

Mosquitoes are widespread vectors of numerous human pathogens and harbor microbiota known to affect host phenotypic traits. However, little research has directly investigated how bacterial communities associated with larvae and adults are connected. We characterized whole-body bacterial communities in mosquito larvae preceding pupation and in newly emerged adults, and investigated whether a significant biotic factor, fungal colonization of the larval hindgut, impacted these microbiomes. Results showed that fungal colonization reduced microbial community variation across individuals and differentially impacted the outcomes of transstadial transmission for certain bacterial genera, revealing downstream effects of the fungus on initial adult microbiomes. The importance of our research is in providing a thorough comparative analysis of whole-body microbiota harbored in larvae and adults of the yellow fever mosquito (Aedes aegypti) and in demonstrating the important role a widespread gut fungus played in a host-associated microbiome.
KEYWORDS: Aedes aegypti, Zancudomyces culisetae, gut fungi, metagenomics, microbial ecology, microbiome, mosquitoes, mycology, transstadial transmission
ABSTRACT
Adult mosquitoes inherit a bacterial community from larvae via transstadial transmission, an understudied process that may influence host-microbe interactions. Microbes contribute to important host life history traits, and analyzing transmitted microbial communities, the interrelationship between larval and adult-associated microbiota, and factors influencing host-microbe relationships provides targets for research. During its larval stage, the yellow fever mosquito (Aedes aegypti) hosts the trichomycete gut fungus Zancudomyces culisetae, and fungal colonization coincides with environmental perturbations in the digestive tract microecosystem. Natural populations are differentially exposed to fungi, thereby potentially harboring distinct microbiota and experiencing disparate host-microbe interactions. This study’s objectives were to characterize larval and initial adult microbiomes, investigate variation in diversity and distribution of microbial communities across individuals, and assess whether larval fungal colonization impacted microbiomes at these developmental stages. Laboratory-based fungal infestation assays, sequencing of 16S rRNA gene amplicons, and bacterial load quantification protocols revealed that initial adult microbiomes varied in diversity and distribution. Larval fungal colonization had downstream effects on initial adult microbiomes, significantly reducing microbial community variation, shifting relative abundances of certain bacterial families, and influencing transstadial transmission outcomes of particular genera. Further, abundances of several families consistently decreased in adults relative to levels in larvae, possibly reflecting impacts of host development on specific bacterial taxa. These findings demonstrated that a prolific gut fungus impacted mosquito-associated microbiota at two developmental stages in an insect connected with global human health.
IMPORTANCE Mosquitoes are widespread vectors of numerous human pathogens and harbor microbiota known to affect host phenotypic traits. However, little research has directly investigated how bacterial communities associated with larvae and adults are connected. We characterized whole-body bacterial communities in mosquito larvae preceding pupation and in newly emerged adults, and investigated whether a significant biotic factor, fungal colonization of the larval hindgut, impacted these microbiomes. Results showed that fungal colonization reduced microbial community variation across individuals and differentially impacted the outcomes of transstadial transmission for certain bacterial genera, revealing downstream effects of the fungus on initial adult microbiomes. The importance of our research is in providing a thorough comparative analysis of whole-body microbiota harbored in larvae and adults of the yellow fever mosquito (Aedes aegypti) and in demonstrating the important role a widespread gut fungus played in a host-associated microbiome.
INTRODUCTION
Studies have investigated mosquito-associated microbiota and factors influencing microbial communities; however, discerning relationships between larval and adult microbiomes still provides targets for research. Phenotypic differences across developmental stages impact microbiomes (1, 2), and larvae and adults harbor disparate microbial communities (3–6). However, certain bacterial taxa are transstadially transmitted (remaining with the host from one life stage to the next) from larvae through pupae to adults (2, 3, 5–7), providing an initial subset of microbes that could influence host-microbe interactions. Unfortunately, the underlying processes driving transstadial transmission are understudied, and the significance of transmitted microbiomes in mosquito biology is unknown. Large quantities of bacteria are expelled from the digestive tract during host metamorphosis (8, 9), suggesting that microbes successfully transfer across developmental stages by inhabiting other anatomical regions. Indeed, certain bacteria in the Malpighian tubules (an excretory and osmoregulatory system in mosquitoes and other insects) transmit transstadially (10, 11) and recolonize the newly formed adult digestive tract (11). Bacteria are found in other regions as well, including the hemocoel (body cavity containing circulatory fluid) (12) and salivary glands (producing secretions used in food ingestion) (13) though the capacity of these microbes to transfer from larvae to adults requires additional analysis. Further, extensive physiological changes occur during mosquito pupation (14), possibly driving changes to the microbiota transferred to adults as well. Transstadial transmission has been studied extensively in various species of ticks, another prevalent arthropodic vector, in which particular bacteria were shown to transmit from larvae to adults and subsequently infect host organisms (15, 16). The known importance of transstadial transmission in other systems encourages further study of these concepts in mosquitoes.
Although mosquito-associated microbiomes are well studied (2, 17–24), variation in the microbiota is found across individuals within populations (23, 25–27), complicating generalizations of microbiome characteristics and identifications of significant microbial interactions. Due to the important connection between microbes and host phenotypic traits (1, 2, 28, 29), understanding processes affecting microbiota and introducing variation throughout the mosquito life cycle is vital to unravel complexities underpinning host-microbiome relationships.
Though often overlooked in studies of host-microbe interactions, fungi are known to interact with bacterial communities across a vast array of study systems (30). Mosquitoes harbor communities of fungi (21, 31) that impact host phenotypic traits and associated microbiota (32–34) and are viable agents for pest control (35), yet very little is known regarding the specific fungus-bacterium-host interactions occurring in this insect. A potential system for studying nonpathogenic fungal-bacterial-host interactions is the Trichomycetes, an ecological group of microfungi including obligate symbionts of certain larval arthropods (36). The yellow fever mosquito (Aedes aegypti), a vector for the human viral pathogens causing yellow, dengue, chikungunya, and Zika fevers, is also a known host of a well-studied trichomycete fungus, Zancudomyces culisetae (37, 38), in certain natural populations (39). Zancudomyces culisetae is particularly noteworthy in the group because it infests a variety of dipteran hosts, including the larval aquatic stages of other mosquito and black fly species. While the true nature of the host-fungus relationship is understudied, it is presumed to be commensalistic. However, a study found that this relationship is dynamic, shifting toward mutualism with a different black fly host under certain environmental conditions (40). The fungal life cycle is tightly interwoven with that of its larval host. Asexual spores (trichospores) in the aquatic environment are ingested by the host and then establish foundational holdfasts to the digestive tract lining. Specific physiological cues in the digestive tract microecosystem mediate sporangiospore extrusion (initial fungal growth) (41, 42), and the subsequent growth and maturation of fungal hyphae introduce spatial disturbances in the larval hindgut (43) (Fig. 1). The fungus extracts nutrients from the host digestive tract lumen (internal region) (36) and releases trichospores that are excreted into the external environment. Importantly, host larvae progress through various developmental stages (instars), periodically shedding the digestive tract and fungi during molting. Subsequent larval instars are then recolonized after each molting period. These morphological and physiological stressors incurred by fungal colonization may impact larval bacterial communities (44) through environmental filtering or competitive exclusion of microbes unable to acclimate to the modified conditions (45, 46). Adult mosquitoes have never been observed to carry Z. culisetae colonization; therefore, any preliminary fungus-bacterium-host interactions in the system likely take place during the larval stage. Shifts in larval microbiota resulting from fungal colonization could impact the microbiomes acquired by newly emerged adults; however, these concepts have never been directly studied. Investigating whether fungal establishment and colonization of the hindgut influence these aspects of mosquito-microbe relationships and the transstadial transmission outcomes of different bacteria is vital as natural mosquito populations may harbor distinct microbiomes contingent on the presence of particular fungi in their local environments.
FIG 1.

Zancudomyces culisetae colonization of a dissected fourth-instar larval mosquito digestive tract (magnified view of area boxed in inset image, bottom right) visualized with phase-contrast microscopy (×100 magnification). Significant regions are labeled as follows: i, midgut; ii, hindgut. The black arrow indicates mature fungal hyphae. The inset shows a larva with the head removed and midgut and hindgut exposed.
To advance the field’s understanding of the interrelationship between bacterial communities (here referred to as microbiome or microbiota) harbored in larvae and adults and factors influencing the outcomes of bacterial transstadial transmission, microbiomes from larvae immediately preceding pupation and from newly emerged adults were analyzed under controlled laboratory conditions. We predicted that, in the absence of larval fungal colonization, initial adult microbiomes would be composed of invariable communities of bacteria adapted to the internal environmental conditions in mosquitoes. Conversely, with the gut fungus, putative morphological and physiological changes would alter larval microbiomes and have downstream effects on the initial adult microbiomes acquired through transstadial transmission. To explore these concepts, laboratory-based experiments with A. aegypti utilizing previously established inoculation assays with Z. culisetae (41, 43, 47, 48), next-generation sequencing of 16S rRNA gene amplicons, and bacterial load quantification protocols were performed to analyze and compare larval and adult mosquito microbiomes with and without fungal colonization in the larval hindgut.
Ultimately, we compare microbiota harbored in the whole bodies of larvae and adults, examine variation in the diversities and distributions of microbiomes across individuals, assess outcomes of transstadial transmission for certain bacterial taxa, investigate downstream effects of larval fungal colonization on initial adult microbiomes, discuss the implications and applications of our findings for future research, and emphasize the significance of fungal interactions within host-associated microbiomes.
RESULTS
Larval microbiomes preceding pupation.
