

Abstract
The ventral pallidum (VP) is a key node in the neural circuits controlling relapse to drug seeking. How this role relates to different VP cell types and their projections is poorly understood. Using male rats, we show how different forms of relapse to alcohol-seeking are assembled from VP cell types and their projections to lateral hypothalamus (LH) and ventral tegmental area (VTA). Using RNAScope in situ hybridization to characterize activity of different VP cell types during relapse to alcohol-seeking provoked by renewal (context-induced reinstatement), we found that VP Gad1 and parvalbumin (PV), but not vGlut2, neurons show relapse-associated changes in c-Fos expression. Next, we used retrograde tracing, chemogenetic, and electrophysiological approaches to study the roles of VPGad1 and VPPV neurons in relapse. We show that VPGad1 neurons contribute to contextual control over relapse (renewal), but not to relapse during reacquisition, via projections to LH, where they converge with ventral striatal inputs onto LHGad1 neurons. This convergence of striatopallidal inputs at the level of individual LHGad1 neurons may be critical to balancing propensity for relapse versus abstinence. In contrast, VPPV neurons contribute to relapse during both renewal and reacquisition via projections to VTA. These findings identify a double dissociation in the roles for different VP cell types and their projections in relapse. VPGad1 neurons control relapse during renewal via projections to LH. VPPV neurons control relapse during both renewal and reacquisition via projections to VTA. Targeting these different pathways may provide tailored interventions for different forms of relapse.
SIGNIFICANCE STATEMENT Relapse to drug or reward seeking after a period of extinction or abstinence remains a key impediment to successful treatment. The ventral pallidum, located in the ventral basal ganglia, has long been recognized as an obligatory node in a 'final common pathway' for relapse. Yet how this role relates to the considerable VP cellular and circuit heterogeneity is not well understood. We studied the cellular and circuit architecture for VP in relapse control. We show that different forms of relapse have complementary VP cellular and circuit architectures, raising the possibility that targeting these different neural architectures may provide tailored interventions for different forms of relapse.
Keywords: extinction, reacquisition, relapse, renewal, ventral pallidum
Introduction
Drug use contributes significantly to the burden of disease (Whiteford et al., 2013). A treatment goal is promoting abstinence but a consistent propensity to relapse impedes this (Jonas et al., 2014). The ventral pallidum (VP) is a key brain region controlling relapse. VP, together with ventral striatum, forms the ventral striatopallidal system (Heimer et al., 1982; de Olmos and Heimer, 1999). Although initially viewed as controlling motor output (Mogenson et al., 1980), the ventral striatopallidal system has broad functions in motivation including evaluating hedonic impact of rewards and mediating the effects of reward predictive cues on decision making (Smith and Berridge, 2005; Smith et al., 2009; Ho and Berridge, 2013; Leung and Balleine, 2013, 2015; Castro et al., 2015; Root et al., 2015; Richard et al., 2016). Lesion and pharmacological studies (McFarland and Kalivas, 2001; McFarland et al., 2004; Tang et al., 2005; Perry and McNally, 2013), circuit-specific manipulations (Stefanik et al., 2013; Mahler et al., 2014; Heinsbroek et al., 2017; Farrell et al., 2019), as well as human lesion (Moussawi et al., 2016) studies identify VP as critical for relapse to drug seeking. This role is shared across different forms of relapse (cue, stress-, context-, and drug-priming reinstatement) to seeking a variety of drugs of abuse (alcohol, psychostimulants, opiates). Hence, VP is an obligatory node in a final common neural circuit for relapse to drug seeking (Kalivas and O'Brien, 2008).
However, the cell-type heterogeneity of VP hinders understanding its role in relapse. VP comprises a variety of different cell types (glutamatergic, GABAergic, and cholinergic) that express different neuropeptides (Root et al., 2015) with diverse electrophysiological characteristics (Napier and Mitrovic, 1999; Root et al., 2010; Kupchik and Kalivas, 2013; Avila and Lin, 2014). Further, VP neurons form multiple output pathways, including to lateral hypothalamus (LH), subthalamic nucleus, lateral habenula, and VTA as well as prefrontal cortex, amygdala, and nucleus accumbens (Haber et al., 1985; Zahm et al., 1987; Root et al., 2015). Recent work has begun to show how this cell-type and projection heterogeneity relates to different behavioral states (Knowland et al., 2017; Faget et al., 2018; Tooley et al., 2018). Yet, how the role for VP in relapse relates to this cell-type and projection heterogeneity remains poorly understood. Here we used an animal model of relapse to address this.
We used renewal (context-induced reinstatement; Crombag and Shaham, 2002; Crombag et al., 2008; Khoo et al., 2017) and reacquisition (Willcocks and McNally, 2011) models of relapse to alcohol-seeking to study the roles of major VP cell populations in relapse. First, we used RNAScope in situ hybridization for c-Fos and Gad1, parvalbumin (PV), or vGlut2 mRNA to assess recruitment of VP cell populations during renewal. Next, we used retrograde tracing, chemogenetic, and electrophysiological approaches to study the causal roles of VPGad1 and VPPV neurons and their projections to the LH and ventral tegmental area (VTA) in relapse. We show that VPGad1 neurons control relapse during renewal via projections to LH, but not VTA, whereas VPPV neurons control relapse during both renewal and reacquisition via projections to VTA, but not LH.
Materials and Methods
Subjects
Subjects were adult male Sprague-Dawley (Animal Resources Centre, Perth, Australia), LE-Tg(Gad1-iCre)3Ottc and LE-Tg(PV-iCre)3Ottc (Optogenetics and Transgenic Technology Core; obtained via Rat Research Resource Centre RRRC#751, RRRC#773). They were housed in ventilated racks, in groups of four, on corn-cob bedding in a climate-controlled colony room maintained on 12 h light/dark cycle (07:00 lights on). Rats in behavioral studies had ad libitum access to food (Gordon's Rat Chow) and water until 2 d before commencement of behavioral training when they received 1 h of access to food and water each day for the remainder of the experiment. All subjects were randomly allocated to experimental conditions. All studies were performed in accordance with the Animal Research Act 1985 (NSW), under the guidelines of the National Health and Medical Research Council Code for the Care and Use of Animals for Scientific Purposes in Australia (2013). The UNSW Animal Care and Ethics Committee approved all procedures.
Surgeries and injections
Stereotaxic surgery was done as described previously (McNally, 2005). Briefly, Rats were anesthetized via intraperitoneal injection with a mixture of 1.3 ml/kg ketamine anesthetic (Ketamil, Troy Laboratories) at a concentration of 100 mg/ml and 0.3 ml/kg of the muscle relaxant xylazine (Xylazil, Troy Laboratories) at a concentration of 20 mg/ml. Rats received a subcutaneous injection of 0.1 ml 50 mg/ml carprofen (Pfizer) before being placed in the stereotaxic frame (Kopf Instruments). They received stereotaxic surgery using the following flat skull coordinates relative to bregma [AP, ML, DV (in mm): VP AAV +0.00, ±2.50, −8.50; LH AAV and tracing −2.60, ±1.80, −8.60; LH optic fibers −2.60, ±3.60, −8.80 (10° angle); AcbSh AAV +1.28, ±0.70, −8.20; VTA AAV −5.80, ±2.25, −8.30 (10° angle)].
Vectors (500–750 nl) and tracers (50 nl) were infused with a 23-gauge, cone-tipped 5 μl stainless steel injector (SGE Analytical Science) over 3 min using an infusion pump (UMP3 with SYS4 Micro-controller, World Precision Instruments). The needle was left in place for 7 min to allow for diffusion and reduce spread up the infusion tract. At the end of surgery, rats received intramuscular injection of 0.2 ml of 150 mg/ml solution of procaine penicillin (Benacillin, Troy Laboratories) and 0.2 ml of 100 mg/ml cephazolin sodium (AFT Pharmaceuticals). All rats were monitored daily for weight and/or behavioral changes.
Viral vectors
pAAV-hSyn-DIO-hM4D(Gi)-mCherry was a gift from Bryan Roth (Addgene viral prep #44362-AAV5; http://n2t.net/addgene:44362; RRID:Addgene_44362; 5.4 × 10e12 vp/ml). pAAV-hSyn-hM4D(Gi)-mCherry was a gift from Bryan Roth (University of North Carolina) (Addgene viral prep #50475-AAV5; http://n2t.net/addgene:50475; RRID:Addgene_50475; 4.1 × 10e12 vp/ml). pAAV-Syn-ChrimsonR-tdT was a gift from Edward Boyden Massachusetts Institute of Technology (Addgene viral prep #59171-AAV5; http://n2t.net/addgene:59171; RRID:Addgene_59171; 2.9 × 10e12 vp/ml). pAAV-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-HGHpA was a gift from Karl Deisseroth (Addgene viral prep #20298-AAV5; http://n2t.net/addgene:20298; RRID:Addgene_20298; 7.4 × 10e12 vp/ml). AAV5-hSyn-DIO-ChR2(H134R)-eYFP AAV5-hSyn-eYFP (4.9 × 10e12 vp/ml), and AAV5-hSyn-DIO-eYFP (4.1 × 10e12 vp/ml) were obtained from UNC Vector core from plasmids as gifts from Karl Deisseroth (Stanford University).
Apparatus
Standard rat operant chambers (ENV-008; Med Associates) were used for all behavioral procedures. The chambers contained two nose-poke holes symmetrically located on one sidewall of the chamber, 3 cm above a grid floor. A recessed magazine was located behind a 4 × 4 cm opening in the center of the same wall between the two nose-pokes. Responding on one (active) nose-poke extinguished the cue light in the nose-poke and triggered a syringe pump to deliver alcoholic beer to the magazine during acquisition training, whereas responding on the other (inactive) nose-poke had no programmed consequences. A computer running MedPC-IV software (Med Associates) controlled all events. Suspended above each chamber was an LED plus fiber-optic rotary joint and LED driver (Doric Lenses) controlled by MedPC-IV. The eight self-administration chambers were divided into two groups of four to serve as distinct contexts for experiments with context as a factor. These chambers differed in their olfactory (rose vs peppermint essence), tactile (grid vs Perspex flooring), and visual (light on vs off) properties. These two contexts were fully counterbalanced to serve as the training (Context A) and extinction (Context B) contexts. Fiber optic cannulae and patch cables were fabricated from 0.39 NA, Ø400 μm core multimode optical fiber and ceramic ferrules (Thorlabs). Plexiglas chambers (Med Associates) with dimensions 43.2 cm (width) × 43.2 cm (length) × 30.5 cm (height) were used for locomotor assessment. Movement was tracked with three 16 beam infrared arrays. Infrared beams were located on both the x- and y-axes for positional tracking.
Behavioral procedures
General behavioral testing procedures.
