

Abstract
C1q, the initiator of the classical complement cascade, mediates synapse elimination in the postnatal mouse dorsolateral geniculate nucleus of the thalamus and sensorimotor cortex. Here, we asked whether C1q plays a role in experience-dependent synaptic refinement in the visual system at later stages of development. The binocular zone of primary visual cortex (V1b) undergoes spine loss and changes in neuronal responsiveness following the closure of one eye during a defined critical period [a process referred to as ocular dominance plasticity (ODP)]. We therefore hypothesized that ODP would be impaired in the absence of C1q, and that V1b development would also be abnormal without C1q-mediated synapse elimination. However, when we examined several features of V1b development in mice lacking C1q, we found that the densities of most spine populations on basal and proximal apical dendrites, as well as firing rates and ocular dominance, were normal. C1q was only transiently required for the development of spines on apical, but not basal, secondary dendrites. Dendritic morphologies were also unaffected. Although we did not observe the previously described spine loss during ODP in either genotype, our results reveal that the animals lacking C1q had normal shifts in neuronal responsiveness following eye closure. Experiments were performed in both male and female mice. These results suggest that the development and plasticity of the mouse V1b is grossly normal in the absence of C1q.
SIGNIFICANCE STATEMENT These findings illustrate that the development and experience-dependent plasticity of V1b is mostly normal in the absence of C1q, even though C1q has previously been shown to be required for developmental synapse elimination in the mouse visual thalamus as well as sensorimotor cortex. The V1b phenotypes in mice lacking C1q are more similar to the mild defects previously observed in the hippocampus of these mice, emphasizing that the contribution of C1q to synapse elimination appears to be dependent on context.
Keywords: C1q, classical complement cascade, neural-immune interactions, ocular dominance plasticity, synapse elimination, visual cortex
Introduction
Synapse formation and elimination occur continuously in subsets of synapses in the brain (Grutzendler et al., 2002; Trachtenberg et al., 2002). Synapse elimination is prominent during development in both mice and humans (Huttenlocher, 1979; Hong and Chen, 2011; Kano et al., 2018), and can be increased by changes in sensory experience (Trachtenberg et al., 2002; Holtmaat et al., 2006; Keck et al., 2008). Excessive synapse elimination has been implicated in neuropsychiatric and neurodegenerative disease (Mucke and Selkoe, 2012; Glausier and Lewis, 2013). Thus, understanding the cellular and molecular mechanisms that regulate this process is of great importance to human health.
Synapse elimination is associated with the induction of ocular dominance plasticity (ODP), a classic model of experience-dependent plasticity in the visual system (Mataga et al., 2004; Coleman et al., 2010; Zhou et al., 2017). In normal mice, the responsiveness of neurons in the binocular zone of primary visual cortex (V1b) is dominated by input from the contralateral eye. However, depriving the contralateral eye of input for 3–4 d by monocular deprivation (MD) during the critical period [∼postnatal day (P)19–P32 in the mouse; Gordon and Stryker, 1996)], causes a shift in V1b responsiveness toward the non-deprived ipsilateral eye. Synapse loss in V1b accompanies this shift (Mataga et al., 2004; Coleman et al., 2010; Zhou et al., 2017). ODP can thus be used as a model to study the mechanisms mediating experience-dependent synapse elimination.
The innate immune molecule C1q is a known regulator of synaptic development, and a candidate for regulating synapse elimination in V1b. C1q is a secreted protein, which in the immune system binds to pathogens and apoptotic cells and initiates a proteolytic cascade referred to as the classical complement cascade (Ricklin et al., 2010; Thielens et al., 2017). In at least some parts of the brain, C1q is thought to mediate synapse elimination by binding to synapses and locally activating the classical complement cascade, which results in engulfment of presynaptic inputs by microglia (Stevens et al., 2007; Schafer et al., 2012). Mice lacking C1q show impaired anatomical and electrophysiological refinement of retinal ganglion cell (RGC) inputs to the dorsolateral geniculate nucleus of the thalamus (dLGN) at 1 month of age, demonstrating sustained defects in connectivity (Stevens et al., 2007). Furthermore, mice lacking C1q show increased densities of dendritic spines in sensorimotor cortex, have increased pyramidal neuron dendritic branching, and are prone to seizures (Chu et al., 2010; Ma et al., 2013). Whether C1q promotes synapse elimination in V1b during development or in response to changing visual experience has, however, not been examined.
Here, we investigated the role of C1q in the development and plasticity of V1b in mice. In mice lacking C1q, we counted spines to examine synapse development and elimination, performed in vivo electrophysiology to measure firing rates, and used electrophysiology and in situ hybridization against the immediate early gene Arc to determine how loss of C1q affects ODP. We found that although C1q is present in V1b during the critical period, it was not required for the development of most spine populations on layer (L)2/3 pyramidal neurons. The dendritic arbors of these neurons were also unaffected by loss of C1q. In vivo electrophysiological recordings furthermore revealed normal spontaneous and visually evoked firing rates in V1b in the absence of C1q. Spine loss following critical period MD has previously been described on the apical dendrites of L2/3 pyramidal neurons (Mataga et al., 2004), but we failed to observe MD-induced spine loss in either genotype. However, we found normal ocular dominance (OD) shifts in mice lacking C1q compared with their wild-type (WT) littermates. Together, these findings thus indicate that in V1b of mice lacking C1q, the development and plasticity of neuronal morphology and eye-specific inputs are largely normal.
Materials and Methods
Mice.
All procedures were approved by the Boston Children's Hospital institutional animal care and use committee in accordance with NIH guidelines for the humane treatment of animals. C1qa−/− mice are on a C57BL/6J background and were a kind gift from M. Botto (Imperial College, London). WT animals used in Figures 1 and 7 were purchased from the The Jackson Laboratory (stock #000664), or derived from C1qa+/−x C1qa+/−crosses. All other animals were derived from C1qa+/−x C1qa+/−crosses.
Figure 1.
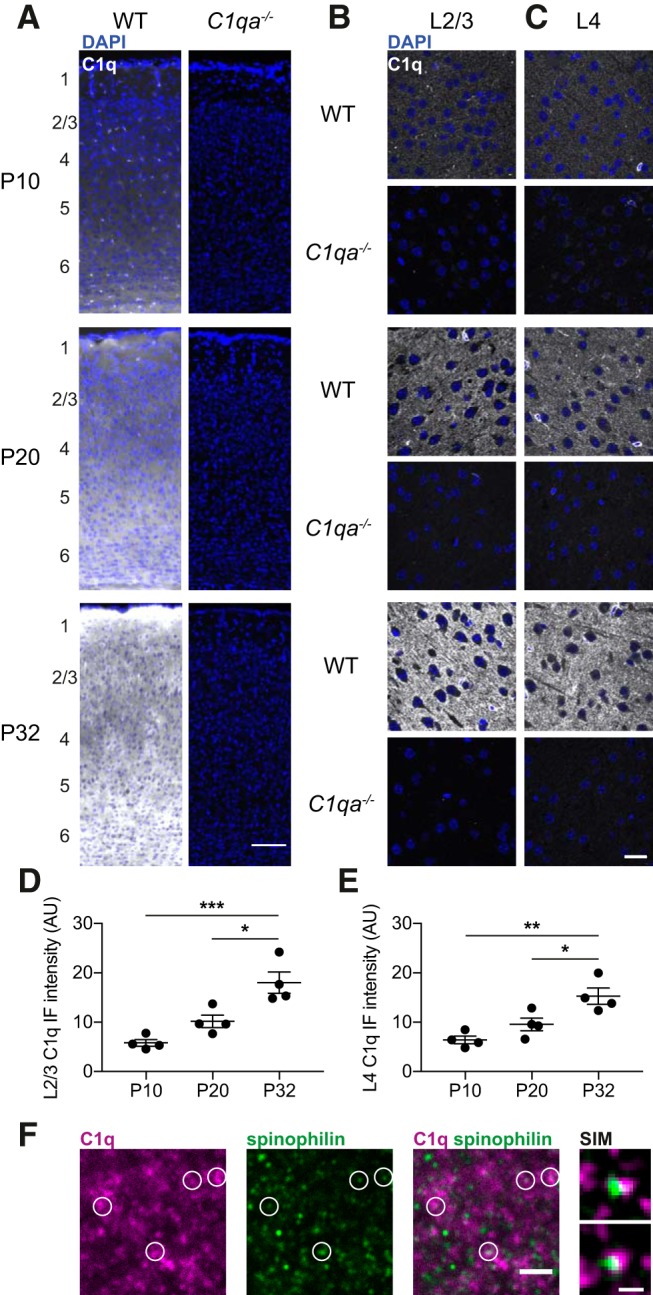
C1q is present in the developing V1b, and levels increase with age. A, Representative images of immunohistochemistry against C1q (white) and DAPI (blue) across cortical layers in V1b at P10 (top), P20 (middle), and P32 (bottom), of WT (left) and C1qa−/− (right) animals. Approximate locations of the cortical layers are noted to the left of the images. Scale bar, 100 μm. B, C, Representative confocal images of immunohistochemistry against C1q (white) and DAPI (blue) in L2/3 (B) and L4 (C) in V1b of WT and C1qa−/− animals, at P10 (top), P20 (middle), and P32 (bottom). The C1q signal is absent in C1qa−/− tissue, demonstrating specificity of the antibody. Scale bar, 20 μm. D, Quantification of C1q confocal immunofluorescence intensities in L2/3 of V1b across ages. C1q levels are significantly higher at P32 than at P20 or P10 (one-way ANOVA, F(2,9) = 17.04, p = 0.0009, n = 4 animals/age. P10 vs P20, p = 0.1538, P10 vs P32, p = 0.0007, P20 vs P32, p = 0.0123, Tukey's multiple comparisons test). E, Quantification of C1q confocal immunofluorescence intensities in L4 of V1b across ages shows significantly higher C1q levels at P32 than at P20 or P10 (one-way ANOVA, F(2,9) = 12.12, p = 0.0028, n = 4 animals/age. P10 vs P20, p = 0.2468, P10 vs P32, p = 0.0023, P20 vs P32, p = 0.0297, Tukey's multiple-comparisons test). F, Left, Representative confocal image of immunohistochemistry against C1q (magenta) and spinophilin (green) in L2/3 of V1b at P32, in a WT animal. Images are representative of n = 3 animals. White circles mark examples of spinophilin puncta that overlap with, or are closely apposed to, C1q puncta. Scale bar, 2 μm. Right, Two examples of super-resolution SIM images of partially overlapping C1q and spinophilin puncta (not from the same sections as the confocal image to the left) from P29–P30 WT mice. Images are representative of n = 3 animals. Scale bar, 0.3 μm. All error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 7.
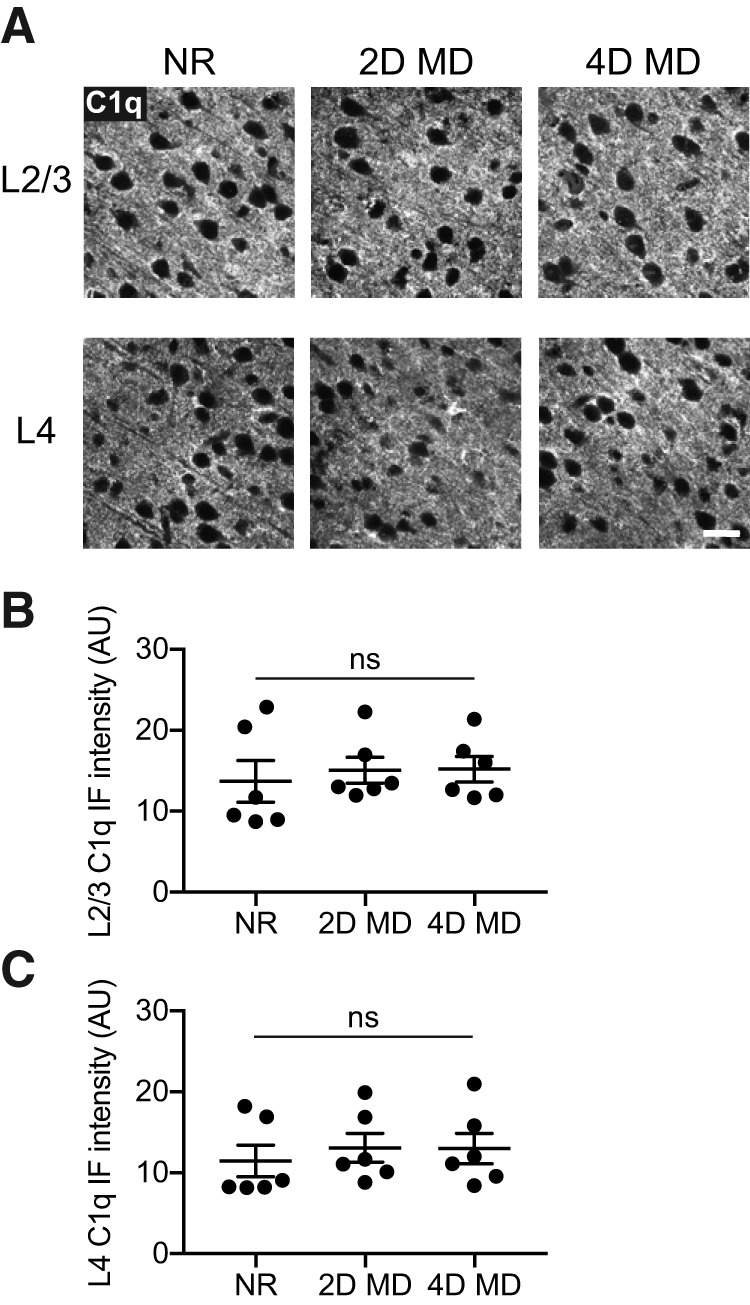
C1q levels in V1b do not change with monocular deprivation. A, Representative images of C1q immunohistochemistry in L2/3 (top) and L4 (bottom) of V1b, from P32 mice that were NR, or from animals that had undergone 2 d (2D MD) or 4 d (4D MD) of MD starting at P28. Images are from the hemisphere contralateral to the deprived eye. Scale bar, 20 μm. B, Quantification of C1q immunofluorescence intensities in L2/3 of V1b in NR, 2D MD, and 4D MD animals. The conditions are not statistically different from one another (one-way ANOVA, F(2,15) = 0.1803, p = 0.8368, n = 6 animals/condition). C, Quantification of C1q immunofluorescence intensities in L4 of V1b in NR, 2D MD, and 4D MD animals. There is no statistically significant difference in immunofluorescence intensities between the conditions (one-way ANOVA, F(2,15) = 0.2338, p = 0.7943, n = 6 animals/condition). All error bars represent SEM. ns, not significant.
