

Abstract
Clinical and preclinical studies show tissue-specific differences in tumorigenesis. Tissue specificity is controlled by differential gene expression. We prioritized genes that encode secreted proteins according to their preferential expression in normal lungs to identify candidates associated with lung cancer. Indeed, most of the lung-enriched genes identified in our analysis have known or suspected roles in lung cancer. We focused on the gene encoding neuron-derived neurotrophic factor (NDNF), which had not yet been associated with lung cancer. We determined that NDNF was preferentially expressed in the normal adult lung and that its expression was decreased in human lung adenocarcinoma and a mouse model of this cancer. Higher expression of NDNF was associated with better clinical outcome of patients with lung adenocarcinoma. Purified NDNF inhibited proliferation of lung cancer cells, whereas silencing NDNF promoted tumor cell growth in culture and in xenograft models. We determined that NDNF is downregulated through DNA hypermethylation near CpG island shores in human lung adenocarcinoma. Furthermore, the lung cancer–related DNA hypermethylation sites corresponded to the methylation sites that occurred in tissues with low NDNF expression. Thus, by analyzing the tissue-specific secretome, we identified a tumor-suppressive factor, NDNF, which is associated with patient outcomes in lung adenocarcinoma.
Keywords: Cell Biology
Keywords: Lung cancer, Tumor suppressors

Ranking secreted proteins according to tissue specificity identifies NDNF as a tumor-suppressive factor in lung cancer.
Introduction
Cancer is a collection of related diseases that develop as a result of the accumulation of somatic mutations and other genetic alterations leading to abnormal cell proliferation and eventually tumorigenesis (1–4). Although cancer can start nearly anywhere in the human body, tissues are substantially different in their susceptibility to certain oncogenic events that cause tumor formation (5–8). Only a handful of genes are associated with tumorigenesis in a wide variety of tissues, whereas alteration of others is linked only to very few cancer types (7, 8). For example, oncogenic KRAS-induced development of cancer is limited to specific tissues in mice, such as the pancreas and lung, but most other tissues are resistant to transformation by this oncogene (7, 9–12). Likewise, BRCA1 functions in all cell types to help preserve genomic stability, but oncogenesis is restricted to only a few tissues, notably ovary and breast (7, 13–16). How and why distinct genetic alterations promote cancer in one tissue but not another remain important and challenging questions in cancer research. Understanding the underlying molecular mechanisms should lead to an improvement in risk prediction, early detection, and prevention of cancers.
Tissue specificity in cancer is not only associated with tumorigenesis but also associated with therapeutic responses and the development of resistance, which are linked to distinct cancer types (7, 8). Traditionally, tumors from the same anatomical site are treated as one tumor entity. In the last decade, large-scale cancer genomics profiling revealed that similar cancer driver mutations initially discovered in a single tumor entity are also present in tumors of other anatomical sites (17–20), which led to the classification of cancers according to molecular alterations shared by tumors across different tissue types (21–23). This new classification supplements the traditional classifications of cancers based on the tissue or organ of origin for the purposes of treatment strategy and led to the proposal of generalized use of targeted drugs across anatomically distinct cancer types after initial proof of efficacy in 1 tumor type (24–28). However, several recent umbrella or basket trials showed that off-label use of targeted therapies across tumors from different tissues was not superior to standard of care (21, 22, 26, 29). Thus, driver mutations may have different effects in tumors in diverse tissues, yielding tissue-specific therapeutic responsiveness because of tissue-specific organization of oncogenic signaling pathways (7). Thus, understanding tissue specificity in cancer will provide new candidates for therapeutic intervention and lead to enhanced therapeutic strategies in precision oncology.
Tissue specificity is ultimately controlled by the differential expression of genes across normal tissues. Genes preferentially expressed in one tissue or a small number of tissues often play critical roles in tissue-specific biological processes, physiological conditions, and pathological states. Furthermore, disease-associated genes tend to be expressed in a limited number of normal tissues where their defective function causes pathology (30–33). Indeed, the identification of genes with tissue-selective expression has provided a deeper molecular understanding in many areas, including, but not limited to, tissue function, pathogenic mechanism, diagnosis, and organ-specific therapeutic applications (31, 33–38). Using the gene expression data sets in public repositories, studies show that many tumor suppressor genes and proto-oncogenes had their highest expression level in the tissue susceptible to their oncogenic effect (39, 40). Thus, it is worthwhile to test whether, and to what extent, tissue-enriched genes possess cancer-promoting or cancer-suppressing functions. We predicted that mutations or alterations in the expression levels of such tissue-enriched genes with tumor-suppressive or oncogenic functions are likely contributors to tumorigenesis or cancer progression in a tissue-specific manner. Therefore, systematic annotation and characterization of the tissue-enriched genes not only should lead to valuable insights about how cancers develop in different tissues but also could be used to identify new therapeutic targets associated with cancers originating from the same tissue.
Here, we focused on genes encoding secreted proteins, which mediate key communication between cells and are essential contributors to tumorigenesis and cancer progression (41–43). We prioritized genes encoding secreted proteins that are preferentially expressed in both mouse and human lung to identify candidates associated with lung cancer. Lung cancer remains a leading cause of cancer-related mortality worldwide and has a 5-year survival rate (18.6%), which is much lower than that of many other cancers (44). Although it may seem counterintuitive to seek cancer-associated genes in normal tissue, by starting with normal tissue, we could identify tissue-enriched (a) tumor-suppressing genes for which reduced expression contributes to cancer, (b) oncogenic genes for which increased expression contributes to cancer, and (c) tumor-enabling genes for which expression is permissive for but is not a driver of the development and progression of cancer.
Our lung secretome analysis led us to investigate the gene encoding neuron-derived neurotrophic factor (NDNF in human, Ndnf in mouse). We experimentally determined that NDNF is preferentially expressed in the normal adult lung but that its expression is decreased in human lung adenocarcinoma and a mouse model of lung adenocarcinoma. Higher expression of NDNF is associated with better clinical outcome of patients with lung adenocarcinoma, indicating that NDNF functions as a tumor suppressor. We experimentally confirmed the cancer-suppressing effects of NDNF, and, with data in The Cancer Genome Atlas (TCGA) database (45), we determined that NDNF is downregulated through DNA hypermethylation at CpG island shores in lung adenocarcinoma. Thus, our study showed that analysis of tissue-enriched genes can identify previously unknown cancer-associated genes and resulted in the identification of a secreted tumor-suppressing protein relevant to lung cancer.
Results
Identification of genes encoding secreted proteins that are preferentially expressed in normal adult mouse and human lung.
Proteins secreted from the cells (the secretome) regulate cell behavior and are clinically relevant in cancer, because they are a rich source of biomarkers and are targets of approved cancer treatments (41–43). Therefore, we focused on genes encoding secreted proteins, which we refer to as sGenes, to identify those preferentially expressed in the normal adult lungs of mice and humans (referred to hereafter as sLungGenes). We collected and curated 2591 orthologous human and mouse sGenes from 2 representative secreted protein databases: the secretome from the Human Protein Atlas (HPA) (46) and the Metazoa (Human/Animal) Secretome and Subcellular Proteome KnowledgeBase (MetazSecKB) (47). For these 2591 sGenes, we performed gene expression analyses using 3 available data sources: HPA database with expression data from 37 adult human tissue types, Genotype-Tissue Expression (GTEx) with expression data from 30 adult human tissue types (48), and Encyclopedia of DNA Elements Consortium (ENCODE) with expression data from 12 tissue types in 10-week-old C57BL/6J mice (49).
For each of the 2591 orthologous human and mouse sGenes, the lung-preferential expression scores (hPLung_HPA, hPLung_GTEx, and mPLung) across the available tissue types from the 3 databases were independently calculated. The preferential expression scores (hPLung_HPA, hPLung_GTEx, and mPLung) were converted to Z scores (Z-hPLung_HPA, Z-hPLung_GTEx, and Z-mPLung) (see Methods and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.129344DS1). We selected the top-ranking sLungGenes by setting the threshold of Z-hPLung (Z-hPLung_HPA and Z-hPLung_GTEx) and Z-mPLung values as 4.0 and 2.0, respectively (Figure 1). We selected the top-ranking sLungGenes overlapped by at least 2 tissue-specific gene expression resources to ensure the reliability of their preferential expression in the lung (Figure 1). For these high-confidence sLungGenes, we performed a literature query in PubMed and found most had literature evidence supporting relevance to lung cancer (29/31) or indicating that their activity restricts cancer cell growth (25/31) (Figure 1 and Supplemental Table 2). These results suggested that the sGenes preferentially expressed in the lung likely contribute to the tissue specificity of lung cancer. We followed up on the one gene that had not been previously reported as relevant to lung cancer, NDNF.