On the first 2 d, the animals received 20 min magazine training sessions in both Context A and Context B. During these sessions, there were 10 noncontingent deliveries of 0.6 ml of the reward (4% alcohol (v/v) de-carbonated beer; Coopers Brewing) at time intervals variable around a mean of 1.2 min. On the next 10 d rats received self-administration training in Context A for 1 h per day. Responding on the active nose-poke extinguished the nose-poke cue light and triggered delivery via syringe pump of 0.6 ml alcoholic beer to the magazine on an FR-1 schedule followed by a 24 s timeout. Responses on the inactive nose-poke were recorded but had no consequences. On the next 4 d, rats received extinction training in Context B for 1 h per day. During this training, responses on the active nose-poke extinguished the cue light and triggered the pump but no beer was delivered. All rats were tested starting 24 h after the last extinction session. We selected these procedures based on our past work that has shown robust context-induced reinstatement and reacquisition under these conditions (Hamlin et al., 2006, 2007, 2008; Gibson et al., 2018).
For renewal, rats were tested twice, once in the extinction context (extinction, Context B) and once in the training context (reinstatement, Context A). Tests lasted 60 min. The order of tests was counterbalanced and tests were identical to self-administration except that the syringe pump was empty. Twenty-four hours after extinction/reinstatement test, rats were tested for the reacquisition of alcohol-seeking in a single session in the training context. Tests lasted 60 min. For chemogenetic manipulation, rats received an intraperitoneal injection 0.1 mg/kg of clozapine (#C6305, Sigma-Aldrich; 1 ml/kg diluted, 5% DMSO and saline) 30 min before tests.
Profile of VP subpopulations during renewal.
There were three groups; Groups AB0 (n = 4), ABB (n = 6), and ABA (n = 6), All rats were trained to respond for alcoholic beer in Context A and extinguished in Context B. Rats were tested once, for 1 h, 24 h after the last extinction session. Group ABB was tested in the extinction context. Group ABA was tested in the training context. Group AB0 was not tested; instead they were simply transported to the laboratory on test and received an equivalent period of handling. This group served as a control for the behavioral and pharmacological training histories. All rats were killed 30 min after test (or at the equivalent time for Group AB0). Their brains were later processed for expression of the cFos mRNA with GAD1, vGlut2 or PV using RNAscope technology. Images from three sets from the VP were obtained from each brain; rostral, medial, and caudal VP. Images were analyzed for single-labeled and double-labeled cells using Adobe Photoshop.
Chemogenetic inhibition of VPGAD1 neurons on renewal, reacquisition and locomotor activity.
There were two groups, VPGAD1 Cre− (n = 8) and VPGAD1 Cre+ (n = 7). Both groups received AAV5-hSyn-DIO-hM4D(Gi)-mCherry, bilaterally in the VP. Rats were trained and extinguished as described. Testing commenced 24 h after extinction. Rats were tested for 1 h in the extinction context (ABB) and for 1 h in the training context (ABA) for expression of extinction and renewal (context-induced reinstatement), respectively. These tests were separated by 24 h and the order of tests was counterbalanced within-subjects. Rats were tested 24 h later for 1 h reacquisition of alcoholic beer seeking in the training context. Our past research has shown no impact of the prior order of ABA and ABB testing on responding during reacquisition.
Chemogenetic inhibition of VPPV neurons on renewal, reacquisition, and locomotor activity.
There were two groups, VPPV Cre− (n = 8) and VPPV Cre+ (n = 7). Both groups received AAV5-hSyn-DIO-hM4D(Gi)-mCherry. All rats were then trained, extinguished, and tested for expression of extinction (ABB), renewal (ABA), reacquisition, and locomotor test as described in the general behavioral testing procedures.
VP output pathways recruited during renewal of alcohol seeking.
To assess recruitment of the VP→LH pathway during renewal we applied the retrograde tracers cholera toxin b subunit (CTb)-488 (ThermoFisher Scientific, catalog #C34775) and CTb-555 (ThermoFisher Scientific, catalog #C34776; counterbalanced) into LH. There were three groups: Group ABB (n = 6; tested in the extinction context), Group ABA (n = 5; tested in the training context), and Group AB0 (n = 3; transported to the laboratory on test, handled, but not tested). The behavioral procedure was the same as above, except all rats were perfused 1 h after the 1 h test (or at the equivalent time for Group AB0). Their brains were later processed for immunohistochemistry for c-Fos protein and CTb tracer.
Chemogenetic disconnection of VPGAD1–LH pathway on renewal, reacquisition, and locomotor activity.
There were three groups: VPGad1 Cre− (n = 8) contralateral disconnection, VPGad1 Cre+ (n = 7) ipsilateral disconnection, and VPGad1 Cre+ (n = 8) contralateral disconnection. All groups received cre-dependent AAV5-hSyn-DIO-hM4D(Gi)-mCherry in one VP and AAV5-hSyn-hM4D(Gi)-mCherry in the ipsilateral or contralateral LH. Hemispheres were counterbalanced across animals. All rats were trained, extinguished, and tested for expression of extinction (ABB), renewal (ABA), reacquisition, and locomotor test as described in the general behavioral testing procedures.
Chemogenetic disconnection of VPGAD1-VTA pathway on renewal, reacquisition, and locomotor activity.
There were two groups, VPGad1 Cre− (n = 7) contralateral disconnection and VPGad1 Cre+ (n = 7) contralateral disconnection. All groups received cre-dependent AAV5-hSyn-DIO-hM4D(Gi)-mCherry in one VP and AAV5-hSyn-hM4D(Gi)-mCitrine in the contralateral VTA. Hemispheres were counterbalanced across animals. All rats were trained, extinguished, and tested for expression of extinction (ABB), renewal (ABA), reacquisition, and locomotor test as described in the general behavioral testing procedures.
Chemogenetic disconnection of VPPV–LH on renewal, reacquisition, and locomotor activity.
There were three groups, VPPV Cre− (n = 6) contralateral disconnection, VPPV Cre+ (n = 7) ipsilateral disconnection, and VPPV Cre+ (n = 7) contralateral disconnection. All groups received AAV5-hSyn-DIO-hM4D(Gi)-mCherry in the VP and AAV5-hSyn-hM4D(Gi)-mCherry in the contralateral or ipsilateral LH, counterbalanced across animals. All rats were trained, extinguished, and tested for expression of extinction (ABB), renewal (ABA), reacquisition, and locomotor activity as described in the general behavioral testing procedures.
Chemogenetic disconnection of VPPV–VTA on renewal, reacquisition, and locomotor activity.
There were three groups, VPPV Cre− (n = 7) contralateral disconnection, VPPV Cre+ (n = 6) ipsilateral disconnection and VPPV Cre+ (n = 9) contralateral disconnection. All groups received AAV5-hSyn-DIO-hM4D(Gi)-mCherry in the VP and AAV5-hSyn-hM4D(Gi)-mCherry in the ipsilateral or contralateral VTA. All rats were trained, extinguished, and tested for expression of extinction (ABB), renewal (ABA), reacquisition, and locomotor activity as described in the general behavioral testing procedures.
Electrophysiology
Brain slices were prepared from Gad1-cre+ rats that had received AAV5-DIO-eYFP to LH, AAV5-hSyn-Chrimson-tdTomato to AcbSh, and AAV5-DIO-hChR2(H134R)-eYFP to VP at least 6 weeks before slice preparation. Rats were deeply anesthetized with Isoflurane (5%), decapitated and their brain rapidly removed and submerged in ice-cold oxygenated (95% O2, 5% CO2) HEPES based artificial CSF [HEPES-aCSF; (in mm) 95 NaCl, 2.5 KCl, 30 NaHCO3, 1.2 NaH2PO4, 20 HEPES, 25 glucose, 5 ascorbate, 2 thiourea, 3 sodium pyruvate, pH adjusted to 7.3–7.4 in NaOH] with low (0.5 mm) CaCl2, and high (10 mm) MgSO4 for 2–3 min. Coronal slices (300 μm) were prepared using a vibratome (model VT1200, Leica), incubated for 10 min in a 30°C in a neural protective recovery HEPES-aCSF (NaCl was replaced by equimolar N-methyl-d-glucamine, pH adjusted to 7.3–7.4 with HCl), and then transferred to a Braincubator (Payo Scientific,#BR26021976) and maintained at 16°C in a HEPES-aCSF holding solution with 2 mm CaCl2, and 2 mm MgSO4.
For recordings, slices were transferred to a recording chamber and continuously perfused (2–3 ml·min−1) with aCSF (30°C) containing the following (in mm): 119 NaCl, 5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1.0 NaH2PO4, 26.2 NaHCO3, 11 glucose (equilibrated with 95% CO2, 5% O2). Targeted whole-cell patch-clamp recordings were made from visually identified neurons using a microscope (Zeiss, Axio Examiner D1) equipped with a 20× water-immersion objective (1.0 NA) a LED fluorescence illumination system (pE-2, CoolLED) and an EMCCD camera (iXon+, Andor Technology). Patch pipettes (2–5 MΩ) were filled with an internal solution containing the following (in mm): 130 potassium gluconate, 10 KCl, 10 HEPES, 4 Mg2-ATP, 0.3 Na3-GTP, 0.3 EGTA, 10 phosphocreatine disodium salt, pH 7.3 with KOH, 280–290 mOsm. Electrophysiological recordings were amplified using a MultiClamp 700B amplifier (Molecular Devices), filtered at 6 kHz, digitized at 20 kHz with a Digidata1440A (Molecular Devices) interface, and controlled using AxoGraph. Electrophysiological data were analyzed off-line using AxoGraph.
Optogenetic stimulation of ChR2 and Chrimson-expressing neurons was elicited using 470 nm or GYR (excitation filter 605 nm, 50 nm FWHM) LED illumination delivered through the objective. Chrimson, like all red-shifted opsins, is excited by both orange (605 nm) and blue (470 nm) light (Klapoetke et al., 2014). To selectively activate ChR2-expressing terminals in dual opsin experiments we used a sequential orange then blue activation protocol adapted from Hooks et al. (2015). First, orange light selectively and transiently activates Chrimson-expressing terminals. Then, after the Chrimson-expressing terminals are inactivated by the orange light, the light switched to blue, selectively activating the ChR2-expressing terminals. Neurons were considered connected if the light reliably evoked a short latency synaptic current > 5 pA (Vm = −50 mV). Light-evoked responses were averaged. The latency of light-evoked responses was calculated from light onset. Picrotoxin (Tocris Bioscience) was dissolved in DMSO, aliquoted and stored frozen (−30°C) as concentrated (1000–4000×) stock solution until needed.
Histology and immunohistochemistry
RNAscope in situ hybridization.
In situ hybridization was performed as previously described (Rubio et al., 2015; Gibson et al., 2018). Thirty minutes post-test session, rats were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.; Virbac) and decapitated. Brains were rapidly extracted and quick frozen on quick freeze (Leica Biosystems). Brains were stored at 80°C until use. Sixteen-micrometer coronal sections of VP were cut using a cryostat and mounted directly onto Super Frost Plus slides (Fisher Scientific). Slides were stored at −80°C until use. The RNAscope Multiplex Fluorescent Reagent Kit (Advanced Cell Diagnostics, #320850) was used to detect GAD1 (316401-C2; GenBank ID: NM_017007.1), vGlut2 (317011-C2; GenBank ID: NM_053427.1), PV (407821-C3; GenBank ID: NM_022499.2), and cFos (403591; GenBank ID: NM_022197.2) mRNA in VP. Slides were fixed in 4% paraformaldehyde for 15 min at 4°C. Slides were then rinsed in PBS followed by dehydration in increasing concentrations of ethanol (50, 70, 100, and 100%) for 10 min at room temperature then air-dried for 10 min and a hydrophobic barrier was drawn around the brain sections. Sections were then protease treated (pretreatment 4) at room temperature for 15 min. Next, sections were then rinsed in distilled water followed by probe application. The target probes used were GAD1 and cFos, vGlut2 and cFos, PV and cFos. Slides were incubated with probes at 40°C for 1 h. Following this, slides were incubated with preamplifier and amplifier probes (AMP1, 40°C for 30 min; AMP2, 40°C for 15 min; AMP3, 40°C for 30 min). Slides were incubated in fluorescently labeled probes by selecting a specific combination of colors associated with each channel, i.e., GAD1 and c-Fos, vGlut2 and c-Fos, PV and cFos. AMP4 Alt B to detect c-Fos/GAD1 and AMP4 Alt C to detect c-Fos/PV in ATTO 550 channel (PV or GAD1) and c-Fos in ATTO 647 channel. Finally, sections were incubated for 20 s with DAPI and coverslipped with PermaFluor mounting medium (ThermoFisher Scientific). Fluorescent images of the VP were taken with an Olympus BX51 microscope using a 40× objective. VP sections were taken anterior, medial and caudal at bregma 0.00 mm, −0.12, −0.24 (rat brain atlas). Photoshop software was used to quantify the number of neurons containing GAD1, vGlut2, PV, c-Fos, and colocalized c-Fos mRNA.
Retrograde tracing.
The retrograde tracers were CTb conjugated to AlexaFluor 488 (CTb-488; catalog #C34775 ThermoFisher Scientific) or AlexaFluor 555 (CTb-555; catalog #C34776, ThermoFisher Scientific). Tracers were targeted to the LH. At least 1 week after surgery, rats were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.; Virbac) and perfused transcardially with 200 ml of 0.9% saline, containing heparin (360 ml/L) and sodium nitrite (12.5 ml/L), followed by 400 ml of 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH7.4. Brains were extracted from the skull and postfixed for 1 h in the same fixative, and then placed in 20% sucrose solution overnight. Brains were frozen and sectioned coronally at 40 μm using a cryostat (Leica, CM1950). Four serially adjacent sets of tissue from the VP and LH from each brain and stored in 0.1% sodium azide in 0.1 m PBS, pH 7.2. Brain tissue was processed for c-Fos immunochemistry as described. Brain slices were mounted onto glass slides, dried for ∼5 min and coverslipped with PermaFluor mounting medium.
eYFP/mCherry/cFos immunohistochemistry.
In all other experiments, rats were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.; Virbac) and perfused transcardially with 200 ml of 0.9% saline, containing heparin (360 ml/L) and sodium nitrite (12.5 ml/L), followed by 400 ml of 4% paraformaldehyde in 0.1 m PB, pH7.4. Brains were extracted from the skull and postfixed for 1 h in the same fixative, and then placed in 20% sucrose solution overnight. Brains were frozen and sectioned coronally at 40 μm using a cryostat (Leica, CM1950). To visualize eYFP, mCherry immunoreactivity (IR; Rabbit anti-eGFP Polyclonal Antibody, catalog #AA11122; RRID:AB_221569; Rabbit anti-mCherry Polyclonal Antibody, catalog #PA5-34974; RRID:AB_2552323; ThermoFisher Scientific) four serially adjacent sets of sections from the regions-of-interest were obtained from each brain and stored in 0.1% sodium azide in 0.1 m PBS, pH 7.2. Sections were washed in 0.1 m PB, followed by 50% ethanol, 50% ethanol with 3% hydrogen peroxidase, then 5% normal horse serum (NHS) in PB (30 min each). Sections were then incubated in rabbit antiserum against eGFP or mCherry (1:2000; ThermoFisher Scientific) in a PB solution containing 2% NHS and 0.2% Triton X-10 (48 h at 4°C). The sections were then washed and incubated in biotinylated donkey anti-rabbit (1:1000; 24 h at 4°C; Biotin Donkey Anti-Rabbit, catalog #711-065-152; RRID:AB_2540016 Jackson ImmunoResearch). Finally, sections were incubated in avidin-biotinylated horseradish peroxidase complex (6 ml/ml avidin and 6 ml/ml biotin; 2 h at room temperature; Vector Laboratories, #PK-6100), washed in PB, and then incubated for 15 min in a diaminobenzidine solution containing 0.1% 3,3-diaminobenzidine, 0.8% d-glucose and 0.016% ammonium chloride. Immunoreactivity was catalyzed by the addition of 0.2 ml/ml glucose oxidase aspergillus (24 mg/ml, 307 U/mg; Sigma-Aldrich). Brain sections were then washed in PB, mounted onto gelatin-coated slides, dehydrated, cleared in histolene, and coverslipped with Entellan (Proscitech), and assessed using an Olympus BX51transmitted light microscope.
For detection of c-Fos, brain sections were then incubated in rabbit anti-c-Fos (1:500; Santa Cruz Biotechnology, catalog #sc-52; RRID:AB_2106783) for 24 h at 4°C. The primary antibodies were diluted in blocking buffer. After washing off unbound primary antibody, sections were incubated overnight at 4°C in biotinylated donkey anti-rabbit IgG (1:500; Jackson ImmunoResearch Laboratories, catalog #711-065-152; RRID:AB_2340593) diluted in blocking buffer. After washing off unbound secondary antibody, sections were incubated for overnight at 4°C in streptavidin, AlexaFluor-350 conjugate (1:300; S11249, Invitrogen). Brain sections were then washed in PB, pH 7.4, and mounted using mounting media. VP sections were delineated according to Paxinos and Watson (2007) and imaged at 20× using a transmitted light microscope (Olympus, BX51). Counts of all neurons IR for c-Fos and CTb-488 or CTb-555 native fluorescence across three VP sections (80 μm apart) were made using Photoshop (Adobe).
Fluorescent images of VP were taken microscope (Olympus, BX51). Counts of all neurons IR for c-Fos and CTb-488 or CTb-555 native fluorescence across three VP sections (80 μm apart) were made using Photoshop (Adobe).
Quantification and statistical analyses
Data in figures are represented as mean ± SEM unless otherwise stated. Group sizes were based on our past experience with these preparations showing that they were sufficient to detect large (d = 0.8) effect sizes in neuroanatomical or behavioral studies with at least 80% power. The criteria for inclusion in final analysis was correct AAV or tracer and/or fiber placements determined after histology. Group numbers used for analyses in each experiment are indicated at two locations: (1) under the subheadings of behavioral procedures above, or (2) in the main results text. Our primary behavioral dependent variables were numbers of active nose-pokes, inactive nose-pokes, and distance traveled (locomotor activity). These data were analyzed by means of ANOVA and analyses involving repeated measures adopted a multivariate approach (Boik, 1981; Harris, 2004). All analyses partitioned variances into main effect and interaction terms using Psy Statistical Package (Bird, 2004). Internal consistency of statistical reporting was verified via statcheck (http://statcheck.io; Nuijten et al., 2016).
Results
Distinct roles for VP cell types in relapse
To identify specific VP cell types recruited during relapse, we used ABA renewal of alcohol seeking (Crombag and Shaham, 2002; Hamlin et al., 2007; Bouton and Todd, 2014). Three groups of rats were trained to respond for alcoholic beer in a distinctive context (Context A). Responses on one nose-poke (active) led to delivery of alcoholic beer to a magazine cup, whereas responses on a second (inactive) did not. Active nose-poking increased across training. Then we extinguished this behavior in a second context where responses did not earn alcohol (Context B). As expected, active nose-poking declined. The mean SEM levels of responding at the end of training and extinction in this and remaining experiments are shown in Table 1. In this and remaining experiments, there were no significant differences between groups in nose-pokes in self-administration or extinction (all p > 0.05). One group was tested for expression of extinction (ABB, n = 6), one for relapse (ABA, n = 6), and a third group remained in their home cages and were not tested (AB0, n = 4). This AB0 group served as a control for the behavioral training and pharmacological histories of the groups tested for renewal (Hamlin et al., 2006, 2007, 2008).
Table 1.
Mean ± SEM active and inactive nose-pokes after training and extinction
| Active: | Acq10 |
E1 |
E4 |
Inactive: | Acq10 |
E1 |
E4 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | ||
| RNAscope | |||||||||||||
| ABA | 56.7 | 2.0 | 33.7 | 4.2 | 6.8 | 1.2 | 9.9 | 1.1 | 3.9 | 1.2 | 1.7 | 0.3 | |
| ABB | 61.8 | 1.0 | 31.2 | 4.7 | 11.3 | 5.0 | 14.5 | 0.5 | 3.4 | 0.8 | 2.1 | 2.9 | |
| ABO | 78.5 | 0.5 | 34.0 | 2.5 | 6.8 | 2.0 | 8.8 | 0.5 | 5.2 | 0.3 | 1.2 | 0.6 | |
| VPGAD1 Inhibition | |||||||||||||
| GAD-CRE− | 161.5 | 13.0 | 30.0 | 4.0 | 7.6 | 1.4 | 1.8 | 0.7 | 4.3 | 0.8 | 0.9 | 0.5 | |
| GAD-CRE+ | 147.9 | 10.1 | 35.0 | 5.6 | 4.4 | 1.3 | 2.1 | 0.7 | 3.7 | 1.0 | 0.9 | 0.7 | |
| VPPV Inhibition | |||||||||||||
| PV-CRE− | 72.9 | 17.8 | 41.9 | 10.0 | 6.1 | 1.3 | 0.6 | 0.4 | 3.8 | 1.0 | 1.3 | 0.6 | |
| PV-CRE+ | 77.1 | 17.7 | 46.1 | 6.9 | 8.1 | 3.6 | 0.8 | 0.3 | 2.9 | 0.7 | 1.0 | 0.7 | |
| VP to LH Tracing VP-LH tracing | |||||||||||||
| ABA | 106.4 | 19.3 | 29.6 | 6.3 | 5.6 | 0.4 | 2.2 | 0.6 | 6.2 | 2.5 | 3.4 | 1.5 | |
| ABB | 88.0 | 17.0 | 34.2 | 3.9 | 6.8 | 1.7 | 1.8 | 0.7 | 4.5 | 1.1 | 3.5 | 0.9 | |
| ABO | 117.3 | 37.6 | 38.3 | 3.8 | 8.3 | 1.2 | 1.0 | 0.5 | 3.3 | 1.0 | 1.7 | 0.3 | |
| VPGAD1 to LH Inhibition | |||||||||||||
| GAD1-CRE−:LH Contra | 72.9 | 11.5 | 75.1 | 46.5 | 8.6 | 2.5 | 1.3 | 0.3 | 3.9 | 0.5 | 1.3 | 0.6 | |
| GAD1-CRE+:LH Ipsi | 68.7 | 14.6 | 38.3 | 5.4 | 7.4 | 1.7 | 1.1 | 0.6 | 3 | 0.7 | 1.7 | 0.4 | |
| GAD1-CRE+:LH Contra | 65.1 | 10.9 | 56.5 | 13.2 | 12 | 2.5 | 0.9 | 0.4 | 4.4 | 0.9 | 1.4 | 0.4 | |
| VPGAD1 to VTA Inhibition | |||||||||||||
| GAD1-CRE−:VTA | 49.7 | 6.7 | 26.3 | 3.1 | 10.6 | 1.6 | 0.71 | 0.3 | 2.6 | 0.8 | 3.1 | 1.2 | |
| GAD1-CRE+:VTA | 75.1 | 26.9 | 34 | 4.4 | 21.9 | 4.4 | 1 | 0.5 | 4.3 | 1.3 | 3.3 | 0.6 | |
| VPPV to LH Inhibition | |||||||||||||
| PV-CRE−:LH Contra | 82.4 | 20.5 | 49.0 | 10.6 | 9.1 | 1.3 | 1.7 | 0.7 | 3.7 | 0.9 | 2.3 | 0.7 | |
| PV-CRE+:LH Ipsi | 74.2 | 8.0 | 39.8 | 7.7 | 14.5 | 3.9 | 1.5 | 0.7 | 4.8 | 1.6 | 2.3 | 1.1 | |
| PV-CRE+:LH Conta | 64.4 | 7.8 | 57.4 | 7.8 | 10.4 | 3.6 | 1.4 | 0.6 | 5.4 | 1.5 | 1.7 | 0.8 | |
| VPPV to VTA Inhibition | |||||||||||||
| PV-CRE−:VTA Contra | 106.1 | 12.8 | 29.3 | 5.0 | 7.3 | 1.5 | 0.6 | 0.3 | 3.4 | 1.4 | 1.9 | 0.7 | |
| PV-CRE+:VTA Ipsi | 160.2 | 48.8 | 25.2 | 2.6 | 14.2 | 3.6 | 2.3 | 0.8 | 4.7 | 1.3 | 1.8 | 0.5 | |
| PV-CRE+:VTA Contra | 84.6 | 16.6 | 22.9 | 5.4 | 6.4 | 0.8 | 1.2 | 0.6 | 3.8 | 0.7 | 2.1 | 0.7 | |
Responding was low for Group ABB tested in the extinction context. In contrast, responding was higher for Group ABA tested in the training context (Context × Nose-poke interaction: F(1,10) = 7.58, p = 0.020; Fig. 1A). This shows renewal of alcohol seeking (i.e., context-induced reinstatement). All rats were perfused 30 min after test and relapse-associated activity in VP GABA (VPGad1), glutamate (VPvGlut2), and PV (VPPV) neurons was assessed via in situ hybridization for c-Fos mRNA. ABA renewal was associated with significant c-Fos expression (i.e., ABA > ABB = AB0) in VPGad1 (F(1,13) = 17.00, p = 0.0012) and VPPV (F(1,13) = 14.61, p < 0.0021) neurons (Fig. 1B; Table 2). In contrast, VPvGlut2 neurons were strongly recruited regardless of test context and their activity, at least as measured by c-Fos mRNA expression, was unrelated to differences in relapse behavior (i.e., ABA = ABB > AB0; F(1,13) = 27.87, p = 0.000149).
