

Short abstract
Non-resolving inflammatory monocytes/macrophages are critically involved in the pathogenesis of chronic inflammatory diseases. However, mechanisms of macrophage polarization are not well understood, thus hindering the development of effective strategies to promote inflammation resolution. In this study, we report that macrophages polarized by subclinical super-low dose LPS preferentially expressed pro-inflammatory mediators such as ccl2 (which encodes the protein monocyte chemo attractant protein-1) with reduced expression of anti-inflammatory/homeostatic mediators such as slc40a1 (which encodes the protein ferroportin-1). We observed significantly elevated levels of the autophagy-associated and pro-inflammatory protein p62 in polarized macrophages, closely correlated with the inflammatory activation of ccl2 gene expression. In contrast, we noted a significant increase of ubiquitinated/inactive nuclear-erythroid-related factor 2 (NRF2), consistent with reduced slc40a1 gene expression in polarized macrophages. Addition of the homeostatic restorative agent phenylbutyrate (4-PBA) effectively reduced cellular levels of p62 as well as ccl2 gene induction by super-low dose LPS. On the other hand, application of 4-PBA also blocked the accumulation of ubiquitinated NRF2 and restored anti-inflammatory slc40a1 gene expression in macrophages. Together, our study provides novel insights with regard to macrophage polarization and reveals 4-PBA as a promising molecule in restoring macrophage homeostasis.
Keywords: Innate polarization, p62, NFR2, LPS, 4-PBA
Introduction
Recent studies suggest that systemic subclinical endotoxemia may be a key contributing factor for non-resolving inflammation associated with chronic inflammatory diseases, including atherosclerosis and obesity.1–3 Chronic inflammation is a key feature of these diseases and is thought to arise from a lack of inflammation resolution, resulting in a failure to return to homeostasis with continued inflammation.4 The polarization of innate immune cells such as monocytes and macrophages to non-resolving pro-inflammatory states may contribute to this failure of inflammation resolution and disease pathogenesis.5–7 These polarized macrophages are often characterized by the expression of pro-inflammatory mediators without anti-inflammatory/resolving mediators.7 Using an in-vivo mouse model of atherosclerosis, we found that subclinical LPS lead to inflammatory polarized monocytes/macrophages and more severe atherosclerosis accompanied by increased plasma levels of the pro-inflammatory chemokine monocyte chemoattractant protein 1 (MCP-1),8–10 which has been shown to be associated with pro-inflammatory M1-like polarized macrophages.7 However, the underlying mechanisms of macrophage polarization by subclinical dose LPS and the effects on anti-inflammatory/resolving mediator expression in macrophages are not well understood.
In addition to MCP-1, we have also previously reported an accumulation of the pro-inflammatory mediator p62 by subclinical LPS.11 Sequestosome1 or p62 is a protein commonly associated with autophagy where it binds to ubiquitinated cargo, targeting it for autophagy, and then is ultimately degraded along with the cargo upon fusion of the autophagosome with the lysosome.12,13 Recent mechanistic studies suggest that compromised autophagy completion in monocytes/macrophages can lead to the accumulation of p62, which may further contribute to the propagation of inflammation14 through the activation of NF-κB.15–17 NF-κB is a pro-inflammatory transcription factor that can be up-regulated downstream of TLR4 signaling to induce the gene expression of a variety of pro-inflammatory mediators, including ccl2 (MCP-1).18,19 Accumulation of p62 has also been implicated in the regulation of the key homeostatic transcription factor nuclear erythroid-related factor 2 (Nrf2).20 Upon activation, Nrf2 can induce the expression of a wide variety of anti-inflammatory/homeostatic genes such as those involved in mediating oxidative stress21 and iron homeostasis,22 including slc40a1 (ferroportin-1 (FPN)).20,23 Up-regulation of FPN has been shown to reduce TLR4-mediated expression of pro-inflammatory cytokines such as TNF-α and IL-6 in mouse macrophages.24,25 Further, up-regulation of FPN has been associated with alternatively activated anti-inflammatory M2-like polarized macrophages.26,27 Thus, further analysis of the connection between these factors (p62, Nrf2, and FPN) may provide a better understanding of the coupled intracellular circuitry responsible for macrophage polarization dynamics.
At the translational level, compounds capable of restoring autophagy completion and reducing cellular stress have promising efficacy in treating inflammatory diseases.28–30 In particular, sodium phenylbutyrate (4-PBA) has gained significant attention as a promising compound in the treatment of diverse inflammatory diseases,31–33where it has been shown to modulate cell stress in part through effects on autophagy and NF-κB activity.33–36 Additionally, we have shown 4-PBA treatment of mouse primary neutrophils was able to enhance lysosomal fusion with the peroxisome.37 However, the effect of 4-PBA on the polarization of macrophages is not well studied. Given its similarities to the beneficial commensal bacteria metabolite butyrate38 and the endogenous cytoprotective ketone body β-hydroxybutyrate,39,40 we were interested in investigating its role as a potential homeostatic restorative agent in macrophage polarization. A clear mechanistic understanding of 4-PBA-mediated macrophage polarization may facilitate the future development of 4-PBA as an effective reagent in treating chronic inflammatory diseases.
Thus, in this study, we aimed (a) to define further the polarization of macrophages by subclinical endotoxin using both primary murine macrophages and immortalized murine macrophage cells, and (b) to examine whether 4-PBA may be able to reverse macrophage polarization caused by subclinical endotoxin. At the mechanistic level, we examined whether the accumulation of p62 as well as ubiquitinated Nrf2 in polarized inflammatory macrophages could be abated by 4-PBA.
Materials and methods
Reagents
Abs used were: p62/SQSTM1 (Cell Signaling Technology, Danvers, MA; #5114; 1:1000 in 5% BSA), Pellino1 (Cell Signaling Technology; #31474; 1:1000)in 5% BSA), Nrf2 (Thermo Fisher Scientific, Waltham, MA; #PA52788, 1:500 in 5% BSA), and GAPDH (Cell Signaling Technology; #2118; 1:1000 in 5% BSA). All blots were detected using an anti-rabbit IgG-HRP-linked Ab (Cell Signaling Technology; #7074; 1:2000 in 5% BSA). Primers were from Integrated DNA Technologies (Coralville, IA) and were against M. musculus MCP-1, forward: 5′-GCT GTA GTT TTT GTC ACC AAG CTC-3′, reverse: 5′-AGT GCT TGA GGT GGT TGT GG-3′, and Mus musculus Slc40a1 (FPN), forward 5′-GAT GGG TCC TTA CTG TCTGCT-3′, reverse: 5′-GAT TGT GAT GCG AGT GGC AG-3′. LPS was from Escherichia coli O111:B4 purified by phenol extraction from Sigma–Aldrich (St. Louis, MO; #L2630) and reconstituted in PBS.
Cell culture
Primary cells
Bone marrow was harvested from C57BL/6 mice aged 6–8 wk and cultured with 10 ng/ml M-CSF (PeproTech, Rocky Hill, NJ; #315-02) and either PBS or 100 pg/ml LPS in RPMI containing 10% FBS, 1% pen/strep, and 1% l-glutamine. The cells were cultured for a total of 5 d, with media changes on d 2 and 4.
Immortalized cells
Wild type immortalized bone marrow–derived macrophages (BMDMs) were a generous gift from Dr. Kate Fitzgerald at the University of Massachusetts Medical School, thawed from liquid nitrogen and passaged three times before use. The cells used for these experiments were all between 13 and 18 passages. For all experiments, WTiMac cells were plated at 1.5 × 106 cells/well in 1% FBS, 1% pen/strep, and 1% l-glutamine DMEM and allowed to adhere overnight. The cells were then treated the following morning with PBS, 1 ng/ml LPS, or 1 mM 4-PBA and cultured for an additional 9 h. All cells were cultured at 37°C, 5% CO2, and 100% humidity.
RNAseq
Total RNAs were extracted from cultured bone-marrow macrophages with or without LPS treatment as described above. The RNA quality, quantity, and purity were assessed by an Agilent Bioanalyzer (Agilent Technologies, Santa Clara CA). Messenger RNA isolation, cDNA library generation, and sequencing were performed according to the Illumina protocol with the Hiseq 2000 (Illumina, San Diego, CA). Sequencing reads were mapped to the mouse reference genome (mm9) using TopHat (iNHouse Communications, London, UK). Fragments per kilobase of transcript per million reads 226 (FPKM) values were calculated for each gene on with Cufflinks v2.2.1 (Cole Trapnell Lab, University of Washington, Seattle, WA). Significant changes between groups were calculated using Cuffdiff v2.2.1 and normalized using the geometric method. The files generated by the Cuffdiff program were then passed to the Cummerbund with an R Core Team package to determine the differentially expressed genes and to visualize the output. Heat maps were generated using R software (The R Foundation, Vienna, Austria). Red indicates high expression, and green indicates low expression.
Quantitative RT-PCR
RNA was isolated using the TRIzol protocol. RNA was reverse transcribed using the high-capacity cDNA reverse transcription kit from Thermo Fisher Scientific (#4368814) and the Mastercycler® personal (Eppendorf, Hamburg, Germany). cDNA was diluted with nuclease-free water before use in the quantitative PCR (qPCR) reaction. SsoAdvanced™ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA; #1725274) was used for the qPCR reaction and run on the Bio-Rad CFX96 Touch Real-Time System. Results were then analyzed using the double delta CT method, using HPRT-1 as the housekeeping control gene, followed by normalization to the control sample (PBS-treated) gene expression. GraphPad Prism v6.0 (GraphPad Software, La Jolla, CA) was used to determine significance and design graphs.
Western blot
Whole-cell lysates were harvested in 2% SDS lysis buffer on ice. Media was aspirated, cells were washed with PBS, scraped in 2% SDS lysis buffer, and transferred to 1.5 ml tubes. Lysates were incubated on ice for 15 min, boiled at 95°C for 5 min, and then stored at –20°C. Protein concentration was determined using the Bio-Rad DC Protein Assay kit (Bio-Rad Laboratories; #5000112). Lysates were run on 10% acrylamide gels, transferred to PVDF membranes (soaked in methanol prior to use), and blocked with 5% nonfat milk in TBST. The blots were washed for 2 × 5 min in TBST before application of primary Ab solution (in 5% BSA). Blots were incubated with primary Ab solution overnight on a 4°C rocker and then detected the following d by incubating with secondary anti-rabbit Ab in 5% BSA for 1 h. Blots were detected using the LAS-3000 detection machine (Fujifilm, Tokyo, Japan) and the WesternBright ECL detecting kit (Advansta, San Jose, CA; VWR #490005-002). Blots were quantified using ImageJ software (National Institutes of Health, Bethesda, MD) and by normalizing each target to its respective housekeeping expression and then normalizing these values to the control (PBS) group. Significance and graphs were determined using GraphPad Prism v6.0.