Figure 1. Identification of genes encoding secreted proteins preferentially expressed in both mouse and human lung.

(A) 3D scatter plot of the lung-preferential expression Z score of 2591 orthologous sGenes in human and mouse. Each dot indicates a cross-species conserved sGene (see Supplemental Table 1). Top-ranking sLungGenes, selected by setting the threshold of Z-hPLung (Z-hPLung_HPA and Z-hPLung_GTEx) as 4.0 and Z-mPLung values as 2.0, were highlighted with a color reflecting their amount of overlap among all 3 data sets as shown in B. (B) Summary of the top-ranking sLungGenes overlapped by at least 2 tissue-specific gene expression resources. sGene, genes encoding secreted proteins; sLungGenes, genes encoding secreted proteins that are preferentially expressed in the adult lung; z-PLung, lung-preferential expression Z score.
Verification of lung-specific expression of NDNF in normal adult mouse and human lung.
NDNF, also known as A930038C07Rik, C4orf31, Epidermacan, and NORD, is the one top-ranking sLungGene without known functions in lung cancer (Figure 1 and Supplemental Table 2). We investigated its expression and potential function in lung cancer.
Previously, mouse Ndnf has been reportedly expressed in the developing and adult central nervous system (50–54), as well as in ischemic skeletal muscle and the heart upon myocardial infarction (55, 56). Human NDNF expression was reported in neocortex, umbilical cord blood, and bone marrow multipotent mesenchymal stromal cells (57, 58). Our analysis of the human and mouse tissue databases indicated preferential expression of Ndnf in the normal adult lung in humans (Figure 1 and Supplemental Figures 1 and 2) and mice (Figure 1 and Supplemental Figure 3), which we validated using several approaches. We measured Ndnf transcripts by quantitative real-time reverse transcriptase PCR (RT-qPCR), which showed that Ndnf was preferentially expressed in the lung compared with other mouse tissues (Figure 2A). Along with the differential expression of Ndnf mRNA, we detected Ndnf proteins by Western blotting predominantly in the adult mouse lung with much lower or barely detectable amounts in the other examined tissues (Figure 2B and Supplemental Figure 4). Furthermore, we detected NDNF mRNA by in situ hybridization in both the normal adult mouse and human lung (Figure 2, C and D, Supplemental Figure 5). NDNF mRNA was not detected in several other human tissues including breast, liver, and spleen (Supplemental Figure 6). Consistent with the information from the HPA and MetazSecKB resources, as well as the previous reports (50, 55, 59), we confirmed that both mouse Ndnf and human NDNF were secreted when ectopically expressed in HEK293T cells (Supplemental Figure 7). Together, we experimentally confirmed that NDNF/Ndnf (human/mouse) encode secreted proteins and are preferentially expressed in the normal adult mouse and human lung.
Figure 2. NDNF is detected predominantly in normal mouse and human lung.

(A) RT-qPCR analysis of Ndnf mRNA levels in the lung and other indicated tissues of 10-week-old C57BL/6N mice (n = 3). (B) Western blot analysis of Ndnf protein levels in the lung and other tissues collected from 10-week-old C57BL/6N mice. A representative image is shown for 1 mouse with quantitative data from n = 3 mice shown below. Band intensity was quantified using ImageJ (NIH), and Ndnf abundance in each tissue was normalized to GAPDH. The relative Ndnf abundances were plotted. Box plots show 25th to 75th percentile; whiskers extend to the minimum and maximum values. One-way ANOVA was used for statistical analysis. ****P < 0.0001. (C and D) RNAscope in situ hybridization detection of NDNF/Ndnf mRNA (red) expression in the normal adult mouse (C) and human (D) lung. Cell nuclei are counterstained with hematoxylin (blue). Note that NDNF/Ndnf mRNA is mainly detected in the alveolar epithelium, whereas much less is found in the bronchial epithelium. Sense probes that served as negative controls are shown in Supplemental Figure 5. Scale bar: 100 μm.
Decreased NDNF expression in lung adenocarcinoma and association with better clinical outcome of patients.
The alveolar epithelium contains lung stem or progenitor cells and constitutes the major cell type responsible for lung adenocarcinoma, which is the most prevalent form of lung cancer and arises from the alveoli throughout the lungs (60–62). Intriguingly, in situ hybridization in the normal human and mouse lung revealed that NDNF/Ndnf were expressed mainly by the alveolar epithelial cells (Figure 2, C and D). We thus evaluated whether NDNF has clinical relevance in lung adenocarcinoma. We examined the expression of NDNF in tumor and normal samples using the lung adenocarcinoma data set in TCGA. When compared with histologically normal lung tissues adjacent to the tumor, expression of NDNF was significantly reduced in lung adenocarcinoma (Figure 3A). Expression of Ndnf in lung adenocarcinoma was also examined using the K-rasLA1 mouse model. The K-rasLA1 mice develop lung adenocarcinoma through somatic activation of a K-ras allele carrying an activating mutation in codon 12 (G12D) (9). Both RT-qPCR analysis and Western blots showed that Ndnf mRNA and protein were decreased in K-rasG12D–induced adenocarcinoma when compared with paired surrounding nontumor lung tissues (Figure 3, B and C). Thus, we found that NDNF was downregulated in human lung adenocarcinoma and in a murine model of this cancer.
Figure 3. NDNF expression is decreased in lung adenocarcinoma and associated with better clinical outcome of patients.

(A) NDNF mRNA in tumor (n = 517) and normal lung tissues (n = 59) from TCGA lung adenocarcinoma database. Box plots show 25th to 75th percentile; whiskers extend to the minimum and maximum values. The 2-tailed Mann-Whitney U test was used for statistical analysis. ****P < 0.0001. (B) Ndnf mRNA in tumor and matched normal lung tissue adjacent to the tumor (NAT) from K-rasLA1 mice (n = 10). Transcript abundance was determined by RT-qPCR analysis. The 2-tailed paired t test was used for statistical analysis. ****P < 0.0001. (C) Western blot analysis of Ndnf in tumor (T) and matched normal lung tissue adjacent to the tumor (NAT) from K-rasLA1 mice (n = 8). (D–F) Kaplan-Meier curves showing the correlation between NDNF expression and clinical outcome, as analyzed for overall survival (OS), progression-free survival (PFS), and postprogression survival (PPS) of lung adenocarcinoma patients. (G–I) Kaplan-Meier curves showing the correlation between NDNF expression and OS of lung adenocarcinoma patients at indicated stages. Number of samples in the high- and low-NDNF groups and a corresponding log-rank P value are indicated on each graph. Kaplan-Meier curves were created using the Kaplan-Meier Plotter (www.kmplot.com) with lung adenocarcinoma patients grouped according to the median expression value of NDNF. Information of lung adenocarcinoma patients involved in the survival analysis is in Supplemental Table 3.
We evaluated whether the expression level of NDNF has any predictive value for survival of lung adenocarcinoma patients by performing Kaplan-Meier analyses (63). We found patients with higher NDNF expression had better overall survival than those with lower expression (Figure 3D, Supplemental Figure 8, and Supplemental Tables 3 and 4). Lung adenocarcinoma patients with high NDNF also showed significantly better progression-free survival and postprogression survival compared with those with low levels of NDNF (Figure 3, E and F). In addition, the higher expression of NDNF was better associated with overall survival of patients with stage I, when compared with stages II and III, lung adenocarcinoma (Figure 3, G–I). Therefore, the aberrant expression of NDNF/Ndnf in human and mouse lung adenocarcinoma, as well as the association of NDNF mRNA level with patient survival, indicated that NDNF may serve as a prognostic biomarker for early-stage lung adenocarcinoma.
Negative correlation between NDNF expression and progression of human lung adenocarcinoma.
To further investigate the relevance of NDNF expression level with human lung adenocarcinoma, we performed a detailed analysis of RNA-Seq data from TCGA. In addition to a significantly lower amount of NDNF in tumor tissues compared with matched normal surrounding tissues (Figure 4A), we found that patients with larger tumors (T3–T4) had substantially less NDNF expression compared with patients with small tumors (T1–T2) (Figure 4B). We observed a similar relationship between NDNF expression and TNM stages: samples at advanced stage (II and III) had lower NDNF expression than samples at early stage (I) (Figure 4C). Moreover, lung adenocarcinoma patients with lymph node metastasis expressed less NDNF than those without lymph node metastasis (Figure 4D).