Figure 1.

Distinct roles for pallidal cell types in relapse. A, ABA renewal of extinguished alcohol seeking. B, ABA renewal is associated with c-Fos mRNA expression in VP Gad1 and PV neurons whereas vGlut2 neurons express c-Fos mRNA independently of test context. Scale bar, 30 μm. C, There was substantial overlap in VP cell populations as shown by coexpression of Gad1 and PV mRNA expression. D, Cre-dependent inhibitory hM4Di was applied to VP of Gad1-Cre rats, with location of hM4Di expression shown for all animals at 10% opacity. E, Chemogenetic inhibition of VP Gad1 neurons reduced relapse during renewal but not reacquisition. F, Cre-dependent inhibitory hM4Di was applied to VP of PV-Cre rats, with location of hM4Di expression shown for all animals at 10% opacity. G, Chemogenetic inhibition of VP PV neurons reduced relapse during renewal and reacquisition. *p < 0.05.
Table 2.
Mean ± SEM counts for PV, vGlut2, Gad1, and c-Fos mRNA expression during renewal
| ABA | c-Fos | PV | Dual | ABA | c-Fos | Gad1 | Dual | ABA | c-Fos | vGlut2 | Dual |
| Mean | 982.7 | 462.5 | 441.7 | Mean | 938.3 | 824.8 | 576.7 | Mean | 858.8 | 495.5 | 419.7 |
| SEM | 18.3 | 4.6 | 6.5 | SEM | 24.9 | 54.7 | 36.8 | SEM | 24.9 | 38.9 | 36.8 |
| ABB | c-Fos | PV | Dual | ABB | c-Fos | Gad1 | Dual | ABB | c-Fos | vGlut2 | Dual |
| Mean | 676.8 | 483.8 | 343.5 | Mean | 752.2 | 848.3 | 413.8 | Mean | 609.7 | 477.5 | 350.3 |
| SEM | 26.4 | 39.7 | 38.9 | SEM | 26.9 | 125.7 | 25.9 | SEM | 40.3 | 50.6 | 29.7 |
| AB0 | c-Fos | PV | Dual | AB0 | c-Fos | Gad1 | Dual | AB0 | c-Fos | vGlut2 | Dual |
| Mean | 662.5 | 446.8 | 289.0 | Mean | 717.8 | 760.8 | 431.0 | Mean | 590.8 | 393.0 | 155.5 |
| SEM | 36.8 | 39.9 | 20.39 | SEM | 12.8 | 78.2 | 23.4 | SEM | 49.7 | 28.3 | 23.5 |
The identities of VP neurons overlap (Root et al., 2015). In particular, VPPV neurons express vesicular transporters for GABA (vGat) or glutamate (vGlut2) and transmit either inhibitory or excitatory signals (Knowland et al., 2017). To understand the degree of overlap between these cell populations in the rat, we measured PV and Gad1 mRNA in 819 VP neurons from a separate group of animals that did not receive any behavioral training or testing (n = 3). Gad1 and PV mRNA were coexpressed in a large number of VP neurons with a minority expressing one but not the other (Fig. 1C). So, the common recruitment of VPGad1 and VPPV neurons during relapse could simply reflect this overlap.
To determine the roles of VPGad1 and VPPV neurons in relapse, we used adeno-associated viral vectors (AAVs) in LE-Tg(Gad1-iCre)3Ottc (Gad1-Cre+) rats (n = 8) or Gad1-Cre− (n = 7) rats (Sharpe et al., 2017) to express the inhibitory hM4Di designer receptor (AAV5-hSyn-DIO-hM4Di-mCherry; Armbruster et al., 2007) in VPGad1 neurons (Fig. 1D). If VPGad1 neurons contribute to relapse, then renewal should be reduced by VPGad1 neuron chemogenetic inhibition. We used a low dose of clozapine (0.1 mg/kg, i.p.) as the hM4Di ligand (Gomez et al., 2017) and have shown previously that this dose of clozapine does not affect relapse behavior in non-DREADD-expressing rats (Gibson et al., 2018). Systemic injection of clozapine reduced renewal in Gad1-Cre+ animals (Context × Nose-poke × Group interaction: F(1,13) = 5.024, p = 0.043; Fig. 1E). There was no effect of VPGad1 chemogenetic inhibition on latency to first nose-poke during renewal (F(1,13) = 0.029, p = 0.867). VP-dependent relapse can also be provoked by contingent re-exposure to alcohol (reacquisition; Khoo et al., 2015) and this underpins a rapid return to drinking in humans (Marlatt and Donovan, 2005). We assessed whether VPGad1 neurons contribute to this second form of relapse by pretreating the two groups with 0.1 mg/kg clozapine and re-training them in a single session of self-administration. However, VPGad1 chemogenetic inhibition had no effect on reacquisition (Nose-poke × Group interaction: F(1,13) = 0.054, p = 0.820; Fig. 1E). There was no effect of VPGad1 chemogenetic inhibition on latency to first nose-poke during reacquisition (F(1,13) = 2.261, p = 0.157). There was also no effect of VPGad1 chemogenetic inhibition on locomotor activity when assessed in a locomotor chamber [Gad1-Cre−: 4705 ± 799 cm; Gad1-Cre+: 4901 ± 318 cm (mean ± SEM); F(1,13) = 0.012, p = 0.9144].
Then we expressed the inhibitory hM4Di designer receptor in VPPV neurons (AAV5-hSyn-DIO-hM4Di-mCherry) of LE-Tg(Pvalb-iCre)2Ottc (PV-Cre+; n = 7) or PV-Cre− (n = 8) rats (Fig. 1F) and chemogenetically silenced these neurons via injection of low-dose clozapine (0.1 mg/kg, i.p.) before test. Similar to VPGad1 neurons, VPPV chemogenetic inhibition significantly reduced relapse during renewal (Context × Nose-poke × Group interaction: F(1,13) = 9.47, p = 0.0088; Fig. 1G). However, in contrast to VPGad1, chemogenetic inhibition of VPPV neurons also reduced relapse during reacquisition (Nose-poke × Group interaction: F(1,13) 4.73, p = 0.0486; Fig. 1G). VPPV chemogenetic inhibition had no effect on latency to first nose-poke during either renewal (F(1,13) = 1.140, p = 0.305) or latency to first nose-poke reacquisition: F(1,13) = 0.081, p = 0.780). There was also no effect of VPPV chemogenetic inhibition on locomotor activity when assessed in a locomotor chamber [PV-Cre−: 4908 ± 496 cm; PV-Cre+: 6555 ± 777 cm (mean ± SEM); F(1,13) 3.362, p = 0.0897].
These results show distinct roles for VP neuronal populations in relapse. PV and Gad1, but not vGlut2, neurons express relapse-associated changes in c-Fos mRNA. Consistent with these changes, chemogenetic inhibition of Gad1 and PV neurons reduces relapse during renewal. However, chemogenetic inhibition of PV neurons additionally reduces relapse during reacquisition.
VPGad1 neurons promote relapse via LH, not VTA
VPGad1 neurons control relapse during renewal but not reacquisition. VP neurons have extensive projections, including to LH, lateral habenula, and VTA. LH is a likely target for VP contributions to relapse because LH has been strongly implicated in renewal of alcohol-seeking after extinction or punishment (Marchant et al., 2009, 2014) and also mediates the effects of VP manipulations on feeding (Stratford and Wirtshafter, 2013). To study whether VP neurons projecting to LH were recruited during renewal we combined retrograde tracing from LH using CTb with c-Fos immunohistochemistry in VP (Fig. 2A). Rats in groups ABA (n = 5), ABB (n = 6), and AB0 (n = 3) received application of CTb-488 or CTb-555 (counterbalanced) to LH (Fig. 2C) then were trained to self-administer alcoholic beer and extinguished before test for expression of extinction (ABB), renewal (ABA), or not tested (AB0). On test there was renewal in Group ABA (Context × Nose-poke × Group interaction: F(1,9) = 92.089, p < 0.00001; Fig. 2B). Renewal was associated with c-Fos protein expression in VP (ABA > ABB=AB0, F(1,11) = 13.763, p = 0.00344), including in VP neurons retrograde labeled from LH (F(1,11) = 56.99, p = 0.00001; Fig. 2D,E; Table 3).
Figure 2.

