

The authors performed a retrospective, observational, cross-sectional study to assess the prevalence and characteristic features of cerebellar heterotopias in 35 patients with CHARGE syndrome with available brain MR imaging studies, as well as to evaluate additional features of cerebellar dysgenesis. Cerebellar heterotopias were identified in 27/35 (77%) patients with CHARGE, characteristic in both location and appearance. Additional features of cerebellar dysgenesis were present in 31/34 evaluable patients (91%), including inferior vermian hypoplasia (90%), anteromedial rotation of the inferior tonsils (90%), and disorganized foliation of the cerebellar hemispheres (74%) or superior vermis (16%). Patients with CHARGE syndrome have a high prevalence of characteristic cerebellar heterotopias.
Abstract
BACKGROUND AND PURPOSE:
Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, and Ear abnormalities and deafness (CHARGE) syndrome is a multisystem developmental disorder associated with a number of well-described clinical and imaging findings, including cerebellar hypoplasia. We observed cerebellar heterotopias on MR imaging in 2 patients with CHARGE, confirmed by postmortem examination. We sought to determine the prevalence and define the characteristics of similar findings on MR imaging for a cohort of patients with CHARGE syndrome.
MATERIALS AND METHODS:
We performed a retrospective, observational, cross-sectional study to assess the prevalence and characteristic features of cerebellar heterotopias in 35 patients with CHARGE syndrome with available brain MR imaging studies, as well as to evaluate additional features of cerebellar dysgenesis.
RESULTS:
Cerebellar heterotopias were identified in 27/35 (77%) patients with CHARGE, characteristic in both location and appearance. Additional features of cerebellar dysgenesis were present in 31/34 evaluable patients (91%), including inferior vermian hypoplasia (90%), anteromedial rotation of the inferior tonsils (90%), and disorganized foliation of the cerebellar hemispheres (74%) or superior vermis (16%).
CONCLUSIONS:
Patients with CHARGE syndrome have a high prevalence of characteristic cerebellar heterotopias and disorganized foliation and abnormal cerebellar morphology, thereby expanding the phenotype of cerebellar dysgenesis in this syndrome.
Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, and Ear abnormalities and deafness (CHARGE) syndrome (Online Mendelian Inheritance in Man, 214800) is a multisystem developmental disorder most commonly caused by heterozygous pathogenic variants in the chromodomain helicase DNA binding protein 7 (CHD7) gene encoding chromodomain helicase DNA-binding protein 7.1 Cardinal features include colobomata, congenital heart disease, choanal atresia, growth retardation and developmental delay, genitourinary abnormalities, and characteristic ear anomalies or sensorineural hearing loss. Diagnostic criteria as initially proposed by Blake et al2 and modified by Amiel et al3 and Verloes4 include the combination of major and minor criteria based on both clinical and radiologic analyses (Table).
Diagnostic criteria for CHARGE syndrome
| Diagnostic Characteristics | Manifestations |
|---|---|
| Major criteria | |
| Ocular coloboma | Coloboma of the iris, retina, choroid, disc; microphthalmos |
| Choanal atresia or stenosis | Unilateral or bilateral, bony or membranous, atresia or stenosis |
| Cranial nerve dysfunction or anomaly | I, Hyposmia or anosmia |
| VII, Facial palsy (unilateral or bilateral) | |
| VIII, Hypoplasia of auditory nerve | |
| IX/X, Swallowing problems with aspiration | |
| CHARGE syndrome ear | Outer ear: short, wide ear with little or no lobe, “snipped off” helix, prominent antihelix that is often discontinuous with tragus, triangular concha, decreased cartilage; often protruding and usually asymmetric |
| Middle ear: ossicular malformations | |
| Cochlea: incomplete partition | |
| Vestibular apparatus: absent or hypoplastic semicircular canals | |
| Minor criteria | |
| Genital hypoplasia | Males: micropenis, cryptorchidism |
| Females: hypoplastic labia | |
| Developmental delay | Delayed milestones, hypotonia |
| Cardiovascular malformation | Conotruncal defects (eg, tetralogy of Fallot), atrioventricular canal defects, aortic arch anomalies |
| Growth deficiency | Short stature, usually postnatal with or without growth hormone deficiency |
| Orofacial cleft | Cleft lip and/or palate |
| Tracheoesophageal fistula | Tracheoesophageal defects of all types |
| Distinctive facial features | Square face with broad prominent forehead, prominent nasal bridge and columella, flat midface |
More recently, several additional radiologic features characteristic of CHARGE syndrome have been reported that may serve to improve the performance of future iterations of diagnostic criteria. Among these, posterior fossa anomalies, including cerebellar hypoplasia5,6 and skull base malformations,7–12 have recently been reported, highlighting increasing recognition of CNS manifestations of the disease.