Figure 4. NDNF transcript abundance negatively correlates with progression of human lung adenocarcinoma.

(A) NDNF mRNA in matched normal lung tissue adjacent to the tumor (NAT) and tumor tissues (n = 57). (B–D) NDNF mRNA in normal adjacent lung tissues and tumor tissues with different primary tumor size and extent (B), different tumor-node-metastasis (TNM) stages (C), and different lymph node metastasis status (D). In A–D, analysis was performed with data in TCGA for lung adenocarcinoma. Box plots show 25th to 75th percentile; whiskers extend to the minimum and maximum values. (E–J) NDNF mRNA expression from human lung adenocarcinoma microarrays. Representative images of NDNF mRNA (red) detected by RNAscope ISH in matched normal adjacent lung tissues (NAT) and tumor tissues with different primary tumor size and extent (E), different TNM stages (G) and different lymph node metastasis status (I). Scale bar, 100 μm. Quantitative analysis of NDNF expression detected by RNAscope ISH (NDNF ISH score; see Methods) from lung adenocarcinoma with different primary tumor size and extent (F), different TNM stages (H), and different lymph node metastasis status (J). Data are shown as mean ± SD. The 2-tailed paired t test (A) or 1-way ANOVA followed by Holm-Šídák multiple-comparisons test (B–D, F, H, and J) was used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We evaluated this negative correlation between NDNF mRNA level and lung adenocarcinoma progression by in situ hybridization using tissue microarrays from 4 independent human lung adenocarcinoma cohorts consisting of 30 normal lung tissue and 207 adenocarcinoma samples covering distinct pathological stages (Supplemental Table 5). Consistent with the results from TCGA database analysis, we observed that NDNF mRNA detected by in situ hybridization (ISH) was markedly lower (for ISH scores, see Methods) in lung adenocarcinoma tumor tissues than in the matched normal adjacent samples (Figure 4, E–J). Specifically, lower levels of NDNF were associated with larger tumor size (Figure 4, E and F), more advanced tumor stage (Figure 4, G and H), and further cancer progression (Figure 4, I and J). We applied χ2 testing, which established the significance of the finding that reduced NDNF levels correlated with the higher clinical grade and TNM classifications in patients with lung adenocarcinoma (Supplemental Table 5). Together, these data demonstrated that decreased NDNF expression occurred in patients with lung adenocarcinoma and was significantly correlated with tumor progression and poor prognosis.
Tumor-suppressive properties of NDNF in mouse and human lung cancer cell lines.
Little is known about the functional role of NDNF in cancer. We explored the potential role of NDNF in tumorigenesis by exposing A549 and LLC1, lung cancer cell lines from human and mouse, respectively, to purified Ndnf protein. The purified Ndnf protein inhibited growth of A549 and LLC1, as indicated by a reduced rate of growth assessed using a Cell Counting Kit-8 (CCK-8) assay (Figure 5A). Likewise, ectopic expression of NDNF/Ndnf caused similar growth inhibition in A549/LLC1 lung cancer cells (Supplemental Figure 9). Exogenous Ndnf also impaired anchorage-dependent and anchorage-independent growth of both cell lines (Figure 5, B and C). We also assessed the effects of reduced NDNF expression in the growth of A549 and LLC1 lung cancer cells. Knockdown of human NDNF in A549 cells with 2 shRNAs, 1 targeting the coding sequence and the other targeting the 3′-UTR of NDNF, increased cell growth compared with that of control cells expressing scrambled shRNA (Figure 5, D–F, and Supplemental Figure 10). Knockdown with 2 shRNAs targeting the mouse Ndnf gene, but not a scrambled sequence, produced similar results in LLC1 cells (Figure 5, D–F, and Supplemental Figure 10). In addition, purified Ndnf protein attenuated the increased growth of A549 and LLC1 cells resulting from NDNF (or Ndnf) knockdown (Figure 5, D–F, and Supplemental Figure 11). Collectively, these results indicated that NDNF plays an important role in limiting the growth and tumorigenic properties of both mouse and human lung cancer cells.
Figure 5. Tumor-suppressive properties of NDNF in mouse and human lung cancer cell lines.

(A–C) Effect of purified Ndnf protein on the growth of human and mouse lung cancer cell lines. Quantitative analysis of cell viability using the CCK-8 assay (A), of colony formation (B), and of growth in soft agar (C) of the A549 and LLC1 cells with or without purified Ndnf (200 ng/mL). Representative images from the colony formation assay and soft agar assay are shown in B and C. (D–F) Effect of shRNA-based knockdown of NDNF on the growth of human A549 cells and knockdown of Ndnf on LLC1 cells. Knockdown efficiency of shRNA targeting human NDNF and mouse Ndnf is shown in Supplemental Figure 10. Quantitative analysis of cell viability using the CCK-8 assay (D), of colony formation (E), and of growth in soft agar (F) of the indicated cells stably expressing shRNA targeting NDNF or Ndnf as appropriate or control scrambled shRNA of cells with or without purified Ndnf (200 ng/mL). In A and D, data are shown as mean ± SD of n > 3 replicates of a single experiment. Data are representative of n ≥ 3 experiments. In B, C, E, and F, data are shown as box-and-whiskers plots of n ≥ 6 replicates from n ≥ 3 experiments. Box plots show 25th to 75th percentile; whiskers extend to the minimum and maximum values. The 2-tailed Mann-Whitney U test (A–C) or 1-way ANOVA followed by Holm-Šídák multiple-comparisons test (D–F) was used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Representative images of the colony formation assay (E) and soft agar assay (F) are shown in Supplemental Figure 11.
Tumor-suppressive properties of NDNF in the xenograft model.
To assess the relevance of NDNF in vivo, we generated subcutaneous xenograft tumors using the human A549 cells or mouse LLC1 cells in immune-compromised mice. We found that downregulation of NDNF expression significantly promoted the growth of A549 tumors (Figure 6A). Not only were the tumor sizes of A549-NDNF-shRNA–derived xenografts notably larger than those of xenografts originating from A549-scramble-shRNA cells at day 32 after injection (Figure 6B), but the tumor weights of A549-NDNF-shRNA–derived xenografts were also much higher than those of xenografts originating from A549-scramble-shRNA cells at day 32 (Figure 6B). Moreover, the percentage of cells positive for Ki67, a marker of proliferating cells, was greater in the NDNF-shRNA–derived xenografts compared with the percentage in the scramble-shRNA–derived xenografts (Figure 6C). NDNF knockdown had similar effects on LLC1-derived tumors (Figure 6, D–F). These results are consistent with the observation that NDNF expression levels negatively correlated with Ki67 expression (Spearman’s r = –0.3915) and with the abundance of transcripts of other proliferation-related genes PCNA, CDC6, CDC45, and CDT1 (Supplemental Figure 12). Collectively, our in vitro and in vivo experiments support a tumor-suppressive role for NDNF in lung cancer.
Figure 6. Tumor-suppressive properties of NDNF in the xenograft model.

Human lung cancer A549 cells (A–C) or mouse lung cancer LLC1 cells (D–F) stably expressing shRNA targeting NDNF gene or control scrambled shRNA were injected into nude mice. (A and D) Tumor volume was measured at the indicated time points. Data are shown as mean ± SD for tumors from n = 6 mice at each time point. (B and E) Mice injected with A549 cells were sacrificed on day 32 after injection and those injected with LLC1 cells on day 23 after injection. Tumors were removed and photographed. Tumor volumes and weights were measured. (C and F) Proliferating cells were detected by Ki67 staining in tumors at day 32 (A549 cell tumors) or day 23 (LLC1 cell tumors). Representative images and quantitative analyses are shown. Scale bar: 20 μm. Data are shown as box-and-whiskers plots of tumors from 5 mice for each cell line from 20 images per tumor. Box plots show 25th to 75th percentile; whiskers extend to the minimum and maximum values. The 2-tailed Mann-Whitney U test was used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Epigenetic silencing of NDNF in lung adenocarcinoma.
Tumor suppressor genes are often rendered dysfunctional through multiple mechanisms during the oncogenic processes. Such processes include mutation, deletion, genetic rearrangement, and epigenetic silencing of transcription (64–66). Using data from cBioPortal (67, 68), we determined that reduced NDNF genomic DNA copy number or mutation of the NDNF gene did not frequently occur in patients with lung adenocarcinoma. Thus, we explored other mechanisms of lung cancer–associated downregulation of NDNF.
Epigenetic gene silencing associated with DNA methylation primarily in the promoter region is an important mechanism of gene inactivation in cancer (69). In lung cancer, hypermethylation inactivation of tumor suppressor genes is common (70, 71). We investigated whether NDNF is epigenetically silenced through promoter methylation in lung adenocarcinoma. We analyzed 422 lung adenocarcinoma tissue samples with both gene expression data from RNA-Seq and matched DNA methylation data from Infinium HumanMethylation450 microarray in TCGA database. Paired normal tissue was available for 21 of these samples. We compared the methylation level across the 16 CpG sites in the NDNF promoter region (Figure 7A). DNA methylation levels at 14 of the 16 CpG sites were significantly higher in tumor tissues than in normal tissues (Figure 7B). Furthermore, an inverse correlation (Spearman’s r = –0.6971) between NDNF expression and the average methylation level across all the 16 CpG sites in the NDNF promoter region was observed (Figure 7C). We also examined the magnitude of differential methylation across the 16 CpG sites. As reported previously (72), we found that the most dramatic DNA methylation alterations (ΔM) in lung adenocarcinoma preferentially occurred at CpG island shores, which are the relatively low CpG density regions flanking the traditional CpG island (Supplemental Figure 13). Consistently, the correlation between NDNF expression level and methylation level at individual CpG sites located on each shore was greater than that within the traditional CpG islands (Supplemental Figure 14 and Supplemental Table 6). These data indicated that downregulation of NDNF can occur by epigenetic silencing in lung adenocarcinoma. Together, our results suggested that DNA methylation at the CpG island shores in the predicted promoter region contributes to NDNF silencing in lung adenocarcinoma.
Figure 7. Cancer-related DNA methylation sites correspond to those associated with tissue-specific expression of NDNF.

(A) Schematic diagram of the human NDNF locus. The 16 available CpG sites in the NDNF promoter region from Infinium HumanMethylation450 microarray are indicated as vertical lines. Black boxes, exons; blue box, CpG island; green text, CpG island shore methylation sites. (B) Methylation levels at the 16 CpG sites in tumor (red) compared with that in normal lung tissue adjacent to the tumor (black) using lung adenocarcinoma data from TCGA database. (C) Scatter plot and correlation between NDNF mRNA abundance and the average methylation level across 16 CpG sites in the NDNF promoter region in lung adenocarcinoma samples (n = 422, red dots) and normal lung tissue adjacent to the tumor (n = 21, black dots) from TCGA database. (D) Methylation level averaged across the 16 CpG sites in normal tissue from bladder, breast, kidney, and prostate (blue) compared with that in normal lung tissue (black). Data are from samples adjacent to tumor tissue from TCGA database (Supplemental Table 8). (E) Scatter plot and correlation between NDNF mRNA abundance and the average methylation level across all the 16 CpG sites in the NDNF promoter region in samples from normal lung, bladder, breast, kidney, and prostate tissues adjacent to tumor from TCGA. Lung samples shown as black dots, all others as blue dots. (F) Scatter plot and correlation between alterations in tissue-specific DNA methylation and alterations in cancer-specific methylation at each of the 16 CpG sites in the NDNF promoter region. Each circle represents a CpG site: green circle, CpG island shore; black circle, CpG island. Tissue-specific DNA methylation alterations were calculated as the difference in DNA methylation (ΔM) between the average methylation for 4 other tissues (bladder, breast, kidney, and prostate) and lung (from the averages in D); cancer-specific methylation alterations were calculated as the difference in DNA methylation between the average for lung adenocarcinoma tissues and the average for normal lung from tissue adjacent to tumors (from averages in B; Supplemental Figure 13). Spearman’s r and P values are indicated. Box plots show 25th to 75th percentile; whiskers extend to the minimum and maximum values. The 2-tailed Mann-Whitney U test was used for statistical analysis. n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Association between cancer-related DNA methylation sites and those for tissue-specific expression of NDNF.
DNA methylation is also a key epigenetic mechanism underlying tissue-specific gene expression (73, 74). Many of the locations of cancer-related methylation changes overlap with those that distinguish gene expression in normal tissues (72, 75–78). Thus, we analyzed histologically normal tissue samples adjacent to tumors across 5 tissue types — lung, bladder, breast, kidney, and prostate — with DNA methylation data in TCGA database. We found that methylation levels at most of the 16 CpG sites in the NDNF promoter region were significantly lower in the lung when compared with the 4 other tissues in combination (Figure 7D) or individually (Supplemental Figure 15). Additionally, we observed an inverse correlation (Spearman’s r = –0.6363) between NDNF mRNA expression and the average methylation level across the 16 CpG sites in the samples from the 5 tissues with both gene expression data from RNA-Seq and matched DNA methylation data in TCGA database (Figure 7E). The greatest differences in tissue-specific DNA methylation (ΔM), as well as stronger correlations between NDNF expression level and the methylation level at individual CpG sites, occurred at CpG island shores (Supplemental Figures 16 and 17 and Supplemental Table 7). The ΔM values between either the combination or each of the other 4 tissues individually and lung were highly correlated with that between lung adenocarcinoma tissues and normal lung tissue across the 16 CpG sites in the NDNF promoter region (Figure 7F and Supplemental Figure 18). Taken together, we demonstrated that lung cancer–related DNA methylation sites, predominantly occurring at CpG island shores, correspond to those associated with tissue-specific expression of NDNF and contribute to NDNF silencing in lung cancer.
Discussion
Tissue-specific differences contribute to tumorigenesis, therapeutic responses, and the development of resistance that are associated with distinct cancer type (5–8). A thorough understanding of these differences will improve prevention and early detection of cancer, as well as inform the development and implementation of targeted cancer therapies. Our approach of analyzing tissue-specific gene expression in normal tissue revealed both lung-enriched genes with well-known protumorigenic functions, such as WNT2, WNT3A, and RSPO4, as well as genes with previously recognized tumor-suppressive functions, such as ADAM metallopeptidase with thrombospondin type 1 motif 8 (ADAMTS8) and WNT inhibitor factor 1 (WIF1). An advantage of starting with normal tissue is the ability to identify both tissue-specific genes that may contribute to cancer through an increase in activity or expression and those that contribute following a decrease in expression. The latter are difficult to identify from studies starting with cancer tissue. For tumor-promoting sLungGenes, their contribution would depend on dysregulated activation of the lung-enriched genes or their products, whereas inactivation of sLungGenes with tumor-suppressive features may result in failure to restrain inappropriate cell proliferation, thus facilitating lung cancer progression. Together, our findings revealed the existence of multiple lung-enriched cancer-associated genes and suggested that the collective functions of these factors may contribute to the tissue specificity of tumorigenesis and inform therapeutic intervention. Because NDNF is one of the few top-ranking sLungGenes without any reported connection to lung cancer, we focused on this gene.
Our analysis showed that NDNF exhibited tumor-suppressive features. Its expression was decreased in human lung adenocarcinoma and in a mouse model of this cancer. Higher expression of NDNF was significantly associated with better clinical outcome of patients with lung adenocarcinoma (Figure 3, D–F). We determined that higher methylation of the NDNF promoter at CpG island shores in lung adenocarcinoma and in nonlung tissues was a mechanism for limiting the expression of NDNF in cancer and physiologically to achieve tissue-specific expression. Exposing lung cancer cells in culture to purified Ndnf reduced cell viability and decreased anchorage-dependent and -independent growth. NDNF or Ndnf knockdown enhanced the viability of human and mouse lung cancer cell lines and promoted their anchorage-dependent and -independent growth. Knockdown of human or mouse NDNF also enhanced growth of tumors derived from these cell lines in xenograft models. Collectively, we demonstrate NDNF was preferentially expressed in both mouse and human lung and functioned as a secreted factor with tumor-suppressive features. Previous studies reported that NDNF promotes endothelial cell and cardiomyocyte survival by activating AKT signaling (55, 56); however, we observed little or no effect of exogenously added Ndnf on AKT signaling in A549 or LLC1 lung cancer cell lines (Supplemental Figure 19). Our analysis of NDNF was limited by the antibody, which only recognized denatured protein and thus was useful for Western blot experiments but not for immunohistochemistry or depletion assays. Future studies and reagents are needed to uncover the mechanisms underlying the tumor-suppressive functions and the physiological roles of NDNF in human and mouse lung.
Of note, most of the top-ranking sLungGenes identified in our study are not exclusively expressed in the adult lung (Supplemental Figures 1–3), and these genes also have reported roles in other types of cancer (Supplemental Table 2). For example, ADAMTS8, which encodes a secreted protease with antiangiogenic properties, is a tumor suppressor that is silenced not only in lung carcinoma (79) but also in many other cancers, including brain tumors (80), breast carcinoma (81), head and neck squamous cell carcinoma (82), pancreatic cancer (83), and gastric or colorectal cancers (84). Likewise, WIF1 has been described as a tumor suppressor in lung cancer (85–88) and in osteosarcoma (89), kidney cancer (90), and pleomorphic adenoma (91). Indeed, a recent study of renal carcinoma cells reported a role for NDNF in suppressing characteristics of the epithelial-mesenchymal transition (92). Thus, our study not only identified potentially tissue-specific cancer genes but also genes associated with cancers in multiple tissues. Tissue-specific cancer functions may arise through tissue-specific combinatorial effects of the cancer-associated genes. Regardless, our study showed that analysis of tissue-specific gene expression in normal tissue can provide insights into the molecular mechanisms of cancer (here, epigenetic gene silencing of NDNF in lung cancer) and inform therapeutic approaches.
Methods
Generation of sLungGenes gene list.
The list of predicted secreted proteins in human was downloaded from the HPA (https://www.proteinatlas.org/humanproteome/tissue/secretome) (46). The list of predicted secreted proteins in mouse was downloaded from the curated database at MetazSecKB (http://proteomics.ysu.edu/secretomes/animal/index.php) (47). Protein names were then converted to gene names according to UniProt database (http://www.uniprot.org) (93), yielding 3010 sGenes in human and 2833 sGenes in mouse. The sGene lists from human and mouse were merged, and 2591 orthologous genes conserved in human and mouse were selected for further analysis.
Gene expression data in adult human tissues were downloaded from 2 independent sources: the HPA database (37 tissue types) and the GTEx database (30 tissue types) (48). Gene expression data from 12 tissue types in 10-week-old mice were downloaded from the ENCODE (49). For each gene, the median expression value (transcripts per million) from multiple samples of each tissue was retrieved to represent the expression value in that tissue.
For each of the orthologous human and mouse sGenes, the lung-preferential expression scores (hPLung_HPA, hPLung_GTEx, and mPLung) across available tissue types from 3 databases were independently calculated using the following formula (94), where X is the gene expression level in each human or mouse tissue. The PLung scores are constrained between 0 and 1, where 0 indicates a gene not expressed and 1 represents a gene that is expressed only in the lung.
(Equation 1)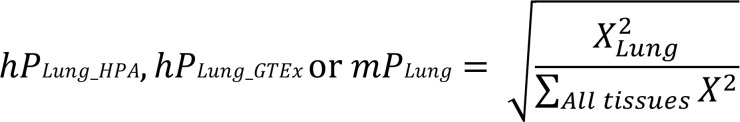
To mitigate possible bias caused by different numbers of human and mouse tissues involved in the preferential expression scores calculation, the lung-preferential expression scores (hPLung_HPA, hPLung_GTEx, and mPLung) were then converted to lung-preferential expression Z scores (Supplemental Table 1) according to the following formula, where X is the lung-preferential expression score of each gene and μ and σ are the mean and standard deviation of the lung-preferential expression scores, respectively.
(Equation 2) 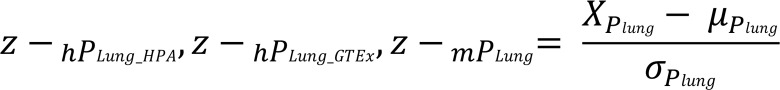
The 3D scatter plot was constructed using plot3D R package (http://CRAN.R-project.org/package=plot3D) by assigning the lung-preferential expression Z scores hPLung_HPA, hPLung_GTEx, and mPLung as the x axis, y axis, and z axis, respectively.
TCGA DNA methylation and gene expression data.
DNA methylation array data, mRNA expression data, and corresponding clinical information (95) of the lung adenocarcinoma data set were downloaded from TCGA data set available in Broad Institute GDAC FireBrowse portal (http://firebrowse.org). The platform for methylation array data was Illumina Infinium HumanMethylation450 BeadChip. The mRNA expression data were Illumina HiSeq rnaseqv2 Level 3 RSEM–normalized data
To study correlations between DNA methylation and NDNF expression in lung adenocarcinoma, DNA methylation array data from tumor tissues of 463 lung adenocarcinoma patients and mRNA expression data of 517 lung adenocarcinoma patients were downloaded. In addition, DNA methylation array data from 32 adjacent tumor tissues and mRNA expression data from 59 adjacent tumor tissues were also downloaded from the lung adenocarcinoma data set. Among these samples, 422 lung adenocarcinoma patients and 21 adjacent tumor tissues with both gene expression data and matched DNA methylation data were used to calculate the correlation between DNA methylation and NDNF expression in lung adenocarcinoma.
To study correlation between DNA methylation and NDNF expression in different tissues, we used DNA methylation array data and mRNA expression data from the adjacent normal tissues of 7 cancer types across 5 tissues (Supplemental Table 8).
Animal studies.
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at George Washington University. All handling, transplantation, and infection procedures were conducted in a laminar flow biosafety hood. C57BL/6N mice were purchased from the Charles River Laboratories. K-rasLA1 mice were originally generated by Tyler Jacks at Massachusetts Institute of Technology (9). Genotypes were confirmed by PCR. For xenograft experiments, female athymic (nu/nu) nude mice (The Jackson Laboratory), 6–8 weeks of age and weighing 20–25 g, were housed in appropriate sterile filter-capped cages and provided food and water ad libitum.
Xenograft studies were performed as follows. Exponentially growing cultures of A549 or LLC1 cells were harvested, washed, and resuspended in DMEM. Viable A549 cells (5 × 106) or LLC1 cells (1 × 105) were transplanted subcutaneously into both flanks of the athymic mice. Tumor size was monitored every 2–3 days and measured using a caliper. The tumor volume was calculated by the formula V (cm3) = (L × W2)/2, where L represents the longest dimension and W the shortest dimension of the tumor. The mice were sacrificed at day 32 or day 23 to collect tumors.
RT-qPCR.
Normal mouse tissues from lung, brain, ovary, heart ventricle, adipose tissue, back skin, spleen, stomach, thymus, bladder, liver, testis, muscle, kidney, pancreas, small intestine, and colon were dissected from 10-week-old C57BL/6N mice. Tumor and matched normal lung tissue adjacent to tumor were dissected out from 20-week-old K-rasLA1 mice. Total RNA was isolated from freshly dissected mouse tissues using the mirVana RNA isolation kit (AM1560, Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. Possible contamination of genomic DNA was excluded by treatments of DNAse I (AM2222, Thermo Fisher Scientific). RNA was reverse-transcribed to cDNA using Maxima Reverse Transcriptase (EP0742, Thermo Fisher Scientific) with random hexamers. All samples within an experiment were reverse-transcribed at the same time; the resulting cDNA was stored in aliquots at –80°C until used.
cDNA was PCR-amplified using iQ SYBR Green Supermix (1708880; Bio-Rad). qPCR was carried out with an ABI PRISM 7700 Sequence Detection System (Applied Biosystems). Reactions were run in triplicate in 3 independent experiments. Expression data were normalized to the geometric mean of housekeeping gene GAPDH and were analyzed using the 2–ΔΔCT method (96). The primer sequences are provided in Supplemental Table 9.
Protein extraction and immunoblotting.
Normal mouse tissues from lung, heart ventricle, liver, thymus, kidney, stomach, pancreas, small intestine, and colon were dissected from 10-week-old C57BL/6N mice (n = 3). Tumor and matched normal lung tissue adjacent to tumor were dissected out from 20-week-old K-rasLA1 mice (n = 8). Freshly dissect mouse tissues were lysed individually in RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) supplemented with phosphatase (PhosSTOP, Roche) and protease (Complete, Roche) inhibitors. The lysate was clarified by centrifugation (18,000 g for 15 minutes at 4°C), and proteins were recovered directly in SDS-PAGE sample buffer. Proteins were separated by SDS-PAGE under reducing conditions and then transferred onto PVDF membranes (MilliporeSigma). After protein transfer, the membranes were blocked and then immunostained with primary antibodies and HRP-conjugated secondary antibodies. Antibodies used were listed in Supplemental Table 9C. Blots were developed using Immobilon Forte Western HRP substrate (MilliporeSigma, WBLUF0100) with GeneSys Imaging System (Syngene).