VPGad1 neurons promote relapse via LH. A, C, CTb-488 or CTb-555 was applied to LH prior training and testing, Scale bar, 200 μm. B, ABA renewal of alcohol seeking. D, Example of c-Fos (blue), CTb (green), and dual labeling (shown by arrows) in VP. E, ABA renewal was associated with c-Fos expression in VP neurons including in VP→LH neurons. F, Cre-dependent inhibitory hM4Di was applied to VP of Gad1-Cre rats and hM4Di also applied to LH of the same animals. Locations of hM4Di expression shown for all animals at 10% opacity. Chemogenetic disconnection of VPGad1→LH pathway reduced renewal but not reacquisition or locomotor activity. G, Cre-dependent inhibitory hM4Di was applied to VP of Gad1-Cre rats and hM4Di also applied to VTA of the same animals. Locations of hM4Di expression shown for all animals at 10% opacity. Chemogenetic disconnection of VPGad1→VTA pathway had no effect on renewal, reacquisition, or locomotor activity. *p < 0.05.
Table 3.
Mean ± SEM counts for CTb and c-Fos during renewal
| ABA | c-Fos | CTb | Dual | ABB | c-Fos | CTb | Dual | ABO | c-Fos | CTb | Dual |
| Mean | 174.8 | 216.8 | 46.2 | Mean | 115.7 | 204.8 | 10.3 | Mean | 91.0 | 219.3 | 10.7 |
| SEM | 9.9 | 16.7 | 4.5 | SEM | 14.5 | 8.8 | 2.6 | SEM | 26.6 | 55.9 | 5.2 |
These findings show that a VP→LH pathway is recruited during renewal. However, these findings do not identify the VP cell types responsible nor do they show a causal role for the VP→LH pathway in renewal. VPGad1 neurons project extensively to LH (Jennings et al., 2013) so we tested whether the role of VPGad1 in renewal depends on LH. We used a three-group chemogenetic approach disconnecting VPGad1 from LH. We expressed the inhibitory hM4Di designer receptor in VPGad1 neurons via AAV5-hSyn-DIO-hM4Di-mCherry in one hemisphere and in either the contralateral (Gad1-Cre+ Contra; n = 8) or ipsilateral (Gad1-Cre+ Ipsi; n = 7) LH via AAV5-hSyn-hM4Di-mCherry (Fig. 2F). This approach disconnects VPGad1 from LH in Group Gad1-Cre+ Contra but not Group Gad1-Cre+ Ipsi. In control Gad1-Cre− rats, we applied the hSyn-DIO-hM4Di to one VP and the hSyn-hM4Di to the contralateral LH (Gad1-Cre− Contra; n = 8). We trained, extinguished, and tested these groups for relapse via renewal and reacquisition. Rats were injected with clozapine (0.1 mg/kg, i.p.) before both tests. Renewal was selectively reduced in Gad1-Cre+ Contra rats (group × nose-poke interaction: F(1,20) = 18.227, p = 0.00037; Fig. 2F). Importantly, this same disconnection had no effect on relapse during reacquisition (F(1,20) = 1.597, p = 0.221; Fig. 2I). There was no effect of chemogenetic disconnection on locomotor activity (F(1,20) = 0.001, p = 0.975), latency to first nose-poke during renewal (F(1,20) = 1.465, p = 0.240) or latency to first nose-poke during reacquisition (F(1,20) = 0.848, p = 0.368).
These effects of disconnecting VPGad1 from LH recapitulated the effects on relapse of silencing VPGad1 neurons, showing that the role of VPGad1 neurons in relapse depends on LH. However, other VP output pathways could also be important. VP inputs to VTA are recruited during different forms of relapse (Mahler and Aston-Jones, 2012; Mahler et al., 2014; Prasad and McNally, 2016), and nonselective chemogenetic disconnection of VP neurons from VTA reduces relapse (Mahler et al., 2014; Prasad and McNally, 2016). Therefore, we used a two-group chemogenetic disconnection design in Gad1-cre rats to assess whether a VPGad1 →VTA pathway contributes to relapse. We expressed the inhibitory hM4Di designer receptor in VPGad1 neurons via AAV5-hSyn-DIO-hM4Di-mCherry in one hemisphere and in the contralateral VTA via AAV5-hSyn-hM4Di-mCherry in Gad1-Cre+ (n = 7) or Gad1-Cre− (n = 7) rats (Fig. 2G). We trained, extinguished, and tested these two groups for relapse via renewal and reacquisition. Rats were injected with clozapine (0.1 mg/kg, i.p.) before both tests. There was no effect of this disconnection on renewal (F(1,12) = 0.264, p = 0.617), reacquisition (F(1,12) = 0.568, p = 0.466), locomotor activity (F(1,12) = 0.781, p = 0.394; Fig. 2G), latency to first nose-poke during renewal (F(1,12) = 1.610, p = 0.229), or latency to first nose-poke during reacquisition (F(1,12) = 1.400, p = 0.260) So, VPGad1 neurons contribute to renewal via projections to LH but not VTA.
Convergence and segregation of ventral striatopallidal inputs to LH
LH comprises a diversity of cell types relevant to appetitive motivation (Bonnavion et al., 2016). In mice, there are dense VP GABA inputs to LH GABA neurons (Jennings et al., 2013). We tested whether VPGad1 neurons also provide inhibitory projections onto rat LH GABA neurons. To do this, we expressed ChR2 in VPGad1 using AAV5-hSyn-DIO-ChR2(H134R)-eYFP and made whole-cell patch-clamp recordings from eYFP-labeled LHGad1 neurons in Gad1-Cre+ rats (Fig. 3A). Photostimulation (470 nm) of VP terminals evoked short latency inhibitory responses (3.3 ± 0.8 ms from light onset; mean ± SD) in eYFP+ (i.e., LHGad1; 53%, 10/19) but not eYFP- neurons (Fig. 3B,C). The current's kinetics, reversal potential, and sensitivity to the GABA-A receptor antagonist picrotoxin (100 μm; n = 4; Fig. 3D,E) indicated that VPGad1 neurons provide monosynaptic GABAergic inputs onto LHGad1 neurons.
Figure 3.

VPGad1 neurons provide monosynaptic inhibitory GABAergic input to LHGad1 neurons. A, Schematic for ChR2 mapping of VP inputs to LHGad1 neurons. B, Representative responses of connected LHGad1 neurons to light pulse trains (40 ms, 470 nm at 12.5 Hz). Trains evoked fast picrotoxin-sensitive IPSCs. GABA-B receptor-mediated slow IPSCs were not observed. C, Amplitude of the light-evoked (470 nm, 10 ms) postsynaptic current (PSC; −50 mV) in eYFP+ (LHGad1), and eYFP− neurons, with connectivity chart. Light evoked a reliable current in 10 of 19 (53%) LHGad1 neurons. D, The light-evoked current (Vm − 50 mV) was blocked (96 ± 3%; mean ± SD; n = 4) by bath application of the GABA-A receptor blocker picrotoxin (100 μm). E, Individual example and summary data (mean ± SEM; n = 10) of the PSC evoked at holding potentials −100 to − 50 mV. The PSC reversal potential matches the Cl− equilibrium potential. F, Schematic for testing convergence of VP and AcbSh inputs to LHGad1 neurons. G, Light-evoked responses in Chrimson and ChR2-expressing neurons under voltage-clamp (top traces) and current-clamp (bottom traces). Chrimson-expressing AcbSh neurons are activated by orange (605 nm) and blue (470 nm) light. Under current-clamp, orange light-evoked APs were restricted to the onset of the light pulse. Rapidly switching from orange to blue light, did not evoke any additional APs. ChR2-expressing VP neurons responded blue, but not orange light stimulation. Currents (Vm − 70 mV) evoked by blue and orange light are plotted above the voltage response evoked by an orange (250 ms)-blue (25 ms) light. Chrimson-expressing AcbSh neurons responded to 605 and 470 nm stimulation. Currents (Vm − 70 mV) evoked by blue and orange light are plotted above voltage responses to orange and orange-blue light. H, Representative currents evoked in LHGad1 neurons by stimulation of AcbSh and VP terminals and connectivity chart. AcbSh inputs converged with VP inputs to LHGad1 neurons but VP inputs also provided additional segregated inputs to LHGad1 neurons. I, Locations of LHGad1 neurons recorded as defined by source of input.
LHGad1 neurons also receive monosynaptic GABA inputs from AcbSh (O'Connor et al., 2015; Gibson et al., 2018). Whereas VP GABA inputs to LH neurons promote relapse, AcbSh inputs prevent relapse (Gibson et al., 2018). These opposing roles in relapse raise the possibility that these two components of the ventral striatopallidal system target the same LHGad1 neurons. However, there is considerable diversity even within LH GABA neurons populations (Mickelsen et al., 2019), so the finding that LHGad1 neurons receive monosynaptic GABAergic inputs from both VPGad1 neurons (Fig. 3) and AcbSh (O'Connor et al., 2015; Gibson et al., 2018) does not mean that the same LHGad1 neurons receive these inputs. To address this, we used two-color channel rhodopsin assisted circuit mapping (Klapoetke et al., 2014). We recorded from eYFP-labeled LHGad1 neurons in Gad1-cre rats after expressing ChR2 in VPGad1 neurons (AAV5-hSyn-DIO-ChR2(H134R)-eYFP) and the red-shifted opsin Chrimson (AAV5-hSyn-Chrimson-tdTomato) in AcbSh neurons (Fig. 3F). This configuration allowed us to activate AcbSh and VP sequential inputs to the same LHGad1 neurons via 605 and 470 nm optical stimulation, respectively (Fig. 3G). Using this approach, we found that many LHGad1 showed optically-evoked currents but others did not (Fig. 3H), consistent with the known heterogeneity of this cell population (Mickelsen et al., 2019). Importantly there was evidence for convergence of ventral striatopallidal inputs, with 37% of LHGad1 neurons receiving monosynaptic input from both AcbSh and VP. Interestingly, although 22% of LHGad1 neurons responded selectively to VPGad1 stimulation, only a minority (7%) responded selectively to AcbSh stimulation (Fig. 3H). Thus, whereas AcbSh inputs converge with VPGad1 inputs onto LHGad1 neurons, there is further segregation of VPGad1 inputs in the LH. There was no obvious relationship between ventral striatopallidal input selectivity and anatomical location of these Gad1 neurons in LH (Fig. 3I).
