < 0 · 0001). Inebilizumab demonstrated a favorable safety and tolerability profile, with an adverse event rate similar to placebo.47
Leronlimab (CytoDyn Inc.)
Leronlimab (PRO140) is a humanized IgG4 antibody directed against CC chemokine receptor 5 (CCR5), which is involved in many pathophysiological processes, such as HIV-1 entry into CD4 + T cells, promotion of tumor invasion and metastases, pathogenesis of nonalcoholic steatohepatitis (NASH) and development of acute graft-versus-host disease (GvHD). Leronlimab was granted FDA’s Fast Track designation, which allows CytoDyn Inc. to submit individual sections of the BLA for review, i.e., a ‘rolling’ review. On March 19, 2019, the company announced the submission of the first of 3 sections (non-clinical data) of the BLA for leronlimab in combination with highly active antiretroviral therapy for the treatment of HIV+ patients.48 The company expects to submit the remaining sections of the BLA by the end of 2019.49 Leronlimab is also being evaluated as monotherapy in HIV. In a dose-escalation study, 229, 201, and 134 patients with HIV received 350 mg, 525 mg, or 700 mg of leronlimab once a week, respectively. The rate of viral load suppression post-10 weeks of monotherapy was 68%, 94%, and 85% with 350 mg, 525 mg, and 700 mg, respectively, and ~ 150 patients exhibited sustained viral suppression at ~ 1 year of monotherapy.50
In addition to HIV, CytoDyn is evaluating the safety and efficacy of leronlimab in Phase 1/2 or Phase 2 clinical trials of patients with metastatic colorectal cancer (mCRC), NASH, metastatic triple-negative breast cancer (TNBC) and acute GvHD. Leronlimab has been granted Fast Track designation for use in combination with carboplatin for the treatment of CCR5+ metastatic TNBC patients, and FDA granted Orphan Drug designation to leronlimab for the prevention of GvHD.50
Sacituzumab govitecan (Immunomedics, Inc.)
Sacituzumab govitecan is an ADC comprising SN38 (active metabolite of the topoisomerase I inhibitor irinotecan) conjugated to an anti-Trop 2 antibody.51 Trop-2 is highly expressed on most epithelial cancers, including TNBC. Immunomedics, Inc. is seeking an accelerated approval based on Phase 1/2 data of sacituzumab govitecan in TNBC. FDA granted sacituzumab govitecan Breakthrough Therapy and Fast Track designations for this indication. In July 2018, the FDA accepted the BLA for sacituzumab govitecan as a treatment for patients with metastatic TNBC who have received at least 2 prior therapies and granted it priority review. In January 2019, the FDA issued a complete response letter detailing issues related to approvability, which were exclusively focused on Chemistry, Manufacturing and Control matters.52 In December 2019, Immunomedics announced the BLA had been resubmitted.53 In April 2019, Immunomedics announced an exclusive license agreement with Everest Medicines to develop, register, and commercialize sacituzumab govitecan in Greater China, South Korea and certain Southeast Asian countries.54
The safety and efficacy of sacituzumab govitecan are being evaluated in the Phase 3 ASCENT (NCT02574455) and TROPICS-02 (NCT03901339) studies in metastatic TNBC and hormone receptor-positive/HER2-negative metastatic breast cancer, respectively. Top-line data from the ASCENT study is expected to be available in mid-2020.53 Sacituzumab govitecan is also being assessed in early-stage clinical studies of patients with solid tumors, including urothelial carcinoma, epithelial cancers, glioblastoma, and non-small cell and small cell lung cancers.
Satralizumab (Chugai Pharmaceutical Co., Ltd)
Satralizumab, a humanized antibody that binds IL-6 receptor in a pH-dependent manner, is undergoing review by EMA.55 The marketing application is being reviewed under EMA’s accelerated assessment program. The drug is expected to suppress relapse of NMOSD, which is a rare, lifelong, and debilitating autoimmune disease characterized by inflammatory lesions in the optic nerves and spinal cord. NMOSD patients frequently experience relapsing disease that leads to accumulating neurological damage and disability. Satralizumab was granted Orphan Drug designations by FDA, EMA and Japan’s MHLW. FDA also granted satralizumab Breakthrough Therapy Designation for the treatment of NMOSD.56
In two Phase 3 studies in NMOSD patients, the primary endpoint was achieved with satralizumab either as an add-on therapy to baseline treatment (SAkuraSky study, NCT02028884) or as monotherapy (SAkuraStar study, NCT02073279). In the SAkuraSky study, the risk of relapse was lower in patients who received satralizumab added to immunosuppressant treatment compared to those who received placebo.57 The largest effect was observed among AQP4-IgG–seropositive patients (n = 55); relapse occurred in 11% of patients who received satralizumab group versus 43% of those in the placebo group (hazard ratio, 0.21; 95% CI, 0.06 to 0.75). In the SAkuraStar study, NMOSD patients were subcutaneously administered satralizumab (120 mg) or placebo at Week 0, 2, and 4, and subsequent treatment was continued at 4-week intervals. As reported by Chugai in September 2019,58 the SAkuraStar study showed that satralizumab significantly reduced the risk of relapse by 55% (hazard ratio = 0.45 [95% confidence interval: 0.23–0.89], p = .0184 [stratified log-rank test]) in the overall population, representative of NMOSD patients (including AQP4-IgG seropositive and seronegative patients). Satralizumab showed a favorable safety profile during the study.
Narsoplimab (Omeros Corporation)
Narsoplimab (OMS721) is a human IgG4 antibody targeting mannan-binding lectin-associated serine protease-2 (MASP-2), a pro-inflammatory protein critical to activation of the lectin complement pathway of the immune system. MASP-2 is associated with complement-mediated diseases, such as thrombotic microangiopathies (TMAs), which includes hematopoietic stem cell transplant (HSCT)-related TMA and atypical hemolytic uremic syndrome (aHUS). Narsoplimab is designed to prevent complement-mediated inflammation and endothelial damage associated with these diseases while not interfering with functions of the other pathways of innate immunity.59 FDA granted narsoplimab Breakthrough Therapy and Orphan Drug designations for HSCT-TMA. Omeros initiated submission of a rolling BLA for narsoplimab for treatment of HSCT-TMA in October 2019.60 Omeros is also preparing an MAA requesting approval for this same indication in the EU, where narsoplimab has Orphan Drug designation.
Narsoplimab is being evaluated in a Phase 2, uncontrolled, 3-stage, dose-escalation cohort study (NCT02222545) in patients with 3 forms of TMA: aHUS, thrombotic thrombocytopenia (TTP), and HSCT-associated TMA. In the study’s first stage, narsoplimab was administered to escalating dose (low, medium and high) cohorts of 3 patients per cohort to identify the optimal dosing regimen. In the second stage, the dose selected in the first stage is administered to expanded cohorts of 40 subjects per cohort with distinct etiologies (aHUS alone in one cohort and TTP or HSCT-TMA in the other cohort). Patients completing the second stage may be eligible for continued treatment in the third stage if the investigator believes the subject is at risk for relapse of TMA, narsoplimab treatment was tolerated, and the patient has no conditions that increase the risk of narsoplimab treatment. Preliminary results indicated that narsoplimab significantly improved 100-day survival compared to a historical control (53% vs 10%) in high-risk HSCT-TMA patients.61
Narsoplimab is also being evaluated in Phase 3 clinical programs for the treatment of aHUS and immunoglobulin A (IgA) nephropathy. FDA has granted narsoplimab Fast Track designation for aHUS and Breakthrough Therapy designation for IgA nephropathy, and FDA and EMA have granted narsoplimab Orphan Drug designations for IgA nephropathy.59
Tafasitamab (Morphosys AG)
Also known as MOR208, tafasitamab is an Fc-engineered IgG1 antibody targeting CD19, which is expressed in B cell malignancies such as DLBCL and chronic lymphocytic leukemia (CLL). The development of tafasitamab has focused on the treatment of patients with relapsed of refractory DLBCL. Fc engineering enhances binding of tafasitamab’s Fc domain to activating Fcγ-receptors, which has been shown to enhance cytotoxicity, relative to a native non-engineered anti-CD19 antibody, in vitro.62
Tafasitamab was granted FDA’s Breakthrough Therapy and Fast Track designations, and has been designated an Orphan Drug by both FDA and EMA for the treatment of relapsed/refractory DLBCL. As of October 2019, MorphoSys had initiated a rolling BLA submission that included pivotal clinical study data, and expected the submission to be complete by the end of 2019. MorphoSys has also confirmed their intention to submit an MAA based on the pivotal Phase 2 L-MIND to EMA.63–65
The pivotal, single-arm Phase 2 L-MIND trial (NCT02399085) is evaluating tafasitamab in combination with lenalidomine for the treatment of relapsed/refractory DLBCL. In this study, patients were administered 12 mg/kg tafasitamab as an intravenous Infusion (IV), weekly (Cycle 1–3) to bi-weekly (Cycle 4 onwards), 4 weeks cycles, until disease progression or unacceptable toxicity or discontinuation due to any other reason. In May 2019, MorphoSys announced that the primary endpoint of L-MIND, ORR, had been met. The ORR was 60% (48/80 patients) and the complete response rate was 43% (34/80 patients).65 In October 2019, MorphoSys announced results from an analysis that compared the effectiveness of lenalidomide monotherapy (retrospective observational matched control) with the efficacy outcomes of the tafasitamab/lenalidomide combination, as investigated in the L-MIND trial. The analysis indicated clinical superiority of the combination of tafasitamab and lenalidomide compared to lenalidomide alone.64
Tafasitamab is also being evaluated in the randomized, 2-arm Phase 2/3 B-MIND trial (NCT02763319) in combination with bendamustine versus rituximab + bendamustine for patients with relapsed/refractory DLBCL who are not eligible for high-dose chemotherapy and autologous stem-cell transplantation. In this study, 330 patients received IV tafasitamab at 12 mg/kg + bendamustine or rituximab at 375 mg/m2 + bendamustine. The primary endpoint is PFS. The secondary outcome measures include ORR, duration of response, OS, disease control rate, time to progression, time to next treatment, number of patients with adverse events, quality of life, and number of patients developing anti-drug antibodies. The estimated primary completion date of the study is March 2020.
REGNEB3 (Regeneron)
REGNEB3 is a mixture of 3 human IgG1 mAbs (REGN3470, REGN3471, and REGN3479) that bind separate epitopes on the Ebola virus glycoprotein (GP). The mAbs were derived from transgenic VelocImmune mice immunized with DNA constructs encoding the GP antigen or recombinant purified GP (Makona strain).66 REGNEB3 was granted Breakthrough Therapy designation by FDA, and Regeneron has initiated submission of a rolling BLA.67 REGN-EB3 was also granted Orphan Drug designation from both the FDA and EMA.
REGNEB3 was included in the Phase 2/3 Pamoja Tulinde Maisha (PALM, NCT03719586) study, which was a 4-arm trial evaluating investigational therapies for Ebola virus infection initiated in November 2018 in the Democratic Republic of the Congo. The study initially included 3 test drugs: 1) mAb114, a single monoclonal antibody developed by the National Institute of Allergy and Infectious Diseases; 2) remdesivir (GS-5734), an antiviral drug developed by Gilead Sciences, Inc.; and 3) ZMapp, an antibody mixture developed by Mapp Biopharmaceutical, Inc. ZMapp was included as the standard of care (i.e., control arm) of the study. The trial protocol was amended to include REGNEB3 after the World Health Organization recommended its addition as a fourth treatment arm. In August 2019, the PALM study was terminated early, after preliminary results indicated that patients receiving REGN-EB3 or mAb114 had a greater chance of survival compared to those in the other 2 study arms. At 28 days, death occurred in 33.5% of patients in the REGN-EB3 group compared with 51.3% in the ZMapp subgroup (P = .002), and 35.1% of patients in the MAb114 group compared with 49.7% in the ZMapp group (P = .007).68
Naxitamab (Y-mAbs Therapeutics, Inc.)
Naxitamab (hu3F8) is a humanized IgG1 antibody targeting disialoganglioside GD2, which is expressed in many tumors, such as neuroblastomas, melanoma, bone and soft-tissue sarcomas and small cell lung cancer. Naxitamab was granted Orphan Drug designations in the EU and US for neuroblastoma, as well as Rare Pediatric Drug and Breakthrough Therapy designations by the FDA. On November 29, 2019, Y-mabs Therapeutics, Inc. announced that it initiated a rolling BLA submission for naxitamab for the treatment of patients with relapsed/refractory high-risk neuroblastoma.69 The BLA submission is based on data from the Phase 1/2 Study 12–230 (NCT01757626) and pivotal Phase 2 Study 201 (NCT03363373).
Data from patients with primary refractory high-risk neuroblastoma enrolled in the Phase 1 Study 12–230 were presented at the International Society of Pediatric Oncology Annual Congress held October 23–26, 2019, in Lyon, France.70 Patients with relapsed/refractory high-risk neuroblastoma received combination therapy of naxitamab with granulocyte-macrophage colony stimulating factor (GM-CSF) in this study. A 50% 2-year PFS and 78% ORR were observed in 28 patients refractory to intensive induction therapy. Patients with relapsed neuroblastoma resistant to salvage therapy (30 evaluable patients of 35 total) had a 36% rate of 2-year PFS and an ORR of 37%.
The Phase 2 Study 201 is designed to evaluate naxitamab as a treatment for high-risk neuroblastoma patients with primary refractory disease or incomplete response to salvage treatment in bone and/or bone marrow. In this study, patients receive GM-CSF for 5 days administered at 250 µg/m2/day in advance of the start of naxitamab administration. GM-CSF is thereafter administered at 500 µg/m2 per day on Days 1 to 5. As standard treatment, naxitamab is administered at 3 mg/kg/day on Days 1, 3, and 5, totaling 9 mg/kg per cycle. Treatment cycles are repeated every 4 weeks (±1 week) until complete response or partial response followed by 5 additional cycles every 4 weeks (±1 week). Subsequent cycles are repeated every 8 weeks (±2 weeks) through 101 weeks from first infusion at the discretion of the investigator. Estimated enrollment is 37 patients, and the primary outcome measure is ORR during naxitamab treatment. The estimated primary completion date of the study is December 2020. In July 2019, Y-mAbs Therapeutics, Inc. reported preliminary data showing that 11 evaluable patients had an ORR of 73%, including 55% complete responses.71
Antibodies to watch in 2020: non-cancer indications
According to the information publicly available as of November 2019, 39 antibody therapeutics are undergoing evaluation in late-stage studies for non-cancer indications, with 2 of these (ublituximab, pamrevlumab) also in late-stage studies for cancer indications (Table 3). Companies developing 7 of these novel antibody therapeutics have indicated that they may submit a BLA or MAA in 2020. The summaries below are listed in the chronological order in which the applications may be submitted: tanezumab, aducanumab, evinacumab, etrolizumab, sutimlimab, anifrolumab, and teplizumab.
Table 3.
Investigational monoclonal antibodies in late-stage clinical studies for non-cancer indications.
| Primary sponsoring company | INN or code name | Molecular format | Target(s) | Most advanced phase | Pivotal Phase 2, Phase 2/3 or Phase 3 indications |
|---|---|---|---|---|---|
| Pfizer; Eli Lilly & Company | Tanezumab | Humanized IgG2 | Nerve growth factor | Phase 3 | Pain due to osteoarthritis of knee or hip* (NCT02528188, NCT02697773, NCT02709486); Cancer pain due to bone metastasis (NCT02609828) |
| Biogen Inc., Eisai, Co., Ltd. | Aducanumab | Human IgG1 | Amyloid beta | Phase 3 | Alzheimer’s disease* (NCT02484547, NCT02477800) |
| Regeneron Pharmaceuticals, Inc. | Evinacumab | Human IgG4 | Angiopoietin-like 3 | Phase 3 | Homozygous familial hypercholesterolemia* (NCT0339978, NCT034097446) |
| Genentech | Etrolizumab | Humanized IgG1 | a4-b7/aE-b7 integrin receptor | Phase 3 | Ulcerative colitis* (NCT02100696, NCT02118584, NCT02165215, NCT02171429, NCT02163759, NCT02136069); Crohn’s disease (NCT02394028, NCT02403323) |
| Bioverativ | Sutimlimab (BIVV009) | Humanized IgG4 | C1s | Phase 3 | Cold agglutinin disease* (NCT03347396, NCT03347422) |
| AstraZeneca | Anifrolumab | Human IgG1 | IFN a, b, ω receptor 1 | Phase 3 | Systemic lupus erythematosus* (NCT02794285, NCT02446899, NCT02446912) |
| Provention Bio | Teplizumab | Humanized IgG1 | CD3 | Phase 3 | Type 1 diabetes* (NCT03875729) |
| Hoffmann-La Roche | Gantenerumab | Human IgG1 | Amyloid beta | Phase 3 | Early or mild Alzheimer’s disease (NCT03443973, NCT03444870, NCT02051608, NCT01224106) |
| Eisai Inc. | BAN2401 | Humanized IgG1 | Amyloid β protofibrils | Phase 3 | Early Alzheimer’s disease (NCT03887455) |
| Novartis Pharmaceuticals Corp. | Ianalumab (VAY736) | Human IgG1 | BLyS/BAFF/TACI/BCMA receptor | Phase 2/3 | Autoimmune hepatitis (NCT03217422) |
| Xenikos | T-Guard | mAb immunotoxin; mixture of 2 | CD3, CD7 | Phase 3 | Steroid-refractory acute graft-versus-host disease (NCT04128319) |
| Mapp Biopharmaceutical/LeafBio | Cosfroviximab, larcaviximab, porgaviximab (Zmapp) | Chimeric IgG1 mixture (3 mAbs) | Ebola virus | Phase 2/3 | Ebola virus infection (NCT03719586) |
| Ridgeback Biotherapeutics | mAb114, VRC-EBOMAB092-00-AB | Human IgG1 | Ebola virus glycoprotein | Phase 2/3 | Ebola virus infection (NCT03719586) |
| Momenta Pharmaceuticals | Nipocalimab (M281) | Human IgG1 | FcRn | Phase 2/3 | Warm autoimmune hemolytic anemia (NCT04119050) |
| UCB Biopharma | Rozanolixizumab | Humanized IgG4 | FcRn | Phase 3 | Myasthenia gravis (NCT03971422) |
| GlaxoSmithKline | Otilimab (GSK3196165, MOR103) | Human IgG1 | GM-CSF | Phase 3 | Rheumatoid arthritis (NCT03980483, NCT03970837, NCT04134728) |
| GC Pharma | Lenvervimab (GC1102) | Humanized IgG1 | Hepatitis B virus surface antigen | Phase 2/3 | Hepatitis B virus-associated liver transplant (NCT03519113) |
| Novartis Pharmaceuticals Corp. | Ligelizumab | Human IgG1 | IgE | Phase 3 | Chronic spontaneous urticaria (NCT03907878, NCT03580356) |
| Dermira, Inc. | Lebrikizumab | Humanized IgG4 | IL-13 | Phase 3 | Atopic dermatitis (NCT04178967) |
| AstraZeneca | Tralokinumab | Human IgG4 | IL-13 | Phase 3 | Atopic dermatitis (NCT03526861, NCT03131648, NCT03160885, NCT03363854, NCT03761537, NCT03587805) |
| UCB Biopharma | Bimekizumab | Humanized IgG1 | IL-17A, F | Phase 3 | Ankylosing spondylitis (NCT03928743, NCT03928704); Plaque psoriasis (NCT03766685, NCT03598790, NCT03410992, NCT03536884, NCT03412747, NCT03370133); Psoriatic arthritis (NCT03895203, NCT03896581, NCT04009499) |
| Allergan | Brazikumab | Human IgG2 | IL-23 | Phase 3 | Crohn’s disease (NCT03961815, NCT03759288) |
| Eli Lilly & Co. | Mirikizumab | Humanized IgG4 | IL-23p19 | Phase 3 | Ulcerative colitis (NCT03519945, NCT03518086, NCT03524092): Psoriasis (NCT03482011, NCT03535194, NCT03556202); Croh’s disease (NCT03926130) |
| Galderma; Maruho Co. Ltd. | Nemolizumab | Humanized IgG2 | IL-31R alpha | Phase 3 | Atopic dermatitis (JapicCTI-173740, NCT03985943) |
| Boehringer Ingelheim | Spesolimab (BI655130) | Humanized IgG1 | IL-36R | Phase 2/3; Phase 3 | Ulcerative colitis (NCT03482635); Pustular psoriasis (NCT03886246) |
| Vitaeris, Inc. | Clazakizumab | Humanized IgG1 | IL-6 | Phase 3 | Prevention of kidney transplant rejection (NCT03744910) |
| R-Pharm | Olokizumab | Humanized IgG4 | IL-6 | Phase 3 | Rheumatoid arthritis (NCT02760368, NCT02760407, NCT02760433, NCT03120949) |
| Takeda | Ontamalimab (SHP647) | Human IgG2 | Mucosal addressin cell adhesion molecule | Phase 3 | Ulcerative colitis (NCT03290781, NCT03259334, NCT03259308); Crohn’s disease (NCT03627091, NCT03566823, NCT03559517) |
| Regeneron Pharmaceuticals, Inc. | Fasinumab | Human IgG4 | Nerve growth factor | Phase 2/3 and Phase 3 | Pain due to osteoarthritis of knee or hip (NCT03161093, NCT03245008, NCT02447276, NCT03304379, NCT02683239); Low back pain (NCT03285646) |
| AstraZeneca | Nirsevimab (MEDI8897) | Human IgG1 | RSV | Phase 2/3 | Respiratory syncytial virus infections (NCT03979313) |
| Aridis Pharmaceuticals, Inc. | Tosatoxumab | Human IgG1 | S. aureus alpha-toxin | Phase 3 | S. aureus ventilator-associated pneumonia (NCT03816956) |
| AstraZeneca | Tezepelumab | Human IgG2 | Thymic stromal lympho-poietin | Phase 3 | Severe uncontrolled asthma (NCT04048343, NCT03347279, NCT03968978, NCT03927157, NCT03706079, NCT03406078) |
| Novo Nordisk | Concizumab | Humanized IgG4 | Tissue factor pathway inhibitor | Phase 3 | Hemophilia A or B with inhibitors (NCT04083781); Hemophilia A or B without inhibitors (NCT04082429) |
| Pfizer | Marstacimab | Human IgG1 | Tissue factor pathway inhibitor | Phase 3 | Hemophilia A or B (NCT03938792) |
| Taisho Pharmaceutical Co., Ltd. | Ozoralizumab | Humanized bispecific nanobody | TNF, albumin | Phase 3 | Rheumatoid arthritis (NCT04077567, JapicCTI-184031) |
| Kodiak Sciences Inc | KSI-301 | Antibody-biopolymer conjugate | VEGF | Phase 2 (pivotal) | Neovascular (wet) age-related macular degeneration (NCT04049266) |
| Hoffmann-La Roche | Faricimab | Bispecific CrossMab | VEGF-A, Ang2 | Phase 3 | Diabetic macular edema (NCT03622593, NCT03622580); Wet macular degeneration (NCT03823287, NCT03823300) |
| TG Therapeutics | Ublituximab | Chimeric IgG1 | CD20 | Phase 3; Phase 2/3 | Multiple sclerosis (NCT03277261, NCT03277248, NCT04130997); Chronic lymphocytic leukemia (NCT02301156, NCT02612311); Non-Hodgkin’s lymphoma (Phase 2/3 NCT02793583) |
| FibroGen | Pamrevlumab | Human IgG1 | Connective tissue growth factor | Phase 3 | Idiopathic pulmonary fibrosis (NCT03955146); Pancreatic cancer (NCT03941093) |
*Indication for which a marketing application may be submitted by the end of 2020.
Table 3 includes data publicly available as of December 5, 2019. Ublituximab and pamrevlumab are included here due to the advanced stage of the non-cancer studies, but they are also in late-stage studies for cancer indications.
Tanezumab (Pfizer, Eli Lilly and Company)
Tanezumab is a humanized IgG2 antibody that selectively targets nerve growth factor. It has a novel mechanism compared to opioids and other analgesics, including nonsteroidal anti-inflammatory drugs (NSAIDs), and, in studies to date, tanezumab has not demonstrated a risk of addiction, misuse or dependence. FDA granted Fast Track designation for tanezumab for the treatment of osteoarthritis (OA) pain and chronic lower back pain.72 Eli Lilly and Pfizer plan to pursue a BLA submission for tanezumab in patients with moderate-to-severe OA by the first quarter of 2020, which may be followed by regulatory filings in the EU and Japan.73
The safety and efficacy of tanezumab have been evaluated in numerous late-stage clinical studies, at least 17 of which included OA patients. Results have recently been reported for the Phase 3 NCT02528188 and NCT02697773 studies of patients with OA of the hips or knees. In the active-controlled NCT02528188 study, patients received tanezumab 2.5 mg or 5 mg every 8 weeks for 56 weeks. The active comparator was orally administered NSAIDs twice daily for 56 weeks. The tanezumab 5 mg treatment arm met 2 of 3 co-primary efficacy endpoints (changes from baseline to Week 16 in the Western Ontario and McMasters Universities Osteoarthritis Index (WOMAC) Pain subscale, the WOMAC Physical Function subscale, and the Patient’s Global Assessment (PGA) of OA), demonstrating a statistically significant improvement in pain and physical function compared to NSAIDs at the 16-week analysis.72 The NCT02697773 study assessed the efficacy and safety of a subcutaneous tanezumab titration dosing regimen in patients with moderate-to-severe OA of the hip or knee.74 Patients were administered either 2.5 mg tanezumab at Day 1 and Week 8 (n = 231); 2.5 mg tanezumab at Day 1 and 5 mg at Week 8; or placebo at Day 1 and Week 8. From baseline to 16 weeks, mean WOMAC Pain scores decreased from 7.1 to 3.6 in the tanezumab 2.5 mg group; 7.3 to 3.6 in the tanezumab 2.5/5 mg group; and 7.3 to 4.4 in the placebo group. Mean WOMAC Physical Function scores decreased from 7.2 to 3.7 in the tanezumab 2.5-mg group, 7.4 to 3.6 in the tanezumab 2.5/5-mg group, and 7.4 to 4.5 in the placebo group. Mean PGA-OA scores decreased from 3.4 to 2.4 in the 2.5-mg group, 3.5 to 2.4 in the 2.5/5-mg group, and 3.5 to 2.7 with placebo. The study results indicated that treatment with tanezumab, compared with placebo, resulted in statistically significant improvements in scores assessing pain and physical function, and in PGA-OA. Tanezumab-treated patients had more joint safety events and total joint replacements in these studies.
Aducanumab (Biogen Inc., Eisai, Co., Ltd.)
Aducanumab (BIIB037) is a human anti-amyloid beta (Aβ) IgG1 mAb studied for the treatment of early Alzheimer’s disease. Aducanumab binds a linear epitope formed by amino acids 3–7 of the Aβ peptide and discriminates between monomers and oligomeric or fibrillar aggregates based on weak monovalent affinity, fast binding kinetics and strong avidity for epitope-rich aggregates.75 In August 2016 aducanumab was accepted into EMA’s PRIME program. In September 2016 the FDA granted aducanumab Fast Track designation, and in April 2017 aducanumab was accepted into MHLW’s Sakigake Designation System. Aducanumab was evaluated in two Phase 3 studies of patients with early Alzheimer’s disease, EMERGE (NCT02484547) and ENGAGE (NCT02477800). Based on discussions with the FDA, Biogen Inc. and Eisai, Co., Ltd. plan to file a BLA in early 2020.76
The placebo-controlled Phase 3 EMERGE and ENGAGE studies were designed to evaluate the efficacy and safety of 2 dosing regimens of aducanumab. These studies were discontinued on March 21, 2019 because a pre-specified futility analysis based on data available as of December 26, 2018, from 1,748 patients who had the opportunity to complete the 18-month study period, predicted that both studies were unlikely to meet their primary endpoint. However, later analysis of data from a total of 3,285 patients, 2,066 of whom had the opportunity to complete the full 18 months of treatment, indicated a different outcome.76
Using the new analysis, the EMERGE study met its primary endpoint, change from baseline in Clinical Dementia Rating–Sum of Boxes (CDR-SB) score at Week 78. Patients administered high-dose aducanumab showed a significant reduction of clinical decline from baseline in CDR-SB scores at 78 weeks (23% versus placebo, P = .01). These patients also showed a consistent reduction of clinical decline as measured by: the Mini-Mental State Examination (15% versus placebo, P = .06), the AD Assessment Scale-Cognitive Subscale 13 Items (27% versus placebo, P = .01), and the AD Cooperative Study-Activities of Daily Living Inventory Mild Cognitive Impairment Version (40% versus placebo, P = .001). Amyloid plaque burden was reduced with low- and high-dose aducanumab compared to placebo at 26 and 78 weeks (P < .001). Additional details on the analysis of the larger dataset from the EMERGE and ENGAGE studies will be presented at the Clinical Trials on Alzheimer’s Disease meeting in December 2019.76
Evinacumab (Regeneron Pharmaceuticals, Inc.)
Evinacumab is a human IgG4 antibody that specifically binds to angiopoietin-like protein 3 (ANGPTL3), which inhibits lipoprotein lipase and endothelial lipase and may play a central role in lipoprotein metabolism.77 Evinacumab is currently being studied in patients with homozygous familial hypercholesterolemia (HoFH) (Phase 3 NCT03399786 and NCT03409744 studies), refractory hypercholesterolemia (Phase 2 NCT03175367 study) and severe hypertriglyceridemia (Phase 2 NCT03452228 study). FDA granted Breakthrough Therapy designation for evinacumab for the treatment of hypercholesterolemia in patients with HoFH. Regeneron has indicated that clinical study data will be submitted to regulatory authorities in 2020.
The placebo-controlled Phase 3 NCT03399786 study assessed the effects of evinacumab on low-density lipoprotein (LDL) cholesterol and other lipid parameters in patients with HoFH. Patients received evinacumab 15 mg/kg IV administered every 4 weeks. Results from the evinacumab group at Week 24 showed a 49% reduction in LDL cholesterol from baseline compared to placebo (47% reduction for evinacumab compared to a 2% increase for placebo, p < .0001), which was the primary study endpoint. A 132 mg/dL absolute change in LDL cholesterol from baseline was observed compared to placebo (135 mg/dL reduction for evinacumab compared to a 3 mg/dL reduction for placebo, p < .0001). Of patients who received evinacumab, 47% achieved LDL cholesterol levels less than 100 mg/dL compared to 23% of those who received placebo. Additionally, patients treated with evinacumab had reduced apolipoprotein B, non-HDL cholesterol and total cholesterol compared to those who were administered placebo. LDL cholesterol reductions were observed from the first assessment at Week 2, and were maintained throughout the 24-week double-blind treatment period.77
Etrolizumab (Roche)
Etrolizumab is a humanized anti-integrin IgG1 antibody designed to selectively control disease in patients with moderate-to-severe inflammatory bowel disease (IBD), such as ulcerative colitis (UC) or Crohn’s disease. Etrolizumab selectively binds the β7 subunit of α4β7 and αEβ7 integrins, which are found on cells that play a key role in inflammation in IBD. The antibody works by preventing inflammatory cells from entering and being retained in the gut. Recently published data suggests that internalization of the etrolizumab-antigen complex may play an important role in the mechanism of action.78
There are currently 6 ongoing Phase 3 clinical trials evaluating the safety and efficacy of etrolizumab in UC patients,79 and 3 of these studies (HICKORY (NCT02100696), LAUREL (NCT02165215) and GARDENIA (NCT02136069)) have primary completion dates in April 2020. Roche anticipates regulatory submissions for etrolizumab for UC in 2020.79
The HICKORY study is evaluating the efficacy and safety of etrolizumab during induction and maintenance of remission compared with placebo in the treatment of patients with moderately to severely active UC who have been previously exposed to tumor necrosis factor (TNF) inhibitors. This study includes open-label and blinded cohorts. Patients who receive etrolizumab will receive 105 mg administered by subcutaneous injection once every 4 weeks. The primary endpoint for the induction phase is the percentage of patients with remission at Week 14, as determined by the Mayo Clinic Score (MCS), and the primary endpoint for the maintenance phase is the percentage of patients with remission at Week 66 among those who had achieved a clinical response at Week 14, as determined by the MCS. Recruitment in the HICKORY study was completed in the first quarter of 2019. First data from the HICKORY study were presented at the European Crohn’s and Colitis Organization meeting in 2017.80
The LAUREL study is evaluating the efficacy and safety of etrolizumab in maintenance of remission in patients with moderately to severely active UC who are naïve to TNF inhibitors and refractory to or intolerant of prior immunosuppressant and/or corticosteroid treatment. In the open-label induction phase of this study, all participants will receive treatment with open-label etrolizumab 105 mg administered by subcutaneous injection once every 4 weeks up to Week 10. Then, participants who achieved a clinical response at Week 10 during the induction phase will receive either etrolizumab 105 mg by subcutaneous injection once every 4 weeks from Week 12 up to Week 62 or placebo. The primary endpoint is the percentage of patients in remission at Week 62 among randomized patients with a clinical response at Week 10, as determined by the MCS. Recruitment in the LAUREL study was completed in the first quarter of 2019.
The GARDENIA study is comparing the efficacy and safety of etrolizumab to infliximab in patients with moderate-to-severe UC who are naïve to TNF inhibitors. In this 2-arm study, patients are administered: 1) 105 mg etrolizumab by subcutaneous injection once every 4 weeks until Week 52 and IV placebo; or 2) 5 mg/kg of infliximab administered by IV infusion at Weeks 0, 2, and 6 and then every 8 weeks until Week 46 and subcutaneous placebo. The primary endpoint is the percentage of patients in sustained remission as determined by the MCS at Weeks 10, 30 and 54. Recruitment in the GARDENIA study was completed in the second quarter of 2019.
Sutimlimab (Sanofi)
Sutimlimab (formerly BIVV009), a humanized IgG4 antibody, binds to C1s, a serine protease within the C1-complex in the classical complement pathway of the immune system, and thereby inhibits the central mechanism of hemolysis in cold agglutinin disease (CAD), a rare autoimmune hemolytic anemia that is difficult to treat. The antibody is designed to selectively inhibit disease processes in the classical complement pathway, but not interfere with functions of the alternative and lectin complement pathways of the immune system. Sutimlimab was granted Breakthrough Therapy designation by the FDA for treatment of hemolysis in patients with primary CAD,81 and Orphan Drug designations by FDA for treatment of CAD and bullous pemphigoid and by EMA for autoimmune hemolytic anemia including CAD. Sanofi may submit regulatory application(s) for sutimlimab for CAD in 2020.82
Two Phase 3 trials, Cardinal (NCT03347396) and Cadenza (NCT03347422), are evaluating the effects of sutimlimab treatment in CAD patients. The Cardinal study is assessing the efficacy and safety of sutimlimab in patients with primary CAD who have a recent history of blood transfusion. Patients receive sutimlimab by IV infusion. Those who complete Part A per protocol through the end of treatment visit (Day 182) will participate in Part B, and continue to receive sutimlimab up to 1 year after last patient out in Part A. The estimated enrollment is 20 patients. The primary outcome measures are the percentage of patients with response up to Week 26 (Part A) and the number of patients with treatment-emergent adverse events and serious adverse events up to ~ 1 year (Part B). On November 21, 2019, Sanofi announced that sutimlimab met the primary endpoint of the CARDINAL study.83
The Cadenza study is assessing the efficacy and safety of sutimlimab in patients with primary CAD without a recent history of blood transfusion. In Part A, participants will be randomized 1:1 to receive an IV infusion of sutimlimab or placebo. In Part B, all patients will undergo blinded cross-over loading doses to allow all patients to receive sutimlimab while maintaining Part A blinding. The primary outcome measures are the same as the Cardinal study. The estimated enrollment for the Cadenza study is 40 patients, and the primary completion date is October 2020.
Anifrolumab (AstraZeneca)
Anifrolumab (MEDI-546) is an anti-type I interferon (IFN) alpha receptor 1 (IFNAR1) human IgG1 antibody engineered with a triple mutation (L234F/L235E/P331S) in the heavy chain to reduce binding to Fc receptors, and thereby reduce potential Fc-mediated effector functions. By binding a subunit of IFNAR1, the antibody blocks the action of IFN-α, IFN-β and IFN-ω. Anifrolumab is being evaluated in Phase 3 clinical studies as a treatment for moderate-to-severe systemic lupus erythematosus (SLE). The FDA has granted anifrolumab Fast Track designation for SLE. AstraZeneca may submit regulatory application(s) for anifrolumab for SLE in the second half of 2020.84
The safety and efficacy of anifrolumab are being evaluated in two Phase 3 “Treatment of Uncontrolled Lupus via the Interferon IFN Pathway” (TULIP) studies, TULIP 1 (NCT02446912) and TULIP 2 (NCT02446899). The TULIP program also includes a Phase 3 long-term extension trial in SLE (TULIP SLE LTE (NCT02794285)) and a Phase 2 trial in lupus nephritis. The TULIP 2 study met its primary endpoint, the number of participants who achieve the British Isles Lupus Assessment Group Based Composite Lupus Assessment (BICLA) response at Week 52.85 In the TULIP 2 study, patients were administered anifrolumab 300 mg or placebo via IV infusion every 4 weeks for a total of 13 doses, and patients in both arms received standard of care. Patients who received anifrolumab achieved a statistically significant and clinically meaningful reduction in disease activity versus placebo. At Week 52, 47.8% of patients who received anifrolumab responded compared with 31.5% of patients on placebo, as measured by the BICLA composite measure.86 The positive BICLA response in TULIP 2 was consistent with a positive pre-specified analysis of the TULIP 1 trial, which evaluated 2 dose levels of anifrolumab vs placebo and did not meet its primary endpoint of SLE Responder Index of ≥4.
Teplizumab (Provention Bio, Inc.)
Teplizumab (PRV-031) is a humanized, Fc receptor non-binding, anti-CD3 IgG1 antibody being developed by Provention Bio, Inc. for the interception and prevention of type 1 diabetes (T1D). FDA granted teplizumab Breakthrough Therapy designation for the prevention or delay of clinical T1D, and EMA has granted teplizumab PRIME designation for the prevention or delay of clinical T1D in individuals at risk of developing the disease.87 Provention Bio expects to meet with FDA in the fourth quarter of 2019 to discuss an expedited development plan that would allow a BLA submission in the fourth quarter of 2020 for teplizumab for the prevention or delay of clinical T1D in individuals at risk for developing the disease.88
FDA has agreed that results from the Phase 2 “At-Risk” study (NCT01030861), with supportive data from prior teplizumab studies in early onset T1D, may be sufficient for a BLA submission. Teplizumab has been evaluated in multiple clinical studies, with more than 800 patients receiving the drug. The “At-Risk” study, which was conducted by Type 1 Diabetes TrialNet (www.trialnet.org), included relatives of patients with type 1 diabetes who did not have diabetes, but were at high risk for development of clinical disease. A single 14-day course of teplizumab significantly delayed the onset and diagnosis of clinical T1D, as compared to placebo, with median times to the diagnosis of type 1 diabetes of 48.4 months in the teplizumab group and 24.4 months in the placebo group.89 Provention Bio is currently evaluating teplizumab in the Phase 3 PROTECT study (NCT03875729) to determine whether teplizumab slows the loss of β cells and preserves β cell function in children and adolescents 8–17 years old who have been diagnosed with T1D in the previous 6 weeks. Estimated enrollment for the PROTECT study is 300 patients, and the estimated primary completion date is May 2022.
Antibodies to watch in 2020: cancer indications
According to the information publicly available as of November 2019, 40 antibody therapeutics are undergoing evaluation in late-stage studies for cancer (Table 4). As previously noted and listed in Tables 3, 2 additional antibodies (ublituximab, pamrevlumab) in late-stage studies for non-cancer indications are also being evaluated in late-stage studies as treatments for cancer. Companies developing 9 of these novel antibody therapeutics have indicated that they may submit a BLA or MAA in late 2019 or in 2020. The summaries below are listed in the chronological order in which the applications may be submitted: belantamab mafodotin, oportuzumab monatox, margetuximab, dostarlimab, spartalizumab, 131I-omburtamab, loncastuximab tesirine, balstilimab, and zalifrelimab.
Table 4.
Investigational monoclonal antibodies in late-stage clinical studies for cancer indications.
| Primary sponsoring company | INN or code name | Molecular format | Target(s) | Most advanced phase | Pivotal Phase 2, Phase 2/3 or Phase 3 indications |
|---|---|---|---|---|---|
| GlaxoSmithKline | Belantamab mafodotin | Humanized IgG1 ADC | B-cell maturation antigen | Phase 2 (pivotal), Phase 3 pending | Multiple myeloma* (NCT03525678, NCT04091126 (pending)) |
| Sesen Bio | Oportuzumab monatox | Humanized scFv immunotoxin | EpCAM | Phase 3 | Bladder cancer* (NCT02449239) |
| MacroGenics | Margetuximab | Chimeric IgG1 | HER2 | Phase 2/3; Phase 3 | Gastric/gastro-esophageal junction adenocarcinoma (NCT04082364); Breast cancer* (NCT02492711) |
| GlaxoSmithKline/Tesaro, Inc. | Dostarlimab (TSR-042) | Humanized IgG4 | PD-1 | Phase 3 | Endometrial cancer* (NCT03981796); Ovarian cancer (NCT03602859) |
| Novartis Pharmaceuticals Corp. | Spartalizumab (PDR001) | Humanized IgG4 | PD-1 | Phase 3 | Melanoma* (NCT02967692) |
| Y-mAbs Therapeutics | 131I-omburtamab | Murine mAb, radiolabeled | B7-H3 | Phase 2/3 | Neuroblastoma central nervous system/leptomeningeal metastases* (NCT03275402) |
| ADC Therapeutics Sarl | Loncastuximab tesirine | Humanized IgG1 ADC | CD19 | Phase 2 (pivotal) | Diffuse large B-cell lymphoma* (NCT03589469) |
| Agenus Inc. | Balstilimab (AGEN2034) | Human IgG4 | PD-1 | Phase 2 (pivotal) | Cervical cancer* (NCT03894215, NCT03104699, NCT03495882) |
| Agenus Inc. | Zalifrelimab (AGEN1884) | Human IgG1 | CTLA-4 | Phase 2 (pivotal) | Cervical cancer* (NCT03894215, NCT03495882) |
| Pfizer | Utomilumab | Human IgG2 | 4-1BB (CD137) | Phase 3 | Diffuse large B-cell lymphoma (NCT02951156) |
| Sanofi | SAR408701 | mAb ADC | CEACAM5 | Phase 3 | Non-small cell lung cancer (NCT04154956) |
| Molecular Templates | MT-3724 | scFv immunotoxin | CD20 | Phase 2 (potentially pivotal) | Diffuse large B-cell lymphoma (NCT03645395) |
| Regeneron Pharmaceuticals, Inc. | REGN1979 | Human IgG4 bispecific | CD20, CD3 | Phase 2 (potentially pivotal) | Follicular lymphoma (NCT03888105) |
| ADC Therapeutics Sarl | Camidanlumab tesirine | Human IgG1 ADC | CD25 (aka IL-2RA) | Phase 2 (pivotal) | Hodgkin lymphoma (NCT04052997) |
| Affimed N.V. | AFM13 | Human bispecific tandem diabody (TandAb) | CD30, CD16A | Phase 2 (pivotal) | Peripheral T Cell Lymphoma (NCT04101331) |
| I-Mab Biopharma, MorphoSys | TJ202, MOR202 | Human IgG1 | CD38 | Phase 3 | Multiple myeloma (NCT03952091) |
| Actinium Pharmaceuticals | Iodine (131I) apamistamab | Murine IgG1, radio-labeled | CD45 | Phase 3 | Ablation of bone marrow prior to hematopoietic cell transplantation in AML patients (NCT02665065) |
| Astellas Pharma Inc | Zolbetuximab | Chimeric IgG1 | Claudin-18.2 | Phase 3 | Gastric/gastro-esophageal junction adenocarcinoma (NCT03653507, NCT03504397) |
| AstraZeneca | Tremelimumab | Human IgG2 | CTLA4 | Phase 3 | Small cell lung cancer (NCT03703297); Urothelial cancer (NCT03682068, NCT02516241); Hepatocellular carcinoma (NCT03298451) |
| Rakuten Aspyrian, Inc. | Cetuximab-IRDye® 700DX | Chimeric IgG1 conjugated to IR700; photoimmunotherapy | EGFR | Phase 3 | Head and neck squamous cell carcinomas (NCT03769506) |
| Five Prime Therapeutics, Zai Lab Limited | Bemarituzumab | Humanized IgG1 | FGFR2b | Phase 3 | Gastric/gastro-esophageal junction adenocarcinoma (NCT03694522) |
| Philogen SpA | Daromun (L19IL2 + L19TNF combination) | scFv conjugates | Fibronectin extra-domain B | Phase 3 | Melanoma (NCT03567889, NCT02938299) |
| ImmunoGen | Mirvetuximab soravtansine | Humanized IgG1 ADC | Folate receptor 1 | Phase 3 | Ovarian cancer, primary peritoneal cancer or fallopian tube cancer (NCT02631876) |
| Immunocore Ltd | Tebentafusp | scFv bispecific immunoconjugate | gp100, CD3 | Phase 2 (pivotal) | Melanoma, uveal (NCT03070392) |
| Synthon Biopharmaceuticals BV | [vic-] trastuzumab duocarmazine | Humanized IgG1 ADC | HER2 | Phase 3 | Breast cancer (NCT03262935) |
| Bio-Thera Solutions | BAT8001 | Humanized IgG1 ADC | HER2 | Phase 3 | Breast cancer (CTR20180157, NCT04185649) |
| Bristol-Myers Squibb | Relatlimab (BMS-986016) | Human IgG4 | LAG-3 | Phase 2/3 | Melanoma (NCT03470922) |
| Akeso | AK105 | Humanized IgG1 | PD-1 | Phase 3 | Non-small cell lung cancer (NCT03866993, NCT03866980) |
| Jiangsu HengRui Medicine Co., Ltd | Camrelizumab | Humanized IgG4 | PD-1 | Phase 3; regulatory review in China | Gastric/gastro-esophageal junction adenocarcinoma (NCT03813784); Nasopharyngeal Carcinoma (NCT03707509); Esophageal cancer (NCT03099382, NCT03691090); Non-small cell lung cancer (NCT03134872, NCT03668496); Hepatocellular carcinoma (NCT03605706, NCT03764293); Hodgkin’s lymphoma |
| Shanghai Henlius Biotech | HLX10 | Humanized mAb | PD-1 | Phase 3 | Esophageal squamous cell carcinoma (NCT03958890); Non-small cell lung cancer (NCT04033354, NCT03952403); Small cell lung cancer (NCT04063163) |
| Macrogenics | INCMGA00012, MGA012 | Humanized IgG4 | PD-1 | Phase 2/3 | Gastric cancer/gastro-esophageal junction adenocarcinoma (NCT04082364); Squamous cell carcinoma of the head and neck (NCT04129320 pending) |
| Biocad | Prolgolimab (BCD-100) | Human IgG1 | PD-1 | Phase 2/3 | Non-small cell lung cancer (NCT03288870) |
| BeiGene | Tislelizumab (BGB-A317) | Humanized IgG4 | PD-1 | Phase 3; regulatory review in China | Non-small cell lung cancer (NCT03594747, NCT03663205, NCT03745222, NCT03358875); Nasopharyngeal cancer (NCT03924986); Gastric/gastroesophageal junction adenocarcinoma (NCT03777657); Urothelial carcinoma (NCT03967977); Esophageal squamous cell carcinoma (NCT03957590, NCT03783442, NCT03430843); Small cell lung cancer (NCT04005716); Hodgkin’s lymphoma |
| Macrogenics | MGD013 | Human IgG4 bispecific DART | PD-1, LAG-3 | Phase 2/3 | Gastric cancer/gastro-esophageal junction adenocarcinoma (NCT04082364) |
| CBT Pharmaceuticals, Inc | TQB2450, CBT-502 | Humanized IgG1 | PD-L1 | Phase 3 | Head and neck cancer (NCT03855384) |
| CStone Pharmaceuticals | CS1001 | Human IgG4 | PD-L1 | Phase 3 | Non-small cell lung cancer (NCT03789604); Gastric/gastro-esophageal junction adenocarcinoma (NCT03802591) |
| Alphamab Oncology | Envafolimab (KN035) | mAb, single domain | PD-L1 | Phase 3 | Biliary tract carcinoma (NCT03478488) |
| EMD Serono | Bintrafusp alfa | Human IgG1 bispecific immunoconjugate | PD-L1, TGFβ | Phase 2/3 | Biliary tract cancer (NCT04066491) |
| Novartis | MBG453 | Humanized IgG4 | TIM-3 | Phase 2 (pivotal) | Myelodysplastic syndromes (NCT03946670) |
| Genmab | Tisotumab vedotin | Human IgG1 ADC | Tissue factor | Phase 2 (pivotal) | Cervical cancer (NCT03438396) |
Belantamab mafodotin (GlaxoSmithKline Plc)
Also known as GSK2857916, belantamab mafodotin is an anti-B cell maturation antigen IgG1 antibody linked to a cytotoxic agent, monomethyl auristatin F. This ADC is in Phase 2 clinical development for patients with relapsed/refractory multiple myeloma. Belantamab mafodotin was granted Breakthrough Therapy designation and PRIME designation from FDA and EMA, respectively, as well as Orphan Drug designations from both agencies. GlaxoSmithKline is on track to submit a BLA for belantamab mafodotin by the end of 2019.90 If a BLA is submitted by December 2019 and receives a priority review, then a first action by FDA would be expected by the end of June 2020.
In August 2019, GlaxoSmithKline announced positive results from the pivotal DREAMM-2 study (NCT03525678) evaluating the efficacy and safety of 2 doses of belantamab mafodotin in patients with multiple myeloma who had 3 or more prior lines of treatment, are refractory to a proteasome inhibitor and an immunomodulatory agent and have failed an anti-CD38 antibody. In this Phase 2 trial, patients were randomized to receive 2.5 or 3.4 mg/kg belantamab mafodotin intravenously every 3 weeks. The primary endpoint was ORR, and key secondary outcome measures were clinical benefit rate, duration of response, time to response, PFS, time to progression, and OS. The 2-arm study met is primary objective, demonstrating a clinically meaningful ORR in patients with relapsed/refractory multiple myeloma. DREAMM-2 study results will be the basis for regulatory submissions for belantamab mafodotin for the treatment of relapsed/refractory multiple myeloma.
Belantamab mafodotin is also being studied in combination with pembrolizumab (Phase 2 DREAMM-4 study, NCT03848845), with pomalidomide and dexamethasone (Phase 1/2 NCT03715478 study) and with lenalidomide + dexamethasone or with bortezomib + dexamethasone (Phase 2 NCT03544281 study) in patients with multiple myeloma. The Phase 3 DREAMM-3 study (GSK ID# 207495), an open-label, randomized study to evaluate the efficacy and safety of belantamab mafodotin compared to pomalidomide plus low-dose dexamethasone in relapsed/refractory multiple myeloma patients is planned.
Oportuzumab monatox (Sesen Bio)
Oportuzumab monatox (Vicinium, VB4-845) is a recombinant fusion protein composed of a humanized scFv targeting epithelial cell adhesion molecule (EpCAM) fused to Pseudomonas exotoxin A. The drug targets EpCAM on the surface of tumor cells and is internalized, leading to the cleavage of the payload exotoxin, which induces apoptosis. Vicinium was granted FDA’s Fast Track designation for the treatment of bacillus Calmette-Guérin (BCG)-unresponsive non-muscle invasive bladder cancer (NMIBC). In November 2019, Sesen Bio announced that the company was on track for anticipated initiation of BLA submission for Vicinium, under an accelerated approval pathway with rolling review, in December 2019.91
Vicinium is currently being evaluated in the single-arm Phase 3 VISTA study (NCT02449239) for the treatment of patients with high-grade NMIBC in situ and high-grade papillary disease previously treated with BCG.92 The trial enrolled a total of 133 patients across 3 cohorts. Cohort 1 includes carcinoma in situ (CIS) patients with or without papillary disease whose cancer was determined to be refractory or recurred within 6 months of their last course of adequate BCG. Cohort 2 includes CIS patients with or without papillary disease whose cancer was determined to be refractory or recurred after 6 months, but less than 12 months, after their last course of adequate BCG. Cohort 3 includes patients with papillary disease without CIS whose cancer was determined to be refractory or recurred within 6 months of their last course of adequate BCG. The primary endpoint is the complete response rate and duration of response in patients with CIS with or without papillary disease (Cohort 1). Secondary efficacy endpoints include time to disease recurrence for patients in Cohort 3, time to cystectomy, PFS, event-free survival, and OS for patients across all cohorts.
In August 2019, Sesen Bio announced updated preliminary results for the VISTA trial.91 The complete response rates at 3, 6, 9 and 12 months in Cohort 1 (82 evaluable patients) were 39%, 26%, 20% and 17%, respectively. For Cohort 2 (7 evaluable patients), the complete response rates at 3, 6, 9 and 12 months were 57%, 57%, 43% and 14%, respectively. Pooled data for all CIS patients (n = 93) showed that, among patients who achieved a complete response at 3 months, 52% had a complete response for a total of 12 months or longer after starting therapy. Regarding secondary endpoints, the median time to disease recurrence for patients in Cohort 3 (n = 40) is 402 days. After 2.5 years, across all 133 patients treated with Vicinium, >75% of patients are estimated to remain cystectomy-free. Moreover, 90% of all 133 patients treated with Vicinium are estimated to remain progression-free for 2 years or greater, 96% of all 133 patients treated with Vicinium are estimated to have an overall survival of 2 years or greater, and 29% of treated patients are estimated to remain event-free at 12 months. The drug was well-tolerated in the VISTA trial, with 95% of adverse events being grade 1 (mild, with mild or no symptoms; no interventions required) or 2 (moderate; minimal intervention indicated; some limitation of activities).91
Margetuximab (Macrogenics, Inc.)