Whole-body microbiomes harbored in fourth-instar larvae with and without fungal colonization were compared to assess fungal effects on larval microbiota. After data processing, 154 amplicon sequence variants (ASVs) were identified from 38 samples. No differences in alpha diversity measures were detected, indicating that fungal colonization did not alter larval microbiome diversity or distribution (Fig. 2; Table 1). Additionally, beta diversity measures were not significantly different (Table 1), demonstrating that the compositions and structures of microbiomes were similar between groups. However, data failed the homogeneity of group dispersions test for unweighted UniFrac distance [F(1, 36) = 4.6534, P = 0.036) (Fig. 3; Table 1), with nonfungal larvae exhibiting greater within-group variation for the metric. Unweighted UniFrac distance does not account for relative abundances of community members and instead calculates composition distance between samples based on shared bacterial taxa and phylogenetic distance, suggesting that nonfungal larvae had high variability of taxa present in microbiomes across individuals. No differences across groups were found for Bray-Curtis dissimilarity [F(1, 6) = 1.042, P = 0.442] (Table 1) and weighted UniFrac distance [F(1, 6) = 0.977, P = 0.424] (Table 1), which account for relative abundances of community members, indicating that variable taxa detected across nonfungal larvae were of low relative abundance. Comparative analyses of the top 15 abundant families found no significant differences in family abundances across treatment groups (see Fig. S3a and Table S1 in the supplemental material).
FIG 2.
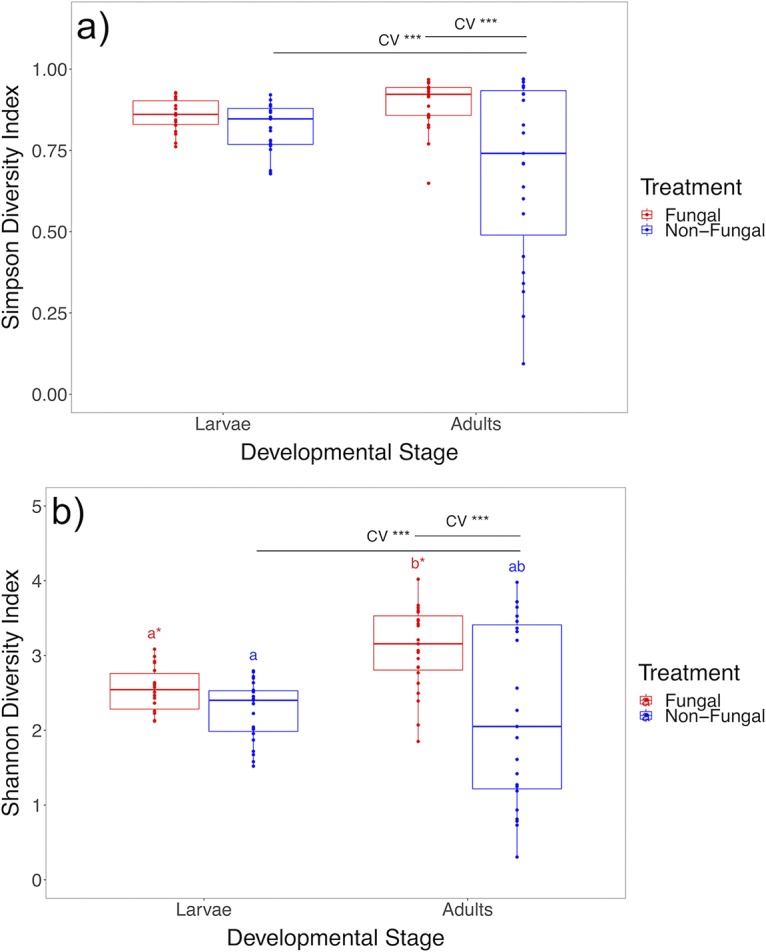
Box plots of microbiome alpha diversity measures across developmental stages and treatments. (a) Comparative analyses of Simpson diversity index values show a significant increase in variation of measures across developmental stages for nonfungal mosquitoes and treatments for newly emerged adults. (b) Comparative analyses of Shannon diversity index values show a significant increase in diversity across developmental stages for fungal mosquitoes, an increase in variation of measures across developmental stages for nonfungal mosquitoes, and treatments for newly emerged adults (n = 87). Upper and lower limits of boxes represent quartiles around the mean, and horizontal lines within boxes represent median values. Significant differences of CV values (CV ***, P < 0.001) were calculated with asymptotic and modified signed-likelihood ratio tests. Significant differences of mean alpha diversity measures were calculated with a linear mixed model (same letter, no significant difference; different letter and asterisk, P < 0.05).
TABLE 1.
Results from comparative statistical analyses for measures of alpha and beta diversity and SCML read counts across groups within each independent data seta
| Data set and metric | DF Numb | DF Denb | Fb | Pb | CV (%) | Asymptotic test |
MSLRT |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| Value | CV P | Value | CV P | |||||||
| Treatment | Nonfungal | Fungal | ||||||||
| Larva type | ||||||||||
| Simpson | 1 | 5.947 | 0.798 | 0.406 | 9.02 | 5.91 | 3.119 | 0.077 | 3.377 | 0.066 |
| Shannon | 1 | 5.925 | 1.941 | 0.214 | 17.65 | 11.77 | 2.783 | 0.095 | 2.874 | 0.090 |
| Bray-Curtis | 1 (1) | 6 (36) | 1.042 (0.537) | 0.442 (0.489) | ||||||
| Unweighted UniFrac | 1 (1) | 6 (36) | 1.085 (4.653) | 0.398 (0.036) | ||||||
| Weighted UniFrac | 1 (1) | 6 (36) | 0.977 (0.297) | 0.424 (0.597) | ||||||
| Jaccard | 1 (1) | 6 (36) | 0.959 (1.116) | 0.791 (0.323) | ||||||
| Adult type | ||||||||||
| Simpson | 1 | 5.975 | 4.039 | 0.091 | 39.64 | 8.42 | 33.292 | <0.001 | 37.175 | <0.001 |
| Shannon | 1 | 5.978 | 3.277 | 0.120 | 54.45 | 17.76 | 18.023 | <0.001 | 19.251 | <0.001 |
| Bray-Curtis | 1 (1) | 6 (31) | 1.700 (2.711) | 0.088 (0.1) | ||||||
| Unweighted UniFrac | 1 (1) | 6 (31) | 1.493 (0.860) | 0.084 (0.383) | ||||||
| Weighted UniFrac | 1 (1) | 6 (31) | 5.180 (2.701) | 0.06 (0.128) | ||||||
| Jaccard | 1 (1) | 6 (31) | 1.427 (0.939) | 0.075 (0.356) | ||||||
| Developmental stage | Larval | Adult | ||||||||
| Nonfungal mosquitoes | ||||||||||
| Simpson | 1 | 5.993 | 1.468 | 0.271 | 9.02 | 39.64 | 31.177 | <0.001 | 33.340 | <0.001 |
| Shannon | 1 | 5.995 | 0.006 | 0.939 | 17.65 | 54.45 | 18.189 | <0.001 | 34.281 | <0.001 |
| Bray-Curtis | 1 (1) | 6 (34) | 3.705 (31.461) | 0.022 (<0.001) | ||||||
| Unweighted UniFrac | 1 (1) | 6 (34) | 6.968 (43.821) | 0.035 (<0.001) | ||||||
| Weighted UniFrac | 1 (1) | 6 (34) | 8.919 (0.052) | 0.036 (0.804) | ||||||
| Jaccard | 1 (1) | 6 (34) | 2.512 (23.523) | 0.037 (<0.001) | ||||||
| SCML | 1 | 6 | 10.183 | 0.019 | ||||||
| Fungal mosquitoes | ||||||||||
| Simpson | 1 | 5.510 | 3.227 | 0.127 | 5.91 | 8.42 | 2.232 | 0.135 | 2.279 | 0.131 |
| Shannon | 1 | 5.510 | 13.903 | 0.011 | 11.77 | 17.76 | 2.860 | 0.091 | 2.960 | 0.085 |
| Bray-Curtis | 1 (1) | 6 (29) | 6.299 (32.067) | 0.028 (<0.001) | ||||||
| Unweighted UniFrac | 1 (1) | 6 (29) | 10.743 (53.22) | 0.03 (<0.001) | ||||||
| Weighted UniFrac | 1 (1) | 6 (29) | 8.859 (0.061) | 0.025 (0.79) | ||||||
| Jaccard | 1 (1) | 6 (29) | 3.737 (32.723) | 0.029 (<0.001) | ||||||
| SCML | 1 | 6 | 6.974 | 0.038 | ||||||
Significant differences of mean alpha diversity measures and SCML calibrated read counts were calculated with linear mixed models, CV values were calculated with asymptotic and modified signed-likelihood ratio tests (MSLRT), beta diversity measures were calculated with nested permutational multivariate analysis of variance, and within-group variations of beta diversity measures were calculated with permutational statistical tests for the homogeneity of group dispersions. Values in boldface are significant (P < 0.05).
Values in parentheses represent results from homogeneity of group dispersion tests. DF Den, denominator degrees of freedom; DF Num, numerator degrees of freedom.
FIG 3.
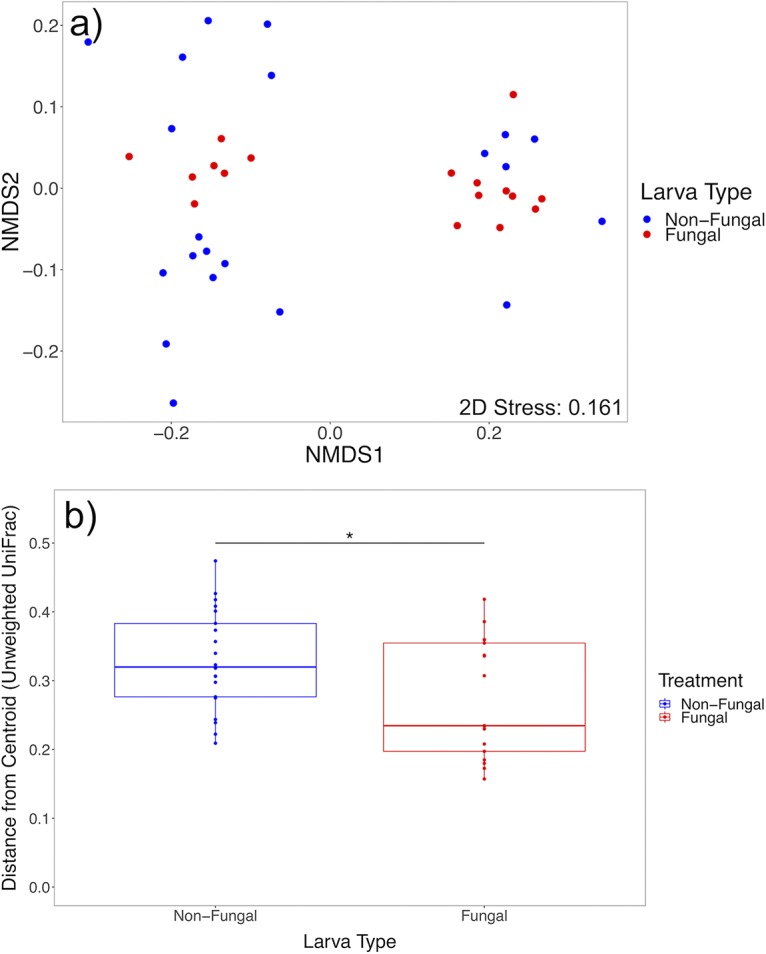
Analyses of unweighted UniFrac distance for larval microbiomes across treatments. (a) Nonmetric multidimensional scaling plot based on unweighted UniFrac distance. (b) Box plots of within-group variation of unweighted UniFrac distance, represented by the distance of each sample’s value from the group’s centroid value, show that nonfungal larvae have higher within-group variation for the metric (n = 38). Upper and lower limits of boxes represent quartiles around the mean, and horizontal lines within boxes represent median values. Significant differences of within-group variation across treatments were calculated with a permutational statistical test for the homogeneity of group dispersions (*, P < 0.05).
Comparisons of larval and adult microbiota.
Larval and adult microbiomes from nonfungal and fungal groups were compared separately to evaluate microbiomes harbored at the two developmental stages and assess transmission outcomes of bacterial taxa. After data processing, 484 ASVs were identified from 36 samples for nonfungal mosquito groups, and 600 ASVs were identified from 31 samples for fungal mosquito groups. No differences in mean alpha diversity measures in these developmental stages were detected in the nonfungal group; however, coefficient of variation (CV) values were significantly higher in adults for Simpson (P < 0.001) (Fig. 2a; Table 1) and Shannon (P < 0.001) (Fig. 2b; Table 1) diversity indices, revealing that adult microbial communities inherited through transstadial transmission had higher variation in diversity and distribution than larval microbiomes. In contrast, no differences in CV values between larvae and adults were observed in fungal mosquitoes for Simpson (P < 0.14) (Fig. 2a; Table 1) or Shannon (P < 0.1) (Fig. 2b; Table 1) diversity indices, and mean measures of Shannon diversity were higher in adults than in larvae (P = 0.011) (Fig. 2b; Table 1), reflecting an increase in community diversity.
In both groups of mosquitoes, differences between larval and adult microbiota were detected for all beta diversity metrics tested (Table 1), demonstrating that the taxonomic composition and structure of adult-associated microbial communities differed from those of larval microbiomes. Similarly, within-group dispersions of Bray-Curtis dissimilarity and unweighted UniFrac distance measures were greater in adults (Table 1), indicating that initially acquired adult microbiomes had higher within-group variation than larvae (Fig. S1). Permutational multivariate analyses of variance are sensitive to differences in variation across group comparisons and may produce false detections of significance; however, nonmetric multidimensional scaling (NMDS) plots show separation between larvae and adults for all metrics tested in both groups (Fig. 4; Fig. S2).
FIG 4.

Nonmetric multidimensional scaling plots of beta diversity measures across developmental stages for each treatment group. (a and b) Bray-Curtis dissimilarity and unweighted UniFrac distance for nonfungal mosquitoes (n = 36), respectively. (c and d) Bray-Curtis dissimilarity and unweighted UniFrac distance for fungal mosquitoes (n = 31), respectively.
Mean relative abundances of the 15 most abundant bacterial families shared in both developmental stages were calculated, plotted, and statistically tested to identify taxa increasing or decreasing in relative abundance in adults relative to that in larvae (Table S1; Fig. 5). In both groups, abundances of Corynebacteriaceae, Family_XI, Moraxellaceae, and Staphylococcaceae increased, indicating that microbiota from these families were consistently acquired by adults. Conversely, abundances of Microbacteriaceae and Rhizobiaceae decreased in both groups, suggesting that these families were uniformly lost during the host metamorphosis with no effect of fungi detected. However, transmission outcomes of the family Burkholderiaceae were affected by larval fungal colonization and increased in relative abundance in nonfungal adults to become the predominant family (P = 0.002), whereas no change was detected in fungal mosquitoes (P = 0.511).
FIG 5.

Mean relative abundances of the 15 most abundant families shared across developmental stages of nonfungal (n = 36) (a) and fungal (n = 31) (b) mosquitoes.
These families were further analyzed at the genus level to evaluate whether genera followed similar trends. Results showed that the majority of genera within each family exhibited similar transmission outcomes regardless of fungal colonization (Fig. 6; Table S2). Significant exceptions to this generalization were the disparate outcomes of transmission for the genera Acidovorax and Delftia, both in the family Burkholderiaceae, which were present at low abundances in both groups of larvae (Fig. 7a). These increased in nonfungal but decreased in fungal adults (Fig. 6 and 7b), indicating that transmission outcomes of these genera were directly affected by the fungus. Furthermore, particular genera shared between treatment groups that successfully transferred had differing relative abundances in initial adult microbiomes. Corynebacterium_1 (Corynebacteriaceae) and Staphylococcus (Staphylococcaceae) exhibited conserved positive log2(fold change) values (Fig. 6) and were universally higher in adults than in larvae. However, their relative abundances were substantially greater in fungal than in nonfungal adults (Fig. 7b) despite low levels in both groups of larvae (Fig. 7a), indicating that fungal colonization impacted the structure of microbiomes and relative abundances of certain genera acquired by adults. In addition, several low-abundance taxa, including genera in Family_XI, were transmitted to adults in both treatment groups but were present in higher abundances in initial fungal adult microbiomes (Fig. 7b; Table S2).
FIG 6.

Log2(fold change) values for genera from families identified to significantly differ in mean relative abundances across developmental stages of nonfungal (blue) and fungal (red) mosquitoes. Only genera present at greater than 1% relative abundances in either developmental stage within each treatment group are shown. Positive values indicate an increase while negative values indicate a decrease in the abundance of each genus in adults relative to levels in larvae.
FIG 7.

Relative abundances of genera from families identified to significantly change in mean relative abundances across developmental stages of nonfungal (blue) and fungal (red) mosquito larvae (a) and adults (b). Only genera present at greater than 1% relative abundances in either developmental stage within each treatment group are shown.
Newly emerged adult microbiomes.
Whole-body microbiomes harbored in newly emerged adults developed from larvae with and without fungal colonization were compared to evaluate downstream effects of larval fungal colonization on the initial microbiota inherited by adults. After data processing, 881 ASVs were identified from 33 samples. No differences for mean alpha diversity measures were detected; however, CV values were higher in nonfungal adults for Simpson (P < 0.001) (Fig. 2a; Table 1) and Shannon (P < 0.001) (Fig. 2b; Table 1) diversity indices, revealing that adults acquired initial microbiomes that varied in diversity and distribution across individuals in the absence of fungal colonization in larvae. In contrast, fungal colonization preceding pupation reduced the variability of newly emerged adult microbiota. No significant effect of larval fungal colonization was detected on adult microbiomes for any of the beta diversity metrics (Table 1); however, all metrics had P values of <0.1, suggesting a possible signal.
Comparisons of mean relative abundances of prominent community members showed that larval fungal colonization affected the prevalences of certain bacterial families present in initial adult microbiomes (Fig. S3b; Table S1). Larval fungal colonization corresponded with an increase in the abundances of Corynebacteriaceae (P = 0.025), found in the food source used in all groups (Fig. S3c), and Moraxellaceae (P = 0.048), found on the eggs and in the food source used in all groups (Fig. S3c and d). Experimental samples were presumed to be equally exposed to microbiota found on the food and eggs over the course of the experiment; however, this assumption was not directly tested.
Bacterial loads in larvae and adults.
Sequencing read counts were calibrated for a subset of experimental larvae and adults from nonfungal and fungal groups using spike-in calibration to microbial load (SCML) (49) to quantify and compare bacterial loads in larvae and adults (Table S2). Larvae contained higher bacterial loads than adults for both groups (P = 0.019 and P = 0.038, respectively), indicating that adults emerged from pupae with lower quantities of bacteria (Fig. 8) regardless of fungal colonization. The majority of reads for all groups belonged to the spike-in bacterium Salinibacter ruber, suggesting that the quantity of 16S copies added during initial PCR was exceedingly high for these samples, which prevented more thorough analyses of bacterial load.
FIG 8.
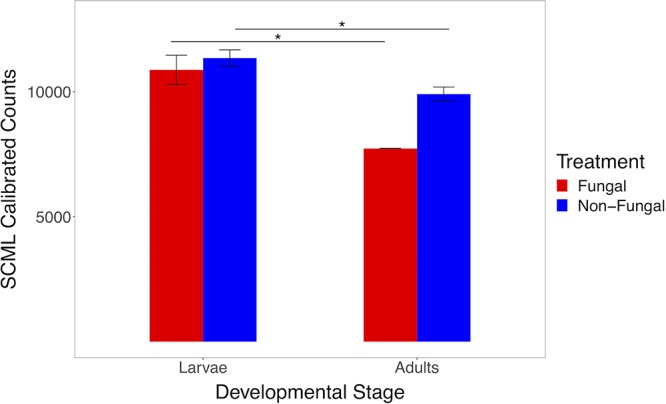
Bar plots of mean read counts calibrated using spike-in calibration to microbial load (SCML) across developmental stages for each treatment group. Comparative analyses show larger bacterial loads in larvae than in adults for both groups. Error bars indicate standard error (n = 42). Statistical differences were calculated with a linear mixed model (*, P < 0.05).
Detection of fungi in experimental larvae and adults.
Polymerase chain reactions using Z. culisetae-specific primers (50) on experimental larvae and adults were performed to assess fungal contamination in nonfungal larvae, fungal colonization in fungal larvae, and the capacity of the fungus to transmit transstadially. No fungal material was detected in nonfungal larvae (see Image S1a in the supplemental material), demonstrating that the experimental protocols performed prevented fungal contamination. In contrast, the majority of fungal larval samples exhibited strong amplification (Image S1b), revealing large amounts of fungal colonization at the time of sample collection and DNA extraction. However, several samples showed little or no amplification, suggesting some variation in the amount of fungal colonization in these samples. Minimal amplification was detected in fungal adults (Image S1c), indicating that the fungus does not transfer from larvae to adults. Several samples exhibited faint amplification, but it is unclear whether these results are indicative of small amounts of fungal material or are artifacts from PCR.
DISCUSSION
This laboratory-based study is offered as an analysis of mosquito-associated microbiota in the presence or absence of larval fungal colonization, adding to the scientific community’s understanding of how larval and adult microbiomes are related and impacted by a prolific gut fungus in an insect associated with global human health. The collection and microbial DNA extraction of adults immediately following emergence from pupae provided a snapshot of the microbiome inherited through transstadial transmission prior to adult microbiome assembly, enabling interpretations of transmission outcomes of certain bacterial taxa. Previous studies identified taxa found in both larvae and adults (2, 3, 5–7) but with analyses and experimental designs not specifically optimized for characterizations and comparisons of entire microbial communities. Our analyses incorporated microbes found throughout the mosquito anatomy, allowing for comparisons of microbial communities harbored in all anatomical regions of the host.
By using a modern protocol to quantify bacterial loads in experimental samples (49), newly emerged adults were found to harbor smaller amounts of bacteria than larvae, supporting previous studies demonstrating that large quantities of microbes are expelled from the digestive tract during and after pupation (8, 9). Due to the detected reduction of bacterial load in newly emerged adults, we infer that taxa increasing in mean relative abundances resulted from overall decreases in other bacteria, rather than from increased proliferation during host metamorphosis. The decreases in the abundances of certain bacteria may reflect the expulsion of microbes from the digestive tract, whereas taxa increasing in mean relative abundances may inhabit other anatomical regions (10–13) prior to and during pupation, resulting in successful transfer from larvae through pupae to newly emerged adults (10, 11). If the described spatial components are involved, studies that isolate and analyze microbes harbored solely in the digestive tract may underestimate the complexities of processes connecting larval and adult microbiomes. Future experiments could characterize microbial communities and quantify bacterial loads contained in distinct anatomical regions to assess bacterial dynamics outside the digestive tract. These studies could help reveal the precise locations of particular bacteria that are successfully transferred to adults and further our understanding of the morphological mechanisms of transstadial transmission that help shape initial adult microbiomes. In addition, conserved decreases of particular taxa may reflect detrimental effects from changing host physiology during pupation (14). The inclusion of analyses investigating pupal microbiota in future studies would help unravel mechanisms driving changes in the relative abundances of target taxa and could identify specific bacteria directly impacted by host development. These data would provide important information regarding the transitional microbiome that exists between larvae and adults and enable a more in-depth assessment of changes in the microbiota over developmental time.
Contrary to our prediction of a consistent community of microbes being inherited by newly emerged adults, comparative analyses of larval and adult microbiota revealed that initial adult microbiomes varied in diversity and distribution. Substantial differences across individual microbiomes acquired by newly emerged adults may suggest that transstadial transmission processes inherently contribute to intrapopulation variation. However, our analyses investigated solely the outcomes of transstadial transmission. The inclusion of temporal analyses of microbiomes across larvae, pupae, and adults is required to test whether transstadial transmission processes truly vary across individuals.
Compositional and structural microbiome variation within natural populations (22, 23, 25–27) complicates identification of significant host-microbe interactions but may be explained, in part, by implicit properties of microbial community assembly. Though the mechanisms driving variation were not directly tested, neutral (stochastic) assembly processes, which explain microbiome variation in other study systems (51–54), may contribute to the inconsistent taxonomic compositions detected in larvae and variably altered microbial communities acquired by newly emerged adults. Future research studying microbiome assemblage in mosquitoes using neutral and deterministic models may clarify processes contributing to taxonomic variation in larval microbiota and whether larval taxonomic composition influences the outcomes of transstadial transmission processes. The inclusion of a pupal data set would provide vital information regarding assembly processes taking place in the transitional microbiome, providing a better evaluation of microbiome assemblage pathways.
While our findings revealed that the diversity and distributions of initial adult microbiomes varied across individuals, we showed that a biotic factor during the larval stage, fungal colonization of the hindgut, reduced this variation and affected the transmission outcomes of certain bacterial taxa. This supported our prediction that larval fungal colonization would have downstream effects on initial adult microbiomes. Fungal colonization impacted transmission outcomes of particular genera, resulting in either a complete obstruction in transference or differential relative abundances in initial adult microbiomes, indicating a distinct influence of the fungus on certain bacteria. Morphological and physiological factors corresponding with fungal establishment may have played significant roles. The establishment of fungi in the hindgut depletes nutrients in the digestive tract lumen (36) and coincides with changes in the gut pH (41, 42), creating physiological stressors to the microbiota. Environmental stress can restrict taxonomic richness in microbial communities (44) through environmental filtering and competitive exclusion of members maladapted to altered environmental conditions (45, 46). These elements may drive the observed reduction in microbiome composition variation in larvae. Furthermore, extensive spatial perturbations from fungal proliferation (43) may result in the redistribution of bacterial communities to other anatomical regions and create novel microbial interactions, similar to those described in another mosquito-fungus system (33). Consequently, certain bacterial taxa may have successfully adapted to the novel conditions, outcompeting others for space and nutrients, resulting in successful transstadial transmission. Future research implementing fluorescence-based assays with transgenic strains of target taxa (10, 11) and including a pupal data set may help identify morphological interactions occurring between fungi and bacteria during larval microbiome assembly and enable the detection of spatial distributions of bacteria across developmental stages.
It is unknown whether fungal colonization in larvae affects microbiota throughout the mosquito life cycle, which shifts in composition and structure during development (22) and is influenced by nutrient intake (3, 17, 55, 56). Future research could examine whether larval fungal colonization has long-lasting effects on adult-associated microbiota by implementing a similar experimental design and rearing adults past initial emergence from pupae. These analyses could reveal alterations to host-microbe interactions involved with blood digestion (1) and egg production (1, 29) throughout host development, which may identify significant relationships between initial adult microbe acquisition, assembly, and life history traits.
Although our study was conducted in a controlled laboratory environment, our findings could also support interpretations of natural host-associated microbiota. Microbiomes may differ across natural populations contingent on the presence or absence of particular fungi in the local environment. The detection of intrapopulation microbiome variation in natural studies may reflect mosquitoes not infested with gut fungi. Future research in mosquitoes, and other systems, analyzing natural host-associated microbiota could also consider fungal data, which may clarify detections of differential microbiomes and levels of intrapopulation variation.
Different populations of mosquitoes exhibit disparate vector competences for certain human viral pathogens (57, 58) and microbes contribute to host-pathogen interactions (59–63), suggesting that shifts in microbiomes may impact the dynamics of mosquito-borne diseases. Alterations to host-associated microbiota incurred by fungal colonization may affect these processes, and focused research could investigate whether gut fungi impact adult vector competence by utilizing infestation protocols and vector competence assays (58, 64). In addition, certain pathogenic fungi have been proposed and implemented as viable agents for pest control (35). However, it is paramount to carefully consider that the use of pathogenic fungi could destroy entire populations of mosquitoes that serve as food sources for other organisms within ecosystems. Zancudomyces culisetae is nonpathogenic and widespread, has a publicly available genome sequence (65), and is now shown to impact microbiomes in larval and adult mosquito stages in a laboratory setting. With further research and development, the Z. culisetae-A. aegypti system could emerge as a model to study nonlethal fungus-bacterium-host interactions, conceivably of considerable human health significance. As with all studies of host-associated microbiota, independent replication under different laboratory conditions and work flows is essential to support the findings of this and other such experiments, as proposed and implemented in human and arthropod microbiome studies (66–68).
This study adds to the growing body of evidence gathered from a broad range of study systems that fungi are important members in microbiomes. Despite the intimate relationship between fungi and bacteria, there has not been enough overlap between research in the fields of microbial ecology and traditional mycology. The unification of these fields is an incompletely realized opportunity for collaboration that could enable a more holistic framework to study host-microbe relationships. Research could incorporate fungal data in analyses by simply targeting fungal DNA sequences in tandem with standard bacterial protocols, contributing to both fields of study and advancing the scientific community’s understanding of the complex processes impacting microbiota. While further exploration is necessary to unravel the morphological and physiological factors involved, it is becoming increasingly apparent that fungi should be accounted for in studies assessing microbiome composition, structure, function, and host-microbe interactions.
MATERIALS AND METHODS
Fungal strain culturing and trichospore collection.
A culture of Z. culisetae (formerly Smittium culisetae; ARSEF 9012 [USDA-ARS Collection of Entomopathogenic Fungal Cultures, Ithaca, NY], COL-18-3) was maintained at room temperature on a 1/10 brain heart infusion agar plate with 3 ml of autoclaved Nanopure Water (Barnstead Thermolyne Corp., Dubuque, IA, USA) overlay containing 2 mg/ml of penicillin and 7 mg/ml of streptomycin to prevent bacterial contamination. Fungal trichospores were harvested by sterile collection and filtration of the overlay through a sheet of Miracloth (EMD Millipore, Burlington, MA, USA) and transfer to 1.5-ml microcentrifuge tubes (Eppendorf, Hamburg, Germany). Trichospores were concentrated by centrifugation at 900 × g for 10 min. Trichospore pellets were combined and resuspended in 1 ml of autoclaved Arrowhead bottled spring water (Nestle, Vevey, Switzerland), and spore concentration was calculated by counting viable spores using a Neubauer Improved C-Chip hemocytometer (SKC, Inc., Covington, GA, USA).
Experimental preparation and mosquito rearing.
Aedes aegypti eggs, derived from the USDA-ARS Gainesville line (Benzon Research, Inc., Carlisle, PA, USA), were stored at room temperature. Histology containers (Fisher Scientific, Pittsburgh, PA, USA) were filled with 350 ml of autoclaved bottled spring water, with four assigned to each of four experimental groups: group A, nonfungal larvae; group B, fungal larvae; group C, nonfungal adults; group D, fungal adults. Approximately 50 eggs were added to each rearing container which was then covered with four layers of autoclaved Miracloth and separately placed in a vacuum chamber (SP Industries, Inc., Warminster, PA, USA) to synchronize egg hatch timing (69). Approximately 400,000 fungal trichospores were added to rearing containers assigned to groups B and D. Mosquitoes were reared at 24 ± 1°C with a 16:8-h light/dark cycle in a low-temperature refrigerated incubator (model no. 3724; Fisher Scientific). Mosquito larvae were fed finely ground Tetramin fish food (Tetra, Melle, Germany), prepared with a mortar and pestle suspended as 0.2 g per 10 ml of autoclaved bottled spring water, with 1 to 2 ml of the fish slurry being added daily to each rearing container. All experimental protocols were performed on a sterilized laboratory workbench next to a Bunsen burner to minimize contamination.
Larval digestive tract visualization.
To visualize fungal colonization rates in groups B and D and check for fungal contamination in groups A and C, at least one third- or fourth-instar larva was collected from 14 of the 16 experimental replicate containers. Eight gut dissections were performed on larvae from treatment group A as well as 14 from group B, 9 from group C, and 15 from group D. Hindguts were removed and visualized with phase-contrast and Nomarski microscopy, and the observed fungal colonization was recorded. No fungal material was found in larvae collected from group A or C.
Mosquito sample collection.
Fourth-instar larvae from groups A and B were individually transferred to 1.5-ml microcentrifuge tubes and surface sterilized according to a previously described larval protocol (2), and microbial DNA extractions were performed. Mosquitoes from groups C and D were reared to pupae, transferred to 1.5-ml microcentrifuge tubes, and surface sterilized according to a previously described adult protocol (2). Surface-sterilized pupae were transferred separately to 15-ml centrifuge tubes (Corning, Inc., Corning, NY, USA) containing 7 ml of autoclaved bottled spring water and reared axenically for 2 to 3 days until adult emergence. The sex of newly emerged adults was visually identified, individual adult females were transferred to 1.5-ml microcentrifuge tubes, and microbial DNA extractions were performed.
Microbial DNA extraction.
Microbial DNA was extracted from live mosquitoes and other experimental sources with a Quick-DNA Fungal/Bacterial Miniprep kit (Zymo Research, Irvine, CA, USA) according to the protocol provided by the manufacturer with the following modifications: lysis buffer was added directly to the 1.5-ml microcentrifuge tubes containing harvested mosquitoes. Mosquitoes were manually ruptured with an autoclaved pellet pestle (DWK Life Sciences, Wertheim, Germany) for approximately 0.5 min for larvae and for 1 to 2 min for adults. Homogenized tube mixtures were transferred to bead tubes supplied with the extraction kit and disrupted using a vortex mixer at the maximum setting for 5 min. The elution buffer was heated to 45°C prior to its application to the spin filters supplied by the extraction kit and soaked on the filter surface for 5 min prior to the final elution step. Extracted microbial DNA was stored at –80°C.
At least four DNA extractions were performed on mosquitoes collected from each replicate container for groups A, C, and D, and at least two DNA extractions were performed on mosquitoes collected from each replicate for group B. Other DNA extractions were carried out to identify potentially significant sources of bacteria in the experimental system on 50 A. aegypti eggs and fish food slurry over 3 days after initial food preparation. Additional DNA extractions were also performed on a suite of negative-control samples, including autoclaved spring water to test if rearing water was sterile prior to the start of the experiment, blank extraction kit reagents to account for possible kit contamination, rearing water from a control container to test for airborne laboratory contamination, and autoclaved water from two 15-ml centrifuge tubes containing surface-sterilized pupae to assess the efficacy of pupal surface sterilization protocols. Two blank PCRs were also carried out to identify potential contamination of PCR reagents. All PCRs were performed using 5Prime HotMasterMix (Quantabio, Beverly, MA, USA).
Amplification of the fungal 18S rRNA gene.
PCRs targeting the fungal 18S rRNA gene using Z. culisetae-specific primers TR3/TR4 (5′-GGCACTGTCAGTGGTGAAATAC-3′ and 5′-GATTTCTCTTACGGTGCCAAGCA-3′) (50) (Tables 2 and 3, Rxn_1) were performed on experimental larvae and adults to identify potential fungal contamination in nonfungal larvae (see Image S1a in the supplemental material), quantify fungal colonization in fungal larvae (Image S1b), and evaluate the capacity of the fungus to transmit transstadially (Image S1c).
TABLE 2.
Primer sequences used in experimental PCRs
| Target and primer | Sequence |
||||
|---|---|---|---|---|---|
| Adapter | Spacer | Linker | Primer | Final | |
| 16S rRNA gene | |||||
| 341f_1 | ACACTGACGACATGGTTCTACA | AG | CCTACGGGNGGCWGCAG | ACACTGACGACATGGTTCTACAAGCCTACGGGNGGCWGCAG | |
| 341f_2 | ACACTGACGACATGGTTCTACA | T | AG | CCTACGGGNGGCWGCAG | ACACTGACGACATGGTTCTACATAGCCTACGGGNGGCWGCAG |
| 341f_3 | ACACTGACGACATGGTTCTACA | CT | AG | CCTACGGGNGGCWGCAG | ACACTGACGACATGGTTCTACACTAGCCTACGGGNGGCWGCAG |
| 341f_4 | ACACTGACGACATGGTTCTACA | GCT | AG | CCTACGGGNGGCWGCAG | ACACTGACGACATGGTTCTACAGCTAGCCTACGGGNGGCWGCAG |
| 785r_1 | TACGGTAGCAGAGACTTGGTCT | CT | GACTACHVGGGTATCTAATCC | TACGGTAGCAGAGACTTGGTCTCTGACTACHVGGGTATCTAATCC | |
| 785r_2 | TACGGTAGCAGAGACTTGGTCT | T | CT | GACTACHVGGGTATCTAATCC | TACGGTAGCAGAGACTTGGTCTTCTGACTACHVGGGTATCTAATCC |
| 785r_3 | TACGGTAGCAGAGACTTGGTCT | AT | CT | GACTACHVGGGTATCTAATCC | TACGGTAGCAGAGACTTGGTCTATCTGACTACHVGGGTATCTAATCC |
| 785r_4 | TACGGTAGCAGAGACTTGGTCT | GAT | CT | GACTACHVGGGTATCTAATCC | TACGGTAGCAGAGACTTGGTCTGATCTGACTACHVGGGTATCTAATCC |
| 18S rRNA gene | |||||
| TR3F | GGCACTGTCAGTGGTGAAATAC | GGCACTGTCAGTGGTGAAATAC | |||
| TR4R | GATTTCTCTTACGGTGCCAAGCA | GATTTCTCTTACGGTGCCAAGCA | |||
TABLE 3.
Reaction setup and thermocycler settings used for experimental PCR
| Parameter | Value of the parameter for: |
|||
|---|---|---|---|---|
| Rxn_1a | Rxn_2 | Rxn_3 | Rxn_4 | |
| Total vol (μl) | 50 | 50 | 50 | 25 |
| Primers | TR3-TR4 | 341f-785r | 341f-785r | Barcodes |
| Master mix (μl) | 20 | 20 | 20 | 10 |
| 10 μM primer (μl) | 2 | 2 | 2 | 0.94 |
| Template DNA (μl) | 2 | 2 | 2 | 1.25 |
| Salinibacter ruber DNA (μl) | 0.59 | |||
| Nuclease-free H2O (μl) | 26 | 26 | 25.41 | 12.81 |
| Initial denaturation | ||||
| Temp (°C) | 95 | 94 | 94 | 94 |
| Duration (min) | 2 | 3 | 3 | 1.5 |
| Denaturation | ||||
| Temp (°C) | 95 | 94 | 94 | 94 |
| Duration (min) | 1 | 0.75 | 0.75 | 0.5 |
| Annealing | ||||
| Temp (°C) | 55 | 60 | 60 | 60 |
| Duration (min) | 1.5 | 1 | 1 | 0.5 |
| Elongation | ||||
| Temp (°C) | 72 | 72 | 72 | 72 |
| Duration (min) | 1.5 | 1.5 | 1.5 | 1.5 |
| Total no. of cycles | 35 | 35 | 35 | 10 |
| Final elongation | ||||
| Temp (°C) | 72 | 72 | 72 | 72 |
| Duration (min) | 10 | 10 | 10 | 5 |
Rxn, reaction.
Amplification and sequencing of the bacterial 16S rRNA gene.