RNAscope ISH.
Human NDNF and mouse Ndnf mRNA were detected in tissue sections using the Advanced Cell Diagnostic RNAscope 2.5 HD Detection Reagent-RED kit (322350, Advanced Cell Diagnostics). The target probe sets were generated and supplied by the manufacturer (Supplemental Table 9). A reference RNAscope hybridization protocol provided by Advanced Cell Diagnostics (http://www.acdbio.com/technical-support/user-manuals) was essentially followed. Briefly, prior to ISH, slides were baked for 1 hour at 60°C. Sections were treated with hydrogen peroxide for 10 minutes at room temperature to inhibit endogenous peroxidases. Target retrieval was carried out at boiling temperature for 15 minutes. The tissue sections were completely dried and treated with a broad-spectrum Protease Plus solution (322330, Advanced Cell Diagnostics) at 40°C for 30 minutes. The probes were then hybridized at 40°C for 2 hours. Probe hybridization was followed by serial amplification steps. All washing steps following hybridization and during amplification consisted of 3 incubations in washing buffer (provided with the kit) for 2 minutes at each step. A final hybridization step using an alkaline phosphatase–labeled probe was followed by incubation with Fast Red (322360, Advanced Cell Diagnostics) substrate that resulted in red precipitates (NDNF/Ndnf mRNA signal). Slides were washed in water and counterstained with 50% hematoxylin (MilliporeSigma) for 2 minutes. Sections were dried at 60°C for at least 15 minutes, submerged in xylene, and covered with EcoMount mounting media (Biocare Medical).
Quantification of NDNF mRNA expression.
The analysis of NDNF mRNA expression detected by RNAscope ISH in the tumor samples and normal lung samples (limited to the alveolar epithelium) was carried out using direct light microscopy in at least 5 fields at original magnification ×200. The ISH scores were determined independently by 2 investigators blinded to the patients’ clinical data. The NDNF mRNA expression detected by RNAscope ISH was scored following a semiquantitative scale according to the Fromowitz standard (97). The staining intensity was scored as 0 (no staining), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). The percentage of positive cells was divided into 4 levels: 1 (0%–25% positive cells), 2 (26%–50% positive cells), 3 (51%–75% positive cells), and 4 (76%–100% positive cells). The multiplication of the intensity and percentage was used to represent the final ISH score ranging from 0 to 12. The total expression of NDNF was determined as either low expression with ISH score less than 6 or high expression with ISH score at least 6, which were taken for quantification in Supplemental Table 5.
Immunofluorescence.
Xenograft tumor tissues were fixed overnight in 4% paraformaldehyde at 4°C. Tissues were cryoprotected in 30% sucrose overnight at 4°C and then embedded in O.C.T. (Tissue-Tek, Sakura Finetek). Tissue sections (6 μm) were dried for 30 minutes at room temperature (RT), and O.C.T. was removed by dilution in PBS. Sections were blocked with PBS, 10% BSA, 5% normal goat serum, and 0.3% Triton X-100 for 60 minutes at RT, followed by primary antibody overnight at 4°C in a humidified chamber. After rinsing 5 times with PBS containing 0.3% Triton X-100, sections were incubated with secondary antibody and DAPI (MilliporeSigma) at RT for 2 hours. After rinsing 5 times with PBS containing 0.3% Triton X-100, sections were mounted (Vector Laboratories) for imaging. The primary and secondary antibodies are listed in Supplemental Table 9. Microscopy was performed on a Carl Zeiss Cell Observer spinning disk confocal microscope. Percentage of Ki67+ cells was calculated by dividing the number of Ki67+ cells by the total cell number recognized by DAPI per field. A total of 20 random fields were scored per tumor for each subcutaneous xenograft (n = 5).
Cell lines and tissue microarrays.
HEK293T, A549, and LLC1 cells were purchased from American Type Culture Collection (ATCC) and maintained in complete medium: DMEM (Gibco, Thermo Fisher Scientific) containing 10% heat-inactivated fetal bovine serum (Gibco, Thermo Fisher Scientific), 100 U/mL of penicillin, and 100 μg/mL of streptomycin. All the cells were grown in a humidified atmosphere containing 5% CO2 at 37°C.
Human lung adenocarcinoma tissue microarrays (BCS04017a, LC1005a, LC1501, and LC1921a) were purchased from US Biomax. Normal human lung (HuFPT131), breast (HuFPT127), spleen (HuFPT082), and liver (HuFPT074) were purchased from US Biomax. Antibodies and plasmids used in this study are listed in Supplemental Table 9.
Ndnf protein purification.
Ndnf protein purification was carried out by GenScript: 293-6E cells were grown in serum-free FreeStyle 293 Expression Medium (12338018, Gibco, Thermo Fisher Scientific) and transiently transfected for expression of mouse Ndnf tagged with Myc and 6 × His (mNdnf-MycHis). The cell culture supernatant collected 5 days after transfection was used for protein purification. Cell culture supernatant was centrifuged at 10,000 g for 30 minutes at 4°C and loaded onto a HisTrap FF Crude histidine-tagged protein purification column (11-0004-58, GE Healthcare) at 1.0 mL/min. After washing and elution, the eluted fractions were pooled, and the buffer was exchanged with PBS (pH 7.2). The purified protein was analyzed by SDS-PAGE and Western blot for yield and purity (>85%). The protein solutions were stored in aliquots at –80°C until used.
Generation of Ndnf antibody.
Rabbit anti-mouse Ndnf polyclonal antibody was generated by immunization of New Zealand rabbits with 6 × His–tagged full-length Ndnf protein (amino acids 20–568) purified from E. coli. Rabbits were inoculated on days 0, 14, 28, and 56 using 0.5 mL of bacterial cell suspension per animal per immunization. Sera were collected at day 0 (before immunization), day 38, day 66, and day 87. After testing the specificity of the antibody by ELISA, polyclonal IgG from the sera was purified by antigen-affinity columns. The eluate was extensively dialyzed against PBS and stored with 1 mg/mL carrier BSA. Immunization and antibody purification were carried out by GenScript.
Cell proliferation assay.
Cell proliferation was determined using CCK-8 (CK04-05, Dojindo) according to the manufacturer’s instructions. Briefly, 1 × 103 cells were seeded on 96-well plates and cultured for 24 hours. One hundred microliters of CCK-8 buffer was added to each well, and plates were incubated for 2 hours. The absorbance was measured at 450 nm using a microplate reader (Epoch, Bio-Tek Instruments). Each assay was repeated with more than 3 replicates each time.
Colony formation assay.
One hundred viable LLC1 cells or 200 A549 cells were seeded in 12-well culture plates. Medium was exchanged every 2 days with fresh complete medium with or without 200 ng/mL purified Ndnf protein. After 14 days, cells were fixed with methanol for 5 minutes and stained with 0.05% Coomassie blue in 20% methanol and 5% acetic acid. The colony numbers, defined as more than 50 cells/colony, were counted. Each assay was repeated with more than 2 replicates each time.
Soft agar assay.
Two thousand viable cells were resuspended in 1 mL of complete medium with or without 200 ng/mL purified Ndnf protein containing 0.3% Noble agar (214220, Difco). Resuspended cells were plated on 12-well plates containing a solidified bottom layer of complete medium (with or without Ndnf as appropriate) containing 0.6% agar. One hundred microliters of complete medium containing 0.25% agar with or without purified Ndnf proteins (200 ng/mL) was added every 3–4 days. After 2 weeks, colonies were visualized by staining with 100 μL of 1 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for 30 minutes. The spheres over 100 μm in diameter were counted. Each assay was repeated with more than 2 replicates each time.
