

Abstract
The impact of pannexin-1 (Panx1) channels on synaptic transmission is poorly understood. Here, we show that selective block of Panx1 in single postsynaptic hippocampal CA1 neurons from male rat or mouse brain slices causes intermittent, seconds long increases in the frequency of sEPSC following Schaffer collateral stimulation. The increase in sEPSC frequency occurred without an effect on evoked neurotransmission. Consistent with a presynaptic origin of the augmented glutamate release, the increased sEPSC frequency was prevented by bath-applied EGTA-AM or TTX. Manipulation of a previously described metabotropic NMDAR pathway (i.e., by preventing ligand binding to NMDARs with competitive antagonists or blocking downstream Src kinase) also increased sEPSC frequency similar to that seen when Panx1 was blocked. This facilitated glutamate release was absent in transient receptor potential vanilloid 1 (TRPV1) KO mice and prevented by the TRPV1 antagonist, capsazepine, suggesting it required presynaptic TRPV1. We show presynaptic expression of TRPV1 by immunoelectron microscopy and link TRPV1 to Panx1 because Panx1 block increases tissue levels of the endovanilloid, anandamide. Together, these findings demonstrate an unexpected role for metabotropic NMDARs and postsynaptic Panx1 in suppression of facilitated glutamate neurotransmission.
SIGNIFICANCE STATEMENT The postsynaptic ion and metabolite channel, pannexin-1, is regulated by metabotropic NMDAR signaling through Src kinase. This pathway suppresses facilitated release of presynaptic glutamate during synaptic activity by regulating tissue levels of the transient receptor potential vanilloid 1 agonist anandamide.
Keywords: anandamide, glutamate transmission, hippocampus, metabotropic NMDAR, pannexin, TRPV1
Introduction
Pannexin-1 (Panx1) is an ion/metabolite channel that is broadly distributed in the brain (Bruzzone et al., 2003; Vogt et al., 2005) and highly expressed in the postsynaptic density (Zoidl et al., 2007). Panx1 opening is implicated in several pathologies, including neuronal death during ischemia (Thompson et al., 2006; Bargiotas et al., 2011; Weilinger et al., 2012), NMDAR excitotoxicity (Thompson et al., 2008; Weilinger et al., 2016), inflammasome activation in the gut (Gulbransen et al., 2012), and inflammation in the vasculature (Lohman et al., 2015). An important role for Panx1 in ATP release during apoptosis has also been proposed (Chekeni et al., 2010). Despite the clear roles of Panx1 opening in pathology, our understanding of the physiological functions of Panx1 in the brain is limited.
We showed recently that Panx1 opening following phosphorylation of its C-terminal regulatory domain by sarcoma (Src) kinase contributes to neuronal death during NMDAR excitotoxicity (Weilinger et al., 2016). In other studies, it was reported that Src phosphorylation of Panx1 in the channel's intracellular loop (at Y198) in vascular smooth muscle can regulate ATP release and may contribute to hypertensive pathologies (DeLalio et al., 2019). Thus, Src kinase appears to be a potent regulator of Panx1 activity. Src also phosphorylates NDMARs in neurons and contributes to chronic pain through potentiation of the receptor (Yu et al., 1997; Salter and Kalia, 2004; Yang et al., 2012). Given the impact of Src activity on both NMDARs and Panx1, we tested here whether the NMDAR-Src-Panx1 signalsome contributes to synaptic activity in the hippocampus. We show that blocking Panx1 in single postsynaptic CA1 neurons generates intermittent facilitation of glutamate release during low-frequency Schaffer collateral stimulation. Moreover, blocking Src directly or preventing ligand binding (both glutamate and glycine) to NMDARs caused similar stimulation-dependent facilitated glutamate release. We further show that blocking Panx1 increased tissue concentrations of the transient receptor potential vanilloid 1 (TRPV1) channel agonist anandamide (AEA) and that the facilitated release was TRPV1-dependent. Our findings show that metabotropic NMDAR-Panx1 opening in the postsynapse is a homeostatic regulatory mechanism that buffers AEA accumulation and suppresses TRPV1-dependent glutamate release. A portion of these data have posted on the BioRxiv preprint server (Bialecki et al., 2018).
Materials and Methods
Animals.
All animal care and use were in accordance with the Canadian Council on Animal Care guidelines and approved by the University of Calgary's Animal Care and Use Committee. Male Sprague Dawley rats, 21–37 d of age, were housed on a 12 h light/dark cycle with access to Purina Laboratory Chow and water ad libitum. Male WT mice (C57BL/6J), conditional pannexin-1 KO mice (Panx1fl/fl-wfs1-Cre) (Weilinger et al., 2012), and TRPV1 KO mice (TRPV1−/− Birder et al., 2002) were bred in-house and kept under the same conditions as the rats. Panx1 KO was achieved by intraperitoneal tamoxifen injections (100 mg/kg) once daily for 5 d (Weilinger et al., 2012). Control conditions for Panx1 KO were littermates that received injections of the vehicle alone (95% corn oil 5% ethanol) once daily for 5 d.
Chemicals and reagents.
All salts were from Millipore Sigma. Capsazepine (CPZ) at a final in vitro concentration of 10 μm) was from Tocris Bioscience. The selective peptide inhibitor of Panx1, 10panx (WRQAAFVDSY), and its scrambled control peptide, sc10panx (FSVYWAQADR), were synthesized by AnaSpec or New England Peptide and used at the final concentration of 100 μm. The C-terminal anti-Panx1 polyclonal antibody (α-panx1; 0.25 ng/μl), which blocks Panx1 when included in the patch pipette (Weilinger et al., 2012, 2016), was from Invitrogen (catalog #488100, rabbit polyclonal). Its control (Weilinger et al., 2016) was an anti-connexin-43 polyclonal antibody (α-Cx43; 0.3 ng/μl) that was from Abcam (catalog #ab11370). All drugs were dissolved in water, DMSO, or ethanol and aliquoted and frozen until use. Drugs were then dissolved into aCSF at their final concentrations. Final concentrations of DMSO or ethanol did not exceed 0.1%. The aCSF was saturated with 95% O2/5% CO2 and contained 120 mm NaCl, 26 mm NaHCO3, 3 mm KCl, 1.25 mm NaH2PO4, 1.3 mm MgCl2, 2 mm CaCl2, 10 mm glucose, and 100 μm picrotoxin.
Acute hippocampal slice preparation.
Rats or mice were anesthetized by isoflurane inhalation in air and decapitated; the brain was extracted, blocked, mounted on a vibrating slicer (VT1200S; Leica Microsystems), and submerged in an ice-cold high-sucrose solution consisting of the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, and 75 sucrose, saturated with 95% O2/5% CO2. Transverse hippocampal slices were cut (370 μm for rats and 300 μm for mice) and placed into a chamber containing aCSF at 33°C for at least 1 h before use.
Electrophysiology.
Slices were transferred to a recording chamber and constantly perfused with aCSF (33°C–35°C) at a rate of 1–2 ml/min. Visualization of hippocampal CA1 pyramidal neurons was achieved with differential interference contrast microscopy with an Olympus BX51Wi microscope. Electrophysiological data were collected with a MultiClamp 700B amplifier and digitized with a Digidata 1440A analog to digital converter (Molecular Devices) at 10 kHz and low-pass Bessel filtered at 1 kHz. Data were recorded using pCLAMP 10, Clampex 10.3, and Axoscope 10.3 (Molecular Devices) software and stored for future analysis with Clampfit 10.3, GraphPad Prism, and Excel (Microsoft). Whole-cell voltage-clamp recordings were performed using borosilicate glass microelectrodes (Sutter Instrument) with a tip resistance of 3–6 mΩ that were pulled using a P-1000 Flaming/Brown Micropipette Puller (Sutter Instrument). Microelectrodes were filled with an intracellular solution containing 108 mm potassium gluconate, 2 mm MgCl2, 8 mm sodium gluconate, 8 mm KCl, 2.5 mm K2-EGTA, 4 mm K2-ATP, and 0.3 mm Na3-GTP at pH 7.25 with 10 mm HEPES. Access resistance (RA) was monitored over the course of the experiments and was <8 mΩ (range 7–8 mΩ) after break-in or the cell was discarded. Cells were also discarded from the dataset if RA reached 10 mΩ at any point in the recording. The holding potential was −70 mV unless otherwise noted.
Panx1 block in single CA1 pyramidal neurons was achieved by inclusion of an anti-Panx1 polyclonal antibody (α-Panx1;0.25 ng/ μl) or its negative control, anti-connexin-43 polyclonal antibody (α-Cx43; 0.3 ng/ml) in the patch pipette, as previously described (Weilinger et al., 2012, 2016). To buffer intracellular Ca2+, we included 10 mm BAPTA in the pipette. In the figures, within cell comparisons are indicated by the straight line joining the data points in the absence (unfilled bars/symbols) and following (filled bars/symbols) Schaffer collateral paired-pulse stimulation (PPS).
In most experiments, within-cell comparisons between no stimulation and Schaffer collateral stimulation were used: After formation of the whole-cell configuration, we allowed 10 min for equilibration of the intracellular solution, followed by 5 min of recording sEPSCs. A stimulating electrode was then placed in the Schaffer collateral pathway, and PPSs (1 ms pulse duration and 50 ms interpulse interval) were applied at 0.05 Hz for 5 min. Stimulation strength was set at 50% of the maximum evoked response for the duration of the recording. Detection and quantification of sEPSC interevent intervals and amplitude were with Clampfit (Molecular Devices) using a template-based protocol. The sEPSC template was the average of all the events during the 5 min baseline recording. During analysis, the user manually approved (or not) each sEPSC event that was initially identified by Clampfit. User-rejected events were very rare when RA remained <10 mΩ. sEPSC peak amplitude and interevent interval for the 5 min baseline were compared with the subsequent 5 min period, and Schaffer collaterals were stimulated.
Amplitudes were determined by single Gaussian fits to the distribution of peak sEPSC amplitudes. We compared the cumulative amplitude distributions of the last 200 sEPSC events from the baseline (i.e., no Schaffer collateral stimulation) to the experimental period (with Schaffer collateral stimulation) using Kolmogorov–Smirnov statistics (K-S; GraphPad, Prism 7). Significance was set as p < 0.001 for comparison of distributions because the datasets are large (Kim and Alger, 2010).
The frequency of sEPSCs was determined as the inverse of the average interevent interval for the 5 min recording segments. Cumulative interevent interval distributions were compared using K-S tests (with p < 0.001). Under each experimental condition, all of the baseline (i.e., prestimulation) events for each neuron were compared with all of the sEPSC events during the 5 min stimulation period. Paired-pulse ratios (PPRs) were calculated as the ratio of the peak amplitudes of the evoked EPSC (eEPSC) peak 2 divided by the amplitude of evoked peak 1; here a between-cells design was necessary (i.e., control conditions were not the same cells as Panx1 blocked). Data were compared using nonparametric statistics with either the Wilcoxon matched-pairs signed rank test (for paired data from within cell experimental design), the Kruskal–Wallis (K-W) test (with Dunn's multiple comparisons) for >2 conditions that shared the same baseline, or Mann–Whitney U test for unpaired populations of neurons. Significance was set at p ≤ 0.05. All results are presented as mean ± SEM with n equal to the number of neurons. Only one neuron was evaluated per hippocampal slice and at least 3 animals were used for each condition.
RNA isolation and reverse transcription.
Hippocampi and DRG tissue from both male and female mice were collected from C57BL/6J or TRPV1−/− strains. Total RNA was extracted from tissue using the RNeasy Plus Micro Kit (QIAGEN). Total RNA (1.0 μg) was reverse-transcribed into cDNA by using Superscript IV VILO Master Mix with ezDNase (Invitrogen). The resulting cDNA was used as a template for PCR amplification and stored at −20°C if not used immediately.
PCR.
PCR amplification was performed using 1.0 μl of cDNA and KAPA HiFi HotStart ReadyMix (Kapa Biosystems) with TRPV1 primers (forward 5′-CATGCTCATTGCTCTCATGG and reverse 5′-GCCTTCCTCATGCACTTCAG) in an Eppendorf Mastercycler Gradient GSX1 Thermal Cycler. The PCR program used consisted of a 5 min incubation at 95°C followed by 35 cycles of 98°C for 20 s, 60°C for 15 s, 72°C for 15 s, and a final incubation of 72°C for 5 min. PCR amplification products were separated on a 1.0% agarose gel and stained with Safe-Red to verify their size using a 100 bp DNA ladder.
Quantitative PCR (qPCR).
qPCR was performed using 1.0 μl of cDNA and PowerUp SYBR Green Master Mix (Applied Biosystems), following the Fast PCR protocol, in a QuantStudio 3 Real-Time PCR System. Mouse β-actin QuantiTect Primer Assay (QIAGEN) was purchased as a control gene, and TRPV1 primers used were as mentioned above for PCR. TRPV1 primer quality was validated through a qPCR standard curve. Assays were run on MicroAmp Fast Optical 96-Well Reaction Plates (Applied Biosystems) in triplicates. Fold expression of TRPV1 mRNA levels in hippocampi compared with DRGs was determined using the 2−ΔΔCt method after normalization to internal control β-actin RNA levels.
Electron microscopy (EM).
Five WT TRPV1 and 5 TRPV1−/− adult mice of either sex were used in this study. TRPV1−/− mice (C57BL/6J background) (Birder et al., 2002) were originally from The Jackson Laboratory (strain B6.129X1-Trpv1tm1Jul/J). Experimental animals were genotyped by PCR under standard buffer conditions using the primer pair 5′-CCT GCT CAA CAT GCT CAT TG-3′ and 5′-TCC TCA TGC ACT TCA GGA AA-3′ for the WT locus. The primer pair 5′-CAC GAG ACT AGT GAG ACG TG-3′and 5′-TCC TCA TGC ACT TCA GGA AA-3′ was used to detect a fragment in the Neo cassette, specific for the mutant TRPV1 locus. All four primers were used together in the reaction mix (94°C/3 min; 35 × [94°C/30 s, 64°C/1 min, 72°C/1 min]; 1 × 72°C/2 min; 1 × 10°C hold).
Homozygous TRPV1−/− and WT littermates (TRPV1+/+) from heterozygous breedings were used for experiments. They were deeply anesthetized by intraperitoneal injection of ketamine/xylazine (80/10 mg/kg body weight) and then transcardially perfused at room temperature with PBS (0.1 m, pH 7.4) for 20 s, followed by the fixative solution made up of 4% formaldehyde (freshly depolymerized from PFA), 0.2% picric acid, and 0.1% glutaraldehyde in PB (0.1 m, pH 7.4) for 10–15 min. Brains were then removed from the skull and postfixed in the fixative solution for ∼1 week at 4°C. Afterward, brains were stored at 4°C in 1:10 diluted fixative solution until used.
Preembedding immunogold method for TRPV1 EM.
Coronal 50-μm-thick hippocampal vibrosections were collected in 0.1 m PB at room temperature. Then, they were preincubated in a blocking solution of 10% BSA, 0.1% sodium azide, and 0.02% saponine prepared in Tris-HCl buffered saline (TBS 1×, pH 7.4) for 30 min at room temperature. Sections were incubated with the primary goat TRPV1 antibody (1:100, VR1 (P-19), sc-1249, Santa Cruz Biotechnology) prepared in the blocking solution but with 0.004% saponin, for 2 d at 4°C. After several washes, tissue sections were incubated with 1.4 nm gold-labeled rabbit antibody to goat IgG (Fab fragment, 1:100, Nanoprobes) prepared in the same solution as the primary antibody for 3 h at room temperature. Tissue was washed overnight at 4°C and postfixed in 1% glutaraldehyde for 10 min. After several washes with 1% BSA in TBS, gold particles were silver-intensified with a HQ Silver Kit (Nanoprobes) for 12 min in the dark. Then, sections were osmicated, dehydrated, and embedded in Epon resin 812. Finally, ultrathin sections were collected on mesh nickel grids, stained with lead citrate, and examined in a Philips EM208S electron microscope. Tissue preparations were photographed by using a digital camera coupled to the electron microscope. Specificity of the immunostaining was assessed by incubation of the TRPV1 antiserum in TRPV1−/− hippocampal tissue in the same conditions as above.
Statistical analysis of TRPV1 in the CA1 hippocampus.
CA1 hippocampal sections (50 μm thick) from TRPV1+/+ and TRPV1−/− mice (n = 5 each) showing good and reproducible silver-intensified gold particles were cut at 80 nm. Electron micrographs (18,000–28,000×) were taken from grids (2 mm × 1 mm slot) with ultrathin sections showing similar labeling intensity indicating that selected areas were at the same depth. Furthermore, to avoid false negatives, only ultrathin sections in the first 1.5 μm from the surface of the tissue block were examined. Positive labeling was considered if at least one immunoparticle was within ∼30 nm from the plasmalemma.
TRPV1 metal particles on axon terminals were visualized and counted in randomly taken electron micrographs from both animal types. The number of positive terminals was normalized to the total number of terminals in the images to identify the proportion of TRPV1-positive profiles in TRPV1+/+ versus TRPV1−/−. Results were expressed as means of independent data points ± SEM. Statistical analyses (Unpaired Student's t test) were performed using GraphPad software 5.0 (GraphPad Software) and significance was set a p < 0.05.
Mass spectrometric quantification of AEA levels.
Hippocampal slices were prepared as described above and underwent whole-cell patch-clamp recording with Schaffer collateral stimulation in aCSF or aCSF with 100 μm 10panx or 10 μm CPZ before lipid extraction as previously described (Qi et al., 2015). In brief, tissue samples were weighed and placed in borosilicate glass culture tubes containing 2 ml of acetonitrile with 5 pmol of [2H8] AEA for extraction. These samples were homogenized with a glass rod, sonicated for 30 min, incubated overnight at −20°C to precipitate proteins, and then centrifuged at 1500 × g for 5 min to remove particulates. Supernatants were removed to a new glass culture tube and evaporated to dryness under N2 gas, resuspended in 300 μl of acetonitrile to recapture any lipids adhering to the tube, and redried again under N2 gas. The final lipid extracts were suspended in 200 μl of acetonitrile and stored at −80°C until analysis. AEA contents within lipid extracts were determined using isotope-dilution, liquid chromatography-tandem mass spectrometry as previously described (Qi et al., 2015).
Results
Block of postsynaptic Panx1 facilitates glutamate release following Schaffer collateral stimulation
To evaluate putative roles of Panx1 in spontaneous and evoked neurotransmission, we selectively inhibited the channels in single postsynaptic CA1 neurons by inclusion of a blocking antibody, α-panx1 (0.25 ng/μl) in the patch pipette (Weilinger et al., 2012, 2016). Properties of sEPSC over a 5 min baseline were compared with those in the subsequent 5 min when PPSs were applied to the Schaffer collaterals (0.05 Hz, 1 ms pulse duration, 50 ms interstim interval), which also allowed for evaluation of evoked responses. All bathing solutions contained 100 μm picrotoxin to block GABAA receptors.
Figure 1A–F illustrates responses of a CA1 neuron to PPS with the control pipette solution, and Figure 1G–L is from a different CA1 neuron with α-panx1 in the pipette. Figure 1A shows 20-s-long recordings before (left) and after (right) PPS of the Schaffer collaterals, and Figure 1B, C shows the instantaneous frequency of sEPSCs for the entire 5 min recoding periods of this neuron. As expected, PPS did not alter the cumulative interevent interval (Fig. 1D; n = 28 neurons from 28 slices, K-S test; p > 0.99; K-S, D = 0.0019) nor the amplitude distribution of sEPSCs (Fig. 1D; n = 28; K-S test; p > 0.99; K-S, D = 0.0003) under control conditions. In contrast, inclusion of α-panx1 in the pipette solution (Fig. 1G) caused a significant leftward shift in the cumulative interevent interval distribution during PPS, indicating higher frequency (Fig. 1H–J; n = 31; K-S test; p < 0.0001; K-S, D = 0.524). The distribution of amplitudes was not changed (Fig. 1K,L; p > 0.999; K-S test, K-S, D = 0.0005).
Figure 1.

Block of Panx1 in single postsynaptic neurons enhances facilitated glutamate release during Schaffer collateral stimulation. A, Sample 20 s traces taken from 5 min recordings (as indicated) from the same CA1 pyramidal neuron without Schaffer collateral stimulation (black) and with PPS (gray traces) using a control pipette solution. B, Time (5 min total) versus instantaneous frequency under control conditions for the cell in A. C, Time (5 min total) versus instantaneous frequency during PPS of the Schaffer collaterals (vertical dashed lines) for the cell in A. D, Cumulative distribution of the interevent interval of sEPSC for the population of neurons. Comparisons were made by K-S tests (at p < 0.001). No change in the distribution of interevent intervals was detected after Schaffer collateral stimulation under control conditions. E, Families of sEPSCs from the cell in A and B. Thick dark lines indicate mean sEPSCs. F, Comparison of the distribution of sEPSC amplitudes before and after PPS delivery to the Shaffer collaterals. Amplitude was determined by fit of single Gaussian to the distribution. The population of distributions were compared by K-S tests and were not different for the 29 neurons evaluated. G, Sample 20 s recordings under the condition where 0.25 ng/μl α-panx1 (anti-pannexin-1 blocking antibody) is included in the patch pipette to selectively block postsynaptic Panx1. Traces represent without (left) and with (right) PPS of the Schaffer collaterals taken at the indicated time from the total 5 min experiment. There is an increase in the frequency of sEPSC after PPS of the Schaffer collaterals. H, I, Instantaneous sEPSC frequency versus time plots for the cell in G showing that PPS with α-panx1 in the pipette (right) evoked increases in sEPSC frequency compared with the nonstimulated control (right). J, Comparison of the cumulative sEPSC frequency distribution for the population of neurons. Significance (p < 0.001) was confirmed by K-S test. K, Families of sEPSCs with α-panx1 in the pipette. L, Comparison of the distribution of sEPSC amplitudes before and after PPS delivery to the Shaffer collaterals with α-panx1 in the pipette. Amplitude was determined by fit of single Gaussian to the distribution, and distributions were compared by K-S tests and were not different.
The mean ± SEM frequency for the 5 min baseline (i.e., without stim and without α-panx1) was 3.1 ± 0.2 Hz, which was significantly increased to 6.2 ± 0.5 Hz with PPS and pipette α-panx1 (Fig. 2A; K-W test; p < 0.0001, K-W statistic = 33.17; all other conditions vs control p > 0.99). The mean ± SEM amplitudes of sEPSCs are shown in Figure 2B, and they did not significantly change from the control level of 16.5 ± 0.4 pA under any condition (K-W test; p = 0.09, K-W statistic = 6.51). As an important control for intracellular α-panx1, we evaluated a polyclonal antibody against α-Cx43 (0.3 ng/μl) in the pipette (Weilinger et al., 2016) and found no change in the cumulative sEPSC frequency distribution (n = 8; K-S, D = 0.0011, p > 0.99; data not shown) or the mean sEPSC frequency when Shaffer collateral PPS was applied (Fig. 2A; n = 8; p = 0.46; Wilcoxon matched pairs W = 1.0).
Figure 2.

Summary of the frequency and amplitude of sEPSCs during Panx1 block and Schaffer collateral stimulation. A, All Shaffer collateral PPS was delivered at 0.05 Hz (20 s inter-PPS interval). Block of Panx1 with either α-panx1 in the pipette or 10panx (100 μm) in the bath augmented sEPSC frequency following afferent stimulation. There is a lack of effect of the respective controls, α-Cx43 and sc10panx. *p < 0.05 (nonparametric Wilcoxon matched-pairs signed rank test). B, Summary of the sEPSC amplitudes under control (n = 12) and with α-panx1 (n = 31) in the pipette in the presence or absence of PPS as indicated. A within-cell design was used as for the frequency analysis, and no significant difference was detected. C, Comparison of different inter-PPS intervals as indicated above each bar. Frequency was determined for the entire 5 min recordings and was significantly increased at 10 and 30 s inter-PPS intervals.
We tested the peptidergic blocker of Panx1, 10panx (100 μm), and its scrambled control (sc10panx; 100 μm) (Thompson et al., 2008) because these are bath-applied and block the channels through a mechanism distinct from α-panx1. 10panx decreased cumulative sEPSC interevent intervals during PPS (n = 6; p = 0.0006; K-S, D = 0.5357) with a significant increase in sEPSC frequency from 3.1 ± 0.4 Hz to 6.7 ± 0.9 Hz (Fig. 2A; n = 6; p = 0.01, Wilcoxon matched pairs, W = 21). The scrambled control, sc10panx, with PPS did not alter the mean sEPSC frequency (Fig. 2A; n = 7, p = 0.38, Wilcoxon matched pairs, W = −12). Together, these data show that 2 distinct Panx1 blockers, one acting intracellularly in single patched cells and the other bath-applied, result in facilitated glutamate release during low-frequency Schaffer collateral stimulation.
Interestingly, the increased sEPSC frequency was not persistent throughout the full 5 min recording; rather, it appeared as intermittent bursts that lasted, on average, for 13 ± 0.5 s (Fig. 1I; n = 28; range 1.4–19.8 s). Bursts were defined to start when the instantaneous frequency exceeded the baseline frequency by a factor of 2.5 and to terminate when it returned to baseline. On average, a CA1 neuron would burst 3.2 ± 0.4 times (range 0–9; median = 3.5) in the 5 min stimulation period. sEPSC bursting events reached a maximum frequency of 28.9 ± 1.3 Hz (n = 31), which was significantly larger compared with the maximum frequency of 14.6 ± 2.3 Hz (n = 28) without α-panx1 in the pipette (Mann–Whitney U test, p < 0.0001, U = 30.5). The appearance of the high-frequency sEPSC bursts was not clearly time-locked with synaptic stimulation, but they only occurred during PPS when Panx1 was blocked (Fig. 2A). We evaluated a range of intervals between PPS stimulations from 10 to 60 s. Significant increases in mean sEPSC frequency were observed with PPS intervals of 10 s (Fig. 2C; Wilcoxon test, p = 0.0234; n = 8; W = 32), 20 s (Figs. 1, 2A; stats reported above), and 30 s (Fig. 2C; Wilcoxon test, p = 0.031; n = 6; W = 21). Bursts were seen for the 40 s PPS interval but, when averaged over the 5 min recordings, did not reach a statistically significant difference.
To rule out the possibility of presynaptic Panx1 block by extracellular accumulation of α-panx1 leaking from the pipette due to positive pressure in the recording electrode, we locally puffed α-panx1 while recording from CA1 neurons with the control pipette solution. Puffing α-panx1 within 100 μm from the soma of patched neurons had no effect on sEPSC frequency (data not shown; 2.3 ± 0.3 Hz without PPS and 3.3 ± 0.5 Hz with PPS, n = 11 paired recordings, p = 0.175, Wilcoxon matched pairs, W = 32). The cumulative distribution of interevent intervals was also not different between the two conditions (K-S test, p > 0.99, K-S statistic = 0.002), confirming that α-panx1 blocks Panx1 channels intracellularly.
Evaluation of Panx1 KO on sEPSC frequency
Responses to postsynaptic delivery of α-panx1 were evaluated in our pyramidal neuron-specific, conditional Panx1 KO mice (Panx1fl/fl-wfs1-Cre) (Weilinger et al., 2012) and compared with WT animals of the same genetic background (C57BL/6J). CA1 neurons from WT mice had significantly decreased interevent intervals (Fig. 3A,B; n = 8; p < 0.0001; K-S, D = 0.1846), resulting in an increase in the mean sEPSC frequency from 3.6 ± 0.4 Hz to 6.9 ± 0.6 Hz (Fig. 3A,B; n = 8; p = 0.016, Wilcoxon matched pairs, W = 28) during PPS with α-panx1 was in the pipette. α-panx1 application to neurons from vehicle-injected (5% ethanol/95% corn oil) Panx1fl/fl-wfs1-Cre animals showed a similar PPS-dependent shift in cumulative interevent intervals (n = 10; p < 0.0001, K-S, D = 0.2352) and increased sEPSC frequency (5.9 ± 0.6 Hz) over the prestim level of 4.0 ± 0.5 Hz following PPS (Fig. 3C; n = 11; p = 0.006; Wilcoxon matched pairs, W = 51).
Figure 3.

Conditional KO of Panx1 increases sEPSC frequency following Schaffer collateral stimulation. A, Traces from a CA1 neuron in hippocampal slice from WT (C57BL/6J) mouse with α-panx1 in the pipette without (left) or with (right) paired-pulse (PPS) Schaffer collateral stimulation. B, Traces are from a CA1 neuron in slice from a tamoxifen-treated Panx1fl/fl-wfs1-Cre mice to KO Panx1. KO of Panx1 when paired with PPS of the Shaffer collaterals increased the frequency of sEPSCs without α-panx1 in the pipette. C, Cumulative interevent intervals for the cells shown in A (left) and B (right). D, Summary of the sEPSC frequency from WT mice or those carrying floxed Panx1 with and without α-panx1 in the pipette and with and without PPS of the Shaffer collaterals as indicated. Treatment with tamoxifen alone or with intracellular α-panx1 (as indicated) resulted in an increase in sEPSC frequency upon PPS of the Shaffer collaterals. These data indicate that Panx1 is responsible for suppressing augmented sEPSC frequency under control conditions and that α-panx1 is selective. *p < 0.05 (Wilcoxon matched-pairs signed rank test).
We predicted that KO of Panx1 by delivery of tamoxifen to the Panx1fl/fl-wfs1-Cre line should be sufficient to cause an increase in PPS-mediated sEPSC frequency in the absence of intracellular α-panx1. PPS stimulation of neurons in slice from tamoxifen-treated Panx1fl/fl-wfs1-Cre mice (without α-panx1 present) had significantly altered cumulative interevent interval distributions (Fig. 3B,C; n = 7; p < 0.0001; K-S, D = 0.2066) and a significantly increased mean sEPSC frequency from 3.5 ± 0.4 to 5.4 ± 0.8 Hz (Fig. 3D; n = 7; p = 0.031; Wilcoxon matched pairs, W = 21). Including α-panx1 in the pipette during recordings from additional neurons from tamoxifen-treated Panx1fl/fl-wfs1-Cre mice, cumulative interevent interval distributions changed upon PPS delivery (data not shown; n = 9; p < 0.0001; K-S, D = 0.1226) and the mean sEPSC frequency increased from 3.6 ± 0.4 to 5.1 ± 0.6 Hz (Fig. 3D; n = 9; p = 0.031; Wilcoxon matched pairs, W = 37). This change in mean sEPSC frequency was not different from that seen in the control cells where Panx1 was knocked out and without α-panx1 (Fig. 3D; comparing stim conditions of Panx1fl/fl + tamox without and with α-panx1; n = 7 and n = 9, respectively; p = 0.68; Mann–Whitney U = 27). Thus, knocking out Panx1 was sufficient to augment sEPSC frequency during PPS, and the lack of an additional effect of α-panx1 in these mice further demonstrates specificity of the α-panx1 antibody.
NMDAR block increases sEPSC frequency during synaptic stimulation
We have reported previously that phosphorylation of Panx1 by Src kinase is a critical component of metabotropic NMDAR signaling during excitotoxicity (Weilinger et al., 2012, 2016). This activation of Panx1 is blocked by the NMDAR glutamate site competitive antagonist d-APV (50 μm) or the glycine site competitive antagonist CGP-78608 (CGP; 1 μm), indicating that binding of both receptor ligands was required for metabotropic NMDAR signaling (Weilinger et al., 2016). We reasoned that, if metabotropic NMDAR-Panx1 signaling is suppressing facilitated glutamate release, then bath application of d-APV plus CGP (without intracellular α-panx1) would increase sEPSC frequency during PPS. Neither d-APV nor CGP applied alone (without concomitant intracellular α-panx1) altered the cumulative interevent interval distributions with or without PPS of the Schaffer collaterals (Fig. 4B; K-S test; p > 0.99; n = 9 and K-S, D = 0.073 for CGP alone; for APV, n = 5, p > 0.99, K-S, D = 0.005). We then bath-applied 50 μm d-APV plus 1 μm CGP for 5 min and then recorded a 5 min baseline without PPS of Schaffer collaterals. After the 5 min baseline 0.05 Hz PPS was applied as described above. α-panx1 was not included in the pipette so that we could evaluate NMDAR signaling upstream of Panx1. As shown in Figure 4A, C, D, application of NMDAR competitive antagonists resulted in the appearance of intermittent increases in sEPSC frequency seen as a significant shift in the cumulative interevent interval distribution (Fig. 4C; d-APV+CGP baseline vs d-APV+CGP PPS, n = 8, p < 0.0001, K-S, D = 0.262). The average frequency in the presence of d-APV+CGP with Schaffer collateral stimulation was 6.8 ± 2.7 Hz, which was significantly greater than the 3.5 ± 1 Hz frequency without stimulation and 3.2 ± 0.8 Hz for the same cells without NMDAR blockers or stimulation (Fig. 4D; n = 8; Freidman test p = 0.03, Freidman stat = 7). This indicates that blocking binding of both NMDAR ligands was necessary to cause facilitated glutamate release similar to that seen with Panx1 block.
Figure 4.

Preventing ligand binding to NMDARs and Src kinase activity induced facilitated glutamate release during Schaffer collateral stimulation. A, Traces from a CA1 neuron in hippocampal slice (rat) with the competitive blockers for both ligand binding sites of the NMDAR. d-APV (50 μm) and CGP (1 μm) were in the bath and α-panx1 in the pipette. PPS of the Schaffer collaterals caused an increase in sEPSC frequency. B, Cumulative sEPSC frequency distributions with either d-APV (top) or CGP (bottom) in the bath. Neither competitive antagonist alone altered the distribution of interevent intervals as determined by K-S analysis. C, In contrast to either competitive antagonist alone, the combined application of d-APV+CGP caused a significant (p < 0.001) shift in the distribution of interevent intervals and in the mean ± SEM. D, sEPSC frequencies. E, Traces from a neuron with Src activation of Panx1 blocked by inclusion of TAT-Panx308 (10 μm) in the pipette before and after Schaffer collateral stimulation. F, Cumulative interevent distributions for the population of neurons with TAT-Panx308 in the pipette. G, Cumulative interevent distributions for the population of neurons with the Src blocker, PP2, in the bath. H, Comparison of the mean sEPSC frequency under conditions of Src block. α-panx1 was not in the patch pipette under any conditions depicted in this figure. *p < 0.05 (Wilcoxon matched pairs test). n.s., Not significant.
In our previous report (Weilinger et al., 2016), we designed an interfering peptide, TAT-Panx308, that prevents Src kinase-mediated phosphorylation of Panx1 at Y308, thereby blocking its activation by metabotropic NMDAR signaling. Here, we predicted that inclusion of TAT-Panx308 in the patch pipette instead of α-panx1 should induce increased sEPSC frequency upon PPS. As shown in Figure 4E, F, H, this was the case since there was a significant leftward shift in the cumulative interevent interval distributions (Fig. 4F; K-S test; p < 0.01; K-S, D = 0.198), and the mean sEPSC frequency was significantly increased from 4.1 ± 0.7 to 8.0 ± 0.9 Hz (Fig. 4H; n = 13; p = 0.002; Wilcoxon matched pairs, W = 81). It follows that, if Src kinase is activating Panx1 to suppress facilitated glutamate release, then directly inhibiting Src with PP2 (10 μm) should promote intermittent increases in sEPSC frequency during PPS. As predicted, bath application of PP2 combined with PPS caused a significant shift in the cumulative interevent interval distribution (Fig. 4G; n = 7, p < 0.0001, K-S test, D = 0.42) and the mean ± SEM frequency increased from 6.9 ± 2.7 to 11.8 ± 3.9 Hz (Fig. 4H; n = 7, p = 0.04, Wilcoxon matched pairs, W = 24).
Metabotropic NMDAR signaling occurs independently of Ca2+ influx through the ionotropic receptor (Nabavi et al., 2013; Dore et al., 2015; Weilinger et al., 2016). To further support the involvement of metabotropic NMDAR signaling, we evaluated the postsynaptic Ca2+ dependence of augmented sEPSC frequency by α-panx1+PPS by including 10 mm BAPTA with α-panx1 in the pipette. Under these conditions, cumulative interevent intervals were shifted left (n = 7, p = 0.0002; K-S, D = 0.157; data not shown) and the mean sEPSC frequency increased from 3.4 ± 0.6 to 5.7 ± 1.0 Hz (Fig. 5A–C; n = 7; p = 0.031; Wilcoxon matched pairs, W = 26), consistent with a role for Ca2+-independent metabotropic NMDAR signaling.
Figure 5.

Chelating postsynaptic Ca2+ does not prevent Panx1-block induced increases in sEPSC frequency. A, Sample traces from the same neuron showing the presence of 10 mm BAPTA and α-panx1 in the pipette did not alter the increase in frequency of sEPSCs when Schaffer collaterals receive PPS (bottom). B, Plots of instantaneous frequency reveal that synaptic stimulation (bottom; vertical lines) causes bursts of sEPSC release when BAPTA and α-panx1 are in the pipette. C, Summary of the paired changes in frequency upon synaptic stimulation. *p < 0.05 (Wilcoxon matched-pairs signed rank test).
The lack of effect of postsynaptic BAPTA indicates that Panx1's influence on sEPSC frequency is independent of postsynaptic Ca2+ and suggests that classic retrograde signaling is not involved. Consistent with this, block of the endocannabinoid CB1 receptor with bath-applied AM251 (3 μm) did not prevent changes in the cumulative interevent interval distributions (n = 6, K-S, D = 0.431; p < 0.0001; data not shown) or the increase in mean sEPSC frequency during PPS (Fig. 6A,C; n = 6; p = 0.031; Wilcoxon matched pairs, W = 21). Additionally, the nNOS inhibitor l-NAME was also ineffective at blocking changes cumulative interevent interval distributions and mean sEPSC frequency increased upon PPS (Fig. 6B,C; n = 8; p = 0.016; Wilcoxon matched pairs, W = 34), suggesting that NO signaling was not involved.
Figure 6.

Blockers of CB1 receptors and nitric oxide synthase do not prevent the Panx1 block induced facilitated glutamate release. A, B, Exemplar recordings from two different CA1 neurons testing the role of the retrograde transmitter receptor, CB1R, and nNOS in synaptic bursting. Both were ruled out because application of their specific antagonists, AM251 and l-NAME, respectively, did not alter the appearance of increased sEPSC frequency. C, Summary of the mean sEPSC frequency. *p < 0.05 (Wilcoxon matched-pairs signed rank test).
Differential effect of blocking Panx1 on evoked and spontaneous synaptic activity suggests a presynaptic origin
The intermittent timing of the increased sEPSC frequency relative to PPS raised the question of whether action potential evoked presynaptic glutamate release was a requirement for bursting because electrical stimulation may induce gliotransmitter release (Jourdain et al., 2007). To address this, we blocked action potential generation in the slice with bath-applied 1 μm TTX. After the whole-cell configuration was formed, we briefly stimulated Schaffer collaterals with single 1 ms pulses to ensure synapses onto the patched neuron were activated. We then perfused TTX onto the slice for a minimum of 5 min to block all action potential-mediated inputs, which prevented eEPSCs as expected (data not shown). We then applied PPS to the TTX-blocked Schaffer collaterals as above (paired-pulse at 0.05 Hz, 1 ms duration, 50 ms interstim interval). Under these conditions, α-panx1 had no effect on sEPSC cumulative distribution (p > 0.01, K-S test; data not shown) or mean frequency; over 20 min in the presence of TTX and intracellular α-panx1, the mean frequency was 0.9 ± 0.1 Hz before, and 0.9 ± 0.14 Hz after Schaffer collateral PPS (n = 5; p = 0.902, Wilcoxon matched pairs, W = 15, holding potential = −60 mV). This indicates that the source of glutamate for the facilitated glutamate release observed when postsynaptic Panx1 is blocked requires action potentials.
The requirement for action potentials prompted us to ask whether the eEPSCs were also being affected by Panx1 block and PPS. We first evaluated the effect of intracellular α-panx1 on the PPR to determine whether blocking postsynaptic Panx1 altered the probability of evoked glutamate release. An increase in PPR would suggest that the active synapses are stronger. Interestingly, we found no difference in PPR between neurons with control intracellular solution (control, n = 28) and a different cohort with intracellular α-panx1 in the pipette (Fig. 7A; n = 31; labeled “all,” p = 0.19, K-W multiple comparison, Z = 1.858). Due to the intermittent nature of the facilitated sEPSCs, we asked whether the PPR was altered during sEPSC burst events. Comparison of the PPR calculated for the evoked responses when the increased sEPSC frequency overlapped with the PPS (n = 12, labeled “within burst” in Fig. 7A) revealed no change compared with control (Fig. 7A; p > 0.99, K-W multiple comparisons, p > 0.99 for control vs within burst, Z = 1.833) or to the total PPR with α-panx1 in the pipette (p = 0.202, K-W multiple comparisons, Z = 0.409).
Figure 7.
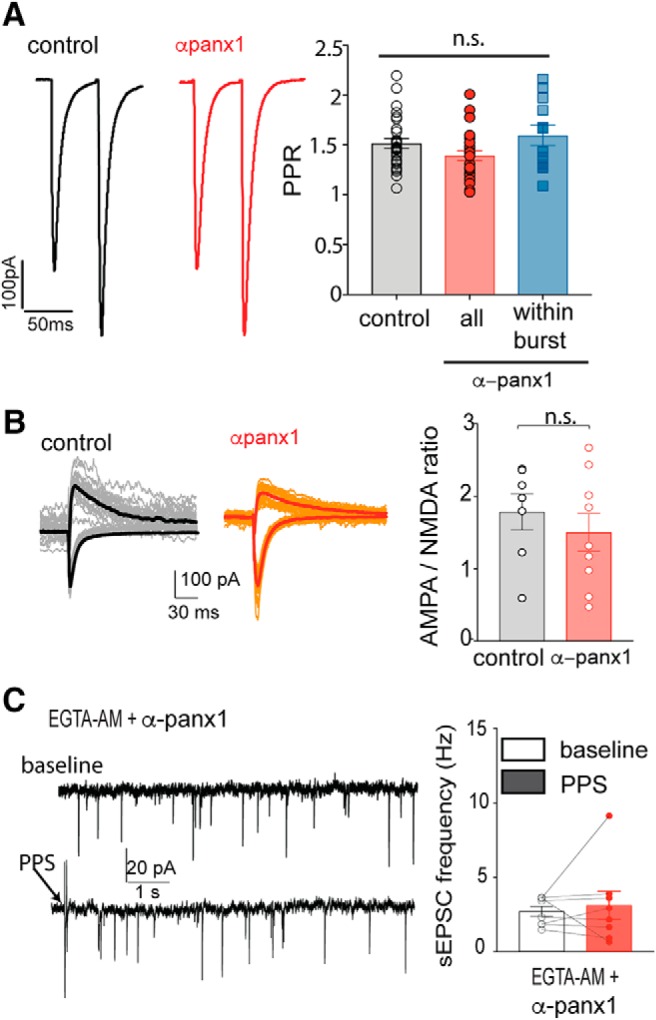
The block of postsynaptic Panx1-induced increase in sEPSCs does not alter evoked EPSCs, AMPAR/NMDAR ratios but requires increased Ca2+. A, PPR is not altered by blocking postsynaptic Panx1. Average (n = 15 pairs) PPRs (at 0.05 Hz, 1 ms duration, 50 ms interstim interval) are shown under control (black) and with α-panx1 in the pipette (red). Right, The population response of the PPR. In the presence of α-panx1 in the pipette (n = 31), the PPR over the 5 min recordings was unchanged compared with the control (n = 28). When the increased sEPSC frequency overlapped with the evoked responses (blue bar; n = 12), the PPR was still unchanged. B, AMPAR/NMDAR ratio is not altered by block of postsynaptic Panx1. Families from both control (normal internal solution; n = 7) and with α-panx1 (n = 9) in the pipette (red) are shown. Downward traces represent the AMPAR component collected at Vm = −70 mV in 1.2 mm Mg2+. Upward traces represent the NMDAR component collected at Vm = 40 mV in 0 Mg2+ and 10 μm DNQX. Right, Calculated ratios. n.s., Not significant. C, Incubation of hippocampal slices with EGTA-AM prevented the increased sEPSC frequency arising from Schaffer collateral stimulation when α-panx1 was in the pipette. Example 10 s traces are shown. Arrow indicates PPS. Right, The plot illustrates a failure of the mean frequency of sEPSCs to increase, suggesting that bursting is Ca2+-dependent.
It has been reported that increased EPSC frequency can arise by potentiating silent synapses, which can be measured as an increase in the AMPAR/NMDAR ratio (Beique et al., 2006). We quantified AMPAR/NMDAR ratios as responses to synaptic stimulation at a holding potential of −80 mV in the presence of 1.2 mm Mg2+ (AMPAR component) and at 40 mV with 0 mm Mg2+ + 10 μm DNQX (NMDAR component). The AMPAR/NMDAR ratio was not altered (n = 7 control and n = 9 with α-panx1; p = 0.606, Mann–Whitney U test, U = 26) by the presence of α-panx1 in the patch pipette (Fig. 7B), suggesting that blocking postsynaptic Panx1 does not potentiate silent synapses.
The increase in sEPSC frequency and block by TTX suggest a presynaptic origin of the glutamate release. We therefore asked whether the sEPSC bursting was dependent on presynaptic Ca2+ rises. Acute hippocampal slices were incubated in membrane-permeable EGTA-AM (50 μm; minimum 15 min loading time). We chose EGTA-AM over BAPTA-AM for bulk loading because EGTA does not block fast-evoked neurotransmission but is sufficient to inhibit increases in Ca2+ that are slower and more prolonged (Ohana and Sakmann, 1998). After EGTA incubation, PPS in the presence of postsynaptic α-panx1 failed to generate sEPSC bursts (n = 8; p = 0.043, K-S, D = 0.079) with no change in sEPSC frequency (Fig. 7C; n = 8, p = 0.25, Wilcoxon matched pairs, W = −18). Together, these data indicate a presynaptic origin of the sEPSC bursts. Furthermore, since no change in the PPR was detected, it suggests that the presynaptic elements being activated are very minor contributors to the evoked responses or are normally silent.
Presynaptic TRPV1 mediates facilitated glutamate release
A recent report demonstrated the presence of functional TRPV1 channels in hippocampal Cajal-Retzius (CR) cells that augment synaptic activity onto GABAergic interneurons (Anstötz et al., 2018). Interestingly, TRPV1-containing CR cells make synaptic connections with CA1 pyramidal neurons (Anstötz et al., 2016). Given that several groups have reported TRPV1 function in the hippocampus, leading to plasticity (Gibson et al., 2008; Chávez et al., 2010) and that TRPV1 in the nucleus tractus solitaris can augment EPSC frequency (Peters et al., 2010), we hypothesized that TRPV1 mediates the increased sEPSC frequency described above when Panx1 is blocked in postsynaptic CA1 neurons.
Since broad TRPV1 expression in the hippocampus is controversial, we used two strategies to confirm expression and determine whether TRPV1 is present in synapses. As a first test, we used PCR and qPCR to verify expression and quantify TRPV1 mRNA levels in hippocampus relative to levels in the DRGs. Hippocampi from TRPV1 KO mice (TRPV1−/−) were also evaluated. As shown in Figure 8A, B, TRPV1 mRNA was detected in hippocampi from WT mice but not TRPV1−/− animals (n = 10 WT and n = 4 TRPV1−/−; p = 0.017; unpaired Student's t test, F = 5117). TRPV1 mRNA was a small fraction of that seen in the DRG (Fig. 8A,B).
Figure 8.

Expression of TRPV1 in the CA1 region is at a portion of presynaptic terminals. A, Demonstration of TRPV1 mRNA by PCR in hippocampus and DRGs. A PCR product is not observed in TRPV1−/− mice. B, Comparison of TRPV1 expression relative to DRG. qPCR levels were first normalized to β-actin levels and then expressed as a fraction of the level in DRG. The hippocampus has a very small, but detectable, amount of TRPV1 mRNA relative to DRG and that TRPV1 mRNA was not detectable in KO mice. C, Sample immuno-EM images of TRPV1 labeling in WT (wt; top) and KO (TRPV1−/−; bottom) mice. Arrows indicate TRPV1 immunogold particles in terminals. Red represents presynaptic terminals. Blue represents CA1 spines. Scale bars, 1 μm. D, Summary of the percentage of labeled terminals in the CA1. B, D, *p < 0.05 (unpaired Student's t test).
We next used immune-EM in WT and TRPV1−/− mice to investigate where TRPV1 is expressed in the CA1. TRPV1 channels have been reported at postsynaptic sites of excitatory and inhibitory synapses in the dentate gyrus of the hippocampus (Canduela et al., 2015; Puente et al., 2015) and GABA sites in the CA1 area (Lee et al., 2015). We found that ∼20% of the excitatory synaptic terminals were TRPV1 positive, and immunogold labeling was almost undetectable in the TRPV1−/− mice (Fig. 8C,D; n = 5 WT and n = 5 TRPV1−/−; p < 0.0001; unpaired Student's t test, F = 17.32).
TRPV1 is a Ca2+-permeable channel that could be responsible for the augmented sEPSC frequency seen with α-panx1+PPS. If so, pharmacological block (or KO) of TRPV1 should prevent the sEPSC frequency increase. In this subset of cells, we blocked Panx1 with intracellular α-panx1 and quantified sEPSC frequency as above. We then bath-applied the TRPV1 blocker, CPZ (10 μm), for 10 min and analyzed the latter 5 min of this period to quantify sEPSC frequency. We then applied PPS in the presence of CPZ to quantify any effect on sEPSCs, allowing for direct within-cell comparisons under the four conditions. As above, intracellular α-panx1 and PPS of the Schaffer collaterals augmented the mean frequency of sEPSCs versus the nonstimulated baseline (Fig. 9A,B; n = 7, p = 0.023; Freidman test of multiple paired comparisons, z = 2.898). Bath application of CPZ did not change mean sEPSC frequency versus control (n = 7; p > 0.99; Freidman test, z = 0.207), but it blocked the PPS-induced increase in frequency (Fig. 9A,B; n = 7) evident as no change in the cumulative interevent distribution (p > 0.99, K-S, D = 0.004), and the mean sEPSC frequency was unchanged versus control (p > 0.99; Freidman test, z = 1.242).
Figure 9.
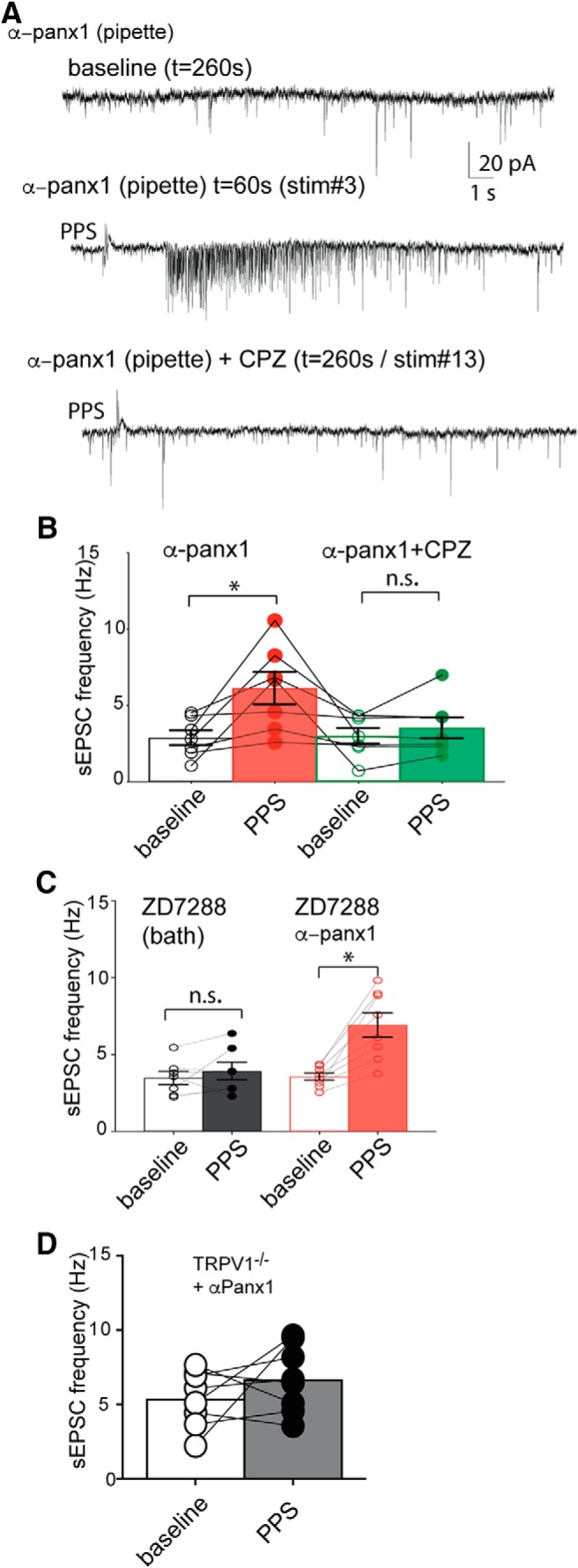
TRPV1 activity is required for Panx1-block-mediated synaptic bursting. A, Sample 20 s traces from the same cell that have α-panx1 in the pipette are shown during baseline (top), PPS stimulation of the Schaffer collaterals (middle), and during PPS stimulation with the TRPV1 antagonist, CPZ, in the bath (bottom). CPZ blocks the PPS-induced increase in sEPSC frequency. B, Summary of the changes in sEPSC frequency in rats with α-panx1 in the pipette. Each neuron was treated with all four conditions, and within-cell comparisons are indicated by the lines joining individual points. The block of the increase in sEPSC frequency with bath application of 10 μm CPZ. C, The Ih blocker, ZD7288 (1 μm), did not induce increased sEPSC frequency when PPS was applied, nor did it prevent the increase when α-panx1 was present. This is an important control for off target effects of CPZ. D, KO of TRPV1 (TRPV1−/−) prevents Schaffer collateral stimulation induced increases in sEPSC frequency. *p < 0.05 (Wilcoxon matched-pairs rank test). n.s., Not significant (p > 0.05).
CPZ is reported to inhibit Ih in pyramidal neurons (Ray et al., 2003), so we evaluated the specific Ih blocker, ZD7288 (10 μm), as a control. In cells from slices perfused with ZD7288 alone (i.e., without α-panx1 in the pipette), there was no detectable increase in sEPSC frequency (Fig. 9C; n = 7; p = 0.375; Wilcoxon matched pairs, W = 12) or change in cumulative interevent intervals distributions (n = 7; K-S, D = 0.002, p > 0.99) when Schaffer collaterals received PPS. Additionally, in a cohort of cells, we evaluated concomitant intracellular α-panx1 and bath ZD7288; PPS still evoked an increase in mean sEPSC frequency (Fig. 9C; n = 8; p = 0.008; Wilcoxon matched pairs, W = 36) and shifted cumulative interevent interval distributions (n = 8; K-S, D = 0.4405, p < 0.0001). Importantly, CPZ did not affect the basal rate of spontaneous events in TRPV1−/− mice compared with WT animals (WT frequency was 2.9 ± 0.5 Hz vs 3.9 ± 0.7 Hz in TRPV1−/− slices; p = 0.45, Mann–Whitney U test, n = 7 and n = 7, respectively), indicating that off-target effects were not detected. Additionally, TRPV1 KO mice did not show an effect of postsynaptic α-panx1 during stimulation (Fig. 9D; n = 8; p = 0.383; Wilcoxon matched pairs) and the cumulative interevent interval distribution was not changed (n = 9; K-S, D = 0.038, p = 0.447), even though WT mice had robust synaptic bursting (Fig. 3).
Panx1 regulates AEA concentration
The above data suggest that inhibiting Panx1 leads to TRPV1 channel opening and presynaptic glutamate release. Given that Panx1 channels are permeable to large metabolites (<1 kDa) (Wang et al., 2007), we tested whether postsynaptic Panx1 opening could buffer endogenous TRPV1 ligands in the synapse. AEA is an important endogenous agonist of TRPV1, and we hypothesized that Panx1 may regulate its concentration in the synapse so that when we block Panx1 AEA accumulates. As shown in Figure 10A, we quantified the tissue concentration of AEA using mass spectrometry (Qi et al., 2015) after whole-cell recordings and PPS. Basal AEA levels were 7.3 ± 0.8 pg/mg (n = 14 hippocampal slices from 6 animals). AEA levels were significantly increased in the presence of 10panx (11.9 ± 1.2 pg/mg, p = 0.002 vs control; one-way ANOVA, n = 13 slices from 6 animals) over basal levels (7.3 ± 0.8 pg/mg, n = 14 hippocampal slices from 6 animals), confirming a role for Panx1 in AEA buffering. It was reported previously that TRPV1 might regulate membrane transport of AEA (Hofmann et al., 2014). However, CPZ (10 μm) did not change tissue AEA levels (with CPZ, AEA = 7.4 ± 0.7 pg/mg; n = 8 slices from 4 animals; p > 0.05, one-way ANOVA). If AEA is the key TRPV1 ligand to trigger sEPSC bursts, inhibiting AEA degradation should resemble Panx1 block because of AEA accumulation. Bath application of the fatty acid amid hydrolase (FAAH) inhibitor, URB597 (1 μm) with PPS of the Schaffer collaterals (without intracellular α-panx1) caused a leftward shift in cumulative interevent interval distributions (n = 10, K-S, D = 0.558, p < 0.0001) and increased the average sEPSC frequency (Fig. 10B; n = 10; p = 0.009; Wilcoxon matched pairs W = 49). Together, we suggest that AEA acts as a ligand for TRPV1 and that Panx1 is regulating the tissue concentration of AEA during synaptic stimulation.
Figure 10.
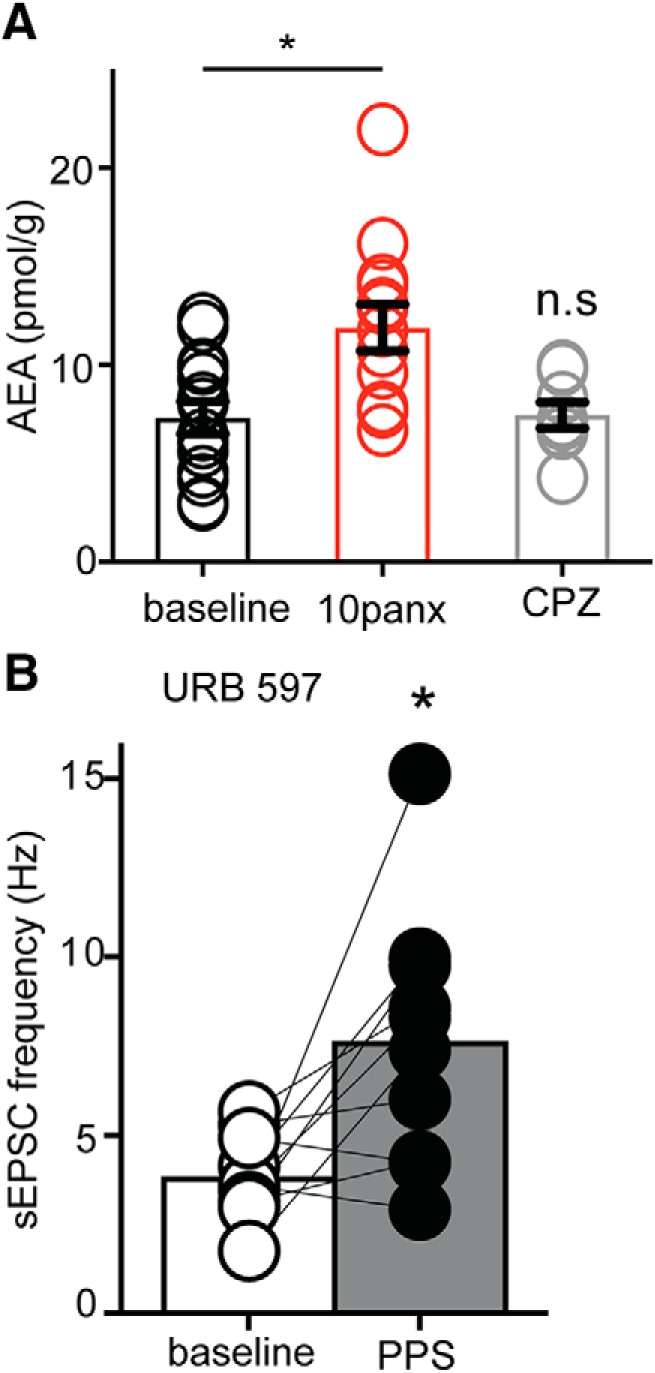
Panx1 regulates tissue levels of the TRPV1 agonist, AEA. A, Quantification of tissue (brain slice) concentrations of AEA by mass spectrometry. The bath-applied Panx1 blocker, 10panx (100 μm), increased AEA concentration, but the TRPV1 blocker, CPZ (10 μm), did not. B, The FAAH antagonist, URB 597 (1 μm), which blocks AEA metabolism, caused an increase in sEPSC frequency upon PPS delivery to Shaffer collaterals. αPanx1 was not included in the pipette. These data suggest that Panx1 regulates tissue AEA levels, which is a key step in activating presynaptic TRPV1 and causing increased sEPSC frequency. *p < 0.05 (Wilcoxon Matched pairs test). n.s., Not significant.
Discussion
Here we report that metabotropic NMDAR-Panx1 suppresses facilitated glutamate release during low-frequency synaptic stimulation because postsynaptic Panx1 maintains low levels of the endovanilloid, AEA, thus limiting activation of presynaptic TRPV1 channels. Panx1 block (or KO) increased the concentration of the TRPV1 agonist, AEA, in hippocampal slices. This increase in AEA activates presynaptic TRPV1 to induce intermittent, seconds-long glutamate release. This new role for Panx1 expands the roles of these ion/metabolite channels in synaptic activity. Our proposed model is presented in Figure 11, and detailed explanations are provided in the following discussion.
Figure 11.

A model of the proposed pathway for suppression of glutamate release by postsynaptic metabotropic NMDAR signaling to Panx1. Left, “Functional Panx1,” the scenario when Panx1 channels are operating normally. This physiological state is indicated as Steps 1–3 (boxed numbers) where the binding of glutamate and glycine/d-serine to NMDARs (Step 1) initiates Src-mediated phosphorylation of Panx1 (Step 2), which facilitates clearance of AEA (Step 3) for degradation by FAAH. Our evidence for this sequence is that blocking ligand binding to NMDRs (with d-APV+CGP; at Step 1) but not postsynaptic increases in Ca2+ (with BAPTA), or blocking Src activation of Panx1 (with TAT-Panx308 or PP2; at Step 2), or inhibiting Panx1 (with α-panx1, 10panx, or Panx1−/−; at Step 3) all augmented sEPSC frequency. Right, “Blocked Panx1,” the sequence of events occurring when Panx1 or metabotropic NMDARs have been blocked (Steps 4–6; dashed arrows). In the current study, direct block of Panx1 increases tissue AEA levels (Step 4), and we propose that AEA acts on presynaptic TRPV1 to increase Ca2+ (Step 5) and facilitate activity-dependent glutamate release (Step 6). Our evidence for this sequence is that facilitated glutamate release was prevented by TRPV1 block (with CPZ or in TRPV1−/−; at Step 4), or by bath-applied EGTA-AM (at Step 5). We also quantified TRPV1 expression in the hippocampus (PCR, qPCR) and found it at presynaptic sites (by immuno-EM) and propose that these are CR cells synapsing onto CA1 neurons because CR cells are the only confirmed site of TRPV1 expression.
NMDARs regulate Panx1 during synaptic activity
The first indication that NMDARs recruited Panx1 to modulate synaptic function came from our work with 0 Mg2+-induced interictal bursting, where blocking Panx1 decreased the frequency and amplitude of interictal bursts (Thompson et al., 2008). Others have reported that Panx1 modulates strength at the CA3-CA1 synapse because Panx1 KO augments LTP (Prochnow et al., 2012; Ardiles et al., 2014) and is suppressive of LTD (Ardiles et al., 2014). In the present study, we have found that Panx1 blockers (or KO) augment glutamate release for tens of seconds in response to low-frequency (0.05 Hz) stimulation of the Schaffer collateral pathway.
Our data support a model (Fig. 11) whereby, under physiological conditions, NMDARs regulate Panx1 activity via Src kinase through a similar pathway to that triggered during excitotoxicity (Weilinger et al., 2016) because, when we blocked Src with PP2, or prevented its action on Panx1 with TAT-Panx308, the facilitated glutamate release following Schaffer collateral stimulation was no longer observed. The excitotoxic activation of Panx1 by metabotropic NMDARs was prevented by blocking ligand binding at either the glutamate (with d-APV) or glycine (with CGP) sites. Interestingly, in the present study, we found that both competitive ligands (for the glutamate and glycine sites) were required to block facilitated glutamate release and that blocking either site alone was not effective. This is, to the best of our knowledge, the first description of simultaneous block of both ligand binding sites being required to prevent NMDAR signaling. In the first description of metabotropic NMDAR signaling, block of the glutamate site and not the glycine site prevented LTD (Nabavi et al., 2013). Thus, there appear to be several metabotropic NMDAR pathways, a notion that is supported by the presence of multiple stable conformational states of the closed receptors (Sasmal and Lu, 2014). We propose that the pathway regulating Panx1 activity to suppress facilitated glutamate release requires occupancy of both NMDAR ligands to promote subsequent Src activation.
Facilitated glutamate release may involve presynaptic unsilencing
Facilitated glutamate release, when Panx1 is blocked, may arise from postsynaptic unsilencing (Beique et al., 2006). However, we found no evidence for a change in the AMPAR:NMDAR ratio, which argues against postsynaptic unsilencing. Our findings are consistent with enhancement of presynaptic glutamate release upon Panx1 block and Schaffer collateral stimulation. The observed response is an asynchronous-like form of vesicle release because it required synaptic stimulation and persisted beyond the duration of the evoked responses. Asynchronous release can occur upon activation of VGCCs that are not closely associated with the exocytotic machinery, which has been demonstrated both experimentally (Fawley et al., 2016) and by modeling (Guerrier and Holcman, 2016, 2018). In our current study, we prevented facilitated release by EGTA-AM treatment of the slice, which is consistent with a Ca2+ source for exocytosis that is further from the synaptic vesicles than the VGCCs (Ohana and Sakmann, 1998; Fawley et al., 2016; Guerrier and Holcman, 2018). However, the duration of the increased sEPSC frequency that we observe (tens of seconds) is substantially longer than the duration of classic asynchronous release (milliseconds) (Kaeser and Regehr, 2014). Although speculative, we suggest that we were recruiting new synaptic elements (i.e., presynaptic unsilencing) instead of activating a Ca2+ influx pathway in the same synapses as the evoked EPSCs. Consistent with this is that no change in the PPR was evident, even when the bursts of sEPSCs overlapped with the eEPSCs. An alternative explanation to presynaptic unsilencing is that relatively few synapses are contributing to the change in sEPSC frequency so that their contribution to the PPR is minimal and statistically undetected.
Presynaptic TRPV1 mediates the facilitated glutamate release
We propose that the facilitated glutamate release observed during Panx1 block and arises from activation of presynaptic TRPV1 (Fig. 11). We detected TRPV1 mRNA in hippocampal slices, observed TRPV1 protein in presynaptic compartments by immuno-EM, prevented Panx1 block/synaptic stimulation induced facilitated glutamate release with the TRPV1 blocker, CPZ, and did not detect increased sEPSC frequency in TRPV1−/− mice. There are numerous reports of functional TRPV1 in the hippocampus (Gibson et al., 2008; Chávez et al., 2010, 2014; Jensen and Edwards, 2012) and a unique splice variant in the hypothalamus (Sharif Naeini et al., 2006). Indeed, in the original systematic mapping of brain TRPV1, it was reported that the vanilloid activated channels are expressed in hippocampus, but only in CR cells (Cavanaugh et al., 2011). CR cells are reelin expressing glutamatergic neurons that make synaptic contacts with interneurons in the molecular layer and pyramidal neurons in the CA1 region (Anstötz et al., 2016). Recently, it was shown that TRPV1 is functional in CR neurons and capsaicin-activation of the channels augmented sEPSC frequency (Anstötz et al., 2018). Thus, it will be of interest to determine whether Panx1 block is leading to activation of TRPV1 in CR to CA1 synapses (Fig. 11) because this would be consistent with a presynaptic unsilencing mechanism. It may also reveal whether there are currently unidentified physiological roles for Panx1's suppression of facilitated glutamate release because CR cells are a component of an intrahippocampal microcircuit (Anstötz et al., 2018).
Capsaicin perfusion onto hippocampal slices led to a persistent augmentation of glutamate release from CR neurons onto interneurons (Anstötz et al., 2018). In contrast, in afferents of the nucleus tractus solitaris, or microglia in the anterior cingulate cortex, TRPV1 activation induces transient (tens of milliseconds) asynchronous glutamate release (Peters et al., 2010; Marrone et al., 2017). So why is the facilitated glutamate release that we observe when Panx1 is blocked intermittent and seconds long instead of very brief (milliseconds) or persistent? It may reflect a sustained increase in TRPV1 ligand (i.e., AEA) concentration when Panx1 is blocked, which could arise from high local (i.e., synaptic) levels or diffusion out of the synapse. While we have focused on AEA in this paper, it is possible that the effective ligand is another endovanilloid. It has been reported that N-aracidonyl-dopamine is a potent ligand for TRPV1 (De Petrocellis et al., 2004); and interestingly, brief application of N-aracidonyl-dopamine can induce prolonged TRPV1 opening and glutamate release lasting tens of seconds (Medvedeva et al., 2008), similar to that reported here.
Several possible mechanisms could account for termination of the augmented sEPSC frequency that we report here. Depletion of the vesicular pool is likely explanation considering the very high instantaneous frequency rates that are reached. Vesicle refilling and availability of AEA may determine when subsequent bursts occur, accounting for the intermittent nature of the response. Other possibilities (that are not mutually exclusive) are TRPV1 endocytosis (Sanz-Salvador et al., 2012) or desensitization (Numazaki et al., 2003).
Panx1 involvement in AEA clearance
The mechanisms that regulate synaptic levels of AEA and other endovanilloids (i.e., transport and clearance) are not well understood (Hillard and Jarrahian, 2005). Our findings suggest that AEA removal from the synapse is facilitated by Panx1 because blocking the channel increased total AEA concentration, presumably by preventing access of AEA to the intracellular enzyme, FAAH, that degrades it (Blankman and Cravatt, 2013). Inhibition of FAAH had a similar effect to blocking Panx1 on sEPSC frequency, indicating that blocking AEA degradation is sufficient to cause Shaffer collateral stimulation-induced bursting. It is notable that, during the revision of this manuscript, a paper from the Fowler group investigated the putative role of Panx1 in AEA transport in human-derived T84 colon cancer cell lines (Alhouayek et al., 2019). While they report no obvious role for Panx1, their experiments were conducted under very different timescales compared with ours, and the T84 cells lack a synaptic structure to mimic that seen in brain slices. Our data published on the BioRxiv preprint server (Bialecki et al., 2018) show that Panx1 expression is directly linked to augmented AEA clearance in hippocampal slices and uptake when ectopically expressed in HEK293T cells. We are now determining whether Panx1 facilitates AEA clearance from brain slices with focus on several possibilities, such as direct transport of AEA by Panx1 or Panx1 regulation of other putative transporters (Hillard et al., 1997; Hillard and Jarrahian, 2005; Fowler, 2013).
In conclusion, we have shown here that NMDARs signal to Panx1 through Src kinase to facilitate uptake of AEA from brain tissue and that, when we block Panx1, AEA levels increase to a level sufficient to activate presynaptic TRPV1 and augment glutamate release. We propose that these TRPV1 channels are expressed in CR cells, the only confirmed site of functional TRPV1 expression (i.e., through measurement of ionic currents) in the hippocampus. Although our work has not yet identified a physiological role of Panx1's regulation of AEA and TRPV1 activity, it is possible that it plays a role in regulation of CR-CA1 excitatory synapses.
Footnotes
This work was supported by Canadian Institutes of Health Research Grant FDN 143329 to M.N.H. and Grant MOP 136812 to R.J.T.; Natural Sciences and Engineering Research Council of Canada to R.J.T.; the Cumming School of Medicine via the Ronald and Irene Ward Foundation, the Gwendolyn McLean Fund, and the Hotchkiss Brain Institute to R.J.T.; and MINECO/FEDER, UE SAF2015-65034-R and The Basque Government BCG IT764-13 to P.G. M.N.H. holds a Canada Research Chair Tier 2.
The authors declare no competing financial interests.