We noted unusual paired foci of signal abnormality on MR imaging in the cerebellar hemispheres of children with CHARGE syndrome, corresponding to neuronal heterotopias on histopathologic analysis in 2 patients who underwent postmortem examination. Imaging-apparent cerebellar heterotopias have not been previously reported in CHARGE syndrome. We subsequently performed a retrospective investigation of the prevalence of cerebellar heterotopias on MR imaging in a larger cohort of children with clinically and/or genetically confirmed CHARGE syndrome. We further sought to correlate the presence of heterotopias with other findings of cerebellar dysgenesis.
MATERIALS AND METHODS
The study was performed with approval from Seattle Children’s Hospital’s institutional review board. From an institutional data base of 43 patients with a clinical and/or genetic diagnosis of CHARGE syndrome, we identified 35 patients 0–18 years of age who had at least 1 brain MR imaging available for analysis. Details regarding the clinical and/or genetic basis for diagnosis were extracted from the medical record by chart review (On-line Table). Clinical diagnoses were subcategorized as “definite CHARGE syndrome” or “probable/possible CHARGE syndrome” as outlined by Lalani et al.13
Using a diagnostic PACS viewer, 2 board-certified pediatric neuroradiologists, with 8 (J.N.W.) and 7 (F.P.) years’ experience, independently reviewed the MR imaging studies and resolved discrepancies by consensus. All available anatomic sequences were evaluated for cerebellar white matter heterotopias, with reviewers noting the presence, location, bilaterality, and signal characteristics. We further evaluated this cohort for additional signs of cerebellar dysgenesis. Based on a prior report of cerebellar abnormalities5,6 and an initial preliminary review of our patient cohort, specific features chosen for analysis included inferior vermian hypoplasia, abnormal rotation of the inferior cerebellar hemispheres, and disorganized cerebellar foliation.
We correlated the presence of cerebellar heterotopias as well as presence of any features of cerebellar dysgenesis with both a definite-versus-probable/possible clinical diagnosis and a clinical-versus-genetic diagnosis of CHARGE syndrome using the Fisher exact test. We correlated the presence or absence of the heterotopias with patient age, magnetic field strength, section thickness, and examination quality using the Fisher exact test for categoric variables and point-biserial correlation for continuous variables.
RESULTS
The cohort of 35 patients with an available MR imaging for analysis included 24 patients with definite CHARGE syndrome, 10 patients with probable/possible CHARGE syndrome, and 1 patient with a negative clinical assessment for CHARGE syndrome but a known pathogenic CHD7 variant (the patient died at 8 months of age, limiting the sensitivity of the clinical criteria for diagnosis). Twenty-two patients underwent genetic analysis for CHD7 variants. Of these, 17 had known or predicted pathogenic variants, 4 were negative for them (2 were definite and 2 were probable/possible CHARGE syndrome based on clinical criteria), and 1 returned a variant of unknown significance (clinically categorized as probable/possible CHARGE syndrome). Among the CHD7 gene variations, 4 were stop-gain mutations; 3, frameshift; 3, canonical splice-site; 2, interstitial deletions; 2, missense (1 known pathogenic and 1 of unknown significance); and 3 were reported pathogenic in the medical records without specific genetic results available for review.
We identified cerebellar white matter heterotopias in 27/35 (77%) patients with CHARGE, with histopathologic correlation in 2 patients on postmortem examination. Histologic analysis in both cases demonstrated neuronal heterotopias, corresponding on gross pathologic sections to the foci of abnormality on MR imaging (Fig 1). The cell rests comprised loose clusters of morphologically normal granular cell neurons admixed with large Purkinje cells in a background of mixed glial cells (Fig 2).
Fig 1.

Radiologic-pathologic correlation in patient 7. Cropped coronal T2-weighted MR image of the right cerebellar hemisphere (A) obtained at 5 days of age shows the subcortical heterotopia (black arrow). Corresponding gross pathologic specimen (B) demonstrates the lesion as a white nodule (black arrow). Whole-mount hematoxylin-eosin–stained microscopic pathologic specimen (C) demonstrates the corresponding neuronal heterotopia (black arrow) (original magnification ×1).
Fig 2.

Photomicrographs demonstrate the histopathology of the heterotopia in patient 7. A, A cluster of disorganized neurons (black arrows) is juxtaposed to an adjacent normal-layered foliar cortex (bracketed by black arrowheads) (hematoxylin-eosin stain, original magnification ×4). B, Heterotopia comprises a dominant population of morphologically normal granular cell neurons in a background of mixed glial cells (hematoxylin-eosin stain, original magnification ×20). C, Purkinje cells (darkly staining cells) are scattered within the granular cell neurons as well as within the adjacent cortex (HuC stain, original magnification ×4). D, Nerve fibers (darkly staining material) surround and traverse the cluster of heterotopic neurons (calretinin stain, original magnification ×4).
The heterotopias were relatively stereotyped in their MR imaging appearance and location, occurring symmetrically in the immediate subcortical white matter of the bilateral inferior cerebellar hemispheres, distinct from the inferior dentate nuclei (Fig 3). They typically comprised a single ellipsoid cellular conglomeration per hemisphere, though occasionally ≥2 clustered heterotopias were noted on a given side (Fig 4B, -D). Signal intensity was similar or identical to that of adjacent gray matter structures, standing out as hyperintense on T1 and hypointense on T2 relative to surrounding unmyelinated white matter, and hypointense on T1 and hyperintense on T2 relative to myelinated white matter (Fig 4). Heterotopias were isointense to the cortex on diffusion-weighted imaging when visible, though conspicuity was limited due to low spatial resolution and axial imaging plane.
Fig 3.
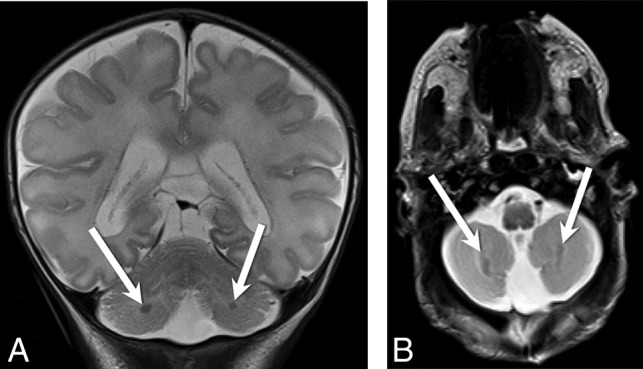
Coronal (A) and axial (B) T2-weighted images from patient 7 demonstrate bilateral cerebellar heterotopias (white arrows, A and B) with characteristic ellipsoid morphology and subcortical location in the bilateral inferior cerebellar hemispheres.
Fig 4.

Coronal T2-weighted images from patient 15 at 3 days (A) and again at 3 years (B) of age demonstrate the expected evolution of the signal intensity of the heterotopias—hypointense relative to surrounding unmyelinated white matter (white arrows, A), becoming hyperintense relative to surrounding myelinated white matter (white arrows, B). Coronal T1-weighted images from patient 7 at 3 days of age (C) and patient 17 at 13 months of age (D) demonstrate similar isointensity of the heterotopias (white arrows, C and D) to gray matter.
Additional findings of cerebellar dysgenesis were identified in a total of 31/34 (91%) patients in our series, with 28/31 (90%) demonstrating multiple features. One patient who was status post medulloblastoma resection was excluded from analysis for features of dysgenesis. Among patients with dysgenesis, the most common findings were abnormal anteromedial rotation of the cerebellar tonsils (28/31, 90%), frequently but not invariably associated with inferior vermian hypoplasia (28/31, 90%) (Figs 5 and 6). Fewer patients demonstrated disorganized foliation of the cerebellar hemispheres (23/31, 74%) or superior vermis (5/31, 16%) (Fig 7). A single patient (1/35, 3%) had a Chiari I malformation. Most (25/26, 96%) patients with identifiable heterotopias had ≥1 additional finding of cerebellar dysgenesis.
Fig 5.

Axial T2-weighted images from patient 22 (A), patient 33 (B), patient 13 (C), and patient 1 (D) demonstrate anteromedial rotation of the inferior cerebellar tonsils. Axial T2-weighted image from a 10-month-old control patient is included for comparison (E).
Fig 7.

Coronal and axial T2-weighted images from patient 11 (A and B), patient 30 (C and D), and patient 22 (E and F) demonstrate a diffusely disorganized pattern of cerebellar foliation of the bilateral cerebellar hemispheres. Coronal (G) and axial (H) T2-weighted images from a 10-month-old control patient are included for comparison.
Fig 6.

Sagittal T1-weighted images in patient 25 (A), patient 19 (B), patient 16 (C), and patient 12 (D) demonstrate a spectrum of inferior vermian hypoplasia. Axial T2-weighted image from a 10-month-old control patient is included for comparison (E).
The presence of heterotopias did not correlate with definite-versus-probable/possible clinical categorization (19/24 [79%] vs 6/10 [60%]; P = .40), or clinical-versus-genetic diagnosis (11/17 [65%] vs 15/18 [83%]; P = .80) of CHARGE syndrome. The presence of cerebellar dysgenesis correlated with definite-versus-probable/possible clinical categorization (23/23 [100%] vs 7/10 [70%]; P = .02), but the correlation did not hold after correction for multiple comparisons. There was no correlation between cerebellar dysgenesis and a clinical-versus-genetic diagnosis (15/17 [88%] vs 16/17 [94%]; P = 1.00) of CHARGE syndrome. Neither the presence of heterotopias nor the presence of cerebellar dysgenesis was more generally correlated with MR imaging field strength (P = .62 for heterotopias, P = 1.00 for dysgenesis), MR imaging section thickness (r = −0.29/P = .11 for heterotopias; r = −0.34/P = .06 for dysgenesis), or age at the time of MR imaging (r = −0.02/P = .920 for heterotopias; r = 0.06/P = .75 for dysgenesis).
DISCUSSION
Microscopic cerebellar heterotopias are relatively common in children on histologic evaluation and are well-described in the pathology literature. Rorke et al14 described 4 histologic subtypes of microscopic heterotopias identified in the cerebella of infants, most (147/200, 74%) of which were found in children without associated somatic or neural malformations. The only reported consistent syndromic association was with Trisomy 13.
Legendre et al15 reported on the neuropathology of 40 aborted fetuses with an antenatal diagnosis of CHARGE syndrome and found “massive Purkinje cell heterotopias” in a minority (8/40, 20%). Lin et al16 also reported a patient with CHARGE syndrome who had a “neuronal heterotopia” on pathologic examination. Neither publication provided specific histopathologic details. Identification of macroscopic cerebellar heterotopias on routine clinical MR imaging has only rarely been reported17–19 and never previously in CHARGE syndrome, to our knowledge.
Histologic analysis in 2 of our patients who underwent postmortem examination confirmed the presence of large neuronal heterotopias, corresponding to the foci of abnormality on MR imaging. The cell rests comprised loose, disorganized clusters of morphologically normal granular cell neurons admixed with normal Purkinje cells in a background of mixed glial cells and neuronal processes. Histologically, these cell rests correspond to the “heterotaxia” subtype described by Rorke et al14 according to the earlier classification of Brun.20
This study demonstrates that macroscopic cerebellar heterotopias are a frequent neuroimaging finding in CHARGE syndrome, present in at least 77% of patients in our series. They were most commonly and easily identified on the T2 sequence in the coronal plane, less commonly on the coronal T1 sequences. Of the 9 patients in whom the finding was not identified, substantial motion artifacts were present on one of the scans, 3 others lacked any sequences obtained in the coronal plane, and severe cerebellar architectural distortion related to Chiari I malformation was present on an additional scan, suggesting that the prevalence of 77% may be an underestimation. Indeed, one of the patients characterized on initial review as being negative for heterotopias with a motion-degraded examination had a subsequent follow-up examination that well-demonstrated bilateral heterotopias.
These cerebellar heterotopias are stereotyped in their MR imaging appearance and location. As expected, signal intensity relative to surrounding white matter evolved with progressive cerebellar myelination. Furthermore, they were consistently noted symmetrically within the immediate subcortical white matter of the bilateral inferior cerebellar hemispheres. Thus, they may prove useful as one of the more objective imaging findings of cerebellar dysgenesis in CHARGE syndrome.
Identification of these heterotopias was not associated with age at scanning, magnet strength, or scan parameters. These findings suggest that lack of awareness of this easily overlooked imaging finding is the primary driver behind lack of identification. In 1 case, however, heterotopias that were clearly identifiable on an MR imaging obtained at 3 days of age were substantially decreased in conspicuity on repeat imaging at 3 months of age, suggesting that transitional myelination in the deep cerebellar white matter that peaks at 3–4 postnatal months may limit detection in some cases.
Cerebellar heterotopias in our series largely occurred in the context of more widespread findings of cerebellar dysgenesis. Recent reports have described inferior vermian hypoplasia and generally disordered foliation in patients with CHARGE syndrome.5,6 Our study both reinforces and expands this assertion. A greater proportion of patients in our series had cerebellar abnormalities than previously reported by Yu et al5 al or Sohn et al6 (91% vs 55% vs 29%, respectively). Furthermore, we report a wider diversity of imaging findings of cerebellar maldevelopment. Although vermian hypoplasia was a common finding, we also noted relatively characteristic abnormalities in cerebellar fissuration and foliation, most notable in the inferior cerebellar hemispheres. These included anteromedial rotation of the inferior cerebellar tonsils, which was commonly but not invariably found in association with inferior vermian hypoplasia and therefore treated as a primary morphologic abnormality, as well as a diffusely disorganized foliar pattern. These findings extend and refine the phenotype of cerebellar dysgenesis seen in patients with CHARGE syndrome.
On the basis of mouse models, Yu et al5 suggested that homeotic transformation of the dorsal first rhombomere related to altered Fgf8 gene expression might be responsible for the cerebellar findings of CHARGE syndrome. The high prevalence of heterotopias in patients with CHARGE syndrome suggests that secondary or unrelated derangements in gene products related to cerebellar cellular migration may also be relevant. The foliation abnormalities we report are consistent with type 2 abnormalities in the classification of cerebellar malformations by Demaerel,21 which he suggested implicated a broader defect in cellular migration and organization.
The close association of cerebellar heterotopias and aberrant foliation is predicted by the classification system of cerebellar malformations proposed by Barkovich et al.18 It is known that cerebellar neuronal migration and proliferation act as a trigger for normal foliation and fissuration.22,23 Mice with pathogenic mutations in genes encoding cellular guidance proteins may demonstrate a phenotype that includes a deficient vermis, cerebellar heterotopias, and dysplastic foