

Abstract
The integrity of the proteome in cardiac myocytes is critical for robust heart function. Proteome integrity in all cells is managed by protein homeostasis or proteostasis, which encompasses processes that maintain the balance of protein synthesis, folding, and degradation in ways that allow cells to adapt to conditions that present a potential challenge to viability (1). While there are processes in various cellular locations in cardiac myocytes that contribute to proteostasis, those in the cytosol, mitochondria and endoplasmic reticulum (ER) have dominant roles in maintaining cardiac contractile function. Cytosolic proteostasis has been reviewed elsewhere (2, 3); accordingly, this review focuses on proteostasis in the ER and mitochondria, and how they might influence each other and, thus, impact heart function in the settings of cardiac physiology and disease.
Keywords: mitochondria, proteostasis, UPR, endoplasmic reticulum, protein folding
ER Proteostasis
Most secreted and membrane proteins are made in the ER, making it a major site for proteostasis (4, 5). Moreover, the specialized ER in cardiac myocytes, which includes the sarco/endoplasmic reticulum, is responsible for contractile calcium handling (6–9), and most of the proteins that are required for this important function of the heart are made at the ER (10, 11). Thus, ER proteostasis in the heart, and in particular in cardiac myocytes, is critical for proper cardiac function. ER proteostasis requires an environment that optimizes a balanced synthesis, folding and degradation of proteins made in this location (Figure 1A). Conditions, including cardiac pathologies can perturb the ER environment in ways that decrease the efficiency of ER protein folding, leading to the accumulation of potentially toxic misfolded proteins, which imbalance and dysregulate proteostasis, leading to activation of the unfolded protein response (UPR) (Figure 1B) (12). Misfolded proteins in the ER are detected by 3 well studied transmembrane proteins, ATF6 (activating transcription factor 6), IRE1 (inositol requiring enzyme 1) and PERK (protein kinase R [PKR]-like ER kinase), each of which exhibits a unique mechanism of activation in response to the accumulation of misfolded proteins in the ER; thus, ATF6, IRE1, and PERK initiate three different but complementary branches of the ER unfolded protein response (UPRER) (Figure 1C) (13). The UPRER can also be activated by other cellular stresses that could impact proteostasis, or may be independent of it, including changes in ER lipid content (14), hypoxia (15, 16), growth stimuli and reactive oxygen species (17). Thus, while ATF6, IRE1 and PERK were originally found to all be activated by overt ER protein misfolding, it is now clear that they are activated differentially by different pathophysiological stresses and, as a result, the downstream signaling events initiated by each stress are different yet complementary, as far as their ultimate effects on cell function.
Figure 1.
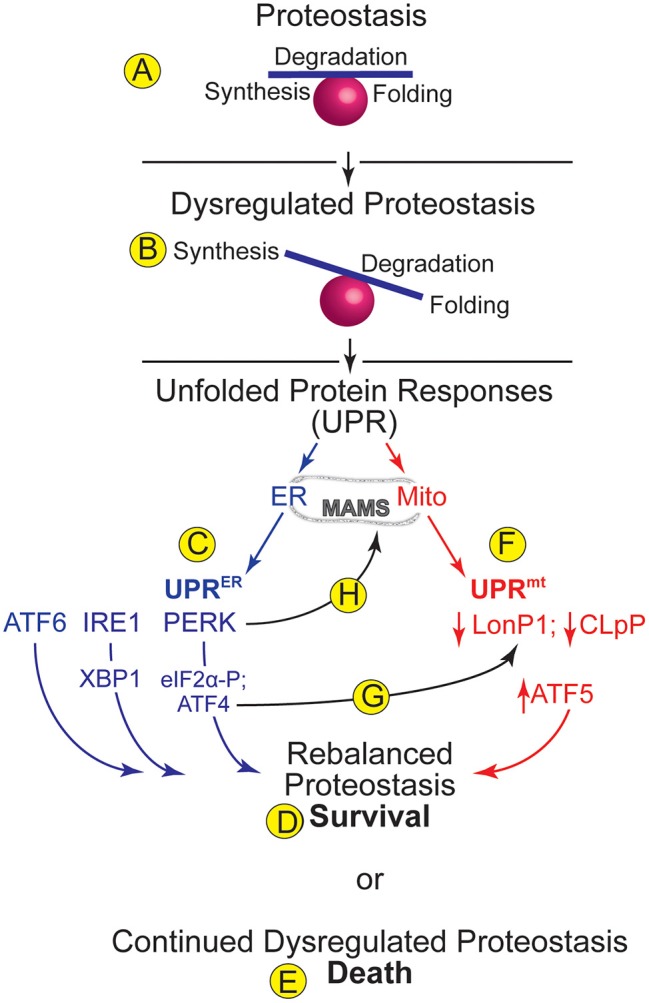
ER and Mitochondrial Proteostasis- (A) Proteostasis encompasses processes such as protein synthesis, degradation, and folding. A balance amongst such processes supports optimal proteome integrity. (B) Dysregulated proteostasis occurs when environmental conditions, including cardiac pathology, cause an imbalance in these processes, which activates adaptive compensatory responses, such as the unfolded protein responses (UPRs) in various organelles. (C) The UPR in the endoplasmic reticulum (ER) is called the UPRER. Increased levels of misfolded proteins in the ER activate three ER transmembrane proteins, ATF6, IRE1, and PERK, which cause increases in the transcription factors ATF6, XBP1, and ATF4, which together regulate genes designed to rebalance ER proteostasis. PERK also phosphorylates eIF2a, which arrests translation of most mRNAs, thus relieving the protein-folding burden on the ER and allowing for cell survival (D). (E) Continued dysregulation of proteostasis leads to chronic activation of the UPRER proximal sensors and cell death. (F) The UPR in mitochondria (mt) is called the UPRmt. The levels of the mitochondrial proteases, LonP1 and CLpP decrease upon dysregulation of mitochondrial proteostasis. Decreased LonP1 and CLpP contribute to increasing the level of the transcription factor, ATF5, which regulates genes designed to rebalance mitochondrial proteostasis. (G) A potential integration point between the UPRER and the UPRmt is the ability of the UPRER-activated transcription factor, ATF4 to increase expression of the UPRmt protease, LonP1. (H) Another potential integration point between the UPRER and the UPRmt is the ability of PERK to tether the ER to mitochondria at contact sites called mitochondrial associated membranes (MAMS).
In terms of the canonical role for ER stress, initially, UPRER signaling is designed to restore proper protein folding to the ER, constituting an adaptive return to proteostasis and cell survival (Figure 1D). This restoration takes place at many levels, including enhanced expression of chaperones to facilitate protein-folding, increases in the rate at which misfolded proteins in the ER are degraded through a process called ER associated degradation (ERAD) (18), and decreases in translation of mRNAs that encode proteins that are not required for the restoration of ER proteostasis (19, 20). However, if these complex initial events of the UPRER are not sufficient to restore proteostasis, then continued dysregulation of proteostasis leads to chronic activation of the proximal sensors and cell death (Figure 1E), and is thus considered maladaptive (21).
ER Proteostasis in Cardiac Pathology
A number of studies have demonstrated important roles for the UPRER in the heart; most of these studies have focused on examining ER proteostasis in cardiac myocytes. For example, the ATF6 branch of the UPRER is mainly adaptive and can protect the heart during pathophysiological maneuvers involving ischemia/reperfusion (I/R) and pressure overload in mice (13, 17, 22–26), the latter of which mimics hypertension and stimulates pathological growth of the heart. The adaptive effects of ATF6 are considered to be largely due to its abilities to serve as a transcription factor following its activation (17, 25, 27). Consistent with this are findings that the genes induced by ATF6 as part of the UPRER are known to participate in adaptive restoration of proteostasis in the heart by inducing canonical adaptive UPR genes, such as those proteins that constitute the ER protein-folding machinery (Figure 2A), thus serving protective roles (28). Surprisingly, upon activation, ATF6 has been shown to induce a number of genes not previously thought to be involved in restoring ER protein folding capacity. For example, the induction of catalase during cardiac I/R (29), was a surprise, since catalase is not an ER protein, nor is it known to be involved in restoration of ER proteostasis. However, in that study it was shown that ATF6 can transcriptionally induce catalase during I/R and, as a result, catalase neutralizes damaging reactive oxygen species that accumulate during reperfusion, which decreases myocardial damage, thus describing catalase as a non-canonical adaptive UPR gene (Figure 2A, ischemia/reperfusion). In another study, it was shown that during acute pressure overload, ATF6 is necessary for the initial growth of the heart, which is an adaptive effect (17). In that study, using mice in which ATF6 was deleted specifically in cardiac myocytes, it was shown that ATF6 transcriptionally induces the small GTP binding protein, Rheb, which is an activator of mTORC1 (Figure 2A, Cardiac Hypertrophy), a well-studied pathway responsible for myocardial growth during development and pathology, thus describing Rheb as a non-canonical adaptive UPR gene induced during cardiac hypertrophy. Another study involved global deletion of ATF6 and showed that after acute pressure overload compensatory hypertrophy was impaired by ATF6 deletion, while ATF6 deletion led to increased hypertrophy and impaired function after chronic pressure overload (25). It is interesting to note that while it was not studied in the context of activating ATF6, Rheb-mediated mTORC1 activation has been shown to suppress mitophagy, which is generally considered adaptive during cardiac pathology (30–32), suggesting that mTORC1 activation via Rheb is not always adaptive in the heart.
Figure 2.

Roles for the UPRER and UPRmt in Cardiac Pathology- (A) In mouse models of cardiac ischemia/reperfusion and pathological cardiac hypertrophy there is evidence for activation of all three arms of the UPRER. (Center) Upon activation each arm of the UPR induces canonical ER stress response genes which support protection for ATF6 and IRE1/XBP1s and damage for PERK/ATF5. However, the ATF6 and IRE1/XBP1s arms of the UPRER also induce non-canonical gene programs that foster protection in the heart (left and right). (B) In mouse models of cardiac pathology the LonP1 and ATF5 aspects of the UPRmt are activated and both are protective in these disease settings.
Other branches of the UPRER have also been implicated in the adaptive responses of the heart of pathological stress. For example, in mouse hearts ischemia/reperfusion activates the IRE1 branch of the UPRER, leading to the formation of an active transcription factor, XBP1, which protects cardiac myocytes from I/R damage, in part, by inducing canonical adaptive UPR genes (Figure 2, IRE1/XBP1s; canonical) (33). In that study it was subsequently shown that XBP1 protects the mouse heart from I/R damage in a non-canonical manner by transcriptionally inducing key genes responsible for the hexosamine biosynthetic pathway, which is required for protein O-GlcNAcylation (Figure 2, I/R non-canonical). Cardiac I/R was shown to increase protein O-GlcNAcylation in the hearts of mice (34), suggesting that O-GlcNAcylation is protective. Moreover, inhibition of O-GlyNAcases increased mitochondrial OXPHOS enzyme activities, implying that this is one way that O-GlcNAcylation might be protective (35); however, the mechanism by which XBP1-mediated protein O-GlcNAcylation results in cardioprotection remains unclear. In terms of heart failure, it was shown that XBP1s stimulates adaptive cardiac growth through activation of mTORC1, which is mediated via FKBP11 (FK506-binding protein 11), a novel transcriptional target of XBP1s, thus describing a non-canonical protective for IRE1/XBP1s in pathological hypertrophy (Figure 2A, IRE1/XBPs cardiac hypertrophy) (36). It has also been shown that in a mouse model of heart failure with preserved ejection fraction (HFpEF), activation of IRE1 is deficient and restoration of activated XBP1 ameliorated the HFpEF phenotype (37). While this study indicates that IRE1 and perhaps the transcription factor, XBP1, protect against the development of HFpEF, the genes that are responsible for this protection have not been identified.
The PERK branch of the UPRER has also been studied in the heart. In many tissues, including the heart, PERK is known to be involved in numerous signaling pathways, one of which leads to activation of the transcription factor, ATF4, which increases expression of the pro-apoptotic transcription factor, C/EBP homologous protein (CHOP) (38). Since apoptosis is a major contributor to the decline in cardiac function observed during heart failure and other cardiac pathologies (39), and since CHOP expression is increased in experimental models of heart disease (40), several studies have focused on the effects of CHOP gene deletion in the mouse heart. For the most part, those studies have shown that PERK-mediated induction of CHOP in the ischemic or hypertrophic heart exacerbates cardiac pathology, in large part by increasing cardiac myocyte dropout by apoptosis (41). However, other studies that examined the effects of PERK deletion suggest that PERK is adaptive in the setting of pressure overload induced heart failure (42) (Figure 2A, PERK/ATF4). Studies such as these demonstrate the complex nature of the UPRER, indicating that depending on the circumstances, the UPRER can be adaptive or maladaptive.
Mitochondrial Proteostasis
Many cardiac physiology and pathology studies have focused on mitochondria, as they play an undeniably central role in energy generation in the metabolically demanding cardiac myocyte. Thus, processes that comprise mitochondrial quality control, which encompass proteostasis, biogenesis, dynamics (fusion and fission) and mitophagy, are critical for maintaining cardiac myocyte viability and heart contractile function (43). Among the features of mitochondrial quality control, relatively little is known about mitochondrial proteostasis in the heart. In non-cardiac cell and tissue types, stresses similar to those occurring during cardiac pathology cause the misfolding of mitochondrial proteins, as well as impaired mitochondrial protein import and decreased translation of mRNAs in mitochondria (44). Mitochondrial ATP production is at risk when mitochondrial proteostasis is dysregulated because it often leads to alterations in the relative quantities of the hundreds of proteins necessary for oxidative phosphorylation (OXPHOS) (45, 46). Moreover, an imbalance between nuclear-encoded and mitochondrial-encoded OXPHOS proteins affects mitochondrial proteostasis in ways that extend lifespan in mice and worms (47). In fact, since the mitochondrial proteome comprises proteins made in the cytosol as well as in mitochondria, the proteostasis balancing act that must be maintained in mitochondria is particularly challenging (46). One important first line of defense against mild mitochondrial damage is carried out by several mitochondrial proteases, which contribute to the mitochondrial unfolded protein response (UPRmt) (Figure 1F). In the mitochondrial matrix, protein turnover is controlled by three AAA proteases: the soluble mitochondrial Lon protease homolog (LonP1) and mitochondrial ATP-dependent CLp protease (CLpP), and the mitochondrial inner membrane-bound m-AAA protease. In the intermembrane space, mitochondrial protein quality is ensured by the membrane-bound ATP-dependent zinc metalloproteinase, YME1L1, the soluble mitochondrial serine protease, HTRA2, the mitochondrial metalloendopeptidase, OMA1, and the mitochondrial presenilins-associated rhomboid-like protein (PARL). These proteases play a variety of roles, such as degradation of misfolded proteins and balancing various mitochondrial constituents, such as OXPHOS proteins. However, most evidence suggests that Lon1 and CLpP are central to the UPRmt, while the other proteases may play roles in other aspects of mitochondrial proteostasis and dynamics (48). Moreover, because of the dire functional consequences of reductions in the quality of the mitochondrial proteome, dysregulation of mitochondrial proteostasis is communicated to various parts of the cell through at least five different pathways, including peptide-derived signaling, mitochondrial backup-signaling, mitochondrial translation control (MTC) loss-induced signaling and the mitochondrial unfolded protein response, UPRmt.