The V3/V4 hypervariable regions of the microbial 16S rRNA gene were amplified with the primer pair 341f/785r (5′-CCTACGGGNGGCWGCAG-3′ and 5′-GACTACHVGGGTATCTAATCC-3′) (70), with attached linker sequences (71) and adapter and spacer sequences provided by the University of Idaho Genomics Resources Core (GRC; University of Idaho, Moscow, ID, USA). Targeted 16S PCRs were performed on extracted experimental DNA samples using four primer pair variants containing spacer sequences of different lengths to mitigate amplification biases (Tables 2 and 3, Rxn_2). Additional 16S PCRs were performed on a subset of experimental samples spiked with DNA from the halophilic bacterium Salinibacter ruber (ATCC product BAA-605D-5) (Tables 2 and 3, Rxn_3), and approximately 1,000,000 16S copies were added to sample template DNA for use in spike-in calibration to microbial load (SCML) quantitative analyses of bacterial loads (49). All PCR products were visualized on 1.5% agarose gels to confirm amplification. Secondary PCRs were performed to attach barcode sequences provided by the University of Idaho GRC to PCR amplicons (Table 3, Rxn_4). Amplicon sequencing was performed with an Illumina MiSeq, version 3 (Illumina, Inc., San Diego, CA, USA), at the University of Idaho GRC, producing 300-bp paired-end reads. Reads were demultiplexed by sample barcode sequences at the sequencing facility.
Data processing and taxonomic assignment of ASVs.
Sequencing reads were processed in the DADA2 pipeline (72) (File S1). Forward and reverse reads were trimmed to 278 and 167 bp, respectively, and at the location of the first occurrence of a base call with a Phred score less than or equal to 15, filtered by discarding reads with any number of N base calls or containing 6 or more estimated errors, and merged with a minimum overlap of 12 bases. Experimental samples with fewer than 100 reads after initial filtering were removed from the pipeline. Chimeric sequences were discarded, and merged reads were dereplicated. DADA2 generates amplicon sequence variants (ASVs) that are analogous and an improvement upon operational taxonomic units (OTUs). Taxonomy was assigned to ASVs using the SILVA, version 132, database (73, 74). A neighbor-joining tree was inferred using the phangorn package in R (75), and a generalized time-reversible with gamma rate variation maximum likelihood tree was fit using the neighbor-joining tree as the starting point. The phylogenetic tree, taxonomically assigned ASVs, read count data, and experimental metadata were combined into a single object using the Phyloseq package in R (76).
Contaminant ASV removal.
Reagents from extraction kits and other laboratory sources add contaminant sequences to experimental samples and can affect characterization of microbiomes if not accounted for (77). Of the negative controls sequenced, three of the four extraction kits had over 100 reads after processing in DADA2, while all others had fewer than 100 reads and were removed from further analyses. Reads from all four kits were pooled, and contaminant ASVs were identified using the decontam package in R (78) with the “prevalence” method and the threshold set to 0.5. All ASVs identified as contaminants were removed prior to downstream analyses.
Data preparation and statistical analyses.
Sequencing data were grouped into separate data sets and independently processed for comparative analyses for larva type (nonfungal and fungal), adult type (nonfungal and fungal), developmental stage of nonfungal (larvae and adults) and fungal (larvae and adults) mosquitoes, and potentially significant sources of bacteria in the experimental system (food and eggs) (File S1).
Alpha diversity measures for Simpson (community diversity and evenness) and Shannon (overall community diversity) diversity indices were calculated using nontransformed reads in Phyloseq (Table S4), and box plots were generated using the ggplot2 package in R (79). Coefficient of variation values (the ratio of the standard deviation to the mean) are used to assess variation of alpha diversity measures within groups (80, 81) and were calculated using the sjstats package in R (82). The R package cvequality (83) was used to test for significant differences of CV values across groups within a data set using an asymptotic test (84) and a modified signed-likelihood ratio test (MSLRT) (85).
Rarefaction curves were generated using the ranacapa package (86) and ggplot2 in R (Fig. S4). Rarefaction read cutoff values were determined independently for each data set to maximize richness captured while minimizing the number of samples cut. Reads were rarefied to 5,251 for the larva type data set, 1,777 for the adult type, 2,560 for nonfungal mosquitoes, and 2,229 for fungal mosquitoes. Singletons were removed, and data sets were further processed by discarding ASVs that were not represented by at least six reads in one sample within a data set after rarefaction.
Bray-Curtis dissimilarity (compositional dissimilarity), unweighted UniFrac distance (qualitative compositional comparison weighted for phylogenetic distance), weighted UniFrac distance (quantitative compositional comparison weighted for phylogenetic distance), and Jaccard similarity index (compositional similarity) were calculated in Phyloseq, and tests for significant differences due to the main effect (treatment or developmental stage) were carried out using permutational multivariate analysis of variance (PERMANOVA) (87) with 999 permutations in the Vegan package in R (88) along with the nested.npmanova function in the BiodiversityR package in R (89). Nested PERMANOVA calculated the correct pseudo F and P values for the main effect for each comparative analysis while accounting for random effects across replicate rearing containers. Nonmetric multidimensional scaling (NMDS) plots were created in Phyloseq in combination with ggplot2. Dispersions of beta diversity are used to measure variation of beta diversity within groups (90) and were calculated for each metric for each group within a data set in Vegan. Permutational statistical tests for the homogeneity of group dispersions (91) were performed with 999 permutations in Vegan to detect significant differences in beta diversity variation across groups, and box plots were created in ggplot2. Mean relative abundances of the 15 most abundant bacterial families shared between groups within each data set (which accounted for greater than 80% of the total reads in all data sets) (Table S1) were calculated, and stacked bar plots were created in Phyloseq in combination with ggplot2. Relative abundances and log2(fold change) values of genera within significant families were calculated (Table S2) and used as proxies to estimate increases or decreases in adults relative to levels in larvae, and taxa found at greater than a mean of 1% in a developmental stage within a treatment were plotted (Fig. 6 and 7) using ggplot2.
SCML calculations.
Sequencing data were grouped into subsets by treatment type for samples spiked with S. ruber DNA. A conversion factor was calculated by dividing the number of S. ruber reads in each sample by the mean number of S. ruber reads within the group. Reads were calibrated by multiplying the total number of reads in a sample by the sample-specific conversion factor (Table S3).
Linear mixed models.
The statistical significance of the main effect on mean alpha diversity measures, shifts of mean relative abundance of each of the 15 most abundant bacterial families shared between groups, and SCML calibrated read counts for each group within a data set was calculated by fitting a linear mixed model accounting for random effects across replicate rearing containers constructed with the lme4 package in R (92). Statistical significance was tested with type II Wald F tests with Kenward-Roger degrees of freedom using the car package in R (93).
Data availability.
Raw sequencing reads from this study were deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA541017.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grants P20GM103408 (Idaho INBRE program) and P20GM109095 (Idaho COBRE), the Robert W. Lichtwardt Student Research Award from the Mycological Society of America, a College of Arts and Sciences Award from Boise State University, and NSF ZyGoLife grant DEB 1441677. All sequencing data collection and preliminary analyses performed by the IBEST Genomics Resources Core at the University of Idaho were supported in part by NIH COBRE grant P30GM103324.
We thank Laura Bond for her advice regarding statistical analyses, Michael Song and Michael Wojahn for advice and assistance with the design of the bioinformatics workflow used in the study, Terry Bricker for academic mentorship, and Laurie Frankel for continued mentorship and support with the scientific writing process. We also thank our colleagues at the University of Idaho GRC for their collaboration and advice: Dan New, Sam Hunter, and Matt Fagnan. Finally, we thank certain members of the tricho-lab at Boise State University and other labs engaged in these studies.