Margetuximab (MGAH22) is a chimeric anti-HER2 IgG1 antibody. Margetuximab and trastuzumab bind the same epitope of HER2 with similar affinities, but 5 amino acid substitutions (L235V, F243L, R292P, Y300L, and P396L) engineered into the margetuximab Fc domain yield increased binding to both isoforms of the stimulatory CD16A (FcγRIIIA) and reduced binding to the inhibitory CD32B (FcγRIIB).93 The FDA granted Fast Track designation for margetuximab as a treatment of patients with metastatic or locally advanced HER2-positive breast cancer who have previously been treated with anti-HER2-targeted therapy. MacroGenics anticipates submitting a BLA for margetuximab to the FDA in the fourth quarter of 2019, based on results of the Phase 3 SOPHIA study (NCT02492711).94
The SOPHIA study is evaluating margetuximab in combination with a chemotherapy agent (capecitabine or eribulin or gemcitabine or vinorelbine) as a potential treatment for HER2-positive metastatic breast cancer. In this study, patients were randomized to receive either margetuximab 15 mg/kg every 21 days plus chemotherapy or trastuzumab 8 mg/kg loading dose then 6 mg/kg every 21 days plus chemotherapy. The primary outcome measures are PFS by independent radiological review, OS and infusion rate sub-study Grade 3 plus safety. Key secondary endpoints include PFS assessed by study investigators and ORR. The trial met the first sequential primary endpoint of prolongation of PFS in patients treated with the combination of margetuximab plus chemotherapy compared to trastuzumab plus chemotherapy. As reported at the American Society of Clinical Oncology annual meeting in June 2019,95 margetuximab prolonged PFS over trastuzumab (median 5.8 vs 4.9 months, hazard ratio [HR], 0.76; 95% CI, 0.59–0.98; P = .033). Treatment effects were more pronounced in patients with CD16A genotypes containing a 158F allele (median PFS 6.9 vs 5.1 months, HR, 0.68; 95% CI, 0.52–0.90; P = .005). In 524 patients with baseline measurable disease (margetuximab, n = 262; trastuzumab, n = 262), ORR was higher with margetuximab (22%; 95% CI, 17.3–27.7%) vs trastuzumab (16%; 95% CI 11.8–21.0%). Safety profiles were comparable in 529 pts who received study therapy. Grade ≥3 AEs and serious AEs occurred in 138 (52%) and 39 (15%) vs 128 (48%) and 46 (17%) patients administered margetuximab vs trastuzumab, respectively.
Margetuximab is also being evaluated as a treatment for gastric or gastroesophageal junction cancer. Sponsored by MacroGenics and Zai Lab, the Phase 2/3 MAHOGANY (NCT04082364) trial will examine the effects of margetuximab in combination with checkpoint inhibitor molecules, including MGA012 (anti-PD-1 mAb) and MGD013 (bispecific PD-1 x LAG-3 DART® molecule) in patients with HER2-positive gastric or gastroesophageal junction cancer. A single-arm cohort (Cohort A, 40 to 100 patients) will evaluate the safety and efficacy of margetuximab plus MGA012. In a 4-arm cohort (Cohort B Part 1, 50 patients per arm), patients will be randomized to margetuximab plus chemotherapy plus MGA012, margetuximab plus chemotherapy plus MGD013, margetuximab plus chemotherapy, or trastuzumab plus chemotherapy. A checkpoint inhibitor (CPI) (MGA012 or MGD013) will be selected from Cohort B Part 1 and evaluated in a randomized 2-arm cohort (Cohort B Part 2, 250 patients per arm) of margetuximab plus chemotherapy plus MGA012 or MGD013, or trastuzumab plus chemotherapy. Initiated in September 2019, the estimated primary completion date of the study is May 2024.
Dostarlimab (GlaxoSmithKline/Tesaro, AnaptysBio)
Dostarlimab (TSR-042) is a humanized IgG4 antibody that binds with high affinity to the PD-1 receptor and effectively blocks its interaction with the ligands PD-L1 and PD-L2. Dostarlimab is being developed by Tesaro (a division of GlaxoSmithKline) for the treatment of solid tumors, including endometrial cancer that could be classified as microsatellite stable (MSS/75%) or microsatellite instability-high (MSI-H/25%). GlaxoSmithKline anticipates potential regulatory submissions for dostarlimab in endometrial cancer in the second half of 2019 based on results of the GARNET clinical study (NCT02715284).96
Described by GlaxoSmithKline as Phase 1/2,97 the GARNET study evaluated the safety and efficacy of dostarlimab as a monotherapy for patients with advanced solid tumors, including women with recurrent or advanced endometrial cancer who progressed on or after a platinum-based regimen. Patients were administered dostarlimab 500 mg once every 3 weeks for 4 doses, followed by 1000 mg once every 6 weeks until disease progression. Data from 125 patients, including 41 MSI-H (33%), and 79 MSS (63%) patients, as well as 5 with an unknown MSI-status (4%), were analyzed. Preliminary results showed that the overall response rate in the full population, MSI-H population, and MSS population were 30%, 49%, and 20%, respectively, while the disease control rate in these populations were 53%, 63% and 47%, respectively. In general, adverse events were Grade 1 or 2, with only 13.6% of patients experiencing Grade 3 or higher adverse events.97
The Phase 3 RUBY study (NCT03981796) is evaluating the efficacy and safety of dostarlimab plus carboplatin-paclitaxel versus placebo plus carboplatin-paclitaxel in patients with recurrent or primary advanced endometrial cancer. An estimated 470 patients will enroll. Initiated in July 2019, the estimated primary completion date of the RUBY study is November 2021.
Tesaro is also evaluating dostarlimab as a treatment for ovarian cancer in the Phase 3 FIRST study (NCT03602859). This study will compare platinum-based therapy with dostarlimab and niraparib versus standard of care platinum-based therapy as first-line treatment of Stage III or IV non-mucinous epithelial ovarian cancer. The primary outcome measure is PFS. An estimated 912 patients are expected to enroll. Initiated in October 2018, the estimated primary completion date is November 2021.
Spartalizumab (Novartis)
Previously called PDR001, spartalizumab is a humanized IgG4 anti-PD1 antibody being tested as a treatment for various types of cancers, including melanoma. Novartis anticipates submission of marketing applications for spartalizumab in combination with dabrafenib and trametinib in metastatic melanoma in 2020.98
Spartalizumab is being evaluated in the 3-part Phase 3 COMBI-i trial (NCT02967692) in combination with dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) versus matching placebo in combination with dabrafenib and trametinib in previously untreated patients with BRAF V600–mutant unresectable or metastatic melanoma. As reported at the American Society of Clinical Oncology on June 3, 2019,99 based on pooled efficacy and safety data from Part 1 (run-in cohort, n = 9) and Part 2 (biomarker cohort, n = 27), the spartalizumab-containing combination showed durable ORR (75%) with complete response in 33% of patients. With > 15 months of follow-up, median PFS was not reached. The safety profile was manageable and reflected the individual toxicities of the individual drugs. In Part 3 of the study, ~ 500 patients with previously untreated unresectable and metastatic BRAF V600 mutated melanoma will be enrolled to compare the efficacy of spartalizumab in combination with dabrafenib and trametinib versus placebo plus dabrafenib and trametinib.
131I-omburtamab (Y-mAbs Therapeutics, Inc.)
131I-Omburtamab (formerly known as 8H9 or burtomab), composed of a murine IgG1 antibody targeting the B7-H3 antigen and iodine-131, is being developed for the treatment of pediatric patients with central nervous system/leptomeningeal metastases (CNS/LM), a rare and usually fatal complication of neuroblastoma. 131I-Omburtamab was granted a Breakthrough Therapy designation by the FDA in this indication. Y-mabs Therapeutics expects to start submission of a rolling BLA in early 2020 that would include pooled data from the Phase 1 Study 03–133 (NCT00089245) and Phase 2/3 Study-101 (NCT03275402), which included patients with CNS/LM from neuroblastoma.71,100
Study 03–133 investigated the maximally tolerated dose (MTD) of intrathecal 131I-omburtamab. To determine the MTD, patients entered cohorts of 3 at each dose level from 10 mCi to 60 mCi and a cohort of 6 at each dose level from 70 mCi to 100 mCi. Data from 93 patients included in the study showed a median OS of 47 months (including an estimated 5-year OS of ~ 43%), as compared to a historical median OS of ~ 6 months.100
The Phase 2/3 Study-101 is assessing the safety and efficacy of 131I-omburtamab as a monotherapy for children (up to 18 years) with CNS/LM from neuroblastoma. The estimated enrollment is 32 patients, who will be administered one treatment cycle of 131I-omburtamab consisting of 2 doses (2 mCi at Week 1 and 50 mCi at Week 2). At Week 6, the patients are evaluated for safety, and, if eligible, receive a second cycle of 131I-omburtamab. The primary endpoints are OS rate at 3 years after the first treatment dose of 131I-omburtamab. The estimated primary completion of Study-101 is December 2019.
Loncastuximab tesirine (ADC Therapeutics SA)
The ADC loncastuximab tesirine (ADCT-402), composed of a humanized IgG1 antibody targeting CD19 conjugated to a pyrrolobenzodiazepine (PBD)-dimer toxin, is in development as a treatment for B cell hematological malignancies. Once bound to CD19-positive cells, loncastuximab tesirine is internalized into the cell, where PBD is released and promotes cell death. Loncastuximab tesirine has been granted FDA’s Orphan Drug designation for the treatment of DLBCL and mantle cell lymphoma. ADC Therapeutics anticipates reporting response rate data from a pivotal Phase 2 trial (NCT03589469) in the second quarter of 2020, and submission of a BLA in the second half of 2020 if the trial is successful.101
Loncastuximab tesirine is currently under investigation in a multi-center, open-label, single-arm pivotal Phase 2 trial (NCT03589469) in patients with relapsed or refractory DLBCL. In this study, the patients receive IV loncastuximab tesirine on Day 1 of each cycle (every 3 weeks) at a dose of 150 μg/kg for 2 cycles and then 75 μg/kg for subsequent cycles for up to 1 year or until disease progression or unacceptable toxicity. The primary endpoint is ORR; secondary outcome measures include duration of response, complete response rate, relapse-free survival, PFS, OS and pharmacokinetic parameters. As of August 2019, interim data showed that the ORR in the first 96 patients treated with loncastuximab tesirine was 41.7%.101
Balstilimab, zalifrelimab (Agenus Inc.)
Agenus Inc. is currently focused on developing balstilimab (AGEN2034) and zalifrelimab (AGEN1884) as treatments for second-line cervical cancer. Balstilimab is a human IgG4 anti-PD-1 antibody and zalifrelimab is a human IgG1 anti-cytotoxic T lymphocyte antigen-4 antibody. Both antigens serve as checkpoints of the immune system. Both antibodies are derived from a proprietary mammalian display technology, Retrocyte Display™.102 Balstilimab is being evaluated as monotherapy in a Phase 1/2 study (NCT03104699) and in combination with zalifrelimab in Phase 1/2 (NCT03495882) and Phase 2 (NCT03894215) studies. Phase 1/2 study results could potentially support BLA submissions as early as 2020 under FDA’s accelerated approval pathway, according to projections by Agenus.103
Initiated in April 2017, the Phase 1/2 NCT03104699 study is an open-label, 3 + 3 dose-escalation trial in patients with metastatic or locally advanced solid tumors, with a consecutive Phase 2 expansion to evaluate efficacy in subjects with recurrent, unresectable, or metastatic (advanced) cervical cancer that has progressed after a platinum doublet. Estimate enrollment is 75 patients, and the primary completion date of the study is September 2019.
Initiated in December 2017, the open-label, multi-arm Phase 1/2 NCT03495882 study is investigating the safety, tolerability, pharmacokinetics, and biological and clinical activity of balstilimab in combination with zalifrelimab in patients with locally advanced, recurrent and/or metastatic solid tumors including cervical cancer. Part 1 of the study uses a standard 3 + 3 dose escalation as follows: 1) balstilimab 1 mg/kg administered every 2 weeks (q2w) in combination with zalifrelimab 1 mg/kg administered every 6 weeks; and 2) balstilimab 3 mg/kg administered every 2 weeks in combination with zalifrelimab 1 mg/kg administered every 6 weeks. In Phase 2, the study will expand in advanced cervical cancer. The study’s estimate enrollment is 60 patients, and the primary completion date is March 2022.
Notable set-backs
In addition to the advances documented here, several notable set-backs occurred in 2019. In particular, the development program for rovalpituzumab tesirine was terminated and Lartruvo® was withdrawn from the market.
Rovalpituzumab tesirine (AbbVie Inc.)
Rovalpituzumab tesirine is an ADC composed of a humanized IgG1 antibody targeting delta-like ligand (DLL3) conjugated to the cytotoxic pyrrolobenzodiazepine dimer D6.5 via a protease-cleavable linker. DLL3 is over-expressed in small cell lung cancer cells, and clinical development of this ADC was focused on small cell lung cancer. Study results, however, showed modest clinical activity. In the Phase 2 TRINITY study (NCT02674568), the ORR was 12.4%, 14.3%, and 13.2% in all, DLL3-high, and DLL3-positive patients, respectively; DLL3-positive and DLL3-high were defined as ≥25% and ≥75% of tumor cells positive for DLL3, respectively. Median OS was 5.6 months in all patients.104 On August 29, 2019 AbbVie announced that the Phase 3 MERU study (NCT03033511), which was evaluating rovalpituzumab tesirine as a first-line maintenance therapy for advanced small cell lung cancer, demonstrated no survival benefit at a preplanned interim analysis for patients receiving the drug as compared with placebo. The MERU trial is being closed, and the research and development program for rovalpituzumab tesirine has been terminated.105
Olaratumab (Eli Lilly and Company)
Olaratumab (Lartruvo®) is an anti-platelet-derived growth factor receptor alpha (PDGFR-α) antibody that specifically binds PDGFR-α and prevents receptor activation. FDA granted Lartruvo® accelerated approval on October 19, 2016, following the release of results from a Phase 1b/2 study in advanced or metastatic soft tissue sarcoma that showed an 11.8-month improvement with olaratumab over placebo for advanced soft tissue sarcoma patients (HR 0.46, 95% CI 0.30–0.71, P = .0003), and a 16.1-month improvement in the leiomyosarcoma subgroup. FDA may grant accelerated approval for drugs for serious conditions that fill an unmet medical need based on whether the drug has an effect on a surrogate or an intermediate clinical endpoint, but subsequent confirmatory trials must verify clinical benefit. During 2017 to 2018, Lartruvo® was granted accelerated, conditional, and full approvals in more than 40 countries.
The confirmatory Phase 3 ANNOUNCE clinical study (NCT02451943), which was a randomized, double-blind, placebo-controlled trial of doxorubicin plus olaratumab versus doxorubicin plus placebo in patients with advanced or metastatic soft tissue sarcoma, was started in September 2015. On April 25, 2019, Eli Lilly and Company announced that the company is withdrawing Lartruvo® (olaratumab) from the market for the treatment of advanced soft tissue sarcoma because the ANNOUNCE clinical trial failed to confirm the drug’s clinical benefit.106 Data from the study was presented at the American Society of Clinical Oncology meeting held in June 2019. Overall, median OS was 20.4 months for patients who received the drug and 19.7 months for patients who participated in the control arm of the study (HR 1.05, 95% CI [0.84, 1.30]; p = .6945). In subset of patients with leiomyosarcoma, median OS was 21.6 months in the investigational versus 21.9 months in the control arm (HR 0.95, 95% CI [0.69, 1.31]; p = .7618).
Outlook for the future
Despite the occasional set-back, our data suggest that the commercial clinical pipeline will yield a substantial number of new antibody therapeutic products during the 2020s. Interestingly, the current data suggests a nascent trend toward use of pivotal/registrational Phase 2 studies, which may decrease the clinical study period for some products, thereby accelerating the rate of approvals.
In the short-term, based on the number of antibody therapeutics already undergoing regulatory review as of December 2019, first approvals in 2020 may set a new record. We anticipate that marketing applications for more than 15 antibody therapeutics currently in late-stage studies will be submitted by the end of 2020, and thus a continuous flow of new products should reach patients in 2021. We do not, however, expect the number of antibody therapeutics in late-stage studies to substantially decrease because we are currently tracking at least 15 antibodies in early-stage studies that may transition to late-stage studies soon. We look forward to documenting progress made with these and other ‘Antibodies to Watch’ in the next installment of this article series.
Abbreviations
- AD
Alzheimer disease
- ADC
antibody-drug conjugate
- AE
adverse events
- aHUS
atypical hemolytic uremic syndrome
- ANGPTL3
angiopoietin-like protein 3
- AQP4
aquaporin-4
- Aβ
amyloid beta
- BCG
bacillus Calmette-Guérin
- BCVA
Best-corrected visual acuity
- BICLA
the British Isles Lupus Assessment Group Based Composite Lupus Assessment
- BLA
biologics license application
- BR
Bendamustine + rituximab
- CAD
cold agglutinin disease
- CAS
Clinical Activity Score
- CCR5
CC chemokine receptor 5
- CDR
complementarity-determining region
- CDR-SB
Clinical Dementia Rating–Sum of Boxes
- Chl
classical Hodgkin lymphoma
- CHMP
Committee for Medicinal Products for Human Use
- CIS
carcinoma in situ
- CLBP
chronic lower back pain
- CLL
chronic lymphocytic leukemia
- CNS
central nervous system
- CPI
checkpoint inhibitor
- CR
complete response
- CTLA-4
cytotoxic T lymphocyte antigen-4
- DLBCL
diffuse large B-cell lymphoma
- DLL3
delta-like ligand 3
- DOR
duration of response
- EC
European Commission
- EMA
European Medicines Agency
- EpCAM
epithelial cell adhesion molecule
- EU
European Union
- FDA
US Food and Drug Administration
- GM-CSF
granulocyte-macrophage colony stimulating factor
- GP
glycoprotein
- GvHD
graft-versus-host disease
- HDL
high density lipoprotein
- HER2
human epidermal receptor 2
- HoFH
homozygous familial hypercholesterolemia
- HSCT
hematopoietic stem cell transplant
- IBD
inflammatory bowel disease
- ICP
immune checkpoint therapy
- IFN
interferon
- IFNAR1
interferon alpha receptor 1
- IgA
immunoglobulin A
- IGF-1R
insulin growth factor 1 receptor
- IV
intravenous
- LDL
low-density lipoprotein
- LM
leptomeningeal metastases
- MAA
marketing authorization application
- mAb
monoclonal antibody
- MASP-2
mannan-binding lectin-associated serine protease-2
- MBC
metastatic breast cancer
- mCRC
metastatic colorectal cancer
- MCS
Mayo Clinic Score
- MHLW
Ministry of Health, Labor and Welfare
- MM
multiple myeloma
- MMAE
monomethyl auristatin E
- MSI
microsatellite instability
- MSS
microsatellite stable
- MTD
maximally tolerated dose
- nAMD
neovascular age-related macular degeneration
- NASH
nonalcoholic steatohepatitis
- NDA
new drug application
- NKTL
natural killer cell/T-cell lymphoma
- NMIBC
non-muscle invasive bladder cancer
- NMOSD
neuromyelitis optica spectrum disorder
- NSAIDs
nonsteroidal anti-inflammatory drugs
- NSCLC
non-small cell lung cancer
- OA
osteoarthritis
- ORR
objective response rate
- OS
overall survival
- PBD
pyrrolobenzodiazepine
- PD-1
programmed cell death 1 protein
- PDGFR-a
platelet-derived growth factor receptor alpha
- PD-L1
programmed death ligand 1
- PDUFA
Prescription Drug User Fee Act
- PFS
progression-free survival
- PGA
Patient’s Global Assessment
- PRIME
Priority Medicines
- rr-cHL
relapsed or refractory cHL
- RRMM
relapsed/refractory multiple myeloma
- rr-NKTL
relapsed or refractory extranodal NKTL
- SC
subcutaneous
- scFv
single-chain variable fragment
- SCLC
small cell lung cancer
- SLE
systemic lupus erythematosus
- sPGA
static Physician Global Assessment
- STS
soft-tissue sarcoma
- T1D
type 1 diabetes
- TBNC
triple-negative breast cancer
- TED
thyroid eye disease
- TMAs
thrombotic microangiopathies
- TNF
tumor necrosis factor
- TTP
thrombotic thrombocytopenia
- UC
ulcerative colitis
- US
United States
- VEGF
human vascular endothelial growth factor
- VH
heavy chain variable domain
- VHH
variable heavy homodimers
- VOC
vaso-occlusive crisis
Acknowledgments
The authors thank Vandna Prasad Rath and Andy Cook, Hanson Wade, for providing access to the Beacon Targeted Therapies database, and Rob Jones, Craic Computing LLC, for providing access to the Therapeutic Antibody Database.
Disclosure Statement
HK is employed by a company that develops antibody therapeutics. MM and JR are employed by The Antibody Society, a non-profit trade association funded by corporate sponsors that develop antibody therapeutics or provide services to companies that develop antibody therapeutics. JR is Editor-in-Chief of mAbs. ZS is an employee and shareholder of Scholar Rock, Inc, and she did not receive compensation for the authorship of this publication. Information in this publication was collected from publicly available sources. Views expressed in this publication are the views of ZS in her personal capacity and do not reflect the views of Scholar Rock, Inc. ZS also serves as Associate Editor of mAbs, for which she does not receive compensation.
References
- 1.Kaplon H, Reichert JM.. Antibodies to watch in 2019. MAbs. 2019;11(2):219–24. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplon H, Reichert JM. Antibodies to watch in 2018. MAbs. 2018;10(2):183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reichert JM. Antibodies to watch in 2017. MAbs. 2017;9(2):167–81. doi: 10.1080/19420862.2016.1269580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reichert JM. Antibodies to watch in 2016. MAbs. 2016;8(2):197–204. doi: 10.1080/19420862.2015.1125583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reichert JM. Antibodies to watch in 2015. MAbs. 2015;7(1):1–8. doi: 10.4161/19420862.2015.988944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reichert JM. Antibodies to watch in 2014: mid-year update. MAbs. 2014;6(4):799–802. doi: 10.4161/mabs.29282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reichert JM. Antibodies to watch in 2014. MAbs. 2014;6(1):5–14. doi: 10.4161/mabs.27333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reichert JM. Antibodies to watch in 2013: mid-year update. MAbs. 2013;5(4):513–17. doi: 10.4161/mabs.24990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reichert JM. Which are the antibodies to watch in 2013? MAbs. 2013;5(1):1–4. doi: 10.4161/mabs.22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reichert JM. Which are the antibodies to watch in 2012? MAbs. 2012;4(1):1–3. doi: 10.4161/mabs.4.1.18719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs. 2011;3(1):76–99. doi: 10.4161/mabs.3.1.13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichert JM. Antibodies to watch in 2010. MAbs. 2010;2(1):84–100. doi: 10.4161/mabs.2.1.10677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.U.S. Food and Drug Administration . Drug approval package: CABLIVI. [cited 2019 Dec 5]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761112Orig1s000TOC.cfm
- 14.European Medicines Agency . Annex I summary of product characteristics. [cited 2019 Dec 5]. Available from: https://www.ema.europa.eu/en/documents/product-information/cablivi-epar-product-information_en.pdf.
- 15.Amgen, Inc . EVENITY™ (romosozumab) receives approval in Japan for the treatment of osteoporosis in patients at high risk of fracture. 2019. January 8. press release. [cited 2019 Dec 5]. Available from: https://www.amgen.com/media/news-releases/2019/01/evenity-romosozumab-receives-approval-in-japan-for-the-treatment-of-osteoporosis-in-patients-at-high-risk-of-fracture/.
- 16.U.S. Food and Drug Administration . FDA approves new treatment for osteoporosis in postmenopausal women at high risk of fracture. 2019. April 9. press release. [cited 2019 Dec 5]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-osteoporosis-postmenopausal-women-high-risk-fracture.
- 17.UCB, Amgen. EVENITY® (romosozumab) receives positive CHMP opinion for the treatment of severe osteoporosis in postmenopausal women at high risk of fracture. 2019. October 18. press release. [cited 2019 Dec 5]. Available from: https://www.ucb.com/stories-media/Press-Releases/article/EVENITY-romosozumab-Receives-Positive-CHMP-Opinion-for-the-Treatment-of-Severe-Osteoporosis-in-Postmenopausal-Women-at-High-Risk-of-Fracture.
- 18.U.S. Food and Drug Administration . SKYRIZI approval letter. [cited 2019 Dec 5]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761105s000ltr.pdf.
- 19.Reich K, Gooderham M, Thaçi D, Crowley JJ, Ryan C, Krueger JG, Tsai TF, Flack M, Gu Y, Williams DA, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet. 2019;394(10198):576–86. doi: 10.1016/S0140-6736(19)30952-3. [DOI] [PubMed] [Google Scholar]
- 20.Langley RG, Blauvelt A, Gooderham M, Papp K, Philipp S, Wu JJ, Igarashi A, Geng Z, Wu T, Camez A, et al. Efficacy and safety of continuous Q12W risankizumab versus treatment withdrawal: results from the phase 3 IMMhance trial. [cited 2019 Dec 5]. Available from: https://server.aad.org/eposters/Submissions/getFile.aspx?id=10093&type=sub.
- 21.Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, Papp KA, Sofen H, Puig L, Foley P, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–61. doi: 10.1016/S0140-6736(18)31713-6. [DOI] [PubMed] [Google Scholar]
- 22.U.S. Food and Drug Administration . Polivy approval letter. [cited 2019 Dec 5]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761121Orig1s000ltr.pdf.
- 23.Novartis . Novartis receives FDA approval for Beovu®, offering wet AMD patients vision gains and greater fluid reductions vs aflibercept. 2019. October 8. press release. [cited 2019 Dec 5]. Available from: https://www.novartis.com/news/media-releases/novartis-receives-fda-approval-beovu-offering-wet-amd-patients-vision-gains-and-greater-fluid-reductions-vs-aflibercept.
- 24.Dugel PU, Koh A, Ogura Y, Jaffe GJ, Schmidt-Erfurth U, Brown DM, Gomes AV, Warburton J, Weichselberger A, Holz FG; HAWK and HARRIER Study Investigators . HAWK and HARRIER: phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology. 2019:S0161-6420(18)33018–5. doi: 10.1016/j.ophtha.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Novartis . Two-year data for Novartis brolucizumab reaffirm superiority versus aflibercept in reducing retinal fluid in patients with nAMD. 2018. October 27. press release. [cited 2019 Dec 5]. Available from: https://www.novartis.com/news/media-releases/two-year-data-novartis-brolucizumab-reaffirm-superiority-versus-aflibercept-reducing-retinal-fluid-patients-namd.
- 26.US Food and Drug Administration . FDA approves first targeted therapy to treat patients with painful complication of sickle cell disease. 2019. November 15. press release. [cited 2019 Dec 5]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-therapy-treat-patients-painful-complication-sickle-cell-disease.
- 27.Novartis . FDA accepts file and accelerates review of Novartis sickle cell disease medicine crizanlizumab (SEG101). 2019. July 16. press release. [cited 2019 Dec 5]. Available from: https://www.novartis.com/news/media-releases/fda-accepts-file-and-accelerates-review-novartis-sickle-cell-disease-medicine-crizanlizumab-seg101.
- 28.Kutlar A, Kanter J, Liles DK, Alvarez OA, Cançado RD, Friedrisch JR, Knight-Madden JM, Bruederle A, Shi M, Zhu Z, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am J Hematol. 2019;94(1):55–61. doi: 10.1002/ajh.25308. [DOI] [PubMed] [Google Scholar]
- 29.BIOCAD . BIOCAD registered the first Russian original therapeutic monoclonal antibody 2019. May 7. press release. [[cited 2019 Dec 5]. Available from: https://biocadglobal.com/post/Russian_original_therapeutic_monoclonal_antibody.