Viral infection.
shRNA-expressing lentiviral plasmids (pLKO.1-puro) targeting Ndnf or NDNF and a control scrambled shRNA (an shRNA sequence not targeting any known gene) plasmid were purchased as bacterial glycerol stocks from MilliporeSigma (Supplemental Table 9). For overexpressing Ndnf or NDNF, mouse or human Ndnf/NDNF cDNA without the 3′-UTR was subcloned into the retroviral vector plasmid pBABE-puro (Addgene, 1764) at unique BamHI and EcoRI sites (Supplemental Table 9). To produce lentiviral particles, this vector was cotransfected with the lentiviral packaging plasmids pLP1, pLP2, and pLP/VSVG (Invitrogen, Thermo Fisher Scientific) into HEK293T cells. Similarly, to produce retroviral particles, overexpressing plasmid was cotransfected with the retroviral packaging plasmid pLP/VSVG (Invitrogen, Thermo Fisher Scientific) into the GP2-293 packaging cell line, which expresses gag and pol proteins (Clontech). Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific) was used as the transfection reagent according to the manufacturer’s instructions. At 48–72 hours after transfection, the virus-containing cell culture medium was harvested and frozen in aliquots, which were then used in transducing A549 or LLC1 cells. Transduced cells were selected by growth in medium with 1 μg/mL puromycin (A1113803, Thermo Fisher Scientific). NDNF/Ndnf silencing efficiency was measured by RT-qPCR as shown in Supplemental Figure 10.
Statistics.
Statistics were performed using GraphPad Prism 8. For comparison of central tendencies, normally distributed data sets were analyzed by unpaired 2-sided t test under the assumption of equal variance; non-normally distributed data sets were analyzed by nonparametric Mann-Whitney U tests. One-way ANOVA followed by Holm-Šídák multiple-comparisons test was used for multiple comparisons. The χ2 test was applied to analyze the relationship between NDNF mRNA levels detected by RNAscope ISH and pathological status. Differences were considered statistically significant when P < 0.05.
Study approval.
All animal studies were approved by the George Washington University IACUC (Washington, DC, USA). No patient samples were directly used in this study.
Author contributions
XZ and Y Zhang designed the overall experiments, analyzed data, and wrote the manuscript. Y Zhang, Xuefeng Wu, CHL, FC, YL, and KHC performed in vitro or animal experiments. Y Zhang, YK, CHL, Y Zhuang, and JG performed bioinformatics analysis of published expression data sets. ZL, MK, YM, NRG, Xiaojun Wu, WZ, AT, WP, ES, and EMS provided intellectual input. All authors provided scientific input and edited and approved the final manuscript.
Supplementary Material
Acknowledgments
We thank Guo-Tao Sun at Henan University, Kaifeng, China, for the pCDNA3.1-hNDNF-MycHis construct. We thank the Stepp lab in the Department of Anatomy and Cell Biology and the Chiappinelli lab in the Department of Microbiology, Immunology, and Tropical Medicine, George Washington University, for helpful discussions. We thank members from the GW Cancer Center for reagents and for comments on the manuscript. This work is supported by NIH, GW Cancer Center, and Katzen Cancer Research Center.
Version 1. 12/19/2019
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(24):e129344.https://doi.org/10.1172/jci.insight.129344.
Contributor Information
Ya Zhang, Email: zhangy15@email.gwu.edu.
Xuefeng Wu, Email: xw2696@cumc.columbia.edu.
Yan Kai, Email: smilekai@gwu.edu.
Chia-Han Lee, Email: gahanleeo@gwmail.gwu.edu.
Fengdong Cheng, Email: jiechen14@email.gwu.edu.
Yixuan Li, Email: yixuanli@gwu.edu.
Yongbao Zhuang, Email: jerrychuang@gwmail.gwu.edu.
Javid Ghaemmaghami, Email: javidmaghami@yahoo.com.
Kun-Han Chuang, Email: 18163221857@163.com.
Zhuo Liu, Email: liuzhuo11@126.com.
Yunxiao Meng, Email: dryxmeng@gmail.com.
Nancy R. Gough, Email: ngough@gwu.edu.
Xiaojun Wu, Email: xwu63@jhmi.edu.
Wenge Zhu, Email: scholarzy@gwu.edu.
Alexandros Tzatsos, Email: atzatsos@gwu.edu.
Weiqun Peng, Email: wpeng@gwu.edu.
Edward Seto, Email: seto@gwu.edu.
Eduardo M. Sotomayor, Email: esotomayor@mfa.gwu.edu.
Xiaoyan Zheng, Email: xzheng@gwu.edu.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13(11):795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349(6255):1483–1489. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- 5.Sieber OM, Tomlinson SR, Tomlinson IP. Tissue, cell and stage specificity of (epi)mutations in cancers. Nat Rev Cancer. 2005;5(8):649–655. doi: 10.1038/nrc1674. [DOI] [PubMed] [Google Scholar]
- 6.Schaefer MH, Serrano L. Cell type-specific properties and environment shape tissue specificity of cancer genes. Sci Rep. 2016;6:20707. doi: 10.1038/srep20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider G, Schmidt-Supprian M, Rad R, Saur D. Tissue-specific tumorigenesis: context matters. Nat Rev Cancer. 2017;17(4):239–253. doi: 10.1038/nrc.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haigis KM, Cichowski K, Elledge SJ. Tissue-specificity in cancer: The rule, not the exception. Science. 2019;363(6432):1150–1151. doi: 10.1126/science.aaw3472. [DOI] [PubMed] [Google Scholar]
- 9.Johnson L, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 10.Guerra C, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4(2):111–120. doi: 10.1016/S1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 11.Eser S, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 12.Young NP, Jacks T. Tissue-specific p19Arf regulation dictates the response to oncogenic K-ras. Proc Natl Acad Sci USA. 2010;107(22):10184–10189. doi: 10.1073/pnas.1004796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408(6811):429–432. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miki Y, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 15.Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002;21(58):8981–8993. doi: 10.1038/sj.onc.1206176. [DOI] [PubMed] [Google Scholar]
- 16.Futreal PA, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266(5182):120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 17.Hoadley KA, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas RK, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39(3):347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 19.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zehir A, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature medicine. 2017;23(6):703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyman DM, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Tourneau C, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–1334. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 23.Hunter DJ, D’Agostino RB., Sr Let’s not put all our eggs in one basket. N Engl J Med. 2015;373(8):691–693. doi: 10.1056/NEJMp1508144. [DOI] [PubMed] [Google Scholar]
- 24.Krzyzanowska MK. Off-label use of cancer drugs: a benchmark is established. J Clin Oncol. 2013;31(9):1125–1127. doi: 10.1200/JCO.2012.46.9460. [DOI] [PubMed] [Google Scholar]
- 25.Conti RM, Bernstein AC, Villaflor VM, Schilsky RL, Rosenthal MB, Bach PB. Prevalence of off-label use and spending in 2010 among patent-protected chemotherapies in a population-based cohort of medical oncologists. J Clin Oncol. 2013;31(9):1134–1139. doi: 10.1200/JCO.2012.42.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullard A. Off-label targeted cancer drugs fail in first randomized trial. Nat Rev Drug Discov. 2015;14(10):669. doi: 10.1038/nrd4758. [DOI] [PubMed] [Google Scholar]
- 27.Redig AJ, Jänne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol. 2015;33(9):975–977. doi: 10.1200/JCO.2014.59.8433. [DOI] [PubMed] [Google Scholar]
- 28.Stenzinger A, Weichert W, Lennerz JK, Klauschen F. Basket Trials: Just the End of the First Quarter. J Clin Oncol. 2015;33(25):2823–2824. doi: 10.1200/JCO.2015.62.1516. [DOI] [PubMed] [Google Scholar]
- 29. Mullard A. Use of personalized cancer drugs runs ahead of the science. Nature. 2015. [Google Scholar]
- 30.Winter EE, Goodstadt L, Ponting CP. Elevated rates of protein secretion, evolution, and disease among tissue-specific genes. Genome Res. 2004;14(1):54–61. doi: 10.1101/gr.1924004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lage K, et al. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci USA. 2008;105(52):20870–20875. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104(21):8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Consortium F. et al. A promoter-level mammalian expression atlas. Nature. 2014;507(7493):462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunham I, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Odom DT, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303(5662):1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang DM, et al. A genome-based resource for molecular cardiovascular medicine: toward a compendium of cardiovascular genes. Circulation. 1997;96(12):4146–4203. doi: 10.1161/01.CIR.96.12.4146. [DOI] [PubMed] [Google Scholar]