Although the UPRmt is beginning to be understood more clearly in mammals (49), much of our knowledge of this process comes from studies of the nematode, Caenorhabditis elegans. In fact, UPRmt activation protects C. elegans against ischemic injury, further supporting potential roles for the UPRmt, in the ischemic mammalian heart (50). A key regulator of the UPRmt is the transcription factor, ATFS-1 in C. elegans, which in mammals is ATF5, a transcription factor that is imported into mitochondria in an ATP-dependent manner when mitochondrial function and protein folding is optimal (46, 51). Under such conditions, LonP1 and CLpP proteases degrade ATF5 (52). However, when dysregulated OXPHOS and other stresses lead to dysregulated mitochondrial proteostasis, LonP1 and CLpP are diverted toward degrading those misfolded proteins to minimize their toxic effects; this diversion leads to the accumulation of intact ATF5 (Figure 1F) (46, 49, 52). Upon accumulation ATF5 is then exported from mitochondria to the nucleus where it acts as a transcription factor that induces genes encoding proteins designed to improve mitochondrial protein folding and rebalance mitochondrial proteostasis (Figure 1F), such as HSPA9, LonP1, and YME1L. ATF5 also serves as a communicator of metabolic stress by temporarily limiting the transcription of OXPHOS genes encoded in nuclear and mitochondrial genomes, while simultaneously increasing nuclear encoded gene transcription of all glycolysis components, and this is thought to maintain cellular ATP levels until mitochondrial dysfunction is resolved (45, 53).
Mitochondrial Proteostasis in Cardiac Pathology
Little is known about the UPRmt in the heart; however, several recent publications have provided initial evidence that the UPRmt is important for optimal cardiac function and recovery from I/R injury, as well as in the setting of pathological cardiac hypertrophy (Figure 2B). For example, using LonP1 transgenic mice, as well as mice that are haploinsufficient for the LonP1 gene, it was shown that this UPRmt protease mitigates cardiac injury during I/R by preventing oxidative damage, in part by rebalancing OXPHOS complex subunit levels in an adaptive manner (54). Moreover, pressure overload in mice was shown to activate the UPRmt. Additionally, pharmacologic boosting of the UPRmt reduced cardiac pathology in this model (55, 56). In the same study it was also shown that hearts from patients with aortic stenosis, which is often associated with left ventricular overload, exhibited increased expression of genes associated with the UPRmt. In another study, mice in which ATF5 was genetically deleted were used to show that the UPRmt protected the heart against I/R in an ATF5-dependent manner (53). Moreover, in the same study RNAseq results demonstrated the induction of numerous genes in an ATF5-dependent manner during pharmacological induction of the UPRmt. While these studies implicate roles for the UPRmt in the setting of cardiac pathology, much remains to be determined about the role of this mitochondrial proteostasis pathway in the heart. Underscoring the need for additional studies is a recent report where it was shown that CLpP, which plays a central role in the UPRmt in C. elegans, and thought to be important for the UPRmt in mammals was not required for the mammalian UPRmt (52). In fact, in that study it was found that CLpP contributes to mitochondrial cardiomyopathy, such that deletion of CLpP increased de novo synthesis of OXPHOS proteins leading to increased ATP and improved cardiac function in mice. On the other hand, a different study, while not in the heart, but done with C2C12 myoblasts, demonstrated that knockdown of CLpP altered mitochondrial morphology and expression of OXPHOS proteins, reduced oxygen consumption, increased reactive oxygen species and impaired myoblast differentiation (57). Interestingly, in this same study it was shown that knocking down CLpP leads to increases in the phosphorylation of EIF2α, which is a hallmark feature of the UPRER.