- 30.Kostareva O, Kolyadenko I, Ulitin A, Ekimova V, Evdokimov S, Garber M, Tishchenko S, Gabdulkhakov A. Fab fragment of VHH-based antibody netakimab: crystal structure and modeling interaction with cytokine IL-17A. Crystals. 2019;9(3):177. doi: 10.3390/cryst9030177. [DOI] [Google Scholar]
- 31.Zydus . Zydus to launch novel biologic for rabies, TwinrabTM. [cited 2019 Dec 5]. Available from: https://zyduscadila.com/public/pdf/pressrelease/Zydus%20to%20launch%20novel%20biologic%20for%20rabies_WHO.pdf.
- 32.U.S. Food and Drug Administration . Monoclonal antibody product development for rabies post-exposure prophylaxis. 2017. July 17. https://www.fda.gov/media/107165/download.
- 33.Alder BioPharmaceuticals, Inc . Alder BioPharmaceuticals reports second quarter 2019 financial and operating results. 2019. August 6. press release. [cited 2019 Dec 5]. Available from: https://investor.alderbio.com/news-releases/news-release-details/alder-biopharmaceuticalsr-reports-second-quarter-2019-financial.
- 34.Horizon Pharma plc . Horizon Pharma plc announces Phase 3 confirmatory trial evaluating teprotumumab (OPTIC) for the treatment of active thyroid eye disease (TED) met primary and all secondary endpoints. 2019. February 28. press release. [cited 2019 Dec 5]. Available from: https://ir.horizontherapeutics.com/news-releases/news-release-details/horizon-pharma-plc-announces-phase-3-confirmatory-trial.
- 35.Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, Harris GJ, Antonelli A, Salvi M, Goldberg RA, et al. Teprotumumab for thyroid-associated ophthalmopathy. N Engl J Med. 2017;376(18):1748–61. doi: 10.1056/NEJMoa1614949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horizon Pharma plc . Horizon therapeutics plc announces the FDA has granted priority review of the teprotumumab biologics license application (BLA) for the treatment of active thyroid eye disease (TED). 2019. September 9. press release. [cited 2019 Dec 5]. Available from: https://ir.horizontherapeutics.com/news-releases/news-release-details/horizon-therapeutics-plc-announces-fda-has-granted-priority.
- 37.Seattle Genetics, Astellas . Seattle Genetics and Astellas announce submission of biologics license application to FDA for enfortumab vedotin for patients with locally advanced or metastatic urothelial cancer. 2019. July 16. press release. [cited 2019 Dec 5]. Available from: http://investor.seattlegenetics.com/news-releases/news-release-details/seattle-genetics-and-astellas-announce-submission-biologics.
- 38.Petrylak DP, Balar AV, O’Donnell PH, McGregor BA, Heath EI, Yu EY, Galsky MD, Hahn NM, Gartner EM, Pinelli J, et al. EV-201: results of enfortumab vedotin monotherapy for locally advanced or metastatic urothelial cancer previously treated with platinum and immune checkpoint inhibitors. J Clin Oncol. 2019. June 20; 37(18_suppl):4505–4505. doi: 10.1200/JCO.2019.37.18_suppl.LBA4505. [DOI] [Google Scholar]
- 39.Moreno L, Perez C, Zabaleta A, Manrique I, Alignani D, Ajona D, Blanco L, Lasa M, Maiso P, Rodriguez I, et al. The mechanism of action of the anti-cd38 monoclonal antibody isatuximab in multiple myeloma. Clin Cancer Res. 2019;25(10):3176–87. doi: 10.1158/1078-0432.CCR-18-1597. [DOI] [PubMed] [Google Scholar]
- 40.Sanofi . Phase 3 trial of isatuximab combination therapy showed 40% reduction in the risk of disease progression or death for patients with relapsed/refractory multiple myeloma. 2019. June 2. press release. [cited 2019 Dec 5]. Available from: http://www.news.sanofi.us/press-releases?item=137220.
- 41.Sanofi . FDA to review isatuximab as a potential treatment for relapsed/refractory multiple myeloma. 2019. July 10. press release. [cited 2019 Dec 5]. Available from: https://www.sanofi.com/en/media-room/press-releases/2019/2019-07-10-07-00-00.
- 42.AstraZeneca and Daiichi Sankyo Company, Limited . Trastuzumab deruxtecan granted FDA priority review for treatment of patients with HER2-positive metastatic breast cancer. 2019. October 17. press release. [cited 2019 Dec 5]. Available from: https://www.astrazeneca.com/media-centre/press-releases/2019/trastuzumab-deruxtecan-granted-fda-priority-review-for-treatment-of-patients-with-her2-positive-metastatic-breast-cancer-17102019.html.
- 43.AstraZeneca and Daiichi Sankyo Company, Limited . Pivotal phase II DESTINY-Breast01 trial met primary endpoint, supporting global regulatory submission plan to start in H2 2019. 2019. May 8. press release. [cited 2019 Dec 5]. Available from: https://www.astrazeneca.com/media-centre/press-releases/2019/trastuzumab-deruxtecan-demonstrated-clinically-meaningful-response-in-patients-with-refractory-her2-positive-metastatic-breast-cancer-a-population-with-high-unmet-need-08052019.html.
- 44.Daiichi Sankyo Company, Limited . Daiichi Sankyo advances [Fam-] Trastuzumab deruxtecan (DS-8201) in Japan with regulatory submission in HER2 positive metastatic breast cancer. 2019. September 9. press release. [cited 2019 Dec 5]. Available from: https://www.daiichisankyo.com/media_investors/media_relations/press_releases/detail/007047.html.
- 45.AstraZeneca and Daiichi Sankyo Company, Limited . AstraZeneca announces two lancet oncology publications of phase I dose-expansion results of trastuzumab deruxtecan in HER2-positive metastatic breast and gastric cancer. 2019. April 29. press release. [cited 2019 Dec 5]. Available from: https://www.astrazeneca.com/media-centre/medical-releases/astrazeneca-announces-two-lancet-oncology-publications-of-phase-i-dose-expansion-results-of-trastuzumab-deruxtecan-in-her2-positive-metastatic-breast-and-gastric-cancer.html#!
- 46.Viela Bio . Viela Bio announces U.S. fda accepts for review inebilizumab biologics license application for neuromyelitis optica spectrum disorder. 2019. August 27. press release. [cited 2019 Dec 5]. Available from: https://vielabio.com/bla_accept/.
- 47.Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, Fujihara K, Paul F, Cutter GR, Marignier R, et al.; N-MOmentum study investigators . Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019:S0140-6736(19)31817–3. doi: 10.1016/S0140-6736(19)31817-3. [DOI] [PubMed] [Google Scholar]
- 48.CytoDyn Inc . CytoDyn reaches historical milestone, submits first of three sections of BLA to FDA for Leronlimab (PRO 140) as a combination therapy for HIV. 2019. March 18. press release. [cited 2019 Dec 5]. Available from: https://www.cytodyn.com/investors/news-events/press-releases/detail/321/cytodyn-reaches-historical-milestonesubmits-first-of.
- 49.CytoDyn Inc . Investor presentation. 2019. September 25.https://content.equisolve.net/_cb938b9f9372eed3b2f00594e8a2df74/cytodyn/db/193/2912/pdf/CytoDyn+-+Investor_presentation+-+Sep-25-2019-Final.pdf.
- 50.CytoDyn Inc . CytoDyn provides update on dose escalating trial with leronlimab for HIV monotherapy for a potential pivotal trial. 2019. August 14. press release. [cited 2019 Dec 5]. Available from: https://www.cytodyn.com/newsroom/press-releases/detail/350/cytodyn-provides-update-on-dose-escalating-trial-with.
- 51.Goldenberg DM, Sharkey RM. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: A case study of anti-TROP-2 sacituzumab govitecan. MAbs. 2019. Aug/Sep;11(6):987–95. doi: 10.1080/19420862.2019.1632115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Immunomedics, Inc . Immunomedics receives complete response letter from FDA for sacituzumab govitecan biologics license application. 2019. January 17. press release. [cited 2019 Dec 5]. Available from: https://www.immunomedics.com/our-company/news-and-events/immunomedics-receives-complete-response-letter-from-fda-for-sacituzumab-govitecan-biologics-license-application/.
- 53.Immunomedics, Inc . Immunomedics resubmits biologics license application to the FDA for sacituzumab govitecan. 2019. December 3. press release. [cited 2019 Dec 5]. Available from: https://www.immunomedics.com/our-company/news-and-events/immunomedics-resubmits-biologics-license-application-to-the-fda-for-sacituzumab-govitecan/.
- 54.Immunomedics, Inc . Immunomedics and everest medicines announce exclusive license agreement for sacituzumab govitecan in East and Southeast Asia excluding Japan. 2019. April 29. press release. [cited 2019 Dec 5]. Available from: https://www.immunomedics.com/our-company/news-and-events/immunomedics-and-everest-medicines-announce-exclusive-license-agreement-for-sacituzumab-govitecan-in-east-and-southeast-asia-excluding-japan/.
- 55.European Medicines Agency . Applications for new human medicines under evaluation by the committee for medicinal products for human use. 2019. October. [cited 2019 Dec 5]. Available from: https://www.ema.europa.eu/en/documents/report/applications-new-human-medicines-under-evaluation-chmp-october-2019_en.pdf.
- 56.Chugai Pharmaceutical Co., Ltd . chugai receives orphan drug designation for satralizumab in neuromyelitis optica and neuromyelitis optica spectrum disorder from the ministry of health, labour and welfare. 2019. September 13. press release. [cited 2019 Dec 5]. Available from: https://www.chugai-pharm.co.jp/english/news/detail/20190913140000_646.html.
- 57.Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, Patti F, Tsai CP, Saiz A, Yamazaki H, et al. Trial of satralizumab in neuromyelitis optica Spectrum disorder. N Engl J Med. 2019;381(22):2114–24. doi: 10.1056/NEJMoa1901747. [DOI] [PubMed] [Google Scholar]
- 58.Chugai Pharmaceutical Co., Ltd . Chugai presents results from second positive global Phase III clinical study of satralizumab in NMOSD at ECTRIMS 2019. 2019. September 12. press release. [cited 2019 Dec 5]. Available from: https://www.chugai-pharm.co.jp/english/news/detail/20190912140300_638.html.
- 59.Omeros Corporation . Narsoplimab (OMS721). Omeros corporation streamlines path to biologics license application for OMS721 in HSTC-TMA following meeting with FDA. 2019. February 14. press release. [cited 2019 Dec 5]. Available from: https://investor.omeros.com/news-releases/news-release-details/omeros-corporation-streamlines-path-biologics-license.
- 60.Omeros Corporation . Omeros corporation initiates BLA submission for narsoplimab for the treatment of HSCT-TMA. 2019. October 28. press release. [cited 2019 Dec 5]. Available from: https://investor.omeros.com/news-releases/news-release-details/omeros-corporation-initiates-bla-submission-narsoplimab.
- 61.Rambaldi A, Khaled S, Smith M, Zecca M, Kwong YM, Claes K, Leung N, Whitaker S. Improved survival following OMS721 treatment of hematopoietic stem cell transplant-associated thrombotic microangiopathies (HCT-TMA). Eur Hematol Assoc Lib. 2018. June 15; 215162:PF724. [cited 2019 Dec 5]. Available from: https://library.ehaweb.org/eha/2018/stockholm/215162/alessandro.rambaldi.improved.survival.following.oms721.treatment.of.html [Google Scholar]
- 62.Kellner C, Zhukovsky EA, Pötzke A, Brüggemann M, Schrauder A, Schrappe M, Kneba M, Repp R, Humpe A, Gramatzki M, et al. The Fc-engineered CD19 antibody MOR208 (XmAb5574) induces natural killer cell-mediated lysis of acute lymphoblastic leukemia cells from pediatric and adult patients. Leukemia. 2013;27(7):1595–98. doi: 10.1038/leu.2012.373. [DOI] [PubMed] [Google Scholar]
- 63.MorphoSys AG. MorphoSys AG reports second quarter 2019 financial results. 2019. August 6. press release. [cited 2019 Dec 5]. Available from: https://www.morphosys.com/media-investors/media-center/morphosys-ag-reports-second-quarter-2019-financial-results.
- 64.MorphoSys AG. MorphoSys AG: primary endpoint met in real-world data study demonstrating clinical superiority of the combination of tafasitamab and lenalidomide compared to lenalidomide alone. 2019. October 29. press release. [cited 2019 Dec 5]. Available from: https://www.morphosys.com/media-investors/media-center/morphosys-ag-primary-endpoint-met-in-real-world-data-study.
- 65.MorphoSys AG. Primary endpoint of L-MIND, a combination study of tafasitamab (MOR208) and lenalidomide, has been met, confirming previously published activity. 2019. May 16. press release. [cited 2019 Dec 5]. Available from: https://www.morphosys.com/media-investors/media-center/morphosys-ag-primary-endpoint-of-l-mind-a-combination-study-of.
- 66.Pascal KE, Dudgeon D, Trefry JC, Anantpadma M, Sakurai Y, Murin CD, Turner HL, Fairhurst J, Torres M, Rafique A, et al. Development of clinical-stage human monoclonal antibodies that treat advanced Ebola virus disease in nonhuman primates. J Infect Dis. 2018;218(suppl_5):S612–S626. doi: 10.1093/infdis/jiy285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Regeneron Pharmaceuticals, Inc . Regeneron reports third quarter 2019 financial and operating results. 2019. November 5. press release. [cited 2019 Dec 5]. Available from: https://investor.regeneron.com/news-releases/news-release-details/regeneron-reports-third-quarter-2019-financial-and-operating.
- 68.Mulangu S, Dodd LE, Davey RT Jr, Tshiani Mbaya O, Proschan M, Mukadi D, Lusakibanza Manzo M, Nzolo D, Tshomba Oloma A, Ibanda A, et al. A randomized, controlled trial of Ebola virus disease therapeutics. N Engl J Med. 2019. November 27. doi: 10.1056/NEJMoa1910993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Y-mabs Therapeutics, Inc . Y-mAbs initiates rolling submission of biologics license application to U.S. FDA for naxitamab for treatment of neuroblastoma. 2019. November 29. press release. [cited 2019 Dec 5]. Available from: http://ir.ymabs.com/node/7156/pdf.
- 70.Y-mabs Therapeutics, Inc . Y-mAbs announces naxitamab update. 2019. October 25. press release. [cited 2019 Dec 5]. Available from: http://ir.ymabs.com/news-releases/news-release-details/y-mabs-announces-naxitamab-update.
- 71.Y-mabs Therapeutics, Inc . Y-mAbs announces second quarter 2019 financial results and recent corporate developments. 2019. August 14. press release. http://ir.ymabs.com/news-releases/news-release-details/y-mabs-announces-second-quarter-2019-financial-results-and.
- 72.Eli Lilly and Company . Pfizer and Lilly announce top-line results from long-term phase 3 study of tanezumab in patients with osteoarthritis. 2019. April 18. press release. https://investor.lilly.com/news-releases/news-release-details/pfizer-and-lilly-announce-top-line-results-long-term-phase-3.
- 73.Eli Lilly and Company . Lilly reports second-quarter 2019 financial results, raises 2019 EPS guidance. 2019. July 30. press release. [cited 2019 Dec 5]. Available from: https://investor.lilly.com/news-releases/news-release-details/lilly-reports-second-quarter-2019-financial-results-raises-2019.
- 74.Schnitzer TJ, Easton R, Pang S, Levinson DJ, Pixton G, Viktrup L, Davignon I, Brown MT, West CR, Verburg KM. Effect of tanezumab on joint pain, physical function, and patient global assessment of osteoarthritis among patients with osteoarthritis of the hip or knee: A randomized clinical trial. JAMA. 2019;322(1):37–48. doi: 10.1001/jama.2019.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arndt JW, Qian F, Smith BA, Quan C, Kilambi KP, Bush MW, Walz T, Pepinsky RB, Bussière T, Hamann S, et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci Rep. 2018. 23;8(1):6412. doi: 10.1038/s41598-018-24501-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Biogen Inc., Eisai, Co., Ltd . Biogen plans regulatory filing for aducanumab in Alzheimer’s disease based on new analysis of larger dataset from phase 3 studies. 2019. October 22. press release. [cited 2019 Dec 5]. Available from: http://investors.biogen.com/news-releases/news-release-details/biogen-plans-regulatory-filing-aducanumab-alzheimers-disease.
- 77.Regeneron Pharmaceuticals, Inc . Regeneron announces positive topline results from phase 3 trial of evinacumab in patients with severe, inherited form of high cholesterol. 2019. August 14. press release. [cited 2019 Dec 5]. Available from: https://newsroom.regeneron.com/news-releases/news-release-details/regeneron-announces-positive-topline-results-phase-3-trial.
- 78.Lichnog C, Klabunde S, Becker E, Fuh F, Tripal P, Atreya R, Klenske E, Erickson R, Chiu H, Reed C, et al. Cellular mechanisms of etrolizumab treatment in inflammatory bowel disease. Front Pharmacol. 2019;10:39. doi: 10.3389/fphar.2019.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roche . Pipeline summary as of July 25, 2019 (slides 5, 72, 73). [cited 2019 Dec 5]. Available from: https://www.roche.com/dam/jcr:9fb4878c-e3a2-4fdc-80f6-62d435df7367/en/pharmahy19.pdf.
- 80.Peyrin-Biroulet L, Feagan BG, Mansfield J, Rubin DT, Arulmani U, Maciuca R, Tyrrell H, Thommes J, Tole S. Etrolizumab treatment leads to early improvement in symptoms and inflammatory biomarkers in anti-TNF-refractory patients in the open-label induction cohort of the phase 3 HICKORY study. European Crohn’s and Colitis Organization meeting OP011. 2017. [cited 2019 Dec 5]. Available from: https://www.ecco-ibd.eu/publications/congress-abstract-s/abstracts-2017/item/op011-etrolizumab-treatment-leads-to-early-improvement-in-symptoms-and-inflammatory-biomarkers-in-anti-tnf-refractory-patients-in-the-open-label-induction-cohort-of-the-phase-3-hickory-study.html.
- 81.Bioveractiv Inc . Bioverativ announces first patient dosed in phase 3 study of BIVV009 for treatment of cold agglutinin disease. 2018. March 6. press release. [cited 2019 Dec 5]. Available from: https://www.sanofigenzyme.com/en/about-us/newsroom/Bioverativ/2018/2018-03-06-07-30-00.
- 82.Sanofi . Sanofi delivered solid growth in Q2 2019. 2019. July 29. press release. [cited 2019 Dec 5]. Available from: https://www.sanofi.com/en/media-room/press-releases/2019/2019-07-29-07-30-00.
- 83.Sanofi . Pivotal data from phase 3 study of sutimlimab in cold agglutinin disease to be presented at ASH 2019 late-breaking session. 2019. November 21. press release. [cited 2019 Dec 5]. Available from: https://www.sanofigenzyme.com/en/about-us/newsroom/2019/2019-11-21-09-22-00.
- 84.AstraZeneca PLC . Year-to-date and Q3 2019 results. 2019. October 24. press release. [cited 2019 Dec 5]. Available from: https://www.astrazeneca.com/content/dam/az/PDF/2019/q3/Year-to-date_and_Q3_2019_Results_announcement.pdf.
- 85.AstraZeneca PLC. Anifrolumab phase III trial meets primary endpoint in systemic lupus erythematosus. 2019. August 29. press release. [cited 2019 Dec 5]. Available from: https://www.astrazeneca.com/media-centre/press-releases/2019/anifrolumab-phase-iii-trial-meets-primary-endpoint-in-systemic-lupus-erythematosus-29082019.html.
- 86.AstraZeneca PLC. Anifrolumab demonstrated superiority across multiple efficacy endpoints in patients with systemic lupus erythematosus in phase III TULIP 2 trial. 2019. November 11. press release. [cited 2019 Dec 5]. Available from: https://www.astrazeneca.com/media-centre/press-releases/2019/anifrolumab-demonstrated-superiority-across-multiple-efficacy-endpoints-in-patients-with-systemic-lupus-erythematosus-in-phase-iii-tulip-2-trial.html.
- 87.Provention Bio, Inc . Provention Bio announces PRV-031 (Teplizumab) granted PRIME designation by the European medicines agency. October 24, 2019. press release. [cited 2019 Dec 5]. Available from: http://investors.proventionbio.com/2019-10-24-Provention-Bio-Announces-PRV-031-Teplizumab-Granted-PRIME-Designation-by-the-European-Medicines-Agency.
- 88.Provention Bio, Inc . Provention Bio reiterates regulatory strategy for PRV-031 and announces completion of follow-on financing. 2019. September 24. press release. [cited 2019 Dec 5]. Available from: http://investors.proventionbio.com/2019-09-24-Provention-Bio-Reiterates-Regulatory-Strategy-for-PRV-031-and-Announces-Completion-of-Follow-on-Financing.
- 89.Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, Gitelman SE, Gottlieb PA, Krischer JP, Linsley PS, et al.; Type 1 Diabetes TrialNet Study Group . An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med. 2019;381(7):603–13. doi: 10.1056/NEJMoa1902226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.GlaxoSmithKline plc. Third quarter 2019. 2019. October 30. press release. [cited 2019 Dec 5]. Available from: https://www.gsk.com/media/5735/q3-2019-results-announcement.pdf.
- 91.Sesen Bio . Sesen bio reports third quarter 2019 financial results and update on regulatory activities related to vicinium. 2019. November 12. press release. [cited 2019 Dec 5]. Available from: https://ir.sesenbio.com/news-releases/news-release-details/sesen-bio-reports-third-quarter-2019-financial-results-and.
- 92.Dickstein R, Wu N, Cowan B, Dunshee C, Franks M, Wolk F, Belkoff L, Castellucci S, Holzbeierlein J, Kulkarni G, et al. VISTA, phase 3 trial of Vicinium, an EPCAM-targeted Pseudomonas exotoxin, in BCG-unresponsive non-muscle invasive bladder cancer. Global Congress on Bladder Cancer 2018; 2018. September 20 – 21; Madrid, Spain. [cited 2019 Dec 5]. Available from: https://sesenbio.com/wp-content/uploads/2019/01/BLADDR_Congress_2018_Posterl.pdf. [Google Scholar]
- 93.Nordstrom JL, Gorlatov S, Zhang W, Yang Y, Huang L, Burke S, Li H, Ciccarone V, Zhang T, Stavenhagen J, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 2011;13(6):R123. doi: 10.1186/bcr3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.MacroGenics provides update on corporate progress and second quarter 2019 financial results. 2019. July 31. press release. [cited 2019 Dec 5]. Available from: http://ir.macrogenics.com/news-releases/news-release-details/macrogenics-provides-update-corporate-progress-and-second-3.
- 95.Rugo HS, Im S-A, Wright GLS, Escriva-de-Romani S, DeLaurentiis M, Cortes J, Wazeen Bahadur S, Haley BB, Oyola RH, Riseberg DA, et al. SOPHIA primary analysis: A phase 3 (P3) study of margetuximab (M) + chemotherapy (C) versus trastuzumab (T) + C in patients (pts) with HER2+ metastatic (met) breast cancer (MBC) after prior anti-HER2 therapies (Tx). American Society of Clinical Oncology annual meeting; 2019. June 4; [cited 2019 Dec 5]. Available from: https://meetinglibrary.asco.org/record/170929/abstract. [Google Scholar]
- 96.GlaxoSmithKline . Second quarter 2019 results. 2019. July 24. press release. [cited 2019 Dec 5]. Available from: https://www.gsk.com/media/5663/q2-2019-results-announcement.pdf.
- 97.GlaxoSmithKline . Data from GARNET study indicates robust activity of dostarlimab in patients with advanced or recurrent endometrial cancer. 2019. March 19. press release. [cited 2019 Dec 5]. Available from: https://us.gsk.com/en-us/media/press-releases/data-from-garnet-study-indicates-robust-activity-of-dostarlimab-in-patients-with-advanced-or-recurrent-endometrial-cancer/.
- 98.Novartis . Q3 2019 results. investor presentation. 2019. October 22. p.20. [cited 2019 Dec 5]. Available from: https://www.novartis.com/sites/www.novartis.com/files/q3-2019-ir-presentation.pdf
- 99.Long GV, Lebbe C, Atkinson V, Mandalà M, Nathan PD, Arance Fernandez AM, Richtig E, Yamazaki N, Robert C, Schadendorf D, et al. The anti–PD-1 antibody spartalizumab (S) in combination with dabrafenib (D) and trametinib (T) in previously untreated patients (pts) with advanced BRAF V600–mutant melanoma: updated efficacy and safety from parts 1 and 2 of COMBI-i. Abstract #9531. 2019 American Society of Clinical Oncology Annual Meeting; May 31-June 4; Chicago, [cited 2019 Dec 5]. Available from: IL.https://ascopubs.org/doi/abs/10.1200/JCO.2019.37.15_suppl.9531. [Google Scholar]
- 100.Y-mAbs Therapeutics, Inc . United States securities and exchange commission form 8-K. 2019. November 1. [cited 2019 Dec 5]. Available from: https://www.sec.gov/Archives/edgar/data/1722964/000110465919058623/a19-21369_38k.htm.
- 101.ADC Therapeutics SA . United States securities and exchange commission form F-1. 2019. September 6. [cited 2019 Dec 5]. Available from: https://www.sec.gov/Archives/edgar/data/1771910/000114036119016305/nt10002526x5_f1.htm.
- 102.Breous-Nystrom E, Schultze K, Meier M, Flueck L, Holzer C, Boll M, Seibert V, Schuster A, Blanusa M, Schaefer V, et al. Retrocyte Display® technology: generation and screening of a high diversity cellular antibody library. Methods. 2014;65(1):57–67. doi: 10.1016/j.ymeth.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 103.Agenus Inc . United States securities and exchange commission form 10-Q for the quarterly period ended June 30, 2019. 2019. August 9. [cited 2019 Dec 5]. Available from: https://investor.agenusbio.com/SEC-Filings.
- 104.Morgensztern D, Besse B, Greillier L, Santana-Davila R, Ready N, Hann CL, Glisson BS, Farago AF, Dowlati A, Rudin CM, et al. Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-expressing, relapsed/refractory small cell lung cancer: results from the phase II TRINITY study. Clin Cancer Res. 2019:clincanres.1133.2019. doi: 10.1158/1078-0432.CCR-19-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.AbbVie Inc . AbbVie discontinues rovalpituzumab tesirine (Rova-T) research and development program. 2019. August 29. press release. [cited 2019 Dec 5]. Available from: https://news.abbvie.com/news/press-releases/abbvie-discontinues-rovalpituzumab-tesirine-rova-t-research-and-development-program.htm.
- 106.Eli Lilly and Company . Lilly to establish an access program for patients as it prepares to withdraw Lartruvo from the global market. 2019. April 25. press release. [cited 2019 Dec 5]. Available from: https://investor.lilly.com/news-releases/news-release-details/lilly-establish-access-program-patients-it-prepares-withdraw.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- U.S. Food and Drug Administration . Drug approval package: CABLIVI. [cited 2019 Dec 5]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761112Orig1s000TOC.cfm
- European Medicines Agency . Annex I summary of product characteristics. [cited 2019 Dec 5]. Available from: https://www.ema.europa.eu/en/documents/product-information/cablivi-epar-product-information_en.pdf.