Monocular deprivation and enucleation.
MD was performed under isoflurane anesthesia. The eyelids were sutured together with a single mattress suture using nylon sutures (Ethicon, catalog #G697G). Monocular enucleation (ME) was performed under isoflurane anesthesia. If the eye to be enucleated had previously been sutured, the sutures were removed and the eyelids opened. The eyelid margins were trimmed. The optic nerve was then cut with scissors and the eye removed, and the eyelids sealed shut with cyanoacrylate.
C1q immunohistochemistry.
Mice were anesthetized with Avertin (240 mg/kg, i.p.) and transcardially perfused with PBS. Brains were dissected out and drop fixed in 4% PFA (Electron Microscopy Sciences, catalog #15710) for 2 h at room temperature. Brains were then washed in PBS, transferred to 30% sucrose in PBS, and kept in sucrose at 4°C for 24–48 h. Brains were embedded in a 2:1 mixture of 30% sucrose in PBS and OCT (Sakura Finetek, catalog #4583), and sectioned at 30 μm on a cryostat onto X-tra slides (Leica Microsystems, catalog #3800200). Tissue sections were dried, washed in PBS, and blocked for 2 h at room temperature with slow agitation in 10% normal goat serum (Sigma-Aldrich, catalog #G9023–10ML) with 1% TX-100 (Sigma-Aldrich, catalog #T8787–100ML) in PBS. Primary antibody (rabbit anti-C1q, 1:500; Abcam, catalog #ab182451; RRID:AB_2732849) was applied overnight, in 5% normal goat serum with 0.5% TX-100 in PBS, at room temperature with slow agitation. Tissue sections were then washed 3× 10 min with PBS and incubated with the appropriate AlexaFluor-conjugated secondary antibody (1:200; Invitrogen/ThermoFisher Scientific) for 2 h, in 5% normal goat serum with 0.1% TX-100 in PBS, at room temperature with slow agitation. Finally, tissue sections were mounted with VECTASHIELD with DAPI (Vector Laboratories, catalog #H-1000). Immunofluorescence intensity was calculated in ImageJ (NIH) from images taken on an SPE confocal microscope (Leica). Layers were identified by examining the density of DAPI-positive nuclei. Epifluorescence images were taken on a VS120 Virtual Slide Microscope (Olympus).
For C1q and spinophilin staining, tissue collection was performed as described in the previous paragraph. Brains were sectioned at 14 μm on a cryostat onto X-tra slides (Leica Microsystems, catalog #3800200). Tissue sections were dried, washed in PBS, and blocked for 2 h at room temperature in 5% donkey serum (Sigma-Aldrich, catalog #D9663-10ML) with 0.3% TX-100 (Sigma-Aldrich, catalog #T8787-100ML) in PBS. Primary antibody (rabbit anti-C1q, 1:500; Abcam, catalog #ab182451; RRID:AB_2732849; sheep anti-spinophilin/PPP1R9B, 1:200; R&D Systems, catalog #AF6465; RRID:AB_10718703) was applied overnight, in 5% donkey serum with 0.3% TX-100 in PBS, at 4°C. Tissue sections were then washed 3× 10 min with PBS and incubated with the appropriate AlexaFluor-conjugated secondary antibody (donkey anti-rabbit, 1:200; Invitrogen/ThermoFisher Scientific; donkey anti-sheep, 1:200; Jackson ImmunoResearch) for 2 h, in 5% donkey serum with 0.3% TX-100 in PBS, at room temperature. Finally, tissue sections were mounted with VECTASHIELD with DAPI (Vector Laboratories, catalog #H-1000). Images were taken on an SP8 confocal microscope (Leica). Layers were identified by examining the density of DAPI-positive nuclei.
For structured illumination microscopy (SIM) imaging, staining was performed in the same way, except Hoechst 33342 (Invitrogen/ThermoFisher Scientific, catalog #H3570) was added to the secondary antibody at 1:10,000, and the sections were mounted with ProLong Glass Antifade Mountant (Invitrogen/ThermoFisher Scientific, catalog #P36984). Images were then acquired with five grating rotations using an ELYRA PS1 Superresolution Microscope (Zeiss), and processed using the Zeiss SIM algorithms.
Colocalization analysis.
Confocal images of C1q and spinophilin immunostaining were acquired on an SP8 confocal microscope (Leica) as described. Images were then processed in Ilastik (Sommer et al., 2011), which is an image processing software that can be trained to recognize various features in images, without being explicitly told which criteria to use for feature recognition. Ilastik was trained to recognize C1q and spinophilin immunostaining using separate training paradigms for each marker, and to output segmented images. The overlap of C1q and spinophilin puncta was then calculated in ImageJ (NIH) using the Analyze Particles function, and compared with the overlap obtained when the segmented C1q channel was rotated by 90°.
Spine counting.
Spines were counted in animals aged P10, P20, and P29–P30. Animals in the oldest group were either normally reared, or had undergone 4 d of MD with MD starting at P25–P26. Spines were visualized using the FD Rapid GolgiStain Kit (FD NeuroTechnologies, catalog #PK401A), according to the manufacturer's instructions. In brief, animals were anesthetized, decapitated, and brains were washed in double-deionized water before immersion in solution A+B. After 4 weeks of incubation, brains were transferred to solution C for 3–7 d. They were then embedded in ice and sectioned coronally at 100 μm on a cryostat. Sections were placed on gelatin-coated slides (FD NeuroTechnologies, catalog #PO101), stained with solutions D+E, dehydrated with ethanol and cleared with xylene. Sections were then mounted under coverslips with Permount (Fisher Scientific, catalog #SP15-500).
Golgi-Cox stained sections were imaged under bright-field on a Virtual Slide Microscope VS120 (Olympus) and on an E800 microscope (Nikon). Using these images, cells were selected for subsequent higher-magnification imaging and spine counts. Cells that fit the following selection criteria were chosen: located in L2/3 of V1b (preferably but not exclusively in deeper L2/3); located superficially in the slice for better imaging quality; no highly clustered cells where it would be difficult to determine to which cell the apical dendrite belonged. For the P10 and P20 age groups cells in both hemispheres were used, whereas cells in the P29–P30 normally reared (NR) and MD groups were only in the hemisphere contralateral to the deprived eye. Depth of cortical layers was determined using the Allen Mouse Brain Atlas (http://portal.brain-map.org/; Sunkin et al., 2012). Identification was performed blind to genotype and experimental paradigm.
Image stacks of Golgi-stained cells that passed the selection process were imaged on a BX63 upright bright-field microscope (Olympus), using a 100× objective for the primary and secondary apical dendrite spine quantification, and a 60× objective for the secondary basal dendrite spine quantification and for Sholl analysis. Light source intensity was adjusted for each cell to account for differences in tissue clarity. When a cell was partially obscured by other stained neurons or by background signal, it was only used for some of the quantifications; the number of cells or mice used thus varies between the quantifications.
For measuring the distance between the soma and the first branch point, and to identify the 25 μm segment distal to the first branch point, cells were reimaged on an SP8 confocal microscope (Leica) by detecting reflected light (Spiga et al., 2011). Measurements were then made in the FilamentTracer module in Imaris v7.7.2. For measuring the length of dendrite segments on secondary branches (apical and basal), dendrite lengths were measured using the Simple Neurite Tracer plugin in ImageJ (Longair et al., 2011). Dendrite segments on secondary branches used for spine quantification generally started at the first branch point, and ended when the dendrite ended, branched again, moved out of the section, or was obscured by other neurites or noise. Spines were then counted manually in ImageJ, with the experimenter blind to genotype and experimental paradigm. For each animal, 2–8 cells were counted.
Sholl analysis.
The arbors of the same neurons used for spine counting were reconstructed manually using the Simple Neurite Tracer plugin in ImageJ (Longair et al., 2011), blind to genotype. Sholl analysis was then performed on the traced apical and basal arbors using the Sholl analysis plugin in ImageJ (Ferreira et al., 2014).
Electrophysiological recordings in primary visual cortex.
Recording methods were adapted from previously published methods (Stephany et al., 2014, 2016, 2018). Mice that were either normally reared, or had undergone 4 d of MD, were anesthetized with isoflurane (2%) and placed in a stereotaxic frame where body temperature was maintained at 37°C using a homeostatically-regulated heat probe (TCAT-2LV, Physitemp). Dexamethasone (4 mg/kg; West-Ward Pharmaceuticals) was administered subcutaneously to reduce cerebral edema. The eyes were flushed with saline, covered with a thin layer of silicone oil (Sigma-Aldrich, catalog #378429), and then covered for the duration of craniotomy. The scalp was resected and a titanium head bar was secured to the skull with cyanoacrylate glue and dental cement (Ortho-Jet Liquid and Ortho-Jet Powder, Lang Dental). A small craniotomy (2 mm in diameter) was made over V1, and a small silver grounding wire (A-M Systems, catalog #782500) was inserted through a burr hole that was made over the contralateral cerebellum. Then, a dose of chlorprothixene (0.5 mg/kg, s.c.; Sigma-Aldrich, catalog #1671) was administered before transferring the mouse to the recording setup.
Recordings were made with silicone multisite electrodes (A1x16-5 mm-50-177-A16, NeuroNexus Technologies) inserted into V1 using a motorized drive (MP-225, Sutter Instruments). Once the electrode was in place, warm agarose (2.5% in ACSF) was placed over the craniotomy to protect the cortex and to reduce mechanical noise. The electrical signal was sampled at 25 kHz on a RZ5P Workstation (Tucker-Davis Technologies), and filtered between 300 and 5000 Hz for spiking activity. Multiunit activity was recorded on all 16 channels (separated by 50 μm) starting 100 μm below the pial surface. In each mouse, there were 3–4 penetrations separated by at least 200 μm across the binocular zone of V1. Subsequently, noise artifacts were removed and single units were isolated from the multiunit activity at each contact site using Offline Sorter (Plexon).
Animals were immobilized using the titanium head bar at a distance of 25 cm from a video monitor (ASUS VG248QE 24 inch LED, Backlit LCD Monitor) with a refresh rate of 144 Hz, and a mean luminance of 40 cd/cm2. Visual responses were driven by custom stimuli generated in MATLAB Psychophysics Toolbox. First, a sparse noise stimulus was presented to both eyes at the beginning of each penetration, and the resulting activity map was used to determine receptive field location. Full luminance (ON) and minimum luminance (OFF) squares covering 4° of visual space were flashed (350 ms) one at a time on a gray background in a pseudorandom order along a 16 × 10 grid subtending 64° of visual space in azimuth, and 40° of visual space in elevation. Activity from sites outside of the binocular zone, as defined by a receptive field >25° in azimuth, were excluded from subsequent analysis. Then, responses were driven by sinusoidal gratings (0.02 cpd, 100% contrast, 2 Hz temporal frequency) drifting in 12 directions separated by 30° for 2 s ON followed by 2 s of a luminance-matched gray screen. Each stimulus condition was repeated at least 10 times in a pseudorandom order, interleaved by a gray screen to measure spontaneous activity. Action potentials (APs) were identified online in recorded traces with Synapse Software (Tucker Davis Technologies). Only waveforms extending beyond 4 SD above the average noise were included in subsequent analysis. For each contact site, the number of APs in response to the grating stimuli was summed and averaged over the number of presentations. If the average number of APs for the grating stimuli was not >50% above the blank, the contact site was discarded.
Single-unit activity was used to calculate the average spontaneous and visually evoked activity. The spontaneous activity was calculated from the average number of APs elicited by the blank, whereas the visually evoked activity was calculated by subtracting the spontaneous activity of that unit from the average number of APs elicited by the grating in the unit's preferred orientation.
The ocular dominance index (ODI) was calculated for the multiunit activity on each contact site within the binocular zone by comparing the number of APs elicited in a given unit when showing the same visual stimulus to each eye independently. The order of presentation (contralateral then ipsilateral vs ipsilateral then contralateral) was alternated between penetrations for each animal. Units were assigned to one of seven ocular dominance (OD) categories (1–7) where units assigned to Category 1 are largely dominated by input from the contralateral eye, and units assigned to Category 7 are largely dominated by input from the ipsilateral eye (Wiesel and Hubel, 1963). To categorize each contact site, the average number of APs elicited by the blank (spontaneous activity) was subtracted from the average number of APs elicited by the gratings (evoked activity) for the contralateral eye (CE) and the ipsilateral eye (IE), across all grating orientations. Next, the ODI, given by ODI = (IE − CE)/(IE + CE) was calculated for each contact site and assigned to OD Categories 1–7 as follows: −1 to −0.6 = 1, −0.6 to −0.4 = 2, −0.4 to −0.1 = 3, −0.1 to 0.1 = 4, 0.1 to 0.4 = 5, 0.4 to 0.6 = 6, 0.6 to 1 = 7. Finally, the sum of the number of cells in each category was used to calculate the contralateral bias index (CBI) for each animal with the following formula: CBI = [(n1 − n7) + (2/3)(n2 − n6) + (1/3)(n3 − n5) + N]/2N where N is the total number of contact sites and nx is the number of contact sites with OD scores equal to x (Gordon and Stryker, 1996).
Measuring binocular zone expansion.
ODP was assayed by measuring binocular zone expansion similar to previous publications (Tagawa et al., 2005; Bochner et al., 2014). For MD experiments, animals underwent MD starting at P25–P28. After 4 d of MD, when the mice were P29–P32, the suture was opened and the deprived eye enucleated. Age-matched littermates (NR in Results) that had not undergone MD were also ME at this time (i.e., at age P29–P32). The enucleated mice were placed in the dark overnight (16 h), then exposed to bright lights for 30 min and finally decapitated under isoflurane anesthesia. Both MD and NR conditions thus received strong stimulation of the remaining eye before kill, thereby making it possible to visualize the binocular region in the hemisphere ipsilateral to the remaining eye, but only the MD mice were given time to adapt to the change in visual input.