Integrating ER and Mitochondrial Function in Cardiac Myocytes
There is some evidence suggesting that there is a potential for integration between the UPRmt and the UPRER. One important piece of this evidence is the physical linkage between mitochondria and the ER at mitochondrial associated membranes, or MAMs (58). Although physical linkages between mitochondria and the ER were reported beginning in the 1960's, the term MAM was christened by Jean Vance, who identified a function for the mitochondrial-ER contact sites in phospholipid transport between these organelles (59). Subsequently, numerous studies of MAMs have identified the proteins that tether the two organelles, including mitofusin2 (60), as well as important physiological roles for their juxta-positioning, which, in the heart, have been centered mostly around the movement of calcium from the ER into mitochondria (61). In this way, MAMs are responsible for coordinating ER calcium flux with a variety of mitochondrial functions, including the ATP generation, as well as apoptosis and mitophagy (62, 63). More recent studies have implicated specific mitochondrial-ER tethering proteins, such as FUNDC1, as having important roles in maintaining normal cardiac contractility in mice (64–66). Studies outside the cardiac context have shown that there are numerous components of the UPRER that are associated with MAM structure and function, including ER chaperones, the IP3 receptor and PERK, which, if deleted decreases calcium movement from the ER to mitochondria (67). Relatedly, PERK deletion in the heart disrupts calcium signaling in cardiac myocytes in mice, in vivo (68).
Integrating ER and Mitochondrial Proteostasis
While studies on MAMs imply that the proteostasis pathways in these organelles must be integrated, to date there have been no studies in the heart that have addressed the molecular details of such integration beyond those involving MAMs. However, studies of molecular integration points between mitochondrial and ER proteostasis pathways have been done in other cell and tissue types, and the results of such studies could begin to inform us about whether such integration might also occur in the heart and, if so, what the functional consequences of this integration might be in terms of cardiac physiology and pathology. One potential molecular integration point between the UPRmt and UPRER that has been studied extensively in non-cardiac cells and tissues is the integrated stress response (IRS). The IRS is an elaborate signaling pathway in eukaryotic cells that is activated in response to an array of stresses including hypoxia, amino acid starvation, glucose deprivation, ER stress and viral infection (42, 69, 70). All of these pathways converge on the activation of kinases, such as PERK, which phosphorylate eIF2α on serine 51. In addition to causing global translational repression, a feature that reduces the protein-folding burden on nearly all of cellular proteostasis, eIF2α phosphorylation leads to the preferential translation of some transcription factors that have upstream ORFs in their 5′ UTRs, such as ATF4 (71, 72) and to subsequent changes in gene expression that are adaptive upon acute ATF4 activation, but can culminate in apoptosis and necrosis upon chronic ATF4 activation. Importantly, the PERK/ATF4 signaling axis, which plays a central role in the IRS and UPRER (38), is also involved in the UPRmt (69). In fact, like ATF4, the ATF5 transcript also has an ORFs in its 5′ UTR, so ATF5 levels also increase upon PERK upon activation of either the UPRER or the UPRmt (73). PERK-mediated increases in ATF4 enhance the expression of the UPRmt component, LonP1 (Figure 1G) (74). Related to this finding, but somewhat perplexing is the observation that chemical inhibition of LonP1 protease activity using CDDO activates the UPRmt, as well as increasing ATF4 mediated gene induction (75), indicating a possible bidirectional regulatory linkage between ATF4 and LonP1. Another finding that could serve as a molecular integration point between the UPRER and UPRmt was shown in chondrocytes, where the ATF6 family member, BBF2H7, induces typical UPRER genes, as well as the regulator of the UPRmt, ATF5 (76). In an examination of the effects of drugs that dysregulate mitochondrial proteostasis in HeLa, 293T, and COS7 cells, as well as maneuvers that cause mitochondrial proteostatic stress, in vivo, it was shown that via activation of the ISR, ATF4 but not ATF5 responds to dysregulated mitochondrial proteostasis and activates the expression of cytoprotective genes (77). Moreover, the PERK/ATF4 signaling axis can affect mitochondrial morphology and functional integrity, presumably having these effects at least partly through regulating mitochondrial proteostasis (78). Thus, it seems possible that through PERK-mediated increases in ATF4 and ATF5, and perhaps through PERKs role as a tether which holds MAMs together (Figure 1H), a function that does not require PERK enzyme activity (79), the UPRER and UPRmt could be integrated and, in some cases co-activated, which could improve both ER and mitochondrial function during stresses that dysregulate proteostasis in these two organelles.