References
- Alhouayek M, Sorti R, Gilthorpe JD, Fowler CJ (2019) Role of pannexin-1 in the cellular uptake, release and hydrolysis of anandamide by T84 colon cancer cells. Sci Rep 9:7622. 10.1038/s41598-019-44057-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstötz M, Huang H, Marchionni I, Haumann I, Maccaferri G, Lübke JH (2016) Developmental profile, morphology, and synaptic connectivity of Cajal-Retzius cells in the postnatal mouse hippocampus. Cereb Cortex 26:855–872. 10.1093/cercor/bhv271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstötz M, Lee SK, Maccaferri G (2018) Expression of TRPV1 channels by Cajal-Retzius cells and layer-specific modulation of synaptic transmission by capsaicin in the mouse hippocampus. J Physiol 596:3739–3758. 10.1113/JP275685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardiles AO, Flores-Muñoz C, Toro-Ayala G, Cárdenas AM, Palacios AG, Muñoz P, Fuenzalida M, Sáez JC, Martínez AD (2014) Pannexin 1 regulates bidirectional hippocampal synaptic plasticity in adult mice. Front Cell Neurosci 8:326. 10.3389/fncel.2014.00326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, Penuela S, von Engelhardt J, Monyer H, Schwaninger M (2011) Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci U S A U A 108:20772–20777. 10.1073/pnas.1018262108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL (2006) Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci U S A 103:19535–19540. 10.1073/pnas.0608492103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialecki J, Weilinger NL, Lohman AW, Vecchiarelli HA, Robinson JHB, Egaña J, Medizabal-Zubiaga J, Werner AC, Grandes P, Teskey GC, Hill MN, Thompson RJ (2018) Postsynaptic pannexin-1 facilitates anandamide uptake to modulate glutamate release and enhance network excitability. bioRxiv. 263640 10.1101/26364 [DOI] [Google Scholar]
- Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, Wang E, Ruiz G, De Groat WC, Apodaca G, Watkins S, Caterina MJ (2002) Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat Neurosci 5:856–860. 10.1038/nn902 [DOI] [PubMed] [Google Scholar]
- Blankman JL, Cravatt BF (2013) Chemical probes of endocannabinoid metabolism. Pharmacol Rev 65:849–871. 10.1124/pr.112.006387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Hormuzdi SG, Barbe MT, Herb A, Monyer H (2003) Pannexins, a family of gap junction proteins expressed in brain. Proc Natl Acad Sci U S A 100:13644–13649. 10.1073/pnas.2233464100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canduela MJ, Mendizabal-Zubiaga J, Puente N, Reguero L, Elezgarai I, Ramos-Uriarte A, Gerrikagoitia I, Grandes P (2015) Visualization by high resolution immunoelectron microscopy of the transient receptor potential vanilloid-1 at inhibitory synapses of the mouse dentate gyrus. PLoS One 10:e0119401. 10.1371/journal.pone.0119401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O'Donnell D, Nicoll RA, Shah NM, Julius D, Basbaum AI (2011) Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci 31:5067–5077. 10.1523/JNEUROSCI.6451-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Chiu CQ, Castillo PE (2010) TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci 13:1511–1518. 10.1038/nn.2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Hernández VM, Rodenas-Ruano A, Chan CS, Castillo PE (2014) Compartment-specific modulation of GABAergic synaptic transmission by TRPV1 channels in the dentate gyrus. J Neurosci 34:16621–16629. 10.1523/JNEUROSCI.3635-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS (2010) Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature 467:863–867. 10.1038/nature09413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLalio LJ, Billaud M, Ruddiman CA, Johnstone SR, Butcher JT, Wolpe AG, Jin X, Keller TC, Keller AS, Rivière T, Good ME, Best AK, Lohman AW, Swayne LA, Penuela S, Thompson RJ, Lampe PD, Yeager M, Isakson BE (2019) Constitutive SRC-mediated phosphorylation of pannexin 1 at tyrosine 198 occurs at the plasma membrane. J Biol Chem 294:6940–6956. 10.1074/jbc.RA118.006982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Chu CJ, Moriello AS, Kellner JC, Walker JM, Di Marzo V (2004) Actions of two naturally occurring saturated N-acyldopamines on transient receptor potential vanilloid 1 (TRPV1) channels. Br J Pharmacol 143:251–256. 10.1038/sj.bjp.0705924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore K, Aow J, Malinow R (2015) Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc Natl Acad Sci U S A 112:14705–14710. 10.1073/pnas.1520023112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawley JA, Hofmann ME, Andresen MC (2016) Distinct calcium sources support multiple modes of synaptic release from cranial sensory afferents. J Neurosci 36:8957–8966. 10.1523/JNEUROSCI.1028-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ. (2013) Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J 280:1895–1904. 10.1111/febs.12212 [DOI] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA (2008) TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron 57:746–759. 10.1016/j.neuron.2007.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier C, Holcman D (2016) Hybrid Markov-mass action law model for cell activation by rare binding events: application to calcium induced vesicular release at neuronal synapses. Sci Rep 6:35506. 10.1038/srep35506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier C, Holcman D (2018) The first 100 nm inside the pre-synaptic terminal where calcium diffusion triggers vesicular release. Front Synaptic Neurosci 10:23. 10.3389/fnsyn.2018.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA, Muruve DA, McKay DM, Beck PL, Mawe GM, Thompson RJ, Sharkey KA (2012) Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med 18:600–604. 10.1038/nm.2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A (2005) Accumulation of anandamide: evidence for cellular diversity. Neuropharmacology 48:1072–1078. 10.1016/j.neuropharm.2004.12.012 [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB (1997) Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem 69:631–638. 10.1046/j.1471-4159.1997.69020631.x [DOI] [PubMed] [Google Scholar]
- Hofmann NA, Barth S, Waldeck-Weiermair M, Klec C, Strunk D, Malli R, Graier WF (2014) TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol Open 3:1164–1172. 10.1242/bio.20149571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen T, Edwards JG (2012) Calcineurin is required for TRPV1-induced long-term depression of hippocampal interneurons. Neurosci Lett 510:82–87. 10.1016/j.neulet.2012.01.006 [DOI] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A (2007) Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci 10:331–339. 10.1038/nn1849 [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG (2014) Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol 76:333–363. 10.1146/annurev-physiol-021113-170338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE (2010) Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci 13:592–600. 10.1038/nn.2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Ledri M, Tóth B, Marchionni I, Henstridge CM, Dudok B, Kenesei K, Barna L, Szabó SI, Renkecz T, Oberoi M, Watanabe M, Limoli CL, Horvai G, Soltesz I, Katona I (2015) Multiple forms of endocannabinoid and endovanilloid signaling regulate the tonic control of GABA release. J Neurosci 35:10039–10057. 10.1523/JNEUROSCI.4112-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman AW, Leskov IL, Butcher JT, Johnstone SR, Stokes TA, Begandt D, DeLalio LJ, Best AK, Penuela S, Leitinger N, Ravichandran KS, Stokes KY, Isakson BE (2015) Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat Commun 6:7965. 10.1038/ncomms8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone MC, Morabito A, Giustizieri M, Chiurchiù V, Leuti A, Mattioli M, Marinelli S, Riganti L, Lombardi M, Murana E, Totaro A, Piomelli D, Ragozzino D, Oddi S, Maccarrone M, Verderio C, Marinelli S (2017) TRPV1 channels are critical brain inflammation detectors and neuropathic pain biomarkers in mice. Nat Commun 8:15292. 10.1038/ncomms15292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva YV, Kim MS, Usachev YM (2008) Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J Neurosci 28:5295–5311. 10.1523/JNEUROSCI.4810-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R (2013) Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci U S A 110:4027–4032. 10.1073/pnas.1219454110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Takeuchi K, Murayama N, Toyooka H, Tominaga M (2003) Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc Natl Acad Sci U S A 100:8002–8006. 10.1073/pnas.1337252100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana O, Sakmann B (1998) Transmitter release modulation in nerve terminals of rat neocortical pyramidal cells by intracellular calcium buffers. J Physiol 513:135–148. 10.1111/j.1469-7793.1998.135by.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Smith SM, Andresen MC (2010) Primary afferent activation of thermosensitive TRPV1 triggers asynchronous glutamate release at central neurons. Neuron 65:657–669. 10.1016/j.neuron.2010.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochnow N, Abdulazim A, Kurtenbach S, Wildförster V, Dvoriantchikova G, Hanske J, Petrasch-Parwez E, Shestopalov VI, Dermietzel R, Manahan-Vaughan D, Zoidl G (2012) Pannexin1 stabilizes synaptic plasticity and is needed for learning. PLoS One 7:e51767. 10.1371/journal.pone.0051767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente N, Reguero L, Elezgarai I, Canduela MJ, Mendizabal-Zubiaga J, Ramos-Uriarte A, Fernández-Espejo E, Grandes P (2015) The transient receptor potential vanilloid-1 is localized at excitatory synapses in the mouse dentate gyrus. Brain Struct Funct 220:1187–1194. 10.1007/s00429-014-0711-2 [DOI] [PubMed] [Google Scholar]
- Qi M, Morena M, Vecchiarelli HA, Hill MN, Schriemer DC (2015) A robust capillary liquid chromatography/tandem mass spectrometry method for quantitation of neuromodulatory endocannabinoids. Rapid Commun Mass Spectrom 29:1889–1897. 10.1002/rcm.7277 [DOI] [PubMed] [Google Scholar]
- Ray AM, Benham CD, Roberts JC, Gill CH, Lanneau C, Gitterman DP, Harries M, Davis JB, Davies CH (2003) Capsazepine protects against neuronal injury caused by oxygen glucose deprivation by inhibiting I(h). J Neurosci 23:10146–10153. 10.1523/JNEUROSCI.23-31-10146.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5:317–328. 10.1038/nrn1368 [DOI] [PubMed] [Google Scholar]
- Sanz-Salvador L, Andrés-Borderia A, Ferrer-Montiel A, Planells-Cases R (2012) Agonist- and Ca2+-dependent desensitization of TRPV1 channel targets the receptor to lysosomes for degradation. J Biol Chem 287:19462–19471. 10.1074/jbc.M111.289751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasmal DK, Lu HP (2014) Single-molecule patch-clamp FRET microscopy studies of NMDA receptor ion channel dynamics in living cells: revealing the multiple conformational states associated with a channel at its electrical off state. J Am Chem Soc 136:12998–13005. 10.1021/ja506231j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif Naeini R, Witty MF, Séguéla P, Bourque CW (2006) An N-terminal variant of Trpv1 channel is required for osmosensory transduction. Nat Neurosci 9:93–98. 10.1038/nn1614 [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Zhou N, MacVicar BA (2006) Ischemia opens neuronal gap junction hemichannels. Science 312:924–927. 10.1126/science.1126241 [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, MacDonald JF, MacVicar BA (2008) Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 322:1555–1559. 10.1126/science.1165209 [DOI] [PubMed] [Google Scholar]
- Vogt A, Hormuzdi SG, Monyer H (2005) Pannexin1 and Pannexin2 expression in the developing and mature rat brain. Brain Res Mol Brain Res 141:113–120. 10.1016/j.molbrainres.2005.08.002 [DOI] [PubMed] [Google Scholar]
- Wang J, Ma M, Locovei S, Keane RW, Dahl G (2007) Modulation of membrane channel currents by gap junction protein mimetic peptides: size matters. Am J Physiol Cell Physiol 293:C1112–C1119. 10.1152/ajpcell.00097.2007 [DOI] [PubMed] [Google Scholar]
- Weilinger NL, Tang PL, Thompson RJ (2012) Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J Neurosci 32:12579–12588. 10.1523/JNEUROSCI.1267-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilinger NL, Lohman AW, Rakai BD, Ma EM, Bialecki J, Maslieieva V, Rilea T, Bandet MV, Ikuta NT, Scott L, Colicos MA, Teskey GC, Winship IR, Thompson RJ (2016) Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat Neurosci 19:432–442. 10.1038/nn.4236 [DOI] [PubMed] [Google Scholar]
- Yang K, Trepanier C, Sidhu B, Xie YF, Li H, Lei G, Salter MW, Orser BA, Nakazawa T, Yamamoto T, Jackson MF, MacDonald JF (2012) Metaplasticity gated through differential regulation of GluN2A versus GluN2B receptors by Src family kinases: metaplasticity gated through differential regulation of NMDA receptors. EMBO J 31:805–816. 10.1038/emboj.2011.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil GJ 2nd, Salter MW (1997) NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275:674–678. 10.1126/science.275.5300.674 [DOI] [PubMed] [Google Scholar]
- Zoidl G, Petrasch-Parwez E, Ray A, Meier C, Bunse S, Habbes HW, Dahl G, Dermietzel R (2007) Localization of the pannexin1 protein at postsynaptic sites in the cerebral cortex and hippocampus. Neuroscience 146:9–16. 10.1016/j.neuroscience.2007.01.061 [DOI] [PubMed] [Google Scholar]