Brains were flash frozen on liquid nitrogen and embedded in OCT (Sakura Finetek, catalog #4583). The brains were sectioned coronally at 16 μm on a cryostat onto X-tra slides (Leica Microsystems, catalog #3800200). Arc in situ hybridization was then performed using RNAscope Fluorescent Multiplex kits (ACD, catalog #320850) with the probe against mouse Arc in color 1 (ACD, catalog #316911), using alt B in the Amp 4 amplification step. The in situ hybridization was performed according to the manufacturer's instructions, except for the protease treatment being shortened to 3–10 min (shorter times for lower quality tissue).
The hemisphere ipsilateral to the remaining eye (contralateral to the deprived eye) was imaged on a TiEclipse microscope (Nikon). The binocular zone of V1 was imaged from bregma −3.15 to −3.87. Sections without signal in L2/3 of V1b were discarded. The length of the Arc-positive zone in L4 and L2/3 was calculated in ImageJ (NIH) as the full-width at half-maximum (FWHM), as has been done previously (William et al., 2017). To measure the FWHM, the half-maximum was first determined. The maximum intensity was defined as the average intensity in a 121.5 × 121.5 μm square placed in the center of V1b of the layer analyzed, while the minimum was defined as the average intensity in a same-sized square placed in the monocular section of V1 (V1m) of the layer analyzed. The half-maximum was calculated as the value halfway between these two intensities. The Arc intensity was then measured across V1 in the following manner. A polygon was drawn around the layer-of-interest in V1b and surrounding areas. The Arc signal was filtered inside this polygon using a mean filter with radius 50 pixels (= 81 μm). A line of width 50 pixels (= 81 μm) was drawn through the filtered region, parallel to the cortical layer and the surface of the brain. The intensity profile along the line was plotted, and the FWHM, and thus the length of the binocular zone, was defined as the length for which the Arc intensity was above the half-maximum. If there were multiple peaks above the half-maximum, the original image was examined to determine whether the binocular zone spanned the peaks or consisted of only one of them. V1b Arc fluorescence intensity was quantified by dividing the maximum intensity by the minimum intensity, using the same measurements as the ones used to determine the FWHM. Analysis was performed blind to genotype and experimental condition.
For ME experiments, mice were monocularly enucleated at P28. After 4 d, at P32, the ME mice were placed in the dark overnight (16 h). Age-matched littermates (NR in Results) that had not previously undergone enucleation were monocularly enucleated at this time and also placed in the dark overnight. The next morning, at P33, mice were then light exposed, and tissue collected and sectioned, as described for the MD animals. For the ME group, Arc transcript was detected using colorimetric in situ hybridization, similar to previous studies (Lein and Shatz, 2000; William et al., 2012). The full-length mouse Arc probe was generously shared with us by C. Shatz (Stanford University), who had received it from P. Whorley (Lyford et al., 1995). DNA was digested with NotI, transcribed from the T7 promoter, and labeled with Digoxigenin using a kit (Sigma-Aldrich, catalog #11175025910). Sections were allowed to thaw at room temperature immediately before use, and then fixed for 10 min in 4% PFA (Electron Microscopy Sciences, catalog #15710) in PBS. Sections were permeabilized with proteinase K for 5 min, fixed for another 5 min in 4% PFA (Electron Microscopy Sciences, catalog #15710), and then acetylated for 10 min. Sections were blocked with yeast RNA for 1 h, then incubated overnight 72°C with an antisense Digoxigenin-labeled Arc probe under coverslips. Coverslips were removed in 5× SSC. Slides were then washed in 0.2× SSC and blocked with 10% normal goat serum (Sigma-Aldrich, catalog #G9023-10ML) at room temperature. Slides were incubated overnight at 4°C in sheep anti-Digoxigenin-AP (1:3000, Fab fragments; Sigma-Aldrich, catalog #11093274910) in 1% normal goat serum (Sigma-Aldrich, catalog #G9023-10ML). Finally, the signal was developed using NBT/BCIP solution (Sigma-Aldrich, catalog #11681451001).
The hemisphere ipsilateral to the remaining eye was imaged under bright-field on a TiEclipse microscope (Nikon). Images were inverted and then analyzed as described for the RNAscope in situs.
Statistics/experimental design.
Both males and females were used for all experiments. Data analysis was performed in GraphPad Prism 8, except for the already mentioned analyses performed using MATLAB. More information about statistical design can be found in the Results and in the figure legends. α = 0.05 unless otherwise stated.
Results
C1q is present in juvenile V1b
C1q is a large complex, consisting of peptides encoded by the three genes C1qa, C1qb, and C1qc. All three genes are highly expressed by microglia and macrophages across the brain, and at negligible levels by other cell types (Fonseca et al., 2017; Saunders et al., 2018; Zeisel et al., 2018; Hammond et al., 2019). We therefore expected C1q protein to be present also in V1b. We performed immunohistochemistry using an antibody raised against the entire C1q complex (Stephan et al., 2013), and examined the staining patterns at P10, P20, and P32; the latter two ages approximately mark the beginning and the end of the critical period for ODP in the mouse (Gordon and Stryker, 1996). At low-magnification, C1q was present across cortical layers at all three ages (Fig. 1A). Because ODP has primarily been studied in L2/3 and in L4, we used confocal imaging to specifically examine these layers. Loss of the C1qa gene is sufficient to disrupt the protein complex and abolish classical complement cascade activation (Botto et al., 1998), and immunoreactivity of the C1q antibody was previously shown to be absent in C1qa−/− animals (Stephan et al., 2013). We therefore used these mice as negative controls for the immunohistochemistry.
At all three ages, C1q protein was present above C1qa−/− levels in both L2/3 and L4 (Fig. 1A–C). The amount of C1q was low at P10, but the immunofluorescence intensities increased with age, including during the critical period (Fig. 1D,E). This finding applied both to L2/3 and L4 (one-way ANOVA with Tukey's multiple-comparisons test. L2/3: F(2,9) = 17.04, p = 0.0009, P10 vs P32, p = 0.0007, P20 vs P32, p = 0.0123; L4: F(2,9) = 12.12, p = 0.0028, P10 vs P32, p = 0.0023, P20 vs P32, p = 0.0297). Thus, C1q protein is present in the developing V1b, with overall C1q levels increasing with age.
Many previous studies have found that C1q localizes to subsets of synapses (Stevens et al., 2007; Stephan et al., 2013; Hong et al., 2016; Lui et al., 2016; Dejanovic et al., 2018). We confirmed these observations in L2/3 of V1b at P29–P32, by costaining for C1q and spinophilin, a protein enriched at spines (Allen et al., 1997). Using confocal imaging, we found that a subset of spinophilin puncta overlapped with, or were closely apposed to, C1q puncta (Fig. 1F, left). To confirm that this colocalization was not solely because of chance, we rotated the C1q channel by 90°, and compared the number of colocalized C1q and spinophilin puncta when the C1q channel was in its original configuration to when it was rotated. We found that the number of colocalized puncta went down slightly but significantly when the C1q channel was rotated (original number of puncta per field-of-view: 2152 ± 116.7, rotated number of puncta per field-of-view: 2007 ± 112.5, error represents SEM; paired two-tailed t test, p = 6.2 × 10−7, n = 30 sections from 3 mice), suggesting that C1q is mildly enriched at spinophilin puncta. We furthermore used super-resolution SIM imaging to examine the C1q and spinophilin staining patterns, and identified examples of colocalization also at this higher resolution (Fig. 1F, right). These results suggest that C1q puncta are present near dendritic spines in V1b, and thus could bind to synaptic structures to promote their elimination by microglia.
C1q is not required for the development of a spine population implicated in ODP
All previous studies on the role of C1q in nervous system development have been performed in mice lacking C1qa. One phenotype described in C1qa−/− mice is increased spine densities in L5 of somatosensory cortex at P23 (Ma et al., 2013). Thus, we next asked whether loss of C1qa impacts spine development in V1b. MD during the critical period induces spine loss on the apical dendrites of V1b L2/3 pyramidal neurons, which is most pronounced in the 25 μm segment distal to the first branch point of the apical dendrite (Mataga et al., 2004). We therefore limited our analysis to this specific segment, and counted spines in WT and C1qa−/− mice at P10, P20, and P29–P30. We labeled neurons using Golgi-Cox staining, and used bright-field microscopy to image pyramidal neurons in L2/3 of V1b (Fig. 2A–C). In z-stacks from P10, P20, and P29–P30 animals, we counted the number of spines in the 25 μm segment following the first branch point. At all three ages, the spine numbers in the C1qa−/− mice were indistinguishable from their WT littermates (Fig. 2D). This finding applied both when spine numbers were analyzed per animal, and when they were analyzed per cell (unpaired two-tailed t test, per mouse: P10: t(7) = 0.7854, p = 0.4580; P20: t(8) = 1.665, p = 0.1345; P29–P30: t(10) = 1.54, p = 0.1546; per cell: P10: t(41) = 0.2109, p = 0.8340; P20: t(52) = 1.504, p = 0.1386; P29–P30: t(46) = 1.511, p = 0.1377; Fig. 2E,F).
Figure 2.
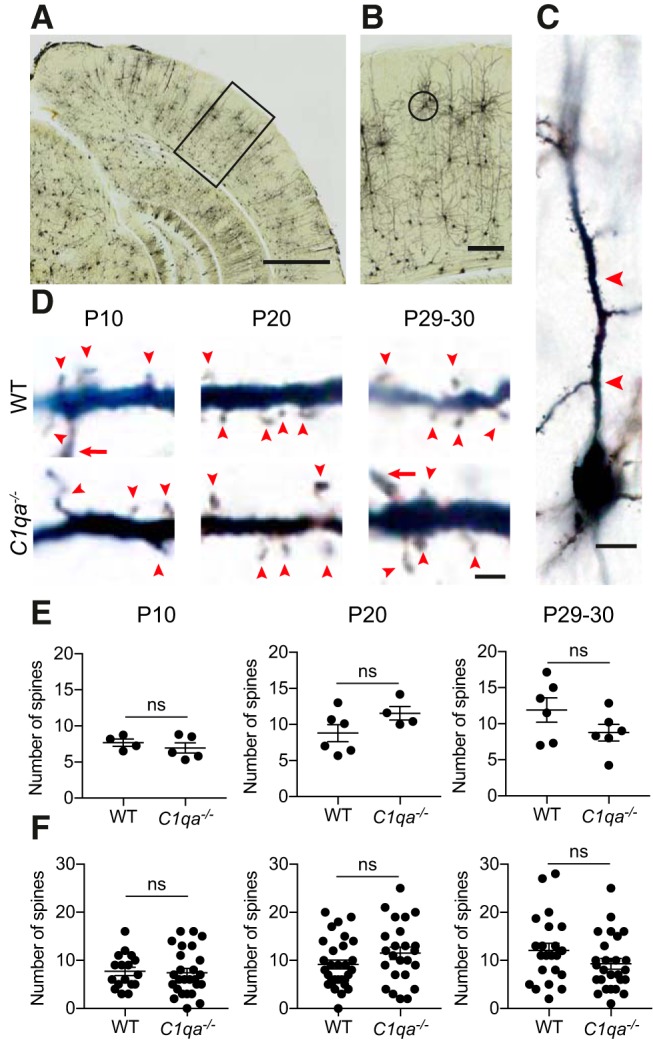
A spine population implicated in ODP develops normally in C1qa−/− mice. A, Bright-field image of a Golgi-Cox stained brain from a critical period mouse. The black box indicates the approximate location of V1b. Scale bar, 1000 μm. B, Enlarged image of the region marked in A. A L2/3 pyramidal neuron used for spine analysis is circled. Scale bar, 200 μm. C, Higher-magnification image of the cell circled in B. The soma is at the bottom of the image, whereas the 25 μm segment used for spine counting is marked with red arrowheads. Scale bar, 10 μm. D, Representative single plane images of L2/3 pyramidal neuron apical dendrites, from P10 (left), P20 (middle), and P29–P30 (right) WT (top in each age group) and C1qa−/− (bottom in each age group) animals. Images show part of the 25 μm segment used for spine counting. Red arrowheads mark spines, whereas a red arrow marks a dendrite branch point. Scale bar, 2 μm. E, Quantification of spine numbers in P10 (left), P20 (middle), and P29–P30 (right) animals, analyzed per mouse. There is no difference between spine numbers in WT and C1qa−/− animals at any of the ages (P10: unpaired two-tailed t test, t(7) = 0.7854, p = 0.4580, n = 4–5 animals/genotype; P20: unpaired two-tailed t test, t(8) = 1.665, p = 0.1345, n = 4–6 animals/genotype; P29–P30: unpaired two-tailed t test, t(10) = 1.54, p = 0.1546, n = 6 animals/genotype). F, Quantification of spine numbers in P10 (left), P20 (middle), and P29–P30 (right) animals, analyzed per cell. There is no difference between spine numbers in WT and C1qa−/− animals at any of the ages (P10: unpaired two-tailed t test, t(41) = 0.2109, p = 0.8340, n = 17 WT, 26 C1qa−/− cells; P20: unpaired two-tailed t test, t(52) = 1.504, p = 0.1386, n = 30 WT, 24 C1qa−/− cells; P29–P30: unpaired two-tailed t test, t(46) = 1.511, p = 0.1377, n = 22 WT, 26 C1qa−/− cells). All error bars represent SEM. ns, not significant.
Spine densities on apical dendrites of L2/3 pyramidal neurons increase with distance from the soma (Mataga et al., 2004). Indeed, we found that the number of spines on a dendrite segment was significantly correlated with the distance between the soma and the first branch point at the two older ages (P10 WT: rs = 0.2616, p = 0.3081; P10 C1qa−/−: rs = 0.5366, p = 0.0047; P20 WT: rs = 0.6199, p = 0.0003; P20 C1qa−/−: rs = 0.5634, p = 0.0041; P29–P30 WT: rs = 0.7321, p = 0.0001; P29–P30 C1qa−/−: rs = 0.7085, p < 0.0001; Fig. 3A). A difference in the position of the first branch point between the genotypes could thus obscure any absolute changes in spine numbers between WT and C1qa−/− mice. We therefore confirmed that the position of the first branch point was unchanged in the C1qa−/− animals, by measuring the distance between the soma and the first branch point in all cells used for spine counting. For all three ages, the average distance in the C1qa−/− animals was the same as in the WT mice (unpaired two-tailed t test, P10: t(7) = 0.2727, p = 0.7929; P20: t(8) = 0.5198, p = 0.6173; P29–P30: t(10) = 0.3156, p = 0.7588; Fig. 3B). Together, these results suggest that the number of spines in this apical dendrite segment develops normally in C1qa−/− mice.
Figure 3.
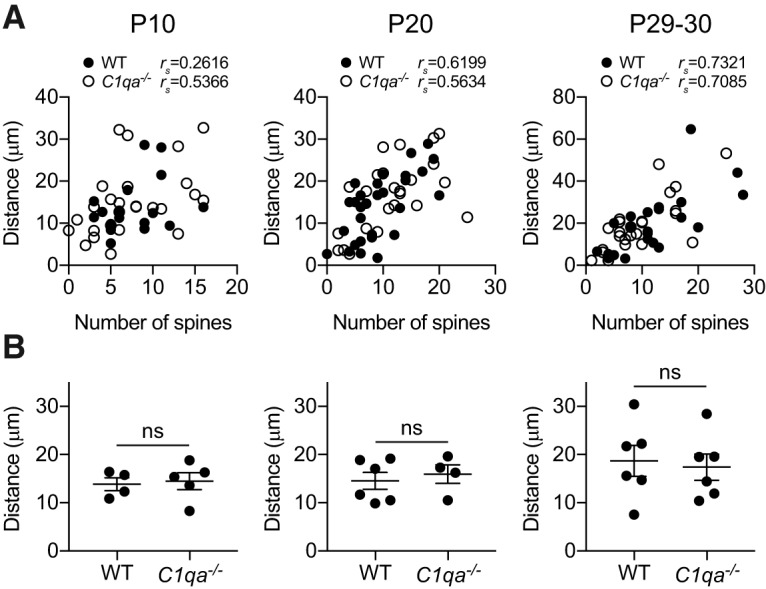
The distance between the soma and first branch point of L2/3 pyramidal neuron apical dendrites is normal in C1qa−/− mice. A, Quantification of the correlation between spine numbers and distance between soma and first branch point in P10 (left), P20 (middle), and P29–P30 (right) animals, analyzed per cell. The spine numbers and distance are significantly correlated at the two older ages, using Spearman's rank correlation coefficient rs (P10 WT: rs = 0.2616, p = 0.3081, n = 17 cells; P10 C1qa−/−: rs = 0.5366, p = 0.0047, n = 26 cells; P20 WT: rs = 0.6199, p = 0.0003, n = 30 cells; P20 C1qa−/−: rs = 0.5634, p = 0.0041, n = 24 cells; P29–P30 WT: rs = 0.7321, p = 0.0001, n = 22 cells; P29–P30 C1qa−/−: rs = 0.7085, p < 0.0001, n = 26 cells). B, Quantification of the distance between the soma and the first branch point in P10 (left), P20 (middle), and P29–P30 (right) animals. This distance is not different between WT and C1qa−/− animals at any of the ages (P10: unpaired two-tailed t test, t(7) = 0.2727, p = 0.7929, n = 4–5 animals/genotype; P20: unpaired two-tailed t test, t(8) = 0.5198, p = 0.6173, n = 4–6 animals/genotype; P29–P30: unpaired two-tailed t test, t(10) = 0.3156, p = 0.7588, n = 6 animals/genotype). All error bars represent SEM. ns, not significant.
C1q is transiently required for the development of spines on apical, but not basal, secondary dendrites
The proximal spines on the apical dendrites of L2/3 pyramidal neurons constitute only one out of many spine populations in V1b. To determine whether other spine populations were also unaffected in mice lacking C1q, we therefore examined spine densities on secondary apical and basal dendrites of V1b L2/3 pyramidal neurons. We again used Golgi-Cox staining to visualize the neurons.
We first examined spine densities on apical dendrite secondary branches at P10, P20, and P29–P30 (Fig. 4A). Surprisingly, these did not behave as the primary apical dendrite segments analyzed in Figure 2. While the C1qa−/− mice showed normal spine densities at both P10 and P29–P30, the densities were increased in the C1qa−/− animals at P20 when the data were quantified per mouse (unpaired two-tailed t test, P10: t(7) = 0.04222, p = 0.9675; P20: t(8) = 2.371, p = 0.0452; P29–P30: t(8) = 0.2614, p = 0.8004; Fig. 4B). The results were the same when data were quantified per cell (unpaired two-tailed t test, P10: t(40) = 0.4146, p = 0.6807; P20: t(47) = 2.251, p = 0.0291; P29–P30: t(32) = 0.8172, p = 0.4199; Fig. 4C). In contrast, the spine densities on the basal secondary dendrites did not change at any of the ages (Fig. 4D). This finding applied both when densities were quantified per mouse (unpaired two-tailed t test, P10: t(7) = 0.6858, p = 0.5149; P20: t(8) = 0.5260, p = 0.6132; P29–P30: t(9) = 0.5023, p = 0.6275; Fig. 4E), as well as per cell (unpaired two-tailed t test, P10: t(41) = 0.7537, p = 0.4553; P20: t(47) = 1.104, p = 0.2750; P29–P30: t(42) = 0.4044, p = 0.6879; Fig. 4F). These results thus suggest that loss of C1qa can cause increased spine numbers, but only in certain spine populations and at certain times. However, before the critical period ends, all spine populations in C1qa−/− mice examined here are indistinguishable from those of their WT littermates.
Figure 4.
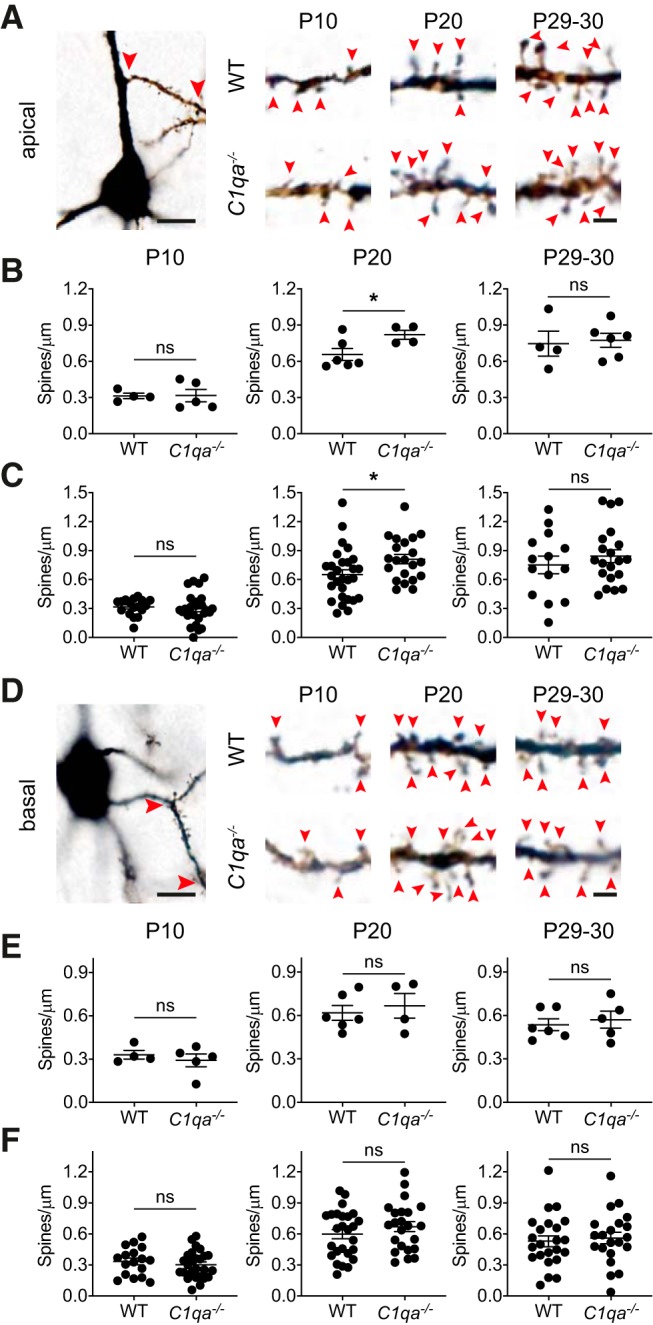
Spine densities in C1qa−/− mice are elevated at P20 on apical secondary dendrites, but are otherwise normal. A, Left, Bright-field image of a L2/3 Golgi-Cox stained pyramidal neuron from a critical period mouse. The soma is at the bottom of the image. An example of a secondary apical dendrite segment used for spine counting is marked with red arrowheads. Scale bar, 10 μm. Right, Representative single plane images of apical dendrite secondary branches from L2/3 pyramidal neurons, from P10 (left), P20 (middle), and P29–P30 (right) WT (top in each age group) and C1qa−/− (bottom in each age group) animals. Red arrowheads mark spines. Scale bar, 2 μm. B, Quantification of spine densities on apical dendrite secondary branches in P10 (left), P20 (middle), and P29–P30 (right) animals, analyzed per mouse. There is no difference between spine densities in WT and C1qa−/− animals at P10 or P29–P30, but P20 C1qa−/− mice show elevated spine densities compared with WT (P10: unpaired two-tailed t test, t(7) = 0.04222, p = 0.9675, n = 4–5 animals/genotype; P20: unpaired two-tailed t test, t(8) = 2.371, p = 0.0452, n = 4–6 animals/genotype; P29–P30: unpaired two-tailed t test, t(8) = 0.2614, p = 0.8004, n = 4–6 animals/genotype). C, Quantification of spine densities on apical dendrite secondary branches in P10 (left), P20 (middle), and P29–P30 (right) animals, analyzed per cell. There is no difference between spine densities in WT and C1qa−/− animals at P10 or P29–P30, but P20 C1qa−/− mice show elevated spine densities compared with WT (P10: unpaired two-tailed t test, t(40) = 0.4146, p = 0.6807, n = 17 WT, 25 C1qa−/− cells; P20: unpaired two-tailed t test, t(47) = 2.251, p = 0.0291, n = 28 WT, 21 C1qa−/− cells; P29–P30: unpaired two-tailed t test, t(32) = 0.8172, p = 0.4199, n = 14 WT, 20 C1qa−/− cells). D, Left, Bright-field image of a L2/3 Golgi-Cox stained pyramidal neuron from a critical period mouse. The soma is in the center of the image. An example of a secondary basal dendrite segment used for spine counting is marked with red arrowheads. Scale bar, 10 μm. Right, Representative single plane images of basal dendrite secondary branches from L2/3 pyramidal neurons, from P10 (left), P20 (middle), and P29–P30 (right) WT (top in each age group) and C1qa−/− (bottom in each age group) animals. Red arrowheads mark spines. Scale bar, 2 μm. E, Quantification of spine densities on basal, secondary branches in P10 (left), P20 (middle), and P29–P30 (right) animals, analyzed per mouse. There is no difference between spine densities in WT and C1qa−/− animals at any of the ages (P10: unpaired two-tailed t test, t(7) = 0.6858, p = 0.5149, n = 4–5 animals/genotype; P20: unpaired two-tailed t test, t(8) = 0.5260, p = 0.6132, n = 4–6 animals/genotype; P29–P30: unpaired two-tailed t test, t(9) = 0.5023, p = 0.6275, n = 5–6 animals/genotype). F, Quantification of spine densities on basal, secondary branches in P10 (left), P20 (middle), and P29–P30 (right) animals, analyzed per cell. There is no difference between spine densities in WT and C1qa−/− animals at any of the ages (P10: unpaired two-tailed t test, t(41) = 0.7537, p = 0.4553, n = 17 WT, 26 C1qa−/− cells; P20: unpaired two-tailed t test, t(47) = 1.104, p = 0.2750, n = 26 WT, 23 C1qa−/− cells; P29–P30: unpaired two-tailed t test, t(42) = 0.4044, p = 0.6879, n = 23 WT, 21 C1qa−/− cells). All error bars represent SEM. *p < 0.05, ns, not significant.
Dendritic morphology is unaffected by loss of C1q
An increase in the number or length of dendrites could cause elevated overall numbers of synapses, even without a dramatic change in spine densities on each neuron. Indeed, increased dendrite branching and length has been reported on L5 pyramidal neurons in sensorimotor cortex of C1qa−/− mice (Ma et al., 2013). We therefore traced the apical and basal dendritic arbors of Golgi-Cox stained neurons in L2/3 of V1b, and performed Sholl analysis on the tracings (Fig. 5A). When we calculated the number of dendrite intersections with concentric shells placed 10, 30, 50, 70, 90, and 110 μm away from the soma, we found no difference between the genotypes at any of the ages, either in the apical or the basal arbor [apical: unpaired two-tailed t test, Holm–Sidak's method with α = 0.05 was used to correct for multiple comparisons; P10: adjusted p = 0.8744 (10 μm), 0.9343 (30 μm), 0.9683 (50 μm), 0.9508 (70 μm), 0.9508 (90 μm), 0.9683 (110 μm); P20: adjusted p = 0.8869 (10 μm), 0.8869 (30 μm), 0.8869 (50 μm), 0.4866 (70 μm), 0.8869 (90 μm), 0.8869 (110 μm); P29–P30: adjusted p = 0.8618 (10 μm), 0.7560 (30 μm), 0.8618 (50 μm), 0.7560 (70 μm), 0.6894 (90 μm), 0.7560 (110 μm); basal: unpaired two-tailed t test, Holm–Sidak's method with α = 0.05 was used to correct for multiple comparisons, P10: adjusted p = 0.6602 (10 μm), 0.6602 (30 μm), 0.8774 (50 μm), 0.6602 (70 μm), 0.6172 (90 μm), NA (110 μm; all values are 0); P20: adjusted p = 0.9969 (10 μm), 0.9969 (30 μm), 0.9969 (50 μm), 0.9969 (70 μm), 0.9969 (90 μm), 0.9969 (110 μm); P29–P30: adjusted p = 0.9720 (10 μm), 0.9720 (30 μm), 0.6167 (50 μm), 0.5629 (70 μm), 0.3536 (90 μm), 0.3119 (110 μm); Fig. 5B–D). Thus, the morphologies of V1b L2/3 pyramidal neuron apical and basal dendritic arbors are not perturbed in C1qa−/− mice.
Figure 5.
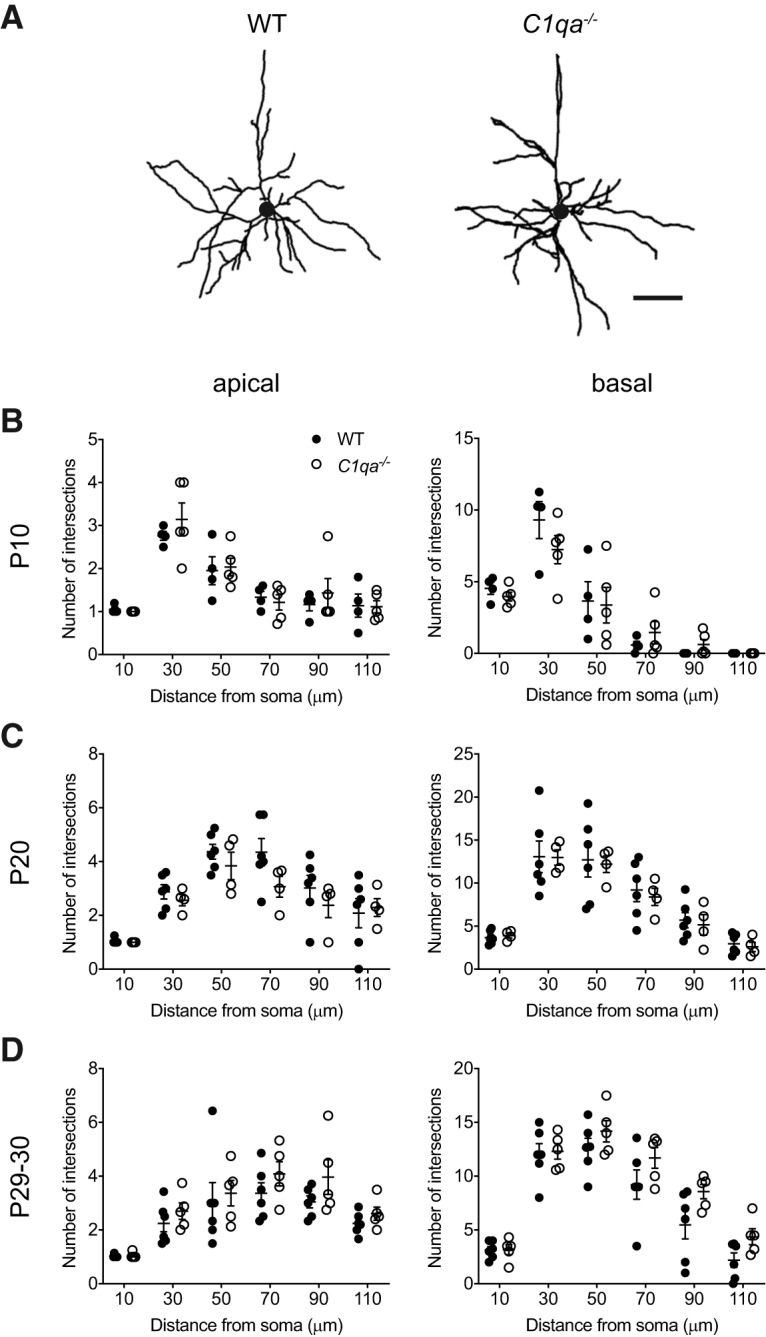
Sholl analysis reveals normal L2/3 pyramidal neuron dendritic arbors in C1qa−/− mice. A, Representative tracings of V1b L2/3 pyramidal neuron dendritic arbors from P29–P30 WT (left) or C1qa−/− (right) mice. Circles in the center of the arbors represent the soma. Scale bar, 50 μm. B–D, Sholl analysis does not find a difference between WT and C1qa−/− V1b L2/3 pyramidal neurons in the number of dendrite intersections at 10, 30, 50, 70, 90, or 110 μm from the soma in either the apical (left) or the basal (right) arbor. This applies at P10 (B), P20 (C), and P29–P30 (D). B, Unpaired two-tailed t test, n = 4–5 mice/genotype. Holm–Sidak's method with α = 0.05 was used to correct for multiple comparisons. Apical: adjusted p = 0.8744 (10 μm), 0.9343 (30 μm), 0.9683 (50 μm), 0.9508 (70 μm), 0.9508 (90 μm), 0.9683 (110 μm); basal: adjusted p = 0.6602 (10 μm), 0.6602 (30 μm), 0.8774 (50 μm), 0.6602 (70 μm), 0.6172 (90 μm), NA (110 μm; all values are 0). C, Unpaired two-tailed t test, n = 4–6 mice/genotype. Holm–Sidak's method with α = 0.05 was used to correct for multiple comparisons. Apical: adjusted p = 0.8869 (10 μm), 0.8869 (30 μm), 0.8869 (50 μm), 0.4866 (70 μm), 0.8869 (90 μm), 0.8869 (110 μm); basal: adjusted p = 0.9969 (10 μm), 0.9969 (30 μm), 0.9969 (50 μm), 0.9969 (70 μm), 0.9969 (90 μm), 0.9969 (110 μm). D, Unpaired two-tailed t test, n = 5–6 mice/genotype. Holm–Sidak's method with α = 0.05 was used to correct for multiple comparisons. Apical: adjusted p = 0.8618 (10 μm), 0.7560 (30 μm), 0.8618 (50 μm), 0.7560 (70 μm), 0.6894 (90 μm), 0.7560 (110 μm); basal: adjusted p = 0.9720 (10 μm), 0.9720 (30 μm), 0.6167 (50 μm), 0.5629 (70 μm), 0.3536 (90 μm), 0.3119 (110 μm). All error bars represent SEM.
Firing rates in V1b of C1qa−/− mice are normal
Although we found mostly normal spine numbers and dendritic morphology in developing C1qa−/− mice, visual processing in these animals may still be different from that of WT mice, as it is possible that other synapse populations in V1b are affected by loss of C1qa. To assess the general properties of visual responsiveness in animals lacking C1qa, we performed in vivo extracellular electrophysiological recordings in P28–P31 WT and C1qa−/− littermates. Neuronal activity was recorded across cortical layers in V1b of anesthetized mice, using multisite silicone probes. Units with receptive fields outside of the binocular zone (further than 25° from the center of the visual field) were excluded from further analysis (see Materials and Methods).
Using these recordings, we examined how loss of C1qa impacts firing rates of single units in V1b. Visual activity was elicited using sinusoidal gratings drifting in 12 different directions interleaved by a gray screen, presented to each eye independently. Each stimulus was presented a minimum of seven times (Fig. 6A). The spontaneous activity was measured as the average firing rate in response to the gray screen, whereas the evoked activity was the average firing rate in response to the direction that elicited the most activity. The average spontaneous activity was the same when compared between eyes and genotypes (one-way ANOVA, F(3,181) = 0.08636, p = 0.9674; Fig. 6B). Similarly, although the evoked firing rate was higher for the contralateral eye, reflecting normal contralateral bias (Gordon and Stryker, 1996), there was no difference in the evoked firing rate between genotypes (one-way ANOVA with Sidak's multiple-comparisons test, F(3,181) = 4.788, p = 0.0031, WT ipsilateral vs WT contralateral, p = 0.0428, C1qa−/− ipsilateral vs C1qa−/− contralateral, p = 0.0424, WT ipsilateral vs C1qa−/− ipsilateral, p = 0.9413, WT contralateral vs C1qa−/− contralateral, p = 0.9165; Fig. 6C). Thus, C1qa does not influence baseline firing rates in V1b.
Figure 6.
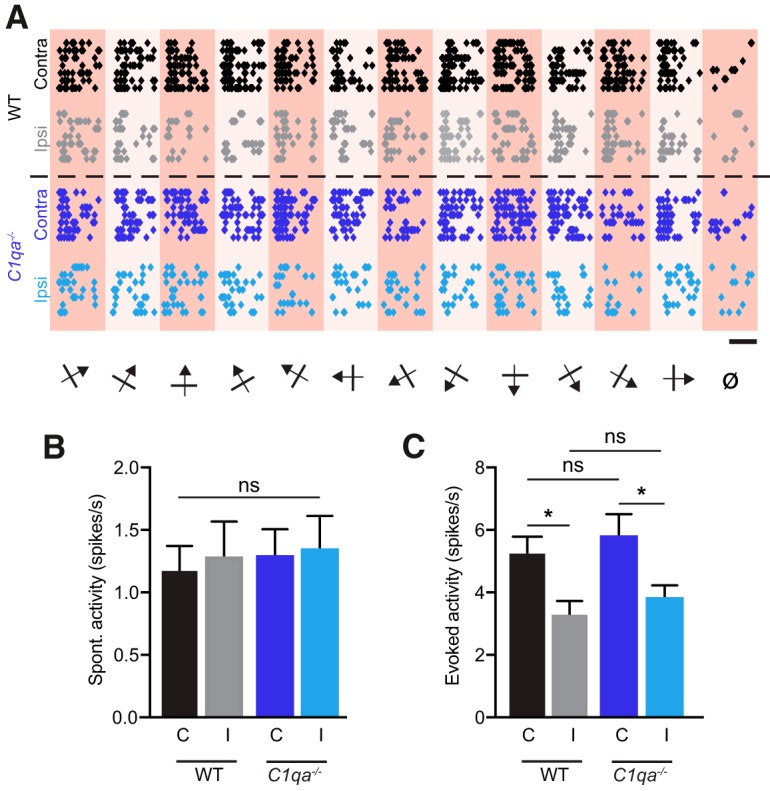
Firing rates are normal in C1qa−/− mice. A, Raster plots for representative single units from WT and C1qa−/− mice in response to drifting sinusoidal gratings in 12 different directions interleaved by a gray screen (ø) shown to each eye independently. Each circle represents one action potential. Scale bar, 1 s. B, C, Spontaneous (B) and evoked (C) activity of all isolated, visually responsive single units from WT and C1qa−/− mice. Spontaneous firing rates are not different across stimulated eye or genotype (one-way ANOVA, F(3,181) = 0.08636, p = 0.9674, n = 42–50 single units/condition, from 6 animals/genotype). Evoked firing rates are higher when the contralateral eye rather than the ipsilateral eye is stimulated, but are not different between the genotypes (one-way ANOVA, F(3,181) = 4.788, p = 0.0031, n = 42–50 single units/condition, from 6 animals/genotype, WT ipsilateral vs WT contralateral, p = 0.0428, C1qa−/− ipsilateral vs C1qa−/− contralateral, p = 0.0424, WT ipsilateral vs C1qa−/− ipsilateral, p = 0.9413, WT contralateral vs C1qa−/− contralateral, p = 0.9165, Sidak's multiple-comparisons test). All error bars represent SEM. *p < 0.05, ns, not significant.
C1q protein levels in V1b are not experience-dependent
Our results suggest that several features of V1b development proceed normally in the absence of C1qa. However, it is possible that the initial development of V1b is independent of C1q, but that C1q instead is required for the synaptic remodeling that occurs in response to visual deprivation. We therefore investigated whether C1q contributes to ODP. We first examined how MD impacts C1q levels in L2/3 and L4 of V1b. An increase could suggest that C1q is upregulated to promote synapse elimination during ODP. We evaluated C1q levels in normally reared P32 mice (NR), as well as in mice that had undergone 2 or 4 d of MD starting at P28. ODP is complete after 4 d of MD (Gordon and Stryker, 1996); 2 d was chosen as an intermediate time point during which remodeling is still in progress. As in Figure 1, we measured C1q levels by quantifying immunofluorescence intensities (Fig. 7A). We found that MD did not impact C1q levels in either layer (one-way ANOVA, L2/3: F(2,15) = 0.1803, p = 0.8368; L4: F(2,15) = 0.2338, p = 0.7943; Fig. 7B,C). Immunofluorescence intensities are a fairly crude measurement of protein levels, so we cannot conclude that MD has no effect on the amounts of C1q in V1b. However, MD does not appear to drive dramatic changes in the levels of C1q.
MD does not reduce spine numbers on L2/3 pyramidal neurons
C1q could contribute to synapse elimination following MD even if C1q levels are unchanged. Subtle changes in C1q localization to synapses or local activation of the classical complement cascade may be sufficient to promote increased synapse engulfment. Alternatively, reduced levels of signals that inhibit synaptic engulfment, such as CD47, could allow increased C1q-dependent synapse phagocytosis to occur (Lehrman et al., 2018). We therefore investigated whether the absence of C1qa prevents MD-induced spine loss on L2/3 pyramidal neuron apical dendrites (Mataga et al., 2004). We executed the experiment as previously described (Mataga et al., 2004). However, one notable difference was that we used Golgi-Cox staining to label the neurons, while the previous study used diolistic labeling. We performed MD on WT and C1qa−/− animals at P25–P26 (in the middle of the critical period) and collected brains 4 d later. The controls consisted of NR WT and C1qa−/− mice aged P29–P30. For all four groups, we counted spines in the 25 μm segment following the first branch point, on the apical dendrites of L2/3 pyramidal neurons. The NR data points were already included in Figure 2, but were collected together with the MD cohort and analyzed blind to both genotype and experimental condition; as described, there was no difference between the genotypes in the number of spines for the NR group. When we compared the WT NR and MD conditions, we surprisingly also found no change in spine numbers (two-way ANOVA with Tukey's multiple-comparisons test, F(1,21) = 3.034, p = 0.0962 for MD; F(1,21) = 0.344, p = 0.5638 for genotype; F(1,21) = 2.906, p = 0.1030 for interaction; WT NR vs WT MD, p > 0.9999; Fig. 8A,B). This finding, in stark contrast to the previously reported 40% reduction (Mataga et al., 2004), held true also when spine numbers were analyzed per cell (two-way ANOVA with Tukey's multiple-comparisons test, F(1,110) = 3.543, p = 0.0625 for MD; F(1,110) = 0.2824, p = 0.5962 for genotype; F(1,110) = 2.695, p = 0.1035 for interaction; WT NR vs WT MD, p = 0.9986; Fig. 8C). In addition, the C1qa−/− mice did not show a spine reduction following MD, and instead displayed a trend toward an increase in spine numbers in the MD condition compared with NR, when spine number were analyzed per mouse (C1qa−/− NR vs C1qa−/− MD, p = 0.0920). When we quantified spine numbers per cell, this trend became statistically significant (C1qa−/− NR vs C1qa−/− MD, p = 0.0411); however, because MD, genotype, and their interaction all lacked statistically significant effects on spine numbers, it is not clear whether the difference between C1qa−/− NR and C1qa−/− MD is relevant. As in Figure 3, we quantified the distance between the soma and first branch point for all cells used for spine counting, and they were the same across all four conditions (two-way ANOVA, F(1,21) = 0.02282, p = 0.8814 for MD; F(1,21) = 0.2156, p = 0.6472 for genotype; F(1,21) = 0.0069, p = 0.9346 for interaction; Fig. 8D). Our observations therefore suggest that MD has no strong effect on spine numbers on this particular part of L2/3 apical dendrites in WT or C1qa−/− animals.