Conclusion
The processes that govern mitochondrial and ER proteostasis are of critical importance for the adaptation of eukaryotic cells to environmental changes that risk proteome integrity. Even though the processes involved in mitochondrial proteostasis have gone relatively unstudied in the heart, it seems likely that in combination with those that regulate ER proteostasis, they are critical for cardiac function and, in particular, cardiac myocyte viability and contractility. In light of this, it is apparent that mitochondrial and ER proteostasis, which are regulated by many processes in addition to the UPRs in these organelles, provide fertile opportunities for future studies that could lead to the design of novel therapeutics for treating cardiac pathologies, ranging from ischemic to hypertrophic and dilated cardiomyopathies. Our hope is that this review has brought such potential intervention points to light amongst the heart research community and that it will spawn investigation into these aspects of proteostasis, with an objective of developing much needed new therapies for treating cardiac pathologies.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by NIH grants 1HL135893, 1HL141463, 1HL149931 to CG and NIH grant 1F31HL140850 and the Inamori Foundation to EB, and the ARCS Foundation, Inc., San Diego Chapter to EB and WS. AA, EB, and WS are Rees-Stealy Research Foundation Phillips Gausewitz, M.D., Scholars of the SDSU Heart Institute.
References
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. (2008) 319:916–9. 10.1126/science.1141448 [DOI] [PubMed] [Google Scholar]
- 2.Henning RH, Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. (2017) 14:637–53. 10.1038/nrcardio.2017.89 [DOI] [PubMed] [Google Scholar]
- 3.Jayaraj GG, Hipp MS, Hartl FU. Functional modules of the proteostasis network. Cold Spring Harb Perspect Biol. (2019) 4:a033951 10.1101/cshperspect.a033951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 5.Reid DW, Nicchitta CV. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat Rev Mol Cell Biol. (2015) 16:221–31. 10.1038/nrm3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bers DM. Cardiac excitation-contraction coupling. Nature. (2002) 415:198–205. 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- 7.Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J Physiol. (1997) 503(Pt1):21–9. 10.1111/j.1469-7793.1997.021bi.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bers DM, Shannon TR. Calcium movements inside the sarcoplasmic reticulum of cardiac myocytes. J Mol Cell Cardiol. (2013) 58:59–66. 10.1016/j.yjmcc.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. (2017) 121:181–95. 10.1161/CIRCRESAHA.117.310230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glembotski CC. Roles for the sarco-/endoplasmic reticulum in cardiac myocyte contraction, protein synthesis, and protein quality control. Physiology. (2012) 27:343–50. 10.1152/physiol.00034.2012 [DOI] [PubMed] [Google Scholar]
- 11.Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: programmed to conserve. J Mol Cell Cardiol. (2013) 55:85–91. 10.1016/j.yjmcc.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. (2007) 101:975–84. 10.1161/CIRCRESAHA.107.161273 [DOI] [PubMed] [Google Scholar]
- 13.Glembotski CC, Rosarda JD, Wiseman RL. Proteostasis and beyond: ATF6 in ischemic disease. Trends Mol Med. (2019) 25:538–550. 10.1016/j.molmed.2019.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tam AB, Roberts LS, Chandra V, Rivera IG, Nomura DK, Forbes DJ, et al. The UPR activator ATF6 responds to proteotoxic and lipotoxic stress by distinct mechanisms. Dev Cell. (2018) 46:327–43.e7. 10.1016/j.devcel.2018.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. (2006) 98:1186–93. 10.1161/01.RES.0000220643.65941.8d [DOI] [PubMed] [Google Scholar]
- 16.Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. (2009) 284:29735–45. 10.1074/jbc.M109.018036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackwood EA, Hofmann C, Santo Domingo M, Bilal AS, Sarakki A, Stauffer W, et al. ATF6 regulates cardiac hypertrophy by transcriptional induction of the mTORC1 activator, Rheb. Circ Res. (2019) 124:79–93. 10.1161/CIRCRESAHA.118.313854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Z, Brodsky JL. Protein quality control in the secretory pathway. J Cell Biol. (2019) 218, 3171–87. 10.1083/jcb.201906047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pavitt GD, Ron D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harb Perspect Biol. (2012) 4:a012278. 10.1101/cshperspect.a012278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Preissler S, Ron D. Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb Perspect Biol. (2019) 11:a033894. 10.1101/cshperspect.a033894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. (2013)1833:3460–70. 10.1016/j.bbamcr.2013.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS. A thrombospondin-dependent pathway for a protective ER stress response. Cell. (2012) 149:1257–68. 10.1016/j.cell.2012.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Xu L, Gillette TG, Jiang X, Wang ZV. The unfolded protein response in ischemic heart disease. J Mol Cell Cardiol. (2018) 117:19–25. 10.1016/j.yjmcc.2018.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang G, Wang X, Gillette TG, Deng Y, Wang ZV. Unfolded protein response as a therapeutic target in cardiovascular disease. Curr Top Med Chem. (2019) 19:1902–17. 10.2174/1568026619666190521093049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Correll RN, Grimes KM, Prasad V, Lynch JM, Khalil H, Molkentin JD. Overlapping and differential functions of ATF6alpha versus ATF6beta in the mouse heart. Sci Rep. (2019) 9:2059. 10.1038/s41598-019-39515-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackwood EA, Azizi K, Thuerauf DJ, Paxman RJ, Plate L, Kelly JW, et al. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun. (2019) 10:187. 10.1038/s41467-018-08129-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, et al. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. (2008) 283:14012–21. 10.1074/jbc.M709776200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Bi X, Zhang G, Deng Y, Luo X, Xu L, et al. Glucose-regulated protein 78 is essential for cardiac myocyte survival. Cell Death Differ. (2018) 25:2181–94. 10.1038/s41418-018-0109-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, et al. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res. (2017) 120:862–75. 10.1161/CIRCRESAHA.116.310266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ranek MJ, Kokkonen-Simon KM, Chen A, Dunkerly-Eyring BL, Vera MP, Oeing CU, et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature. (2019) 566:264–9. 10.1038/s41586-019-0895-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA. (2011) 108:9572–7. 10.1073/pnas.1106291108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. (2015) 116:1477–90. 10.1161/CIRCRESAHA.116.303790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell. (2014) 156:1179–92. 10.1016/j.cell.2014.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen RV, Andreadou I, Hausenloy DJ, Botker HE. The Role of O-GlcNAcylation for protection against ischemia-reperfusion injury. Int J Mol Sci. (2019) 20:E404. 10.3390/ijms20020404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma J, Liu T, Wei AC, Banerjee P, O'Rourke B, Hart GW. O-GlcNAcomic profiling identifies widespread o-linked beta-N-Acetylglucosamine modification (O-GlcNAcylation) in oxidative phosphorylation system regulating cardiac mitochondrial function. J Biol Chem. (2015) 290:29141–53. 10.1074/jbc.M115.691741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Deng Y, Zhang G, Li C, Ding G, May HI, et al. Spliced X-box binding protein 1 stimulates adaptive growth through activation of mTOR. Circulation. (2019) 140:566–579. 10.1161/CIRCULATIONAHA.118.038924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. (2019) 568:351–6. 10.1038/s41586-019-1100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. (2003) 11:619–33. 10.1016/S1097-2765(03)00105-9 [DOI] [PubMed] [Google Scholar]
- 39.Rutkowski DT, Kaufman RJ. All roads lead to ATF4. Dev Cell. (2003) 4:442–4. 10.1016/S1534-5807(03)00100-X [DOI] [PubMed] [Google Scholar]
- 40.Toth A, Nickson P, Mandl A, Bannister ML, Toth K, Erhardt P. Endoplasmic reticulum stress as a novel therapeutic target in heart diseases. Cardiovasc Hematol Disord Drug Targets. (2007) 7:205–18. 10.2174/187152907781745260 [DOI] [PubMed] [Google Scholar]
- 41.Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M, et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. (2010) 122:361–9. 10.1161/CIRCULATIONAHA.109.917914 [DOI] [PubMed] [Google Scholar]
- 42.Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang H, et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension. (2014) 64:738–44. 10.1161/HYPERTENSIONAHA.114.03811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Picca A, Mankowski RT, Burman JL, Donisi L, Kim JS, Marzetti E, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. (2018) 15:543–54. 10.1038/s41569-018-0059-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andreasson C, Ott M, Buttner S. Mitochondria orchestrate proteostatic and metabolic stress responses. EMBO Rep. (2019) 20:e47865. 10.15252/embr.201947865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell. (2015) 58:123–33. 10.1016/j.molcel.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. (2018) 19:109–20. 10.1038/nrm.2017.110 [DOI] [PubMed] [Google Scholar]
- 47.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. (2013) 497:451–7. 10.1038/nature12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. (2015) 350:aad0116. 10.1126/science.aad0116 [DOI] [PubMed] [Google Scholar]
- 49.Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol. (2016) 26:2037–43. 10.1016/j.cub.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaufman DM, Crowder CM. Mitochondrial proteostatic collapse leads to hypoxic injury. Curr Biol. (2015) 25:2171–6. 10.1016/j.cub.2015.06.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neupert W, Brunner M. The protein import motor of mitochondria. Nat Rev Mol Cell Biol. (2002) 3:555–65. 10.1038/nrm878 [DOI] [PubMed] [Google Scholar]
- 52.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. (2012) 337:587–90. 10.1126/science.1223560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang YT, Lim Y, McCall MN, Huang KT, Haynes CM, Nehrke K, et al. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am J Physiol Heart Circ Physiol. (2019) 317:H472–8. 10.1152/ajpheart.00244.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venkatesh S, Li M, Saito T, Tong M, Rashed E, Mareedu S, et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J Mol Cell Cardiol. (2019) 128:38–50. 10.1016/j.yjmcc.2018.12.017 [DOI] [PubMed] [Google Scholar]