- 39.Muir B, Nunney L. The expression of tumour suppressors and proto-oncogenes in tissues susceptible to their hereditary cancers. Br J Cancer. 2015;113(2):345–353. doi: 10.1038/bjc.2015.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nunney L. Lineage selection and the evolution of multistage carcinogenesis. Proc Biol Sci. 1999;266(1418):493–498. doi: 10.1098/rspb.1999.0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paltridge JL, Belle L, Khew-Goodall Y. The secretome in cancer progression. Biochim Biophys Acta. 2013;1834(11):2233–2241. doi: 10.1016/j.bbapap.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 42.Stastna M, Van Eyk JE. Secreted proteins as a fundamental source for biomarker discovery. Proteomics. 2012;12(4-5):722–735. doi: 10.1002/pmic.201100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zanders ED. Cell surface and secreted proteins. Human Drug Targets: A Compendium for Pharmaceutical Discovery. 2016:23-133. [Google Scholar]
- 44.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: a cancer journal for clinicians. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas Research N, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics. 2013;45(10):1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uhlén M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 47.Meinken J, Walker G, Cooper CR, Min XJ. MetazSecKB: the human and animal secretome and subcellular proteome knowledgebase. Database (Oxford) 2015;2015:bav077. doi: 10.1093/database/bav077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carithers LJ, Moore HM. The Genotype-Tissue Expression (GTEx) Project. Biopreserv Biobank. 2015;13(5):307–308. doi: 10.1089/bio.2015.29031.hmm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis CA, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46(D1):D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuang XL, Zhao XM, Xu HF, Shi YY, Deng JB, Sun GT. Spatio-temporal expression of a novel neuron-derived neurotrophic factor (NDNF) in mouse brains during development. BMC Neurosci. 2010;11:137. doi: 10.1186/1471-2202-11-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poorthuis RB, et al. Rapid Neuromodulation of Layer 1 Interneurons in Human Neocortex. Cell Rep. 2018;23(4):951–958. doi: 10.1016/j.celrep.2018.03.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tasic B, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 2016;19(2):335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gong S, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425(6961):917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 54.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohashi K, et al. Neuron-derived neurotrophic factor functions as a novel modulator that enhances endothelial cell function and revascularization processes. J Biol Chem. 2014;289(20):14132–14144. doi: 10.1074/jbc.M114.555789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joki Y, et al. Neuron-derived neurotrophic factor ameliorates adverse cardiac remodeling after experimental myocardial infarction. Circ Heart Fail. 2015;8(2):342–351. doi: 10.1161/CIRCHEARTFAILURE.114.001647. [DOI] [PubMed] [Google Scholar]
- 57.Schira J, et al. Characterization of regenerative phenotype of unrestricted somatic stem cells (USSC) from human umbilical cord blood (hUCB) by functional secretome analysis. Mol Cell Proteomics. 2015;14(10):2630–2643. doi: 10.1074/mcp.M115.049312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song HF, et al. Aged human multipotent mesenchymal stromal cells can be rejuvenated by neuron-derived neurotrophic factor and improve heart function after injury. JACC Basic Transl Sci. 2017;2(6):702–716. doi: 10.1016/j.jacbts.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manabe R, et al. Transcriptome-based systematic identification of extracellular matrix proteins. Proc Natl Acad Sci U S A. 2008;105(35):12849–12854. doi: 10.1073/pnas.0803640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014;507(7491):190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin C, et al. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS ONE. 2012;7(12):e53817. doi: 10.1371/journal.pone.0053817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu X, et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci U S A. 2012;109(13):4910–4915. doi: 10.1073/pnas.1112499109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE. 2013;8(12):e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim Biophys Acta. 2016;1865(2):275–288. doi: 10.1016/j.bbcan.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 65.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 66.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 67.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 70.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4(9):707–717. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 71.Yanagawa N, Tamura G, Oizumi H, Takahashi N, Shimazaki Y, Motoyama T. Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Sci. 2003;94(7):589–592. doi: 10.1111/j.1349-7006.2003.tb01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Irizarry RA, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith ZD, Meissner A DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 74.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293(5532):1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 75.Holm K, et al. An integrated genomics analysis of epigenetic subtypes in human breast tumors links DNA methylation patterns to chromatin states in normal mammary cells. Breast Cancer Res. 2016;18(1):27. doi: 10.1186/s13058-016-0685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Doi A, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41(12):1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dmitrijeva M, Ossowski S, Serrano L, Schaefer MH. Tissue-specific DNA methylation loss during ageing and carcinogenesis is linked to chromosome structure, replication timing and cell division rates. Nucleic Acids Res. 2018;46(14):7022–7039. doi: 10.1093/nar/gky498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Y, Breeze CE, Zhen S, Beck S, Teschendorff AE. Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer. Epigenetics Chromatin. 2016;9:10. doi: 10.1186/s13072-016-0058-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dunn JR, et al. METH-2 silencing and promoter hypermethylation in NSCLC. Br J Cancer. 2004;91(6):1149–1154. doi: 10.1038/sj.bjc.6602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dunn JR, et al. Expression of ADAMTS-8, a secreted protease with antiangiogenic properties, is downregulated in brain tumours. Br J Cancer. 2006;94(8):1186–1193. doi: 10.1038/sj.bjc.6603006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Porter S, et al. Dysregulated expression of adamalysin-thrombospondin genes in human breast carcinoma. Clin Cancer Res. 2004;10(7):2429–2440. doi: 10.1158/1078-0432.CCR-0398-3. [DOI] [PubMed] [Google Scholar]
- 82.Demircan K, et al. Increased mRNA expression of ADAMTS metalloproteinases in metastatic foci of head and neck cancer. Head Neck. 2009;31(6):793–801. doi: 10.1002/hed.21045. [DOI] [PubMed] [Google Scholar]
- 83.Masui T, et al. Expression of METH-1 and METH-2 in pancreatic cancer. Clin Cancer Res. 2001;7(11):3437–3443. [PubMed] [Google Scholar]
- 84.Chen J, et al. Downregulation of ADAMTS8 by DNA hypermethylation in gastric cancer and its clinical significance. Biomed Res Int. 2016;2016:5083841. doi: 10.1155/2016/5083841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poggi L, Casarosa S, Carl M. An eye on the Wnt inhibitory factor Wif1. Front Cell Dev Biol. 2018;6:167. doi: 10.3389/fcell.2018.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mazieres J, et al. Wnt inhibitory factor-1 is silenced by promoter hypermethylation in human lung cancer. Cancer Res. 2004;64(14):4717–4720. doi: 10.1158/0008-5472.CAN-04-1389. [DOI] [PubMed] [Google Scholar]
- 87.Liu YL, Yang HP, Zhou XD, Gong L, Tang CL, Wang HJ. The hypomethylation agent bisdemethoxycurcumin acts on the WIF-1 promoter, inhibits the canonical Wnt pathway and induces apoptosis in human non-small-cell lung cancer. Curr Cancer Drug Targets. 2011;11(9):1098–1110. doi: 10.2174/156800911798073041. [DOI] [PubMed] [Google Scholar]
- 88.Kim J, et al. Wnt inhibitory factor inhibits lung cancer cell growth. J Thorac Cardiovasc Surg. 2007;133(3):733–737. doi: 10.1016/j.jtcvs.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 89.Kansara M, et al. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J Clin Invest. 2009;119(4):837–851. doi: 10.1172/JCI37175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kawakami K, et al. Functional significance of Wnt inhibitory factor-1 gene in kidney cancer. Cancer Res. 2009;69(22):8603–8610. doi: 10.1158/0008-5472.CAN-09-2534. [DOI] [PubMed] [Google Scholar]
- 91.Ramachandran I, et al. Wnt inhibitory factor 1 suppresses cancer stemness and induces cellular senescence. Cell Death Dis. 2014;5:e1246. doi: 10.1038/cddis.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xia L, et al. NDNF inhibits the migration and invasion of human renal cancer cells through epithelial-mesenchymal transition. Oncol Lett. 2019;17(3):2969–2975. doi: 10.3892/ol.2019.9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.UniProt Consortium T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018;46(5):2699. doi: 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xiao SJ, Zhang C, Zou Q, Ji ZL. TiSGeD: a database for tissue-specific genes. Bioinformatics. 2010;26(9):1273–1275. doi: 10.1093/bioinformatics/btq109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 97.Fromowitz FB, et al. Ras p21 expression in the progression of breast cancer. Hum Pathol. 1987;18(12):1268–1275. doi: 10.1016/S0046-8177(87)80412-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.