For protein versus transcript analyses, the un-normalized (to PBS) protein quantification values (from ImageJ analysis) were divided by the corresponding treatment’s un-normalized transcript expression values (the raw double delta CT values). This ratio was then analyzed using GraphPad Prism v6.0 for statistical significance using either Student’s t-test (for PBS vs. LPS analyses) or ANOVA (for analyses including PBS, 4-PBA, LPS, and 4-PBA+LPS treatment groups).
Results
Differential selection and inhibition of pro- versus anti-inflammatory genes by super-low dose LPS
We previously reported that innate monocytes/macrophages preferentially express pro-inflammatory mediators such as MCP-1 when persistently stimulated with subclinical doses of LPS.8 In order to provide a comprehensive profile of macrophage polarization in this context, we performed RNAseq with primary murine BMDMs treated with either PBS or super-low dose (SLD) LPS (100 pg/ml) for 5 days. We used the 0111:B4 LPS, well characterized by several independent groups as a specific agonist for TLR4, which requires the presence of cell-surface TLR4 for signaling.41–43 We used a 5-day culture system of primary murine BMDM well defined in our group, which maintains > 90% cell survival and allows the study of innate immune cell programming by persistent challenge with LPS.8,44 As shown in Figure 1a, macrophages persistently challenged with SLD LPS, similar to those observed in humans with subclinical endotoxemia,1,2 exhibited enhanced expression of inflammatory mediators such as ccl2, ccl5, Nos2, and ccl7,7,45–47 and reduced expression of key homeostatic mediators such as Lrrc32 (GARP)48–50 and slc40a1 (FPN). Lrrc32 encodes the protein GARP, which is involved in the tethering of latent TGF-β to the cell membrane of regulatory T cells48 and has been shown to promote adaptive immune tolerance in B cells.51 Building on our previous studies involving up-regulation of MCP-1 by SLD LPS8 and since FPN has been shown to be associated with anti-inflammatory M2-like macrophage polarization,26,27 we decided to focus on the effects of SLD LPS on ccl2 and slc40a1 to explore macrophage polarization by SLD LPS further.
Figure 1.

SLD LPS differentially induces selected genes such as Ccl2 (MCP1) and reduces genes such as slc40a1 (Fpn). (a) RNA-seq data. Heat map of representative genes up-regulated (red) or down-regulated (green) in bone marrow–derived macrophages (BMDMs) cultured with or without LPS (100 pg/ml) as described in the Materials and methods. (b) and (c) Effect of SLD LPS (100 pg/ml) on ccl2 and slc40a1 transcript expression in BMDMs. (d) and (e) Effect of SLD LPS (1 ng/ml) on ccl2 and slc40a1 transcript expression in immortalized murine macrophages (WTiMac). BMDMs were cultured with SLD LPS (100 pg/ml) for 5 d, while WTiMacs were treated with SLD LPS (1 ng/ml) for 9 h. Target gene expression was determined using the ΔΔCT method and HPRT-1 as the housekeeping gene. All results were then normalized to the control (PBS) expression. GraphPad Prism was used to determine statistical significance of these normalized expression data. Results are representative of at least three separate trials for both BMDM and immortalized macrophage data. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Chronic SLD LPS polarizes macrophages to a pro-inflammatory state through up-regulation of ccl2 and inhibition of slc40a1 transcript expression
We further confirmed the differential expression of ccl2 and slc40a1 using quantitative RT-PCR (qRT-PCR) in primary murine macrophages treated with SLD LPS (100 pg/ml) for 5 days, where we saw both increased ccl2 and decreased slc40a1 transcript expressions (Figure 1b and c). Furthermore, we found this phenomenon could be faithfully recapitulated using a previously developed immortalized macrophage cell line model where macrophages were incubated in the presence of SLD LPS (1 ng/ml) for 9 h to account for the reduced sensitivity and increased proliferative capacity of immortalized cells (Figure 1d and e). Given the relative ease of handling and abundance of materials available with the cell line, we decided to perform mechanistic studies using the cell line system to examine the underlying biochemical circuitry.
Chronic SLD LPS exposure leads to accumulation of pro-inflammatory mediator p62
To explore further the possible mechanisms behind the shift toward a pro-inflammatory phenotype with simultaneous down-regulation of homeostatic mediators, we examined the effects of SLD LPS treatment on the pro-inflammatory protein p62. We have previously published an increase in the autophagy-related protein p62 (also known as Sequestosome1) in response to treatment with SLD LPS.11 In addition to its role in autophagy, p62 can also have a role in the regulation of inflammation through its interaction with TRAF6, leading to up-regulation of NF-κB,15–17 a pro-inflammatory transcription factor activated downstream of TLR4 and responsible for the expression of various pro-inflammatory mediators, including ccl2.18,19 We found that treatment with SLD LPS led to a significant accumulation of p62, as seen by Western blot (Figure 2a). Activation of NF-κB has also been shown to up-regulate p62 transcription to form a feed-forward loop.52 Thus, to determine whether the observed accumulation of p62 by SLD LPS treatment was due to effects at the protein level or the transcript level, we performed qRT-PCR to assess p62 transcript levels (Figure 2b), and then compared the p62 protein expression to the p62 transcript expression by dividing protein expression by transcript expression to yield a p62 protein to transcript ratio (Figure 2c). Here, we saw that while SLD LPS treatment resulted in a significant increase in p62 transcript expression (Figure 2b), the p62 protein accumulation was significantly greater than the increased transcript expression, as indicated by the higher p62 protein to transcript ratio in macrophages treated with SLD LPS compared to those treated with PBS (Figure 2c). This suggested that the accumulation of p62 in macrophages treated with SLD LPS was primarily due to increased protein accumulation.