- 55.Smyrnias I, Gray SP, Okonko DO, Sawyer G, Zoccarato A, Catibog N, et al. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J Am Coll Cardiol. (2019) 73:1795–806. 10.1016/j.jacc.2018.12.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glembotski CC, Arrieta A, Blackwood EA. Unfolding the roles of mitochondria as therapeutic targets for heart disease. J Am Coll Cardiol. (2019) 73:1807–10. 10.1016/j.jacc.2018.12.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deepa SS, Bhaskaran S, Ranjit R, Qaisar R, Nair BC, Liu Y, et al. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic Biol Med. (2016) 91:281–92. 10.1016/j.freeradbiomed.2015.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Brito OM, Scorrano L. An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. (2010) 29:2715–23. 10.1038/emboj.2010.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. (1990) 265:7248–56. [PubMed] [Google Scholar]
- 60.de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal. (2008) 10:621–33. 10.1089/ars.2007.1934 [DOI] [PubMed] [Google Scholar]
- 61.Lu X, Ginsburg KS, Kettlewell S, Bossuyt J, Smith GL, Bers DM. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ Res. (2013) 112:424–31. 10.1161/CIRCRESAHA.111.300501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dorn GW, II, Maack C. SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol. (2013) 55:42–9. 10.1016/j.yjmcc.2012.07.015 [DOI] [PubMed] [Google Scholar]
- 63.Seidlmayer LK, Mages C, Berbner A, Eder-Negrin P, Arias-Loza PA, Kaspar M, et al. Mitofusin 2 is essential for IP3-mediated SR/Mitochondria metabolic feedback in ventricular myocytes. Front Physiol. (2019) 10:733. 10.3389/fphys.2019.00733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, et al. Binding of FUN14 domain containing 1 With inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. (2017) 136:2248–66. 10.1161/CIRCULATIONAHA.117.030235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, et al. Hyperglycemia-driven inhibition of AMP-activated protein kinase alpha2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes In vivo. Circulation. (2019) 139:1913–36. 10.1161/CIRCULATIONAHA.118.033552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu S, Zou MH. Mitochondria-associated endoplasmic reticulum membranes in the heart. Arch Biochem Biophys. (2019) 662:201–12. 10.1016/j.abb.2018.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carreras-Sureda A, Pihan P, Hetz C. The unfolded protein response: at the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front Oncol. (2017) 7:55. 10.3389/fonc.2017.00055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu Z, Cai H, Zhu H, Toque H, Zhao N, Qiu C, et al. Protein kinase RNA-like endoplasmic reticulum kinase (PERK)/calcineurin signaling is a novel pathway regulating intracellular calcium accumulation which might be involved in ventricular arrhythmias in diabetic cardiomyopathy. Cell Signal. (2014) 26:2591–600. 10.1016/j.cellsig.2014.08.015 [DOI] [PubMed] [Google Scholar]
- 69.Rainbolt TK, Saunders JM, Wiseman RL. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab. (2014) 25:528–37. 10.1016/j.tem.2014.06.007 [DOI] [PubMed] [Google Scholar]
- 70.Lu Z, Xu X, Fassett J, Kwak D, Liu X, Hu X, et al. Loss of the eukaryotic initiation factor 2alpha kinase general control nonderepressible 2 protects mice from pressure overload-induced congestive heart failure without affecting ventricular hypertrophy. Hypertension. (2014) 63:128–35. 10.1161/HYPERTENSIONAHA.113.02313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. (2002) 110:1383–8. 10.1172/JCI0216784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. (2013) 15:481–90. 10.1038/ncb2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. (2016) 17:1374–95. 10.15252/embr.201642195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hori O, Ichinoda F, Tamatani T, Yamaguchi A, Sato N, Ozawa K, et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J Cell Biol. (2002) 157:1151–60. 10.1083/jcb.200108103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Munch C, Harper JW. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature. (2016) 534:710–3. 10.1038/nature18302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Izumi S, Saito A, Kanemoto S, Kawasaki N, Asada R, Iwamoto H, et al. The endoplasmic reticulum stress transducer BBF2H7 suppresses apoptosis by activating the ATF5-MCL1 pathway in growth plate cartilage. J Biol Chem. (2012) 287:36190–200. 10.1074/jbc.M112.373746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quiros PM, Prado MA, Zamboni N, D'Amico D, Williams RW, Finley D, et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol. (2017) 216:2027–45. 10.1083/jcb.201702058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MC, et al. The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep. (2018) 22:2827–36. 10.1016/j.celrep.2018.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. (2012) 19:1880–91. 10.1038/cdd.2012.74 [DOI] [PMC free article] [PubMed] [Google Scholar]