We declare that we have no competing financial interests.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Gaio ADO, Gusmão DS, Santos AV, Berbert-Molina MA, Pimenta PFP, Lemos F. 2011. Contribution of midgut bacteria to blood digestion and egg production in Aedes aegypti (Diptera: Culicidae) (L.). Parasit Vectors 4:105. doi: 10.1186/1756-3305-4-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coon KL, Vogel KJ, Brown MR, Strand MR. 2014. Mosquitoes rely on their gut microbiota for development. Mol Ecol 23:2727–2739. doi: 10.1111/mec.12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Gilbreath TM, Kukutla P, Yan G, Xu J. 2011. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6(9):e24767. doi: 10.1371/journal.pone.0024767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gimonneau G, Tchioffo M, Abate L, Boissiere A, Awono-Ambene P, Nsango S, Christen R, Morlais I. 2014. Composition of Anopheles coluzzii and Anopheles gambiae microbiota from larval to adult stages. Infect Genet Evol 28:715–724. doi: 10.1016/j.meegid.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 5.Duguma D, Hall MW, Rugman-Jones P, Stouthamer R, Terenius O, Neufeld JD, Walton WE. 2015. Developmental succession of the microbiome of Culex mosquitoes ecological and evolutionary microbiology. BMC Microbiol 15:140. doi: 10.1186/s12866-015-0475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bascuñán P, Niño-Garcia JP, Galeano-Castañeda Y, Serre D, Correa MM. 2018. Factors shaping the gut bacterial community assembly in two main Colombian malaria vectors. Microbiome 6:148. doi: 10.1186/s40168-018-0528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen S, Bagdasarian M, Walker ED. 2015. Elizabethkingia anophelis: molecular manipulation and interactions with mosquito hosts. Appl Environ Microbiol 81:2233–2243. doi: 10.1128/AEM.03733-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moll RM, Romoser WS, Modrzakowski MC, Moncayo AC, Lerdthusnee K. 2001. Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (Diptera: Culicidae) metamorphosis. J Med Entomol 38:29–32. doi: 10.1603/0022-2585-38.1.29. [DOI] [PubMed] [Google Scholar]
- 9.Moncayo AC, Lerdthusnee K, Leon R, Robich RM, Romoser WS. 2005. Meconial peritrophic matrix structure, formation, and meconial degeneration in mosquito pupae/pharate adults: histological and ultrastructural aspects. J Med Entomol 42:939–944. doi: 10.1093/jmedent/42.6.939. [DOI] [PubMed] [Google Scholar]
- 10.Chavshin AR, Oshaghi MA, Vatandoost H, Yakhchali B, Raeisi A, Zarenejad F. 2013. Escherichia coli expressing a green fluorescent protein (GFP) in Anopheles stephensi: a preliminary model for paratransgenesis. Symbiosis 60:17–24. doi: 10.1007/s13199-013-0231-5. [DOI] [Google Scholar]
- 11.Chavshin AR, Oshaghi MA, Vatandoost H, Yakhchali B, Zarenejad F, Terenius O. 2015. Malpighian tubules are important determinants of Pseudomonas transstadial transmission and longtime persistence in Anopheles stephensi. Parasit Vectors 8:36. doi: 10.1186/s13071-015-0635-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown LD, Thompson GA, Hillyer JF. 2018. Transstadial transmission of larval hemocoelic infection negatively affects development and adult female longevity in the mosquito Anopheles gambiae. J Invertebr Pathol 151:21–31. doi: 10.1016/j.jip.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma P, Sharma S, Maurya RK, Das De T, Thomas T, Lata S, Singh N, Pandey KC, Valecha N, Dixit R. 2014. Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasit Vectors 7:235. doi: 10.1186/1756-3305-7-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briegel H. 2003. Physiological bases of mosquito ecology. J Vector Ecol 28:1–11. [PubMed] [Google Scholar]
- 15.Bhattacharyulu Y, Chaudhri RP, Gill BS. 1975. Transstadial transmission of Theileria annulata through common ixodid ticks infesting Indian cattle. Parasitology 71:1–7. doi: 10.1017/s0031182000053087. [DOI] [PubMed] [Google Scholar]
- 16.De Waal DT, Potgieter FT. 1987. The transstadial transmission of Babesia caballi by Rhipicephalus evertsi evertsi. Onderstepoort J Vet 54:655–656. [PubMed] [Google Scholar]
- 17.Rani A, Sharma A, Rajagopal R, Adak T, Bhatnagar RK. 2009. Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi—an Asian malarial vector. BMC Microbiol 9:96. doi: 10.1186/1471-2180-9-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gusmão DS, Santos AV, Marini DC, Bacci M, Berbert-Molina MA, Lemos F. 2010. Culture-dependent and culture-independent characterization of microorganisms associated with Aedes aegypti (Diptera: Culicidae) (L.) and dynamics of bacterial colonization in the midgut. Acta Trop 115:275–281. doi: 10.1016/j.actatropica.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Zouache K, Raharimalala FN, Raquin V, Tran-Van V, Raveloson LHR, Ravelonandro P, Mavingui P. 2011. Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiol Ecol 75:377–389. doi: 10.1111/j.1574-6941.2010.01012.x. [DOI] [PubMed] [Google Scholar]
- 20.Duguma D, Rugman-Jones P, Kaufman MG, Hall MW, Neufeld JD, Stouthamer R, Walton WE. 2013. Bacterial communities associated with Culex mosquito larvae and two emergent aquatic plants of bioremediation importance. PLoS One 8:e72522. doi: 10.1371/journal.pone.0072522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandler JA, Liu RM, Bennett SN. 2015. RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Front Microbiol 6:185. doi: 10.3389/fmicb.2015.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muturi EJ, Kim CH, Bara J, Bach EM, Siddappaji MH. 2016. Culex pipiens and Culex restuans mosquitoes harbor distinct microbiota dominated by few bacterial taxa. Parasit Vectors 9:18. doi: 10.1186/s13071-016-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muturi EJ, Ramirez JL, Rooney AP, Kim CH. 2017. Comparative analysis of gut microbiota of mosquito communities in central Illinois. PLoS Negl Trop Dis 11:e0005377. doi: 10.1371/journal.pntd.0005377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickson LB, Ghozlane A, Volant S, Bouchier C, Ma L, Vega-Rúa A, Dusfour I, Jiolle D, Paupy C, Mayanja MN, Kohl A, Lutwama JJ, Duong V, Lambrechts L. 2018. Diverse laboratory colonies of Aedes aegypti harbor the same adult midgut bacterial microbiome. Parasit Vectors 11:207. doi: 10.1186/s13071-018-2780-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osei-Poku J, Mbogo CM, Palmer WJ, Jiggins FM. 2012. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol Ecol 21:5138–5150. doi: 10.1111/j.1365-294X.2012.05759.x. [DOI] [PubMed] [Google Scholar]
- 26.Coon KL, Brown MR, Strand MR. 2016. Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Mol Ecol 25:5806–5826. doi: 10.1111/mec.13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muturi EJ, Lagos-Kutz D, Dunlap C, Ramirez JL, Rooney AP, Hartman GL, Fields CJ, Rendon G, Kim CH. 2018. Mosquito microbiota cluster by host sampling location. Parasit Vectors 11:468. doi: 10.1186/s13071-018-3036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chouaia B, Rossi P, Epis S, Mosca M, Ricci I, Damiani C, Ulissi U, Crotti E, Daffonchio D, Bandi C, Favia G. 2012. Delayed larval development in Anopheles mosquitoes deprived of Asaia bacterial symbionts. BMC Microbiol 12:S2. doi: 10.1186/1471-2180-12-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coon KL, Brown MR, Strand MR. 2016. Gut bacteria differentially affect egg production in the anautogenous mosquito Aedes aegypti and facultatively autogenous mosquito Aedes atropalpus (Diptera: Culicidae). Parasit Vectors 9:375. doi: 10.1186/s13071-016-1660-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deveau A, Bonito G, Uehling J, Paoletti M, Becker M, Bindschedler S, Hacquard S, Hervé V, Labbé J, Lastovetsky OA, Mieszkin S, Millet LJ, Vajna B, Junier P, Bonfante P, Krom BP, Olsson S, van Elsas JD, Wick LY. 2018. Bacterial-fungal interactions: ecology, mechanisms and challenges. FEMS Microbiol Rev 42:335–352. doi: 10.1093/femsre/fuy008. [DOI] [PubMed] [Google Scholar]
- 31.Muturi EJ, Bara JJ, Rooney AP, Hansen AK. 2016. Midgut fungal and bacterial microbiota of Aedes triseriatus and Aedes japonicus shift in response to La Crosse virus infection. Mol Ecol 25:4075–4090. doi: 10.1111/mec.13741. [DOI] [PubMed] [Google Scholar]
- 32.Angleró-Rodríguez YI, Talyuli OAC, Blumberg BJ, Kang S, Demby C, Shields A, Carlson J, Jupatanakul N, Dimopoulos G. 2017. An Aedes aegypti-associated fungus increases susceptibility to dengue virus by modulating gut trypsin activity. Elife 6:e28844. doi: 10.7554/eLife.28844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei G, Lai Y, Wang G, Chen H, Li F, Wang S. 2017. Insect pathogenic fungus interacts with the gut microbiota to accelerate mosquito mortality. Proc Natl Acad Sci U S A 114:5994–5999. doi: 10.1073/pnas.1703546114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramirez JL, Dunlap CA, Muturi EJ, Barletta ABF, Rooney AP. 2018. Entomopathogenic fungal infection leads to temporospatial modulation of the mosquito immune system. PLoS Negl Trop Dis 12:e0006433. doi: 10.1371/journal.pntd.0006433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lovett B, Bilgo E, Millogo SA, Ouattarra AK, Sare I, Gnambani EJ, Dabire RK, Diabate A, Leger RJ. 2019. Transgenic Metarhizium rapidly kills mosquitoes in a malaria-endemic region of Burkina Faso. Science 364:894–897. doi: 10.1126/science.aaw8737. [DOI] [PubMed] [Google Scholar]
- 36.Lichtwardt RW. 1986. The trichomycetes: fungal associates of arthropods. Springer-Verlag, New York, NY. [Google Scholar]
- 37.Williams MC, Lichtwardt RW. 1972. Infection of Aedes aegypti larvae by axenic cultures of the fungal genus Smittium (trichomycetes). Am J Bot 59:189–193. doi: 10.2307/2441400. [DOI] [Google Scholar]
- 38.Lichtwardt RW. 1984. Species of Harpellales living within the guts of aquatic diptera larvae. Mycotaxon 19:529–550. [Google Scholar]
- 39.Alencar YB, Ríos-Velásquez CM, Lichtwardt RW, Hamada N. 2003. Trichomycetes (Zygomycota) in the digestive tract of arthropods in Amazonas, Brazil. Mem Inst Oswaldo Cruz 98:799–810. doi: 10.1590/s0074-02762003000600016. [DOI] [PubMed] [Google Scholar]
- 40.McCreadie J, Beard C, Adler P. 2005. Context-dependent symbiosis between black flies (Diptera: Simuliidae) and trichomycete fungi (Harpellales: Legeriomycetaceae). Oikos 108:362–370. doi: 10.1111/j.0030-1299.2005.13417.x. [DOI] [Google Scholar]
- 41.Horn BW. 1989. Ultrastructural changes in trichospores of Smittium culisetae and S. culicis during in vitro sporangiospore extrusion and holdfast formation. Mycologia 81:742–753. doi: 10.2307/3759879. [DOI] [Google Scholar]
- 42.Horn BW. 1990. Physiological changes associated with sporangiospore extrusion from trichospores of Smittium culisetae. Exp Mycol 14:113–123. doi: 10.1016/0147-5975(90)90070-A. [DOI] [Google Scholar]