Figure 2.

Chronic SLD LPS leads to accumulation of pro-inflammatory mediator p62. Immortalized BMDMs were treated for 9 h with 1 ng/ml LPS or PBS (control). (a) SLD LPS results in accumulation of p62 protein. Quantified blot results (right). (b) SLD LPS results in accumulation of p62 transcript. Target gene expression was determined using the ΔΔCT method and with HPRT-1 as the housekeeping gene. All results were then normalized to the control (PBS) expression and analyzed for statistical significance using GraphPad Prism. (c) Raw (non-normalized to PBS) p62 protein quantification values from (a) were divided by raw (non-normalized to PBS) p62 transcript expression values obtained in (b) to yield the protein/transcript ratio. Blot is representative of at least three separate experiments, and at least three separate experimental blots were used for quantification and statistical analysis. At least three separate sample sets were used to determine p62 transcript expression. *P < 0.05; **P < 0.01.
4-PBA reduced SLD LPS–induced p62 accumulation and MCP-1 transcript expression
Accumulation of p62 is often associated with decreased autophagic flux and lysosomal fusion.12,53 We have shown in the past that SLD LPS can lead to a disruption in lysosomal fusion,11 and more recently we have shown that the homeostatic modulator 4-PBA was able to induce peroxisome–lysosome fusion in mouse neutrophils.37 Thus, we sought to determine if 4-PBA could reduce the levels of p62 caused by SLD LPS (Figure 3). We found that 4-PBA was able to reduce SLD LPS–induced p62 accumulation significantly, as seen by Western blot (Figure 3a). Further, when we performed the same transcript (Figure 3b) and protein versus transcript ratio (Figure 3c) analyses as in Figure 2, we saw that while 4-PBA was able to induce p62 transcript expression significantly, it significantly reduced the p62 protein accumulation by SLD LPS (Figure 3c). This reduction in p62 protein was also correlated with reduced ccl2 transcript expression in 4-PBA-treated macrophages, as measured by qRT-PCR (Figure 3d). In total, this suggests that 4-PBA is capable of reducing the SLD LPS–induced pro-inflammatory mediators p62 and ccl2, possibly due to its effect on restoring lysosomal fusion.
Figure 3.

Sodium phenylbutyrate (4-PBA) reduces SLD LPS–induced p62 accumulation and ccl2 expression. Immortalized BMDMs were treated for 9 h with PBS (control), 4-PBA (1 mM), LPS (1 ng/ml), or 4-PBA+LPS and analyzed for p62 protein and transcript expression as well as ccl2 expression. (a) 4-PBA reduced the accumulation of p62 caused by SLD LPS treatment. Quantified blot data on the right. Blot was quantified as described in the Materials and methods and is representative of at least three separate experiments. At least three separate blots were used for quantification and statistical analysis. **P < 0.01; ***P < 0.001. (b) p62 transcript expression. (c) p62 blot quantification from (a) divided by p62 transcript expression in (b) as described in Figure 2 and in the Materials and Methods. (d) ccl2 transcript expression. Transcript data were obtained as previously described in Figure 1 and as described in the Materials and methods section. Data from at least three separate quantitative RT-PCR experiments were used for statistical analysis and to produce the graph. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Chronic SLD LPS results in accumulation of ubiquitinated Nrf2
Next, we decided to examine possible mechanisms behind reduced anti-inflammatory mediator expression in macrophages treated with SLD LPS by examining the levels of the homeostatic protein nuclear-erythroid-related factor 2 (Nrf2). Nrf2 is a transcription factor that aids in maintaining cellular homeostasis by up-regulating a variety of genes involved in regulating oxidative stress and iron homeostasis, including slc40a1.23 Through Western blot analysis, we saw an increase in the ubiquitinated (or inactive) form of Nrf2 (as indicated by its high (100 kDa) molecular weight) with chronic exposure to SLD LPS (Figure 4), suggesting that SLD LPS may polarize macrophages by inhibiting the activity of the homeostatic modulator Nrf2.
Figure 4.
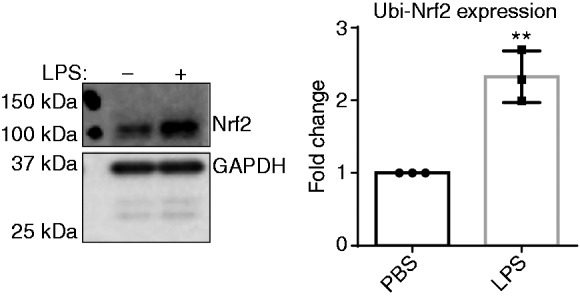
Chronic SLD LPS results in accumulation of ubiquitinated Nrf2. Immortalized macrophages were treated with either PBS (control) or LPS (1 ng/ml) for 9 h. LPS treatment resulted in accumulation of high molecular mass ubiquitinated Nrf2. Blot was quantified using ImageJ, as described in the Materials and methods section and is representative of at least three separate experiments, and at least three separate experiments were used for blot quantification and statistical analysis. **P < 0.01.
4-PBA restores Nrf2 and slc40a1 defects caused by chronic SLD LPS treatment
Next, we assessed whether 4-PBA could restore the disruptions in anti-inflammatory mediators caused by SLD LPS. As shown in Figure 5, 4-PBA was partially able to reduce the levels of ubiquitinated Nrf2 to control (PBS-treated) levels when applied in combination with SLD LPS (Figure 5a). This was also correlated with significant up-regulation of slc40a1 (FPN) gene expression (Figure 5b). Thus, in combination with the data in Figure 3, this suggests that 4-PBA may be able to restore cellular homeostasis by reducing inflammatory effects of chronic SLD LPS exposure while simultaneously restoring the expression of homeostatic modulators in murine macrophages.
Figure 5.

4-PBA restores the defects in Nrf2 and slc40a1 expression caused by SLD LPS. Immortalized macrophages were treated with PBS (control), 4-PBA (1 mM), LPS (1 ng/ml), or 4-PBA+LPS for 9 h. (a) 4-PBA reduced the accumulation of ubiquitinated Nrf2 caused by SLD LPS. Blot is representative of at least three separate experiments, and at least five blots were used for quantification and analysis. Blot was quantified using ImageJ, as described in the Materials and methods section. *P < 0.05; **P < 0.01. (b) 4-PBA restored slc40a1 transcript expression that had been reduced by SLD LPS. At least six separate quantitative RT-PCR experiments were used in the statistical analysis and generation of the pictured graph. Transcript expression was determined as previously described in Figure 1 and in the Materials and methods section. ***P < 0.001; ****P < 0.0001.
Discussion
In summary, our work highlights the simultaneous up-regulation of pro-inflammatory mediators (p62 and ccl2) and down-regulation of anti-inflammatory mediators (Nrf2 and slc40a1) by chronic SLD LPS exposure, providing further insight into the mechanisms of macrophage polarization observed in our previous studies8,54 (Figure 6). While most studies tend to focus on the effects of relatively high doses of LPS leading to robust expression of pro-inflammatory mediators, here we show that SLD LPS (such as those observed in patients with chronic inflammatory disease)1–3 are able to influence both pro- and anti-inflammatory responses in macrophages, giving unique insight into how SLD LPS may propagate non-resolving chronic inflammation.
Figure 6.
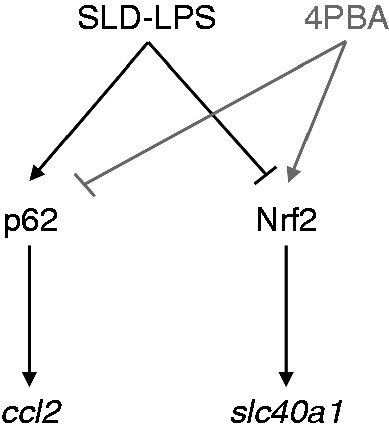
Summary of SLD LPS effects on macrophage polarization with remediation by 4-PBA. Chronic SLD LPS treatment leads to accumulation of pro-inflammatory mediators p62 and ccl2 coupled with simultaneous inhibition of homeostatic mediators Nrf2 and slc40a1. Addition of 4-PBA is able to reduce SLD LPS–induced p62 and restore Nrf2 leading to decreased ccl2 and increased slc40a1 expression.
In addition, we have shown a novel effect of the short-chain fatty acid derivative 4-PBA in being able to reverse the macrophage polarization induced by chronic SLD LPS exposure by altering both the pro- and anti-inflammatory response (Figure 6). While the beneficial effects of 4-PBA have been investigated in a variety of disease settings and cell types, including adipose tissue, osteoclasts, hepatocytes, and neurons,31,33,36,55 data concerning 4-PBA’s effects on macrophage polarization are limited. Previous studies have shown the ability of 4-PBA to decrease ccl2 (MCP-1) expression in various cell types,36,56 but we are the first to show that 4-PBA can reduce the expression of the inflammatory mediators p62 and ccl2 in murine macrophages. Further, we have shown that in the same system, 4-PBA is also capable of restoring levels of the homeostatic transcription factor Nrf2 and its related gene product, slc40a1 (FPN), an effect never before observed in macrophages. However, the mechanism behind 4-PBA’s restorative ability still requires further study.