VPPV neurons promote relapse via VTA, not LH
VPPV neurons control both renewal and reacquisition (Fig. 1F). So, next we examined whether the role for VPPV neurons in relapse also depends on LH. We used the three-group chemogenetic approach disconnecting VPPV from LH. We expressed the inhibitory hM4Di designer receptor in VPPV neurons via AAV5-hSyn-DIO-hM4Di-mCherry in one hemisphere and in either the contralateral (PV-Cre+ Contra; n = 7) or ipsilateral (PV-Cre+ Ipsi; n = 6) LH via AAV5-hSyn-hM4Di-mCherry (Fig. 4A,B). This approach disconnects VPPV from LH in Group PV-Cre+ Contra but not PV-Cre+ Ipsi. In Group control PV-Cre− rats, we applied the hSyn-DIO-hM4Di to one VP and the hSyn-hM4Di to the contralateral LH (PV-Cre− Contra; n = 7). We trained, extinguished, and tested these groups for relapse via renewal and reacquisition. Rats were injected with clozapine (0.1 mg/kg, i.p.) before both tests. In both renewal and reacquisition tests there was relapse (Renewal Context × Nose-poke interaction: F(1,17) = 78.12, p < 0.00001; Reacquisition main effect of Nose-poke: F(1,17) = 58.27, p < 0.00001; Fig. 4C) but there was no effect of chemogenetic VPPV–LH disconnection (Renewal Context × Group × Nose-poke interaction: F(1,17) = 1.43, p = 0.248; Reacquisition Group × Nose-poke interaction: F(1,17) = 1.81, p = 0.1961). There was also no effect of this disconnection on latency to first nose-poke during renewal (F(1,17) = 1.438, p = 0.247), latency to first nose-poke during reacquisition (F(1,17) = 1.005, p = 0.330), or on locomotor activity (F(1,17) = 0.274, p = 0.6074; Fig. 4C). Therefore, the role of VPPV neurons in relapse is independent of LH and dissociable from VPGad1 neurons.
Figure 4.

VPPV neurons promote relapse via VTA. A, Cre-dependent inhibitory hM4Di was applied to VP of PV-Cre rats and hM4Di applied to LH of the same animals. B, Locations of hM4Di expression shown for all animals at 10% opacity. C, Chemogenetic disconnection of VPPV→LH pathway had no effect on renewal, reacquisition, or locomotor activity. D, Cre-dependent inhibitory hM4Di was applied to VP of PV-Cre rats and hM4Di also applied to VTA of the same animals. E, Locations of hM4Di expression shown for all animals at 10% opacity. F, Chemogenetic disconnection of VPPV→VTA pathway reduced renewal and reacquisition but not locomotor activity. *p < 0.05.
A VP→VTA pathway is a second obvious candidate for VPPV contributions to relapse. VPPV provide monosynaptic input to VTA dopamine and GABAergic neurons (Knowland et al., 2017; Faget et al., 2018). Moreover, as just noted, VP inputs to VTA are recruited during different forms of relapse (Mahler and Aston-Jones, 2012; Mahler et al., 2014; Prasad and McNally, 2016), and nonselective chemogenetic disconnection of VP neurons from VTA reduces relapse (Mahler et al., 2014; Prasad and McNally, 2016). To determine whether a VPPV→VTA pathway mediates relapse, we used the three group chemogenetic disconnection design (PV-Cre+ Contra, n = 9; PV-Cre− Contra, n = 7; PV-Cre+ Ipsi, n = 6; Fig. 4D,E). We trained and extinguished rats before testing for relapse via renewal and reacquisition. Rats were injected with clozapine (0.1 mg/kg, i.p.) before tests. During both tests there was relapse (Renewal Context × Nose-poke interaction: F(1,19) = 45.60, p < 0.00001; Reacquisition main effect of Nose-poke: F(1,19) = 44.71, p < 0.00001; Fig. 4F). However now, unlike VPPV–LH disconnection, both forms of relapse were reduced by VPPV–VTA disconnection (Renewal Context × Group × Nose-poke interaction: F(1,19) = 8.13, p = 0.01021; Reacquisition Group × Nose-poke interaction: F(1,19) = 5.49, p = 0.03016; Fig. 4F). There was also significant effect of VPPV→VTA chemogenetic inhibition on latency to first nose-poke during renewal (F(1,19) = 7.054, p = 0.016) and reacquisition (F(1,19) = 8.162, p = 0.010) but there was no effect of this disconnection manipulation on locomotor activity (F(1,19) = 2.67, p = 0.11872; Fig. 4F). Thus, chemogenetically disconnecting the VPPV–VTA pathway reduced renewal and reacquisition and recapitulated the effects on relapse of silencing VPPV neurons.
Discussion
We studied the cellular and circuit architecture for relapse control in ventral striatopallidal pathways. VP has been viewed as an obligatory node in a “final common pathway” for relapse. Different forms of relapse (cue, stress, prime, context) have different subcortical substrates, but these converge in dorsomedial prefrontal cortex to be routed through the ventral striatopallidal system generating relapse behavior (Kalivas and Volkow, 2005; Kalivas and O'Brien, 2008). This view has proved useful in understanding the role of VP in relapse. Although our findings confirm this key role for VP, they show that this role is more complex still and that different forms of relapse are assembled from different VP neuronal populations and their projections.
VP cell types in relapse
The VP comprises different cell types whose roles in relapse have been poorly understood. VPGad1 and VPPV neuronal populations were both recruited during ABA renewal (context-induced reinstatement) of alcohol seeking. However, chemogenetic manipulations of these neuronal populations and their projections dissociated their roles in relapse.
Chemogenetic inhibition of VPGad1 neurons reduced renewal but not reacquisition of alcohol-seeking. This role in renewal depends, in part, on projections to LH because VP inputs to LH were recruited during renewal and chemogenetic disconnection of VPGad1 neurons from LH, but not VTA, reduced renewal but not reacquisition. So, the role of VPGad1 neurons in renewal depends on VPGad1–LH interactions. The precise relationship between VPGad1 neurons and diverse LH cell types remains to be fully mapped but we could show that VPGad1 neurons provide inhibitory monosynaptic GABAergic input to LHGad1 neurons. This is consistent with previous monosynaptic input mapping studies (Jennings et al., 2013).
In contrast, VPPV neurons control relapse during both renewal and reacquisition. This role for VPPV neurons is independent of LH and depends on VTA. The relapse phenotype observed after silencing VPPV neurons was not observed after silencing VPPV–LH interactions but was recapitulated after silencing VPPV–VTA interactions. VPPV neurons provide monosynaptic inputs to VTA GABA as well as dopamine neurons (Knowland et al., 2017) and both of these VTA cell types are important for relapse (Mahler et al., 2014; Gibson et al., 2018). However, the nature of VP input to these cell types differs. VPPV input to VTA GABAergic neurons is inhibitory whereas VPPV input to VTA dopamine neurons is predominantly excitatory (Knowland et al., 2017). So, different VP inputs to GABA and dopamine neurons could be linked to specific forms of relapse, but this awaits further investigation. Regardless, given the considerable overlap between VPGad1 and VPPV neurons, the differences we described here between these two cell populations was surprising and show functional as well as anatomical dissociations between the roles of these VP cell types in relapse.
Finally, there was no evidence for selective recruitment of VPvGlut2 neurons during relapse. Rather, VPvGlut2 neurons were recruited during test in either the extinction context (where relapse did not occur) or the training context (where relapse did occur). Therefore, the role of VPvGlut2 neurons in promoting or preventing relapse remains to be determined. These neurons have been implicated in aversion and behavioral avoidance via their projections to the lateral habenula (Knowland et al., 2017; Faget et al., 2018) and this same projection acts to constrain reward seeking (Tooley et al., 2018). One common feature of tests in both the extinction and training contexts here was that the reinforcer was absent and these tests were under extinction conditions. So, recruitment of VPvGlut2 neurons during testing here may reflect reward omission and constrain reward seeking accordingly. Further investigation of the role of VPvGlut2 neurons in relapse is needed. Indeed, the role of non-reinforcement in shaping neural circuits for relapse is worth investigation. Tests for relapse inevitably involve extinction conditions because the drug reinforcer is omitted. These extinction conditions could support further extinction learning that could have been affected by the manipulations here.
Circuit interactions in control over extinction and relapse
Contexts associated with drug self-administration promote relapse whereas contexts associated with extinction protect against relapse. Specifically, extinction imposes context-specific inhibition on responding (Bouton and Todd, 2014). In this way, there is competition between extinction and self-administration memories for control over behavior (Bouton and Todd, 2014). The nature of this competition, at circuit or cellular levels, has been unknown. Here we identify LHGad1 neurons as one possible locus for this competition.
Contextual control over renewal and extinction depend on glutamatergic inputs to AcbSh from prefrontal cortex, basolateral amygdala, ventral hippocampus, and paraventricular thalamus (Hamlin et al., 2009; Marchant et al., 2015; Khoo et al., 2017; Fig. 5). Within AcbSh, dopamine 1 receptor (D1)-expressing neurons projecting to LHGad1 neurons control extinction (Gibson et al., 2018) whereas AcbSh D1 inputs to VTAGad1 neurons control renewal (Gibson et al., 2018; Fig. 5). AcbSh D1 inputs to VTA also collateralize extensively to VP (Pardo-Garcia et al., 2019). Here, we show considerable convergence between VP and AcbSh synaptic inputs to the same LHGad1 neurons. LHGad1 neurons are essential to using appetitive associations to guide behavior (Nieh et al., 2016; Sharpe et al., 2017). Thus, this convergence of AcbSh→LHGad1 and VP→LHGad1 pathways onto the same LHGad1 neurons provides one cellular locus for competition in behavioral control between these pathways, with VPGad1 inputs to LH promoting relapse and AcbSh GABAergic inputs to the same LHGad1 neurons promoting extinction (Gibson et al., 2018; Fig. 5). In this way, LHGad1 neurons may serve a critical integrative role in promoting versus preventing relapse.
Figure 5.
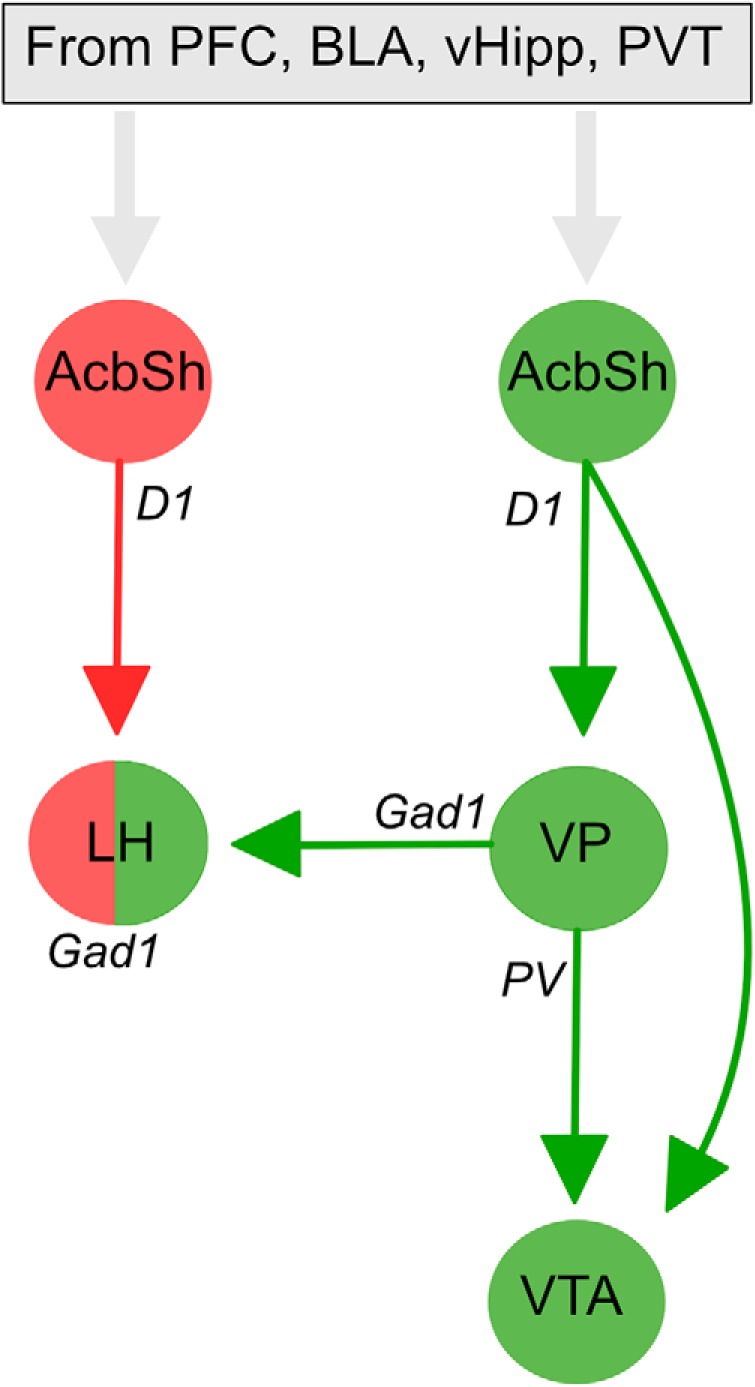
Circuit interactions for contextual control over renewal and reinstatement. Glutamatergic inputs from prefrontal cortex (PFC), basolateral amygdala (BLA), ventral hippocampus (vHipp), and paraventricular thalamus target segregated dopamine 1 receptor expressing neuronal output pathways to promote renewal (via VP and VTA) or extinction (via LH). Distinct VP cell types mediate renewal via their projections to VTA and LH. There is convergence of these renewal and extinction circuits at the level of individual LHGad1 neurons that may determine behavioral propensity to relapse.