- 43.McCreadie JW, Beard CE. 2003. The microdistribution of the trichomycete Smittium culisetae in the hindgut of the black fly host Simulium vittatum. Mycologia 95:998–1003. doi: 10.1080/15572536.2004.11833015. [DOI] [PubMed] [Google Scholar]
- 44.Karl PJ, Hatch AM, Arcidiacono SM, Pearce SC, Pantoja-Feliciano IG, Doherty LA, Soares JW. 2018. Effects of psychological, environmental and physical stressors on the gut microbiota. Front Microbiol 9:2013. doi: 10.3389/fmicb.2018.02013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fredrickson AG, Stephanopoulos G. 1981. Microbial competition. Science 213:972–979. doi: 10.1126/science.7268409. [DOI] [PubMed] [Google Scholar]
- 46.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. 2010. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol 8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams MC. 1983. Spore longevity of Smittium culisetae (Harpellales, Legeriomycetaceae). Mycologia 75:171–174. doi: 10.1080/00275514.1983.12021649. [DOI] [Google Scholar]
- 48.Vojvodic S, McCreadie JW. 2007. The effect of temperature and host species on the development of the trichomycete Smittium culisetae (Zygomycota). Mycologia 99:412–420. doi: 10.3852/mycologia.99.3.412. [DOI] [PubMed] [Google Scholar]
- 49.Stämmler F, Gläsner J, Hiergeist A, Holler E, Weber D, Oefner PJ, Gessner A, Spang R. 2016. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome 4:28. doi: 10.1186/s40168-016-0175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rizzo AM, Pang K. 2005. New primers for detection of Smittium spp. (Trichomycetes, Zygomycota) in insect hosts. Fungal Divers 19:129–136. [Google Scholar]
- 51.McCafferty J, Mühlbauer M, Gharaibeh RZ, Arthur JC, Perez-Chanona E, Sha W, Jobin C, Fodor AA. 2013. Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. ISME J 7:2116. doi: 10.1038/ismej.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venkataraman A, Bassis CM, Beck JM, Young VB, Curtis JL, Huffnagle GB, Schmidt TM. 2015. Application of a neutral community model to assess structuring of the human lung microbiome. mBio 6:e02284-14. doi: 10.1128/mBio.02284-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeng Q, Sukumaran J, Wu S, Rodrigo A. 2015. Neutral models of microbiome evolution. PLoS Comput Biol 11:e1004365. doi: 10.1371/journal.pcbi.1004365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K, Bohannan B. 2016. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J 10:655. doi: 10.1038/ismej.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oliveira JHM, Gonçalves RLS, Lara FA, Dias FA, Gandara ACP, Menna-Barreto RFS, Edwards MC, Laurindo FRM, Silva-Neto MAC, Sorgine MHF, Oliveira PL. 2011. Blood meal-derived heme decreases ROS levels in the midgut of Aedes aegypti and allows proliferation of intestinal microbiota. PLoS Pathog 7:e1001320. doi: 10.1371/journal.ppat.1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Terenius O, Lindh JM, Eriksson-Gonzales K, Bussière L, Laugen AT, Bergquist H, Titanji K, Faye I. 2012. Midgut bacterial dynamics in Aedes aegypti. FEMS Microbiol Ecol 80:556–565. doi: 10.1111/j.1574-6941.2012.01317.x. [DOI] [PubMed] [Google Scholar]
- 57.Charan SS, Pawar KD, Severson DW, Patole MS, Shouche YS. 2013. Comparative analysis of midgut bacterial communities of Aedes aegypti mosquito strains varying in vector competence to dengue virus. Parasitol Res 112:2627–2637. doi: 10.1007/s00436-013-3428-x. [DOI] [PubMed] [Google Scholar]
- 58.Gonçalves CM, Melo FF, Bezerra JMT, Chaves BA, Silva BM, Silva LD, Pessanha JEM, Arias JR, Secundino NFC, Norris DE, Pimenta P. 2014. Distinct variation in vector competence among nine field populations of Aedes aegypti from a Brazilian dengue-endemic risk city. Parasit Vectors 7:320. doi: 10.1186/1756-3305-7-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramirez JL, Souza-Neto J, Cosme RT, Rovira J, Ortiz A, Pascale JM, Dimopoulos G. 2012. Reciprocal tripartite interactions between the Aedes aegypti midgut microbiota, innate immune system and dengue virus influences vector competence. PLoS Negl Trop Dis 6:e1561. doi: 10.1371/journal.pntd.0001561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dennison NJ, Jupatanakul N, Dimopoulos G. 2014. The mosquito microbiota influences vector competence for human pathogens. Curr Opin Insect Sci 3:6–13. doi: 10.1016/j.cois.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jupatanakul N, Sim S, Dimopoulos G. 2014. The insect microbiome modulates vector competence for arboviruses. Viruses 6:4294–4313. doi: 10.3390/v6114294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hegde S, Rasgon JL, Hughes GL. 2015. The microbiome modulates arbovirus transmission in mosquitoes. Curr Opin Virol 15:97–102. doi: 10.1016/j.coviro.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carissimo G, Pondeville E, McFarlane M, Dietrich I, Mitri C, Bischoff E, Antoniewski C, Bourgouin C, Failloux A-B, Kohl A, Vernick KD. 2015. Antiviral immunity of Anopheles gambiae is highly compartmentalized, with distinct roles for RNA interference and gut microbiota. Proc Natl Acad Sci U S A 112:E176–E185. doi: 10.1073/pnas.1412984112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dickson LB, Jiolle D, Minard G, Moltini-Conclois I, Volant S, Ghozlane A, Bouchier C, Ayala D, Paupy C, Moro CV, Lambrechts L. 2017. Carryover effects of larval exposure to different environmental bacteria drive adult trait variation in a mosquito vector. Sci Adv 3:e1700585. doi: 10.1126/sciadv.1700585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, White MM, Kvist S, Moncalvo JM. 2016. Genome-wide survey of gut fungi (Harpellales) reveals the first horizontally transferred ubiquitin gene from a mosquito host. Mol Biol Evol 33:2544–2554. doi: 10.1093/molbev/msw126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sinha R, Abnet CC, White O, Knight R, Huttenhower C. 2015. The microbiome quality control project: baseline study design and future directions. Genome Biol 16:276. doi: 10.1186/s13059-015-0841-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sinha R, Abu-Ali G, Vogtmann E, Fodor AA, Ren B, Amir A, Schwager E, Crabtree J, Ma S, Abnet CC, Knight R, White O, Huttenhower C. 2017. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat Biotechnol 35:1077–1086. doi: 10.1038/nbt.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frankel-Bricker J, Song MJ, Benner MJ, Schaack S. 2019. Variation in the microbiota associated with Daphnia magna across genotypes, populations, and temperature. Microb Ecol doi: 10.1007/s00248-019-01412-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foggie T, Achee N. 2009. Standard operating procedures: rearing Aedes aegypti for the HITSS and Box Laboratory Assays. Training manual v. 1.0 Uniformed Services University of Health Sciences, Bethesda, MD. [Google Scholar]
- 70.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takahashi S, Tomita J, Nishioka K, Hisada T, Nishijima M. 2014. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS One 9:e105592. doi: 10.1371/journal.pone.0105592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schliep KP. 2011. phangorn: phylogenetic analysis in R. Bioinformatics 27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McMurdie PJ, Holmes S. 2013. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW. 2014. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Davis NM, Proctor D, Holmes SP, Relman DA, Callahan BJ. 2018. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wickham H. 2011. Ggplot2. Wiley Interdiscip Rev Comput Stat 3:180–185. doi: 10.1002/wics.147. [DOI] [Google Scholar]
- 80.Flores GE, Caporaso JG, Henley JB, Rideout JR, Domogala D, Chase J, Leff JW, Vázquez-Baeza Y, Gonzalez A, Knight R, Dunn RR, Fierer N. 2014. Temporal variability is a personalized feature of the human microbiome. Genome Biol 15:531. doi: 10.1186/s13059-014-0531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Galloway-Peña JR, Smith DP, Sahasrabhojane P, Wadsworth WD, Fellman BM, Ajami NJ, Shpall EJ, Daver N, Guindani M, Petrosino JF, Kontoyiannis DP, Shelburne SA. 2017. Characterization of oral and gut microbiome temporal variability in hospitalized cancer patients. Genome Med 9:21. doi: 10.1186/s13073-017-0409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lüdecke D. 2017. sjstats: statistical functions for regression models. https://cran.r-project.org/web/packages/sjstats/index.html.
- 83.Marwick B, Krishnamoorthy K. 2016. cvequality: tests for the equality of coefficients of variation from multiple groups. R software package version 0.1.3. https://github.com/benmarwick/cvequality.
- 84.Feltz CJ, Miller GE. 1996. An asymptotic test for the equality of coefficients of variation from k populations. Stat Med 15:647–658. doi: 10.1002/(SICI)1097-0258(19960330)15:6<647::AID-SIM184>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 85.Krishnamoorthy K, Lee M. 2014. Improved tests for the equality of normal coefficients of variation. Comput Stat 29:215–232. doi: 10.1007/s00180-013-0445-2. [DOI] [Google Scholar]
- 86.Kandlikar GS, Gold ZJ, Cowen MC, Meyer RS, Freise AC, Kraft NJB, Moberg-Parker J, Sprague J, Kushner DJ, Curd EE. 2018. ranacapa: an R package and Shiny web app to explore environmental DNA data with exploratory statistics and interactive visualizations. F1000Research 7:1734. doi: 10.12688/f1000research.16680.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anderson MJ. 2017. Permutational multivariate analysis of variance (PERMANOVA). https://onlinelibrary.wiley.com/doi/pdf/10.1002/9781118445112.stat07841.
- 88.Dixon P. 2003. VEGAN, a package of R functions for community ecology. J Veg Sci 14:927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- 89.Kindt R. 2016. BiodiversityR: package for community ecology and suitability analysis. https://CRAN.R-project.org/package=BiodiversityR.
- 90.Anderson MJ, Ellingsen KE, McArdle BH. 2006. Multivariate dispersion as a measure of beta diversity. Ecol Lett 9:683–693. doi: 10.1111/j.1461-0248.2006.00926.x. [DOI] [PubMed] [Google Scholar]
- 91.Anderson MJ. 2006. Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62:245–253. doi: 10.1111/j.1541-0420.2005.00440.x. [DOI] [PubMed] [Google Scholar]
- 92.Bates D, Maechler M, Bolker B, Walker S, Maechler M, Walker S. 2015. Package lme4: linear mixed-effects models using ‘Eigen’ and S4. https://CRAN.R-project.org/package=lme4.
- 93.Fox J, Weisberg S, Price B, Adler D, Bates D, Baud-Bovy G. 2018. car: companion to applied regression. https://CRAN.R-project.org/package=car. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing reads from this study were deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA541017.