4-PBA has been shown to act as an ammonia scavenger in urea cycle disorders,57 a histone deacetylase inhibitor (HDACi),31,58,59 as well as a chemical chaperone in the context of ER stress.60,61 More recently, it has also been implicated in peroxisome biogenesis62,63 and regulation of the cellular process autophagy.35,36,55,64,65 These studies suggest a wide range of possible activities by 4-PBA that are highly dependent on the cell type, dosage, and treatment regime. However, as alluded to previously, very few of these studies have been conducted in the context of macrophage polarization by LPS. Here, we suggest that 4-PBA may be acting to restore homeostasis in SLD LPS–treated macrophages in part by facilitating lysosomal fusion, as indicated by its ability to reduce p62 protein levels. p62 has been shown to associate with ubiquitinated cargo and become incorporated into the autophagosome, where it is then degraded upon fusion of the autophagosome with the lysosome.53 Thus, its accumulation has been linked to impaired autophagic flux/autophagic completion.53 Here, we have shown that independent of its effects on p62 transcript expression, 4-PBA is able to reduce p62 protein accumulation, as indicated by the decreased p62 protein to transcript ratio. This coupled with data from our recently published study in neutrophils, where 4-PBA was able to restore peroxisome–lysosome fusion and reduce pro-inflammatory mediator expression,37 suggests that 4-PBA may serve as a lysosomal fusion enhancer to restore cellular homeostasis.
Examining this further in the context of the present study on 4-PBA’s effects on macrophage polarization, induction of lysosomal fusion by 4-PBA may also restore anti-inflammatory mediators in part due to effects on the p62–Keap1–Nrf2 axis. Nrf2 is a transcription factor that under basal conditions is retained in the cytosol through interactions with its regulatory protein Keap1.66 Upon cell stress conditions (such as oxidative or xenobiotic stress), this interaction is disrupted, allowing for Nrf2 translocation into the nucleus and the up-regulation of various homeostatic genes.67 Recently, multiple groups have shown that p62 may play a vital role in this process by binding to Keap1 and targeting it for lysosomal degradation.68,69 Thus, by enhancing lysosomal fusion, 4-PBA may be able to enhance active levels of Nrf2 and reduce levels of p62(and Keap1). Further studies on the possible effects of 4-PBA on lysosomal fusion with relation to this p62–Keap1–Nrf2 axis are needed to elucidate better the possible connections between these pathways and the modulatory effects of 4-PBA.
Given the effects of 4-PBA on mediating the gene expression of ccl2 and slc40a1, another possible mechanism by which 4-PBA may act to restore macrophage homeostasis and which may warrant further investigation is its possible HDAC inhibitor activity. Indeed, 4-PBA has been shown to influence the activation of NF-κB-targeted genes directly through HDACi activity,31,70 which may correlate with the observed decrease in ccl2 expression and also possibly the reduction in p62, since p62 has also been shown to be under the transcriptional control of NF-κB.52 However, we did not see a reduction in p62 transcript expression with 4-PBA, and studies investigating 4-PBA’s possible effects on slc40a1 or other Nrf2-regulated genes at the transcript level are lacking. Thus, studies assessing acetylation status along with single-cell resolution genomics to examine gene expression may aid in further exploring this mechanism, as well as provide more extensive knowledge of the possible wide range of genes affected by SLD LPS macrophage polarization and its resolution by 4-PBA.
Our current study complements the emerging interest of innate immune programming and memory dynamics, which may bear relevance in the pathogenesis of non-resolving chronic diseases.48,71 Mechanistic insights regarding the underlying signaling circuitries are limited. Our data suggest that differential and competitive regulation of the pro-inflammatory modulator p62 and anti-inflammatory modulator Nrf2 might be an intriguing motif responsible for the dynamic programming of innate leukocytes. However, future extensive and systematic studies are clearly needed, with integrated approaches employing mutant cells and selective inhibitors, to define the complex network of intra-cellular signaling processes better. Our data also suggest that macrophages polarized by subclinical dose LPS may be programmed to express a unique subset of genes, as shown by the RNAseq analysis. In addition to the polarized induction of MCP1 and FPN, there are likely other novel genes differentially affected by subclinical dose LPS. For example, we noticed that subclinical dose LPS preferentially express selective chemokines, and suppress anti-inflammatory mediators such as GARP. GARP is important for the processing of mature TGF-β, and the suppression of GARP may likely further reinforce the steady pro-inflammatory state.48 Future studies of these additional mediators based on our findings are needed to define a clear profile of differentially polarized macrophages relevant to unique states of inflammatory diseases.
In summary, we have shown the regulation of both pro- and anti-inflammatory mediators by chronic SLD LPS exposure in the same system using murine macrophages. Further, we have shown for the first time the reduction of the inflammatory mediator p62 and the induction of the anti-inflammatory mediators Nrf2 and slc40a1 by 4-PBA in SLD LPS–treated macrophages. Together, these findings suggest a novel regulatory network in the polarization of macrophages by chronic SLD LPS that may provide a unique scaffold and insight for the development of future studies in treatment of chronic inflammatory diseases. Further, given 4-PBA’s structural similarity to both of the well-studied homeostatic modulators butyrate and β-hydroxybutyrate, 4-PBA may be a compound with promising therapeutic potential in the treatment of chronic inflammatory diseases.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was funded through the National Institutes of Health Grant Number RO1AI136386.
References
- 1.Wiedermann CJ, Kiechl S, Dunzendorfer S, et al. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. J Am Coll Cardiol 1999; 34: 1975–1981. [DOI] [PubMed] [Google Scholar]
- 2.Stoll LL, Denning GM, Weintraub NL. Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler Thromb Vasc Biol 2004; 24: 2227–2236. [DOI] [PubMed] [Google Scholar]
- 3.Sun L, Yu Z, Ye X, et al. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care 2010; 33: 1925–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathan C, Ding A. Nonresolving inflammation. Cell 2010; 140: 871–882. [DOI] [PubMed] [Google Scholar]
- 5.Murray PJ. Macrophage polarization. Annu Rev Physiol 2017; 79: 541–566. [DOI] [PubMed] [Google Scholar]
- 6.Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 2018; 233: 6425–6440. [DOI] [PubMed] [Google Scholar]
- 7.Parisi L, Gini E, Baci D, et al. Macrophage polarization in chronic inflammatory diseases: killers or builders? J Immunol Res 2018; 2018: 8917804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geng S, Chen K, Yuan R, et al. The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat Commun 2016; 7: 13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aiello RJ, Bourassa PA, Lindsey S, et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 1999; 19: 1518–1525. [DOI] [PubMed] [Google Scholar]
- 10.Deshmane SL, Kremlev S, Amini S, et al. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 2009; 29: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker B, Geng S, Chen K, et al. Alteration of lysosome fusion and low-grade inflammation mediated by super-low-dose endotoxin. J Biol Chem 2015; 290: 6670–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjørkøy G, Lamark T, Pankiv S, et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452: 181–197. [DOI] [PubMed] [Google Scholar]
- 13.Liu WJ, Ye L, Huang WF, et al. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 2016; 21: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ning H, Liu D, Yu X, et al. Oxidized low-density lipoprotein-induced p62/SQSTM1 accumulation in THP-1-derived macrophages promotes IL-18 secretion and cell death. Exp Ther Med 2017; 14: 5417–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanz L, Diaz–Meco MT, Nakano H, et al. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1–TRAF6 pathway. EMBO J 2000; 19: 1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008; 13: 343–354. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura K, Kimple AJ, Siderovski DP, et al. PB1 domain interaction of p62/sequestosome 1 and MEKK3 regulates NF-kappaB activation. J Biol Chem 2010; 285: 2077–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sutcliffe AM, Clarke DL, Bradbury DA, et al. Transcriptional regulation of monocyte chemotactic protein-1 release by endothelin-1 in human airway smooth muscle cells involves NF-κB and AP-1. Br J Pharmacol 2009; 157: 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JB, Wang TTY. Methyl (E)-(3-(3,4-dihydroxyphenyl)acryloyl)tryptophanate can suppress MCP-1 expression by inhibiting p38 MAP kinase and NF-κB in LPS-stimulated differentiated THP-1 cells. Eur J Pharmacol 2017; 810: 149–155. [DOI] [PubMed] [Google Scholar]
- 20.Katsuragi Y, Ichimura Y, Komatsu M. Regulation of the Keap1-Nrf2 pathway by p62/SQSTM1. Curr Opin Toxicol 2016; 1: 54–61. [Google Scholar]
- 21.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 2009; 284: 13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal 2018; 29: 1756–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harada N, Kanayama M, Maruyama A, et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch Biochem Biophys 2011; 508: 101–109. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Johnson EE, Shi HN, et al. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J Immunol 2008; 181: 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Harrington L, Trebicka E, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest 2009; 119: 3322–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corna G, Campana L, Pignatti E, et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010; 95: 1814–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Recalcati S, Locati M, Marini A, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol 2010; 40: 824–835. [DOI] [PubMed] [Google Scholar]
- 28.Wang C, Niederstrasser H, Douglas PM, et al. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat Commun 2017; 8: 2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim H, Lim Y-M, Kim KH, et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat Commun 2018; 9: 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sciarretta S, Yee D, Nagarajan N, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol 2018; 71: 1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardian G, Browne SE, Choi DK, et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J Biol Chem 2005; 280: 556–563. [DOI] [PubMed] [Google Scholar]
- 32.Huang A, Young TL, Dang VT, et al. 4-phenylbutyrate and valproate treatment attenuates the progression of atherosclerosis and stabilizes existing plaques. Atherosclerosis 2017; 266: 103–112. [DOI] [PubMed] [Google Scholar]
- 33.Guo Q, Xu L, Li H, et al. 4-PBA reverses autophagic dysfunction and improves insulin sensitivity in adipose tissue of obese mice via Akt/mTOR signaling. Biochem Biophys Res Commun 2017; 484: 529–535. [DOI] [PubMed] [Google Scholar]
- 34.Dasgupta S, Zhou Y, Jana M, et al. Sodium phenylacetate inhibits adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. J Immunol 2003; 170: 3874–3882. [DOI] [PubMed] [Google Scholar]
- 35.Zeng M, Sang W, Chen S, et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett 2017; 271: 26–37. [DOI] [PubMed] [Google Scholar]
- 36.Park H-J, Son H-J, Sul O-J, et al. 4-Phenylbutyric acid protects against lipopolysaccharide-induced bone loss by modulating autophagy in osteoclasts. Biochem Pharmacol 2018; 151: 9–17. [DOI] [PubMed] [Google Scholar]
- 37.Geng S, Zhang Y, Lee C, et al. Novel reprogramming of neutrophils modulates inflammation resolution during atherosclerosis. Sci Adv 2019; 5: eaav2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulthess J, Pandey S, Capitani M, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity 2019; 50: 432–445.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013; 339: 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab 2014; 25: 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorenz E, Patel DD, Hartung T, et al. Toll-like receptor 4 (TLR4)-deficient murine macrophage cell line as an in vitro assay system to show TLR4-independent signaling of Bacteroides fragilis lipopolysaccharide. Infect Immun 2002; 70: 4892–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Cousart S, Hu J, et al. Characterization of interleukin-1 receptor-associated kinase in normal and endotoxin-tolerant cells. J Biol Chem 2000; 275: 23340–23345. [DOI] [PubMed] [Google Scholar]
- 43.Maitra U, Deng H, Glaros T, et al. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol 2012; 189: 1014–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan R, Geng S, Chen K, et al. Low-grade inflammatory polarization of monocytes impairs wound healing. J Pathol 2016; 238: 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruytinx P, Proost P, Van Damme J, et al. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol 2018; 9: 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Girkin J, Hatchwell L, Foster P, et al. CCL7 and IRF-7 mediate hallmark inflammatory and IFN responses following rhinovirus 1B infection. J Immunol 2015; 194: 4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore BB, Murray L, Das A, et al. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol 2006; 35: 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran DQ, Andersson J, Wang R, et al. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 2009; 106: 13445–13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang R, Zhu J, Dong X, et al. GARP regulates the bioavailability and activation of TGFβ. Mol Biol Cell 2012; 23: 1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metelli A, Salem M, Wallace CH, et al. Immunoregulatory functions and the therapeutic implications of GARP-TGF-β in inflammation and cancer. J Hematol Oncol 2018; 11: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wallace CH, Wu BX, Salem M, et al. B lymphocytes confer immune tolerance via cell surface GARP-TGF-β complex. JCI Insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016; 164: 896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen K, Yuan R, Geng S, et al. Toll-interacting protein deficiency promotes neurodegeneration via impeding autophagy completion in high-fat diet-fed ApoE−/− mouse model. Brain Behav Immun 2017; 59: 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nissar AU, Sharma L, Mudasir MA, et al. Chemical chaperone 4-phenyl butyric acid (4-PBA) reduces hepatocellular lipid accumulation and lipotoxicity through induction of autophagy. J Lipid Res 2017; 58: 1855–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang F, Liu Y, Ren H, et al. ER-stress regulates macrophage polarization through pancreatic EIF-2alpha kinase. Cell Immunol 2019; 336: 40–47. [DOI] [PubMed] [Google Scholar]
- 57.Lichter-Konecki U, Diaz GA, Merritt JL, 2nd, et al. Ammonia control in children with urea cycle disorders (UCDs); phase 2 comparison of sodium phenylbutyrate and glycerol phenylbutyrate. Mol Genet Metab 2011; 103: 323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lea MA, Randolph VM, Hodge SK. Induction of histone acetylation and growth regulation in eryrthroleukemia cells by 4-phenylbutyrate and structural analogs. Anticancer Res 1999; 19: 1971–1976. [PubMed] [Google Scholar]
- 59.Daosukho C, Chen Y, Noel T, et al. Phenylbutyrate, a histone deacetylase inhibitor, protects against Adriamycin-induced cardiac injury. Free Radic Biol Med 2007; 42: 1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roy D, Kumar V, James J, et al. Evidence that chemical chaperone 4-phenylbutyric acid binds to human serum albumin at fatty acid binding sites. PLoS One 2015; 10: e0133012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Almeida SF, Picarote G, Fleming JV, et al. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem 2007; 282: 27905–27912. [DOI] [PubMed] [Google Scholar]
- 62.Wei H, Kemp S, McGuinness MC, et al. Pharmacological induction of peroxisomes in peroxisome biogenesis disorders. Ann Neurol 2000; 47: 286–296. [PubMed] [Google Scholar]
- 63.Vijayan V, Srinu T, Karnati S, et al. A new immunomodulatory role for peroxisomes in macrophages activated by the TLR4 ligand lipopolysaccharide. J Immunol 2017; 198: 2414–2425. [DOI] [PubMed] [Google Scholar]
- 64.Besio R, Iula G, Garibaldi N, et al. 4-PBA ameliorates cellular homeostasis in fibroblasts from osteogenesis imperfecta patients by enhancing autophagy and stimulating protein secretion. Biochim Biophys Acta 2018; 1864: 1642–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chiang CK, Wang CC, Lu TF, et al. Involvement of endoplasmic reticulum stress, autophagy, and apoptosis in advanced glycation end products–induced glomerular mesangial cell injury. Sci Rep 2016; 6: 34167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999; 13: 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kobayashi EH, Suzuki T, Funayama R, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nature Commun 2016; 7: 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013; 51: 618–631. [DOI] [PubMed] [Google Scholar]
- 69.Taguchi K, Fujikawa N, Komatsu M, et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci U S A 2012; 109: 13561–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park JS, Lee EJ, Lee JC, et al. Anti-inflammatory effects of short chain fatty acids in IFN-gamma-stimulated RAW 264.7 murine macrophage cells: involvement of NF-kappaB and ERK signaling pathways. Int Immunopharmacol 2007; 7: 70–77. [DOI] [PubMed] [Google Scholar]
- 71.Morris MC, Gilliam EA, Li L. Innate immune programing by endotoxin and its pathological consequences. Front Immunol 2014; 5: 680. [DOI] [PMC free article] [PubMed] [Google Scholar]