However, there was also selective segregation of VP inputs from AcbSh inputs. Whereas AcbSh inputs tended to converge with VP inputs to LHGad1 neurons, VP inputs were additionally segregated from AcbSh inputs to LHGad1 neurons. Whether these VP inputs represent parallel, independent channels of VP-derived information flow to LHGad1 neurons or whether these inputs interact during relapse and other forms of appetitive motivation awaits investigation, but it is consistent with recent work showing the considerable diversity of LH GABA neurons (Mickelsen et al., 2019). Regardless, our results show opposing behavioral functions for VP and AcbSh GABAergic inputs to LH.
Methodological considerations
Our conclusion that VPGad1 and VPPV neurons serve complementary roles in relapse rests on multiple lines of evidence. Regardless, at least four methodological issues are worth considering. First, Mahler et al. (2014) showed differences in relapse across the anterior–posterior axis of the VP, with anterior VP more important than posterior VP in controlling reinstatement of cocaine-seeking. Our manipulations did not distinguish between these VP subregions. Indeed, related to this, our DREADD expression occasionally encroached on nearby regions (e.g., LPOA, basal forebrain). So, it remains to be determined how the role for different VP cell types in relapse relates to this distinction or to other VP subdivisions (e.g., dorsomedial vs ventrolateral; Zahm et al., 1987).
Second, consistent with its location in the ventral basal ganglia, VP has a critical role in motor function and locomotor activity (Austin and Kalivas, 1990). This raises the possibility that the effects on relapse reported here were secondary to changes in locomotor activity. We assessed whether relapse prevention was secondary to any locomotor deficits and there was no evidence to support this possibility.
Third, consistent with recent findings (Gomez et al., 2017), we used low-dose clozapine as the hM4Di actuator. This raises concerns about specificity of our chemogenetic manipulations. However, neither hM4Di expression nor clozapine injection alone affected relapse behavior. Rather, the impact of chemogenetic manipulations was selective and depended not just on the behavior measured but also the specific cell-type and region in which hM4Di was expressed. The same low-dose clozapine injection in hM4Di-expressing animals had no effect on relapse, caused a reduction of renewal but not reacquisition, or caused a reduction of both renewal and reacquisition, depending on the cells and pathways studied. This cell-type- and circuit-specificity are difficult to reconcile with nonspecific effects of DREADD expression or clozapine injection.
Finally, consistent with our past work, rats had access to food and water for 1 h per day. Under these conditions, rats could be responding for alcohol-rewarding effects, caloric effects, or a combination thereof. This is an important constraint on the generality of our findings. The generality of the mechanisms we have discovered to other forms of relapse to seeking other drug rewards awaits investigation.
Conclusions
We studied the cellular and circuit architecture for relapse control in ventral striatopallidal pathways, showing how different forms of relapse are assembled from VP cell populations and their projections. Our findings add to an emerging literature showing that the functional diversity in the ventral striatopallidal system can be resolved by analyses of different families of VP neurons and their projection targets (Richard et al., 2016; Knowland et al., 2017; Faget et al., 2018; Tooley et al., 2018). In the context of relapse to drug seeking, we report a double dissociation between the roles of VP Gad1 and PV neurons in relapse. Our findings raise the possibility that treatments targeting these different ventral striatopallidal pathways may provide one approach to tailored interventions for different forms of relapse.
Footnotes
This work was supported by the National Health and Medical Research Council (GNT1098436, GNT1138062), the Australian Research Council (DE170100509), UNSW Major Research Equipment and Infrastructure Initiative, and UNSW School of Psychology. Data reported in the paper are archived in the UNSW Long Term Data Archive (ID: D0235271).
The authors declare no competing financial interests.
References
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL (2007) Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104:5163–5168. 10.1073/pnas.0700293104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin MC, Kalivas PW (1990) Enkephalinergic and GABAergic modulation of motor activity in the ventral pallidum. J Pharmacol Exp Ther 252:1370–1377. [PubMed] [Google Scholar]
- Avila I, Lin SC (2014) Distinct neuronal populations in the basal forebrain encode motivational salience and movement. Front Behav Neurosci 8:421. 10.3389/fnbeh.2014.00421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird KD. (2004) Analysis of variance via confidence intervals. London: Sage. [Google Scholar]
- Boik RJ. (1981) A priori tests in repeated measures designs: effects of non-sphericity. Psychometrika 46:241–255. 10.1007/BF02293733 [DOI] [Google Scholar]
- Bonnavion P, Mickelsen LE, Fujita A, de Lecea L, Jackson AC (2016) Hubs and spokes of the lateral hypothalamus: cell types, circuits and behaviour. J Physiol 594:6443–6462. 10.1113/JP271946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton ME, Todd TP (2014) A fundamental role for context in instrumental learning and extinction. Behav Processes 104:13–19. 10.1016/j.beproc.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro DC, Cole SL, Berridge KC (2015) Lateral hypothalamus, nucleus accumbens, and ventral pallidum roles in eating and hunger: interactions between homeostatic and reward circuitry. Front Syst Neurosci 9:90. 10.3389/fnsys.2015.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombag HS, Shaham Y (2002) Renewal of drug seeking by contextual cues after prolonged extinction in rats. Behav Neurosci 116:169–173. 10.1037/0735-7044.116.1.169 [DOI] [PubMed] [Google Scholar]
- Crombag HS, Bossert JM, Koya E, Shaham Y (2008) Context-induced relapse to drug seeking: a review. Philos Trans R Soc Lond B Biol Sci 363:3233–3243. 10.1098/rstb.2008.0090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Olmos JS, Heimer L (1999) The concepts of the ventral striatopallidal system and extended amygdala. Ann N Y Acad Sci 877:1–32. 10.1111/j.1749-6632.1999.tb09258.x [DOI] [PubMed] [Google Scholar]
- Faget L, Zell V, Souter E, McPherson A, Ressler R, Gutierrez-Reed N, Yoo JH, Dulcis D, Hnasko TS (2018) Opponent control of behavioral reinforcement by inhibitory and excitatory projections from the ventral pallidum. Nat Commun 9:849. 10.1038/s41467-018-03125-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell MR, Ruiz CM, Castillo E, Faget L, Khanbijian C, Liu S, Schoch H, Rojas G, Huerta MY, Hnasko TS, Mahler SV (2019) Ventral pallidum is essential for cocaine relapse after voluntary abstinence in rats. Neuropsychopharmacology 44:2174–2185. 10.1038/s41386-019-0507-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GD, Prasad AA, Jean-Richard-dit-Bressel P, Yau JOY, Millan EZ, Liu Y, Campbell EJ, Lim J, Marchant NJ, Power JM, Killcross S, Lawrence AJ, McNally GP (2018) Distinct accumbens shell output pathways promote versus prevent relapse to alcohol seeking. Neuron 98:512–520.e6. 10.1016/j.neuron.2018.03.033 [DOI] [PubMed] [Google Scholar]
- Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, Ellis RJ, Richie CT, Harvey BK, Dannals RF, Pomper MG, Bonci A, Michaelides M (2017) Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357:503–507. 10.1126/science.aan2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Groenewegen HJ, Grove EA, Nauta WJ (1985) Efferent connections of the ventral pallidum: evidence of a dual striato pallidofugal pathway. J Comp Neurol 235:322–335. 10.1002/cne.902350304 [DOI] [PubMed] [Google Scholar]
- Hamlin AS, Blatchford KE, McNally GP (2006) Renewal of an extinguished instrumental response: neural correlates and the role of D1 dopamine receptors. Neuroscience 143:25–38. 10.1016/j.neuroscience.2006.07.035 [DOI] [PubMed] [Google Scholar]
- Hamlin AS, Newby J, McNally GP (2007) The neural correlates and role of D1 dopamine receptors in renewal of extinguished alcohol-seeking. Neuroscience 146:525–536. 10.1016/j.neuroscience.2007.01.063 [DOI] [PubMed] [Google Scholar]
- Hamlin AS, Clemens KJ, McNally GP (2008) Renewal of extinguished cocaine-seeking. Neuroscience 151:659–670. 10.1016/j.neuroscience.2007.11.018 [DOI] [PubMed] [Google Scholar]
- Hamlin AS, Clemens KJ, Choi EA, McNally GP (2009) Paraventricular thalamus mediates context-induced reinstatement (renewal) of extinguished reward seeking. Eur J Neurosci 29:802–812. 10.1111/j.1460-9568.2009.06623.x [DOI] [PubMed] [Google Scholar]
- Harris RJ. (2004) ANOVA: an analysis of variance primer. Itasca, Illinois: F. E. Peacock. [Google Scholar]
- Heimer L, Switzer RD, Van Hoesen GW (1982) Ventral striatum and ventral pallidum: components of the motor system? Trends Neurosci 5:83–87. 10.1016/0166-2236(82)90037-6 [DOI] [Google Scholar]
- Heinsbroek JA, Neuhofer DN, Griffin WC, Siegel GS, Bobadilla AC, Kupchik YM, Kalivas PW (2017) Loss of plasticity in the D2-accumbens pallidal pathway promotes cocaine seeking. J Neurosci 37:757–767. 10.1523/JNEUROSCI.2659-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CY, Berridge KC (2013) An orexin hotspot in ventral pallidum amplifies hedonic liking for sweetness. Neuropsychopharmacology 38:1655–1664. 10.1038/npp.2013.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks BM, Lin JY, Guo C, Svoboda K (2015) Dual-channel circuit mapping reveals sensorimotor convergence in the primary motor cortex. J Neurosci 35:4418–4426. 10.1523/JNEUROSCI.3741-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD (2013) The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science 341:1517–1521. 10.1126/science.1241812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas DE, Amick HR, Feltner C, Bobashev G, Thomas K, Wines R, Kim MM, Shanahan E, Gass CE, Rowe CJ, Garbutt JC (2014) Pharmacotherapy for adults with alcohol use disorders in outpatient settings. JAMA 311:1889–1900. 10.1001/jama.2014.3628 [DOI] [PubMed] [Google Scholar]