Figure 8.
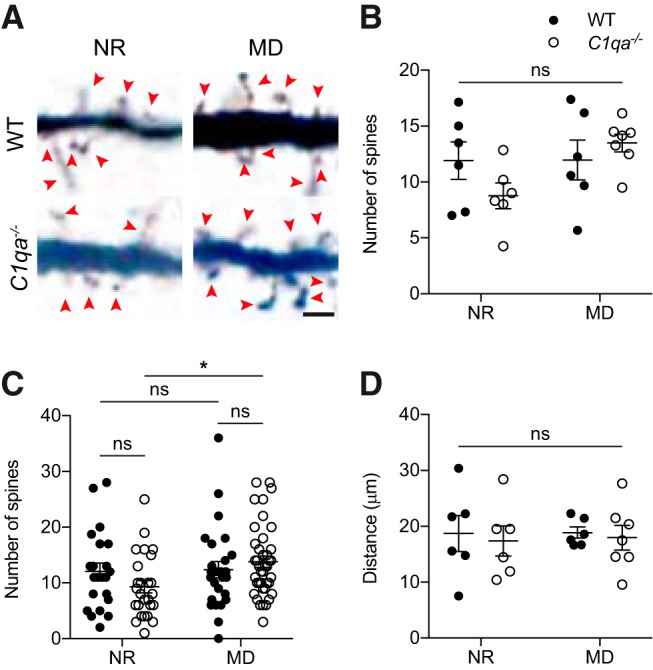
MD does not reduce spine numbers on L2/3 pyramidal neuron apical dendrites. A, Representative single plane images of L2/3 pyramidal neuron apical dendrites from WT and C1qa−/− littermates that were either NR, or that had undergone 4 d of MD. All cells come from the hemisphere contralateral to the deprived eye. Images show part of the 25 μm segment used for spine counting. Red arrowheads mark spines. Scale bar, 2 μm. B, Quantification of spine numbers in WT and C1qa−/− NR and MD animals, per mouse. Data points in the NR condition are the same as the P29–P30 group in Figure 2E. MD does not impact spine numbers, and there is no difference between spine numbers in WT and C1qa−/− animals in either condition (two-way ANOVA, F(1,21) = 3.034, p = 0.0962 for MD; F(1,21) = 0.344, p = 0.5638 for genotype; F(1,21) = 2.906, p = 0.1030 for interaction, n = 6–7 animals/group; WT NR vs C1qa−/− NR, p = 0.4049, WT NR vs WT MD, p > 0.9999, C1qa−/− NR vs C1qa−/− MD, p = 0.0920, WT MD vs C1qa−/− MD, p = 0.8510, Tukey's multiple-comparisons test). C, Quantification of spine numbers in WT and C1qa−/− NR and MD animals, per cell. Data points in the NR condition are the same as the P29–P30 group in Figure 2F. MD does not impact spine numbers in the WT group, but slightly increases spine numbers in the C1qa−/− mice (two-way ANOVA, F(1,110) = 3.543, p = 0.0625 for MD; F(1,110) = 0.2824, p = 0.5962 for genotype; F(1,110) = 2.695, p = 0.1035 for interaction, n = 22–40 cells/group; WT NR vs C1qa−/− NR, p = 0.4774, WT NR vs WT MD, p = 0.9986, C1qa−/− NR vs C1qa−/− MD, p = 0.0411, WT MD vs C1qa−/− MD, p = 0.8323, Tukey's multiple-comparisons test). D, Quantification of the distance between the soma and the first branch point in WT and C1qa−/− NR and MD animals. Data points in the NR condition are the same as the P29–P30 group in Figure 3B. There is no difference in distance between WT and C1qa−/− animals in either condition (two-way ANOVA, F(1,21) = 0.02282, p = 0.8814 for MD; F(1,21) = 0.2156, p = 0.6472 for genotype; F(1,21) = 0.0069, p = 0.9346 for interaction, n = 6–7 animals/group; WT NR vs C1qa−/− NR, p = 0.9808, WT NR vs WT MD, p > 0.9999, C1qa−/− NR vs C1qa−/− MD, p = 0.9982, WT MD vs C1qa−/− MD, p = 0.9925, Tukey's multiple-comparisons test). All error bars represent SEM. *p < 0.05, ns, not significant.
C1qa is not required for binocular zone expansion following MD and ME
Our failure to detect a spine loss following MD in either genotype makes this experiment difficult to interpret. We therefore proceeded to determine whether ODP can occur in the absence of C1q, using two distinct techniques. We first measured how loss of C1qa impacts ODP using in situ hybridization against the immediate early gene Arc (Tagawa et al., 2005). Acute light stimulation results in Arc labeling of V1b. During the critical period, 4 d of ME or MD of the contralateral eye, before light stimulation, causes a widening of the binocular Arc zone (William et al., 2012; Bochner et al., 2014). This observation suggests that Arc in situs can be used as a proxy to measure ODP. Indeed, both enhanced and reduced ODP detected using this method has been confirmed using alternative techniques (Syken et al., 2006; Kanold et al., 2009; William et al., 2012, 2017; Djurisic et al., 2013; Bochner et al., 2014). We therefore used the Arc in situ method to examine ODP in mice lacking C1qa.
We performed MD on WT and C1qa−/− mice during the critical period, with MD starting at P25–P28. After 4 d of MD, the animals were light stimulated and their brains collected. We then performed RNAscope fluorescence in situ hybridization against Arc, and quantified the length of the binocular zone in L2/3 and L4 by measuring the FWHM (William et al., 2017; Fig. 9A). The MD group was compared with age-matched NR control mice that had undergone the same light stimulation paradigm as the MD animals. In both layers, MD induced a robust expansion of the binocular zone in WT as well as in C1qa−/− animals (two-way ANOVA with Tukey's multiple-comparisons test, L2/3: F(1,21) = 35.19, p < 0.0001 for MD; F(1,21) = 2.717, p = 0.1142 for genotype; F(1,21) = 0.02172, p = 0.8843 for interaction; WT NR vs WT MD, p = 0.0014, C1qa−/− NR vs C1qa−/− MD, p = 0.0032; L4: F(1,21) = 69.53, p < 0.0001 for MD; F(1,21) = 0.9028, p = 0.3528 for genotype; F(1,21) = 1.029, p = 0.3219 for interaction; WT NR vs WT MD, p = 0.0002, C1qa−/− NR vs C1qa−/− MD, p < 0.0001; Fig. 9B,C). Furthermore, the C1qa−/− binocular zone lengths were not different from those of WT animals in either the NR or the MD condition (L2/3: WT NR vs C1qa−/− NR, p = 0.6052, WT MD vs C1qa−/− MD, p = 0.7042; L4: WT NR vs C1qa−/− NR, p > 0.9999, WT MD vs C1qa−/− MD, p = 0.5039). These results suggest that the binocular zone is not grossly abnormal in C1qa−/− mice under baseline conditions, and that in the absence of C1qa the binocular zone expands normally following MD.
Figure 9.

Binocular zone expansion is normal in critical period C1qa−/− mice. A, Representative images of RNAscope Arc in situ hybridization in V1b of critical period WT or C1qa−/− mice that were either NR or had undergone 4 d of MD. Approximate locations of cortical layers are noted at the far left of the images. Red stars mark the extent of the binocular zone in L2/3 and L4, as measured using the FWHM. Scale bar, 300 μm. B–D, During the critical period, binocular zone expansion as measured by the FWHM of the Arc-positive signal increases in L2/3 following MD (B), in L4 following MD (C), and in L4 following ME (D) compared with NR mice. However, loss of C1qa has no effect on the width of the binocular zone under any of these conditions. B, Two-way ANOVA, F(1,21) = 35.19, p < 0.0001 for MD; F(1,21) = 2.717, p = 0.1142 for genotype; F(1,21) = 0.02172, p = 0.8843 for interaction, n = 6–7 animals/group; WT NR vs C1qa−/− NR, p = 0.6052, WT NR vs WT MD, p = 0.0014, C1qa−/− NR vs C1qa−/− MD, p = 0.0032, WT MD vs C1qa−/− MD, p = 0.7042, Tukey's multiple-comparisons test. C, Two-way ANOVA, F(1,21) = 69.53, p < 0.0001 for MD; F(1,21) = 0.9028, p = 0.3528 for genotype; F(1,21) = 1.029, p = 0.3219 for interaction, n = 6–7 animals/group; WT NR vs C1qa−/− NR, p > 0.9999, WT NR vs WT MD, p = 0.0002, C1qa−/− NR vs C1qa−/− MD, p < 0.0001, WT MD vs C1qa−/− MD, p = 0.5039, Tukey's multiple-comparisons test. D, Two-way ANOVA, F(1,16) = 29.77, p < 0.0001 for ME; F(1,16) = 0.5278, p = 0.4780 for genotype; F(1,16) = 0.8672, p = 0.3656 for interaction, n = 5 animals/group; WT NR vs C1qa−/− NR, p = 0.6521, WT NR vs WT ME, p = 0.0018, C1qa−/− NR vs C1qa−/− ME, p = 0.0258, WT ME vs C1qa−/− ME, p = 0.9989, Tukey's multiple-comparisons test. E–G, During the critical period, binocular zone Arc signal intensity, normalized to the signal intensity in V1m, increases in L2/3 following MD (E), in L4 following MD (F), but not in L4 following ME (G), compared with NR mice. Loss of C1qa has no effect on the Arc signal strength under any of these conditions. The lack of an effect of ME on signal intensity is likely due to the use of colorimetric, rather than RNAscope, in situ hybridization for this experiment. E, Two-way ANOVA, F(1,21) = 82.53, p < 0.0001 for MD, F(1,21) = 0.02663, p = 0.8719 for genotype, and F(1,21) = 0.2541, p = 0.6195 for interaction, n = 6–7 animals/group. WT NR vs C1qa−/− NR, p = 0.9952, WT NR vs WT MD, p < 0.0001, C1qa−/− NR vs C1qa−/− MD, p < 0.0001, WT MD vs C1qa−/− MD, p = 0.9625, Tukey's multiple-comparisons test. F, Two-way ANOVA, F(1,21) = 55.72, p < 0.0001 for MD; F(1,21) = 0.3273, p = 0.5733 for genotype; F(1,21) = 0.3465, p = 0.5624 for interaction, n = 6–7 animals/group; WT NR vs C1qa−/− NR, p > 0.9999, WT NR vs WT MD, p < 0.0001, C1qa−/− NR vs C1qa−/− MD, p = 0.0006, WT MD vs C1qa−/− MD, p = 0.8366, Tukey's multiple-comparisons test. G, Two-way ANOVA, F(1,16) = 0.9614, p = 0.3414 for ME; F(1,16) = 0.6366, p = 0.4366 for genotype; F(1,16) = 0.4253, p = 0.5236 for interaction, n = 5 animals/group; WT NR vs C1qa−/− NR, p = 0.9996, WT NR vs WT ME, p = 0.6626, C1qa−/− NR vs C1qa−/− ME, p = 0.9954, WT ME vs C1qa−/− ME, p = 0.7374, Tukey's multiple-comparisons test. All error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant.
When examining the Arc in situ images, we noticed that the fluorescence intensity of the Arc signal in the binocular zone appeared brighter in the MD than in the NR animals. This observation is consistent with an increase in open-eye responsiveness at the center of V1b following MD (Tagawa et al., 2005). We therefore quantified the Arc fluorescence intensity in the center of V1b, normalizing the signal to the fluorescence intensity in V1m to correct for variability in the background signal. In both L2/3 and L4, the quantification of the Arc fluorescence intensity was consistent with the quantification of the binocular zone length. MD led to an increase in Arc signal in both genotypes compared with the NR animals (two-way ANOVA with Tukey's multiple-comparisons test, L2/3: F(1,21) = 82.53, p < 0.0001 for MD, F(1,21) = 0.02663, p = 0.8719 for genotype; F(1,21) = 0.2541, p = 0.6195 for interaction; WT NR vs WT MD, p < 0.0001, C1qa−/− NR vs C1qa−/− MD, p < 0.0001; L4: F(1,21) = 55.72, p < 0.0001 for MD; F(1,21) = 0.3273, p = 0.5733 for genotype; F(1,21) = 0.3465, p = 0.5624 for interaction; WT NR vs WT MD, p < 0.0001, C1qa−/− NR vs C1qa−/− MD, p = 0.0006; Fig. 9E,F). Additionally, in both NR and MD animals there was no significant difference between genotypes (L2/3: WT NR vs C1qa−/− NR, p = 0.9952, WT MD vs C1qa−/− MD, p = 0.9625; L4: WT NR vs C1qa−/− NR, p > 0.9999, WT MD vs C1qa−/− MD, p = 0.8366). Thus, ODP in C1qa−/− animals is normal using two methods for quantifying the visually induced Arc signal.
To confirm these observations with an even more dramatic visual manipulation, we measured binocular zone expansion following ME in WT and C1qa−/− animals (Bochner et al., 2014). In this experiment, we enucleated, rather than sutured shut, the contralateral eye at P28. After 4 d of ME, the mice were light stimulated and their brains collected. Age-matched NR animals were also light stimulated before tissue collection. We detected Arc transcript using standard colorimetric, rather than RNAscope, in situ hybridization. When we examined the length of the binocular zone in L4 by measuring the FWHM, we again observed a robust expansion in both genotypes, and no difference between WT and C1qa−/− animals in either condition (two-way ANOVA with Tukey's multiple-comparisons test, F(1,16) = 29.77, p < 0.0001 for ME; F(1,16) = 0.5278, p = 0.4780 for genotype; F(1,16) = 0.8672, p = 0.3656 for interaction; WT NR vs C1qa−/− NR, p = 0.6521, WT NR vs WT ME, p = 0.0018, C1qa−/− NR vs C1qa−/− ME, p = 0.0258, WT ME vs C1qa−/− ME, p = 0.9989; Fig. 9D). We also measured the signal intensity of the Arc signal in the four conditions, but did not see any differences across the groups (two-way ANOVA with Tukey's multiple-comparisons test, F(1,16) = 0.9614, p = 0.3414 for ME; F(1,16) = 0.6366, p = 0.4366 for genotype; F(1,16) = 0.4253, p = 0.5236 for interaction; WT NR vs C1qa−/− NR, p = 0.9996, WT NR vs WT ME, p = 0.6626, C1qa−/− NR vs C1qa−/− ME, p = 0.9954, WT ME vs C1qa−/− ME, p = 0.7374; Fig. 9G). This contrast to the RNAscope results is consistent with RNAscope providing a highly sensitive and quantitative measurement of transcript levels (Wang et al., 2012, 2013). Together, these experiments thus suggest that critical period ODP is normal C1qa−/− mice.
In vivo electrophysiology reveals normal ODP in C1qa−/− mice
The Arc in situ method is an indirect measurement of ocular dominance. Therefore, we used in vivo extracellular recordings to measure ODP directly from neuronal activity in WT and C1qa−/− mice. We recorded from P28–P31 WT and C1qa−/− animals that had either been reared normally or undergone 4 d of MD, and evaluated the eye dominance from the multiunit activity on all visually responsive contact sites. To calculate the relative contribution of each eye to the firing rate measured at each contact site along the silicone probe, we averaged the response across all directions of the sinusoidal grating stimulus for the contralateral and ipsilateral eye, independently (see Materials and Methods). In the NR mice, we found no difference in eye-dominance between WT and C1qa−/− littermates (Figs. 10A–C). Both genotypes had a normal contralateral bias shown by ODI histograms with high proportions of units in the lower (contra-dominated) OD categories (χ2 test, χ2(5) = 7.502, p = 0.1859, α = 0.0125 to account for multiple comparisons; Fig. 10A), similar ODI distributions (Kolmogorov–Smirnov test, D = 0.1272, p = 0.0646, α = 0.0125 to account for multiple comparisons; Fig. 10B), and average CBIs ∼0.65 (two-way ANOVA with Tukey's multiple-comparisons test, F(1,23) = 32.57, p < 0.0001 for MD; F(1,23) = 0.06254, p = 0.8047 for genotype; F(1,23) = 0.3021, p = 0.5879 for interaction; WT NR vs C1qa−/− NR, p = 0.9491; Fig. 10C). C1qa−/− animals thus have normal ocular dominance under baseline conditions.
Figure 10.

ODP is largely normal in critical period C1qa−/− mice. A, OD histograms for NR and 4D MD WT and C1qa−/− animals. MD shifts the histograms in both WT and C1qa−/− mice, but the histograms are not affected by loss of C1qa (WT NR vs C1qa−/− NR: χ2 test, χ2(5) = 7.502, p = 0.1859, n = 197–230 multiunits/genotype, from 6 animals/genotype. Note: Categories 6 and 7 were combined for calculating the χ2 test because there was only one unit in Category 7. WT NR vs WT MD: χ2 test, χ2(6) = 114.5, p < 0.0001, n = 197–297 multiunits/genotype, from 6 to 8 animals/genotype. C1qa−/− NR vs C1qa−/− MD: χ2 test, χ2(6) = 52.64, p < 0.0001, n = 230–239 multiunits/genotype, from 6 to 7 animals/genotype. WT MD vs C1qa−/− MD: χ2 test, χ2(6) = 10.16, p = 0.1179, n = 239–297 multiunits/genotype, from 7 to 8 animals/genotype. To account for multiple comparisons, the Bonferroni correction is used in this panel to set α = 0.0125). The extreme C1qa−/− MD mouse is excluded from this panel. B, MD shifts the distribution of ODIs in both WT and C1qa−/− mice, but the distributions are not affected by loss of C1qa (WT NR vs C1qa−/− NR: Kolmogorov–Smirnov test, D = 0.1272, p = 0.0646, n = 197–230 multiunits/genotype, from 6 animals/genotype. WT NR vs WT MD: Kolmogorov–Smirnov test, D = 0.4620, p < 0.0001, n = 197–297 multiunits/genotype, from 6 to 8 animals/genotype. C1qa−/− NR vs C1qa−/− MD: Kolmogorov–Smirnov test, D = 0.2994, p < 0.0001, n = 230–239 multiunits/genotype, from 6 to 7 animals/genotype. WT MD vs C1qa−/− MD: Kolmogorov–Smirnov test, D = 0.1336, p = 0.0178, n = 239–297 multiunits/genotype, from 7 to 8 animals/genotype. To account for multiple comparisons, the Bonferroni correction is used in this panel to set α = 0.0125). The extreme C1qa−/− MD mouse is excluded from this panel. C, Quantification of CBI in WT and C1qa−/− mice. The CBI is not different between the two genotypes (two-way ANOVA, F(1,23) = 32.57, p < 0.0001 for MD; F(1,23) = 0.06254, p = 0.8047 for genotype; F(1,23) = 0.3021, p = 0.5879 for interaction, n = 6–8 animals/group; WT NR vs C1qa−/− NR, p = 0.9491, WT NR vs WT MD, p = 0.0009, C1qa−/− NR vs C1qa−/− MD, p = 0.0078, WT MD vs C1qa−/− MD, p = 0.9959, Tukey's multiple-comparisons test). The gray circle marks the excluded C1qa−/− MD data point. All error bars represent SEM. **p < 0.01, ***p < 0.001, ns, not significant.
Next we examined ODP in WT and C1qa−/− mice following 4 d of MD during the critical period starting at P25–P26. We found that the WT MD mice had robust OD shifts compared with the WT NR controls (OD categories: χ2 test, χ2(6) = 114.5, p < 0.0001, α = 0.0125 to account for multiple comparisons; ODI distributions: Kolmogorov–Smirnov test, D = 0.4620, p < 0.0001, α = 0.0125 to account for multiple comparisons; CBI: WT NR vs WT MD, p = 0.0009). Among the C1qa−/− animals, the results were less clear: the C1qa−/− MD animals showed a significant OD shift compared with the C1qa−/− NR group by OD categories and ODI distribution, but not by CBI (OD categories: χ2 test, χ2(6) = 39.74, p < 0.0001; ODI distributions: Kolmogorov–Smirnov test, D = 0.2450, p < 0.0001; CBI: C1qa−/− NR vs C1qa−/− MD, p = 0.0860; data not shown). Additionally, the C1qa−/− MD mice were also significantly different from the WT MD animals by OD categories and ODI distribution, but again not by CBI (OD categories: χ2 test, χ2(6) = 26.14, p = 0.0002; ODI distributions: Kolmogorov–Smirnov test, D = 0.1929, p < 0.0001; CBI: WT MD vs C1qa−/− MD, p = 0.7962; data not shown). These results could indicate that although the C1qa−/− animals undergo an OD shift in response to MD, they do not shift to the same degree as WT mice.
However, we noticed that one C1qa−/− MD animal (Fig. 10C, gray) had an unusually high CBI value of 0.8158, which was almost 2 SD above the mean of the C1qa−/− MD group, as well as of the WT and C1qa−/− NR groups. Because we were concerned that this one extreme animal could be skewing our results, we examined the dataset without this mouse. Among the remaining seven C1qa−/− MD animals, OD was significantly different from that of the C1qa−/− NR mice (OD categories: χ2 test, χ2(6) = 52.64, p < 0.0001, α = 0.0125 to account for multiple comparisons; ODI distributions: Kolmogorov–Smirnov test, D = 0.2994, p < 0.0001, α = 0.0125 to account for multiple comparisons; CBI: C1qa−/− NR vs C1qa−/− MD, p = 0.0078). Furthermore, these C1qa−/− MD animals were not different from the WT MD animals (OD categories: χ2 test, χ2(6) = 10.16, p = 0.1179, α = 0.0125 to account for multiple comparisons; ODI distributions: Kolmogorov–Smirnov test, D = 0.1336, p = 0.0178, α = 0.0125 to account for multiple comparisons; CBI: WT MD vs C1qa−/− MD, p = 0.9959). Under the assumption that the high CBI value is atypical for C1qa−/− MD mice, our results thus suggest that C1qa−/− animals undergo normal ODP.
Discussion
In this study, we investigated the contribution of C1q, the initiator of the classical complement cascade, to V1b development and plasticity. We found that C1q protein was present in V1b during the critical period, but that bulk levels were not affected by MD. When we quantified spine numbers on apical and basal dendrites of L2/3 pyramidal neurons, we observed mostly normal spine numbers in C1qa−/− mice during development. The dendritic arbors of these neurons were also unaffected by loss of C1qa. We furthermore found that neuronal firing rates in V1b were unchanged in C1qa−/− animals compared with WT mice. Moreover, we did not observe a reduction in spine numbers in WT or C1qa−/− mice following critical period MD, and when we measured ODP, we detected robust plasticity in C1qa−/− mice using both in situ hybridization against the immediate early gene Arc, and in vivo electrophysiology. Together, our results suggest that V1b is generally normal in the absence of C1qa.
In most experiments performed in this study, the C1qa−/− animals were similar to their WT counterparts. These observations are in agreement with previous work performed in the hippocampus, where C1qa−/− mice were generally found to have no or mild synaptic phenotypes, depending on the experiment and the age of the animal (Stephan et al., 2013). In addition, this previous study found that whole-brain C1q levels increase with age, similar to what we report here. The phenotype that we do observe in the C1qa−/− animals is that they have increased spine densities on apical secondary dendrites at P20. Previous dLGN phenotypes in C1qa−/− mice have been attributed to impaired microglial phagocytosis of retinogeniculate synapses (Stevens et al., 2007; Schafer et al., 2012), and it is conceivable that the elevated spine densities on P20 apical secondary dendrites in the mice lacking C1qa could be due to a lack of synapse engulfment. It is, however, only a transient phenotype, as spine densities return to normal at P29–P30.
In contrast to V1b and the hippocampus, sensorimotor cortex and the dLGN show reduced synapse elimination in the absence of C1qa. Sensorimotor cortex of juvenile C1qa−/− animals have increased spine and bouton numbers, as well as increased frequencies of mEPSCs and seizures, and refinement in the P30 dLGN is impaired in C1qa−/− mice (Stevens et al., 2007; Chu et al., 2010; Ma et al., 2013). These differences in phenotypes are surprising for two reasons: first that the phenotypes seem to be quite variable depending on the brain region examined, and second that the dLGN shows improper refinement at the same time as V1b is largely normal, even though V1b receives input from the dLGN (Stevens et al., 2007). There are possible explanations for why V1b does not appear to inherit the refinement defects of the dLGN. Defects in eye-specific segregation can represent changes in axon arborization that do not necessarily result in changed synaptic connectivity. Furthermore, although electrophysiological studies performed in the part of the dLGN that is targeted by the contralateral eye found that loss of C1qa prevents the elimination of weak inputs in the dLGN, at least some RGC inputs seem to strengthen normally in P30 C1qa−/− mice (Stevens et al., 2007). dLGN relay neurons are only driven by strong RGC inputs (Liu and Chen, 2008). It is therefore possible that the presence of many weak inputs, as is found in the dLGN of C1qa−/− mice at P30, does not impact the transmission of visual information to V1b. Why different brain regions show different phenotypes is less clear. It does not appear to be dependent on the overall levels of C1q protein. C1q levels are generally low in cortex and high in the hippocampus (Stephan et al., 2013), indicating that both regions with high and low levels of C1q, such as the hippocampus and V1b, respectively, can have mild phenotypes. It is possible that expression or localization of downstream complement proteins required for microglial engulfment is context-dependent. In addition, negative regulators of C1q activity, such as complement inhibitors or signaling pathways that block phagocytosis, including CD47, may be heterogeneously distributed across brain regions (Ricklin et al., 2010; Lehrman et al., 2018). Such heterogeneity could explain why phenotypes in C1qa−/− mice are so diverse.
Several previous studies have described synapse loss following critical period MD (Mataga et al., 2004; Coleman et al., 2010; Zhou et al., 2017). Here we attempted to replicate the finding that MD induces a reduction in spine numbers on the apical dendrites of L2/3 pyramidal neurons (Mataga et al., 2004). Surprisingly, we found no loss of spines in WT animals following MD. Why we fail to replicate published work is not clear. One explanation may be the use of different techniques to label spines: whereas the previous study used diolistic labeling to mark the neurons, we used Golgi-Cox staining. It is possible that the two methods label spines differently, so that spines detected using Golgi-Cox staining are not visualized with diolistic labeling, or vice versa. Alternatively, the techniques may be labeling different subpopulations of neurons. Finally, there may be strain-specific differences in responses to ODP between the two transgenic lines. Although we lack a conclusive answer to the contradictory results, it seems reasonable to assume from our results that MD does not induce a robust spine loss on the apical dendrites of all mouse L2/3 pyramidal neurons.
Our results suggest that neither C1q, nor spine loss on proximal apical dendrites of L2/3 pyramidal neurons, is required for ODP, but this does not mean that synapse loss in general has no impact on ODP. Several previous studies have described loss of different synaptic populations following brief (3–4 d) and chronic MD in mice and rats (Coleman et al., 2010; Montey and Quinlan, 2011; Zhou et al., 2017). Furthermore, microglial engulfment has been implicated in ODP. Levels of the microglial lysosomal marker CD68 increase in V1b following MD (Schecter et al., 2017), and microglial engulfment of postsynaptic markers is elevated in V1b during ODP (Sipe et al., 2016). Additionally, the microglial gene P2ry12 contributes to ODP (Sipe et al., 2016), although the microglial chemokine receptor CX3CR1 does not (Lowery et al., 2017; Schecter et al., 2017). Thus, in the circumstances where MD-induced spine loss has been previously described, microglia could contribute to ODP by eliminating synapses through other mechanisms than C1q. Future work will be necessary to determine whether microglial synapse engulfment does indeed help promote synapse loss during ODP, and whether the many observed examples of MD-induced synapse loss are dependent on microglia.
Although the phenotypes that we observe in the C1qa−/− animals in this study are mild, the classical complement cascade has previously been compellingly implicated in several brain disorders involving excess synapse elimination. One connection is to schizophrenia: the downstream classical complement cascade member C4A is genetically linked to that disease (Sekar et al., 2016). Whether C1q contributes to the role of C4A in schizophrenia is, however, not yet clear. By far the strongest connection between C1q and disease instead lies in neurodegeneration. C1q is significantly upregulated and mediates pathological synapse loss in models of Alzheimer's and neurodegenerative disease (Hong et al., 2016; Lui et al., 2016; Vasek et al., 2016; Dejanovic et al., 2018). Depending on the context, C1q can thus have significant effects on brain function. Elucidating the circumstances that determine the impact of C1q on synapses will be highly helpful for understanding its role in disease.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS; R01-NS-071008). We thank Daniel Tom, Ryan Carelli, Michael Blanchard, and Michelle Ocana of the Neurobiology Imaging Facility at Harvard Medical School (NINDS P30 Core Center Grant NS072030); Cassandra-Victoria Innocent of the Intellectual and Developmental Disabilities Research Center (IDDRC) Cellular Imaging Core at Boston Children's Hospital (U54 HD090255) for help with imaging; Allie Muthukumar for technical help with SIM imaging; the Harvard Center for Biological Imaging for infrastructure and support; Nick Andrews of the Neurodevelopmental Behavioral Core at Boston Children's Hospital (CHB IDDRC, 1U54HD090255) for providing a dark room; Carla Shatz for scientific advice and for generously sharing a mouse Arc probe; Tim Hammond and Chinfei Chen for critical reading of the paper; and Dorathy Vargas for help with mouse colony maintenance. We also acknowledge NIH SIG award (1S10RR029237-01), which was used to acquire the ELYRA microscope at the Harvard Center for Biological Imaging.
B.S. serves on the scientific advisory board of Annexon and is a minor shareholder of Annexon. The remaining authors declare no competing financial interests.
References
- Allen PB, Ouimet CC, Greengard P (1997) Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci U S A 94:9956–9961. 10.1073/pnas.94.18.9956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochner DN, Sapp RW, Adelson JD, Zhang S, Lee H, Djurisic M, Syken J, Dan Y, Shatz CJ (2014) Blocking PirB up-regulates spines and functional synapses to unlock visual cortical plasticity and facilitate recovery from amblyopia. Sci Transl Med 6:258ra140. 10.1126/scitranslmed.3010157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto M, Dell' Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ (1998) Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet 19:56–59. 10.1038/ng0598-56 [DOI] [PubMed] [Google Scholar]
- Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA (2010) Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A 107:7975–7980. 10.1073/pnas.0913449107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JE, Nahmani M, Gavornik JP, Haslinger R, Heynen AJ, Erisir A, Bear MF (2010) Rapid structural remodeling of thalamocortical synapses parallels experience-dependent functional plasticity in mouse primary visual cortex. J Neurosci 30:9670–9682. 10.1523/JNEUROSCI.1248-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, Foreman O, Carano RAD, Chih B, Klumperman J, Bakalarski C, Hanson JE, Sheng M (2018) Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron 100:1322–1336.e7. 10.1016/j.neuron.2018.10.014 [DOI] [PubMed] [Google Scholar]
- Djurisic M, Vidal GS, Mann M, Aharon A, Kim T, Ferrao Santos A, Zuo Y, Hübener M, Shatz CJ (2013) PirB regulates a structural substrate for cortical plasticity. Proc Natl Acad Sci U S A 110:20771–20776. 10.1073/pnas.1321092110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjöström PJ, van Meyel DJ (2014) Neuronal morphometry directly from bitmap images. Nat Methods 11:982–984. 10.1038/nmeth.3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, Tenner AJ (2017) Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation 14:48. 10.1186/s12974-017-0814-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glausier JR, Lewis DA (2013) Dendritic spine pathology in schizophrenia. Neuroscience 251:90–107. 10.1016/j.neuroscience.2012.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JA, Stryker MP (1996) Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci 16:3274–3286. 10.1523/JNEUROSCI.16-10-03274.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB (2002) Long-term dendritic spine stability in the adult cortex. Nature 420:812–816. 10.1038/nature01276 [DOI] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, Marsh SE, Saunders A, Macosko E, Ginhoux F, Chen J, Franklin RJM, Piao X, McCarroll SA, Stevens B (2019) Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50:253–271.e6. 10.1016/j.immuni.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K (2006) Experience-dependent and cell-type-specific spine growth in the neocortex. Nature 441:979–983. 10.1038/nature04783 [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YK, Chen C (2011) Wiring and rewiring of the retinogeniculate synapse. Curr Opin Neurobiol 21:228–237. 10.1016/j.conb.2011.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. (1979) Synaptic density in human frontal cortex: developmental changes and effects of aging. Brain Res 163:195–205. 10.1016/0006-8993(79)90349-4 [DOI] [PubMed] [Google Scholar]
- Kano M, Watanabe T, Uesaka N, Watanabe M (2018) Multiple phases of climbing fiber synapse elimination in the developing cerebellum. Cerebellum 17:722–734. 10.1007/s12311-018-0964-z [DOI] [PubMed] [Google Scholar]
- Kanold PO, Kim YA, GrandPre T, Shatz CJ (2009) Co-regulation of ocular dominance plasticity and NMDA receptor subunit expression in glutamic acid decarboxylase-65 knock-out mice. J Physiol 587:2857–2867. 10.1113/jphysiol.2009.171215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T, Mrsic-Flogel TD, Vaz Afonso M, Eysel UT, Bonhoeffer T, Hübener M (2008) Massive restructuring of neuronal circuits during functional reorganization of adult visual cortex. Nat Neurosci 11:1162–1167. 10.1038/nn.2181 [DOI] [PubMed] [Google Scholar]
- Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ, Heller MD, Umemori H, Chen C, Stevens B (2018) CD47 protects synapses from excess microglia-mediated pruning during development. Neuron 100:120–134.e6. 10.1016/j.neuron.2018.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Shatz CJ (2000) Rapid regulation of brain-derived neurotrophic factor mRNA within eye-specific circuits during ocular dominance column formation. J Neurosci 20:1470–1483. 10.1523/JNEUROSCI.20-04-01470.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Chen C (2008) Different roles for AMPA and NMDA receptors in transmission at the immature retinogeniculate synapse. J Neurophysiol 99:629–643. 10.1152/jn.01171.2007 [DOI] [PubMed] [Google Scholar]
- Longair MH, Baker DA, Armstrong JD (2011) Simple neurite tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27:2453–2454. 10.1093/bioinformatics/btr390 [DOI] [PubMed] [Google Scholar]
- Lowery RL, Tremblay ME, Hopkins BE, Majewska AK (2017) The microglial fractalkine receptor is not required for activity-dependent plasticity in the mouse visual system. Glia 65:1744–1761. 10.1002/glia.23192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, Coppola G, Sloan SA, Hsieh CL, Kim CC, Bigio EH, Weintraub S, Mesulam MM, Rademakers R, Mackenzie IR, Seeley WW, et al. (2016) Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 165:921–935. 10.1016/j.cell.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, Worley PF (1995) Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 14:433–445. 10.1016/0896-6273(95)90299-6 [DOI] [PubMed] [Google Scholar]
- Ma Y, Ramachandran A, Ford N, Parada I, Prince DA (2013) Remodeling of dendrites and spines in the C1q knockout model of genetic epilepsy. Epilepsia 54:1232–1239. 10.1111/epi.12195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataga N, Mizuguchi Y, Hensch TK (2004) Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron 44:1031–1041. 10.1016/j.neuron.2004.11.028 [DOI] [PubMed] [Google Scholar]
- Montey KL, Quinlan EM (2011) Recovery from chronic monocular deprivation following reactivation of thalamocortical plasticity by dark exposure. Nat Commun 2:317. 10.1038/ncomms1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2:a006338. 10.1101/cshperspect.a006338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11:785–797. 10.1038/ni.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA (2018) Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174:1015–1030.e16. 10.1016/j.cell.2018.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecter RW, Maher EE, Welsh CA, Stevens B, Erisir A, Bear MF (2017) Experience-dependent synaptic plasticity in V1 occurs without microglial CX3CR1. J Neurosci 37:10541–10553. 10.1523/JNEUROSCI.2679-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE; Schizophrenia Working Group of the Psychiatric Genomics Consortium (2016) Schizophrenia risk from complex variation of complement component 4. Nature 530:177–183. 10.1038/nature16549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe GO, Lowery RL, Tremblay MÈ, Kelly EA, Lamantia CE, Majewska AK (2016) Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun 7:10905. 10.1038/ncomms10905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Straehle C, Köthe U, Hamprecht FA (2011) Ilastik: interactive learning and segmentation toolkit. 2011 IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Chicago, IL, March. [Google Scholar]
- Spiga S, Acquas E, Puddu MC, Mulas G, Lintas A, Diana M (2011) Simultaneous Golgi-Cox and immunofluorescence using confocal microscopy. Brain Structure and Function 216:171–182. 10.1007/s00429-011-0312-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH, Huang EJ, Rowitch DH, Berns DS, Tenner AJ, Shamloo M, Barres BA (2013) A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33:13460–13474. 10.1523/JNEUROSCI.1333-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephany CÉ, Chan LLH, Parivash SN, Dorton HM, Piechowicz M, Qiu S, McGee AW (2014) Plasticity of binocularity and visual acuity are differentially limited by nogo receptor. J Neurosci 34:11631–11640. 10.1523/JNEUROSCI.0545-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephany CÉ, Ikrar T, Nguyen C, Xu X, McGee AW (2016) Nogo receptor 1 confines a disinhibitory microcircuit to the critical period in visual cortex. J Neurosci 36:11006–11012. 10.1523/JNEUROSCI.0935-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephany CÉ, Ma X, Dorton HM, Wu J, Solomon AM, Frantz MG, Qiu S, McGee AW (2018) Distinct circuits for recovery of eye dominance and acuity in murine amblyopia. Curr Biol 28:1914–1923.e5. 10.1016/j.cub.2018.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Sunkin SM, Ng L, Lau C, Dolbeare T, Gilbert TL, Thompson CL, Hawrylycz M, Dang C (2012) Allen brain atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res 41:D996–D1008. 10.1093/nar/gks1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syken J, Grandpre T, Kanold PO, Shatz CJ (2006) PirB restricts ocular-dominance plasticity in visual cortex. Science 313:1795–1800. 10.1126/science.1128232 [DOI] [PubMed] [Google Scholar]
- Tagawa Y, Kanold PO, Majdan M, Shatz CJ (2005) Multiple periods of functional ocular dominance plasticity in mouse visual cortex. Nat Neurosci 8:380–388. 10.1038/nn1410 [DOI] [PubMed] [Google Scholar]
- Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ (2017) C1q: a fresh look upon an old molecule. Mol Immunol 89:73–83. 10.1016/j.molimm.2017.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420:788–794. 10.1038/nature01273 [DOI] [PubMed] [Google Scholar]
- Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, Funk K, DeMasters BK, Jiang X, Bowen JR, Mennerick S, Robinson JK, Garbow JR, Tyler KL, Suthar MS, Schmidt RE, et al. (2016) A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 534:538–543. 10.1038/nature18283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y (2012) RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14:22–29. 10.1016/j.jmoldx.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Portier BP, Gruver AM, Bui S, Wang H, Su N, Vo HT, Ma XJ, Luo Y, Budd GT, Tubbs RR (2013) Automated quantitative RNA in situ hybridization for resolution of equivocal and heterogeneous ERBB2 (HER2) status in invasive breast carcinoma. J Mol Diagn 15:210–219. 10.1016/j.jmoldx.2012.10.003 [DOI] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH (1963) Single-cell responses in striate cortex of kittens deprived of vision in one eye. J Neurophysiol 26:1003–1017. 10.1152/jn.1963.26.6.1003 [DOI] [PubMed] [Google Scholar]
- William CM, Andermann ML, Goldey GJ, Roumis DK, Reid RC, Shatz CJ, Albers MW, Frosch MP, Hyman BT (2012) Synaptic plasticity defect following visual deprivation in Alzheimer's disease model transgenic mice. J Neurosci 32:8004–8011. 10.1523/JNEUROSCI.5369-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- William CM, Saqran L, Stern MA, Chiang CL, Herrick SP, Rangwala A, Albers MW, Frosch MP, Hyman BT (2017) Activity-dependent dysfunction in visual and olfactory sensory systems in mouse models of down syndrome. J Neurosci 37:9880–9888. 10.1523/JNEUROSCI.1045-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G, Codeluppi S, Furlan A, Lee K, Skene N, Harris KD, Hjerling-Leffler J, Arenas E, Ernfors P, Marklund U, Linnarsson S (2018) Molecular architecture of the mouse nervous system. Cell 174:999–1014.e22. 10.1016/j.cell.2018.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Lai B, Gan WB (2017) Monocular deprivation induces dendritic spine elimination in the developing mouse visual cortex. Sci Rep 7:4977. 10.1038/s41598-017-05337-6 [DOI] [PMC free article] [PubMed] [Google Scholar]