- Kalivas PW, O'Brien C (2008) Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology 33:166–180. 10.1038/sj.npp.1301564 [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND (2005) The neural sasis of addiction: a pathology of motivation and choice. Am J Psychiatry 162:1403–1413. 10.1176/appi.ajp.162.8.1403 [DOI] [PubMed] [Google Scholar]
- Khoo AT, Gibson GD, Prasad AA, McNally GP (2015) Role of the striatopallidal pathway in renewal and reacquisition of alcohol seeking. Behav Neurosci 129:2–7. 10.1037/bne0000036 [DOI] [PubMed] [Google Scholar]
- Khoo SY, Gibson GD, Prasad AA, McNally GP (2017) How contexts promote and prevent relapse to drug seeking. Genes Brain Behav 16:185–204. 10.1111/gbb.12328 [DOI] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Boyden ES, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, Wang J, Xie Y, Yan Z, Zhang Y, Chow BY, Surek B, Melkonian M, Jayaraman V, Constantine-Paton M, et al. (2014) Independent optical excitation of distinct neural populations. Nat Methods 11:338–346. 10.1038/nmeth.2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowland D, Lilascharoen V, Pacia CP, Shin S, Wang EH, Lim BK (2017) Distinct ventral pallidal neural populations mediate separate symptoms of depression. Cell 170:284–297.e18. 10.1016/j.cell.2017.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Kalivas PW (2013) The rostral subcommissural ventral pallidum is a mix of ventral pallidal neurons and neurons from adjacent areas: an electrophysiological study. Brain Struct Funct 218:1487–1500. 10.1007/s00429-012-0471-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung BK, Balleine BW (2013) The ventral striato-pallidal pathway mediates the effect of predictive learning on choice between goal-directed actions. J Neurosci 33:13848–13860. 10.1523/JNEUROSCI.1697-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung BK, Balleine BW (2015) Ventral pallidal projections to mediodorsal thalamus and ventral tegmental area play distinct roles in outcome-specific Pavlovian-instrumental transfer. J Neurosci 35:4953–4964. 10.1523/JNEUROSCI.4837-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Aston-Jones GS (2012) Fos activation of selective afferents to ventral tegmental area during cue-induced reinstatement of cocaine seeking in rats. J Neurosci 32:13309–13326. 10.1523/JNEUROSCI.2277-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Vazey EM, Beckley JT, Keistler CR, McGlinchey EM, Kaufling J, Wilson SP, Deisseroth K, Woodward JJ, Aston-Jones G (2014) Designer receptors show role for ventral pallidum input to ventral tegmental area in cocaine seeking. Nat Neurosci 17:577–585. 10.1038/nn.3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NJ, Hamlin AS, McNally GP (2009) Lateral hypothalamus is required for context-induced reinstatement of extinguished reward seeking. J Neurosci 29:1331–1342. 10.1523/JNEUROSCI.5194-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NJ, Rabei R, Kaganovsky K, Caprioli D, Bossert JM, Bonci A, Shaham Y (2014) A critical role of lateral hypothalamus in context-induced relapse to alcohol seeking after punishment-imposed abstinence. J Neurosci 34:7447–7457. 10.1523/JNEUROSCI.0256-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant NJ, Kaganovsky K, Shaham Y, Bossert JM (2015) Role of corticostriatal circuits in context-induced reinstatement of drug seeking. Brain Res 1628:219–232. 10.1016/j.brainres.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlatt GA, Donovan DM (2005) Relapse prevention. New York: Guildford. [Google Scholar]
- McFarland K, Kalivas PW (2001) The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 21:8655–8663. 10.1523/JNEUROSCI.21-21-08655.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Davidge SB, Lapish CC, Kalivas PW (2004) Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci 24:1551–1560. 10.1523/JNEUROSCI.4177-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally GP. (2005) Facilitation of fear extinction by midbrain periaqueductal gray infusions of RB101 (S), an inhibitor of enkephalin-degrading enzymes. Behav Neurosci 119:1672–1677. 10.1037/0735-7044.119.6.1672 [DOI] [PubMed] [Google Scholar]
- Mickelsen LE, Bolisetty M, Chileski BR, Fujita A, Beltrami EJ, Costanzo JT, Naparstek JR, Robson P, Jackson AC (2019) Single-cell transcriptomic analysis of the lateral hypothalamic area reveals molecularly distinct populations of inhibitory and excitatory neurons. Nat Neurosci 22: 642–656. 10.1038/s41593-019-0349-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogenson GJ, Jones DL, Yim CY (1980) From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol 14:69–97. 10.1016/0301-0082(80)90018-0 [DOI] [PubMed] [Google Scholar]
- Moussawi K, Kalivas PW, Lee JW (2016) Abstinence from drug dependence after bilateral globus pallidus hypoxic ischemic injury. Biol Psychiatry 80:e79–e80. 10.1016/j.biopsych.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier TC, Mitrovic I (1999) Opioid modulation of ventral pallidal inputs. Ann N Y Acad Sci 877:176–201. 10.1111/j.1749-6632.1999.tb09268.x [DOI] [PubMed] [Google Scholar]
- Nieh EH, Vander Weele CM, Matthews GA, Presbrey KN, Wichmann R, Leppla CA, Izadmehr EM, Tye KM (2016) Inhibitory input from the lateral hypothalamus to the ventral tegmental area disinhibits dopamine neurons and promotes behavioral activation. Neuron 90:1286–1298. 10.1016/j.neuron.2016.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuijten MB, Hartgerink CH, van Assen MA, Epskamp S, Wicherts JM (2016) The prevalence of statistical reporting errors in psychology (1985–2013). Behav Res Methods 48:1205–1226. 10.3758/s13428-015-0664-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor EC, Kremer Y, Lefort S, Harada M, Pascoli V, Rohner C, Lüscher C (2015) Accumbal D1R neurons projecting to lateral hypothalamus authorize feeding. Neuron 88:553–564. 10.1016/j.neuron.2015.09.038 [DOI] [PubMed] [Google Scholar]
- Pardo-Garcia TR, Garcia-Keller C, Penaloza T, Richie CT, Pickel J, Hope BT, Harvey BK, Kalivas PW, Heinsbroek JA (2019) Ventral pallidum is the primary target for accumbens D1 projections driving cocaine seeking. J Neurosci 39:2041–2051. 10.1523/JNEUROSCI.2822-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinates. 6th Edition Elsevier. [DOI] [PubMed] [Google Scholar]
- Perry CJ, McNally GP (2013) A role for the ventral pallidum in context-induced and primed reinstatement of alcohol seeking. Eur J Neurosci 38:2762–2773. 10.1111/ejn.12283 [DOI] [PubMed] [Google Scholar]
- Prasad AA, McNally GP (2016) Ventral pallidum output pathways in context-induced reinstatement of alcohol seeking. J Neurosci 36:11716–11726. 10.1523/JNEUROSCI.2580-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard JM, Ambroggi F, Janak PH, Fields HL (2016) Ventral pallidum neurons encode incentive value and promote cue-elicited instrumental actions. Neuron 90:1165–1173. 10.1016/j.neuron.2016.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Fabbricatore AT, Ma S, Barker DJ, West MO (2010) Rapid phasic activity of ventral pallidal neurons during cocaine self-administration. Synapse 64:704–713. 10.1002/syn.20792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Melendez RI, Zaborszky L, Napier TC (2015) The ventral pallidum: subregion-specific functional anatomy and roles in motivated behaviors. Prog Neurobiol 130:29–70. 10.1016/j.pneurobio.2015.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio FJ, Liu QR, Li X, Cruz FC, Leão RM, Warren BL, Kambhampati S, Babin KR, McPherson KB, Cimbro R, Bossert JM, Shaham Y, Hope BT (2015) Context-induced reinstatement of methamphetamine seeking is associated with unique molecular alterations in Fos-expressing dorsolateral striatum neurons. J Neurosci 35:5625–5639. 10.1523/JNEUROSCI.4997-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe MJ, Marchant NJ, Whitaker LR, Richie CT, Zhang YJ, Campbell EJ, Koivula PP, Necarsulmer JC, Mejias-Aponte C, Morales M, Pickel J, Smith JC, Niv Y, Shaham Y, Harvey BK, Schoenbaum G (2017) Lateral hypothalamic GABAergic neurons encode reward predictions that are relayed to the ventral tegmental area to regulate learning. Curr Biol 27:2089–2100.e5. 10.1016/j.cub.2017.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Berridge KC (2005) The ventral pallidum and hedonic reward: neurochemical maps of sucrose “liking” and food intake. J Neurosci 25:8637–8649. 10.1523/JNEUROSCI.1902-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Tindell AJ, Aldridge JW, Berridge KC (2009) Ventral pallidum roles in reward and motivation. Behav Brain Res 196:155–167. 10.1016/j.bbr.2008.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanik MT, Kupchik YM, Brown RM, Kalivas PW (2013) Optogenetic evidence that pallidal projections, not nigral projections, from the nucleus accumbens core are necessary for reinstating cocaine seeking. J Neurosci 33:13654–13662. 10.1523/JNEUROSCI.1570-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford TR, Wirtshafter D (2013) Lateral hypothalamic involvement in feeding elicited from the ventral pallidum. Eur J Neurosci 37:648–653. 10.1111/ejn.12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XC, McFarland K, Cagle S, Kalivas PW (2005) Cocaine-induced reinstatement requires endogenous stimulation of mu-opioid receptors in the ventral pallidum. J Neurosci 25:4512–4520. 10.1523/JNEUROSCI.0685-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooley J, Marconi L, Alipio JB, Matikainen-Ankney B, Georgiou P, Kravitz AV, Creed MC (2018) Glutamatergic ventral pallidal neurons modulate activity of the habenula-tegmental circuitry and constrain reward seeking. Biol Psychiatry 83:1012–1023. 10.1016/j.biopsych.2018.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, Charlson FJ, Norman RE, Flaxman AD, Johns N, Burstein R, Murray CJ, Vos T (2013) Global burden of disease attributable to mental and substance use disorders: findings from the global burden of disease study 2010. Lancet 382:1575–1586. 10.1016/S0140-6736(13)61611-6 [DOI] [PubMed] [Google Scholar]
- Willcocks AL, McNally GP (2011) The role of context in re-acquisition of extinguished alcoholic beer-seeking. Behav Neurosci 125:541–550. 10.1037/a0024100 [DOI] [PubMed] [Google Scholar]
- Zahm DS, Zaborszky L, Alheid GF, Heimer L (1987) The ventral striatopallidothalamic projection: II. The ventral pallidothalamic link. J Comp Neurol 255:592–605. 10.1002/cne.902550410 [DOI] [PubMed] [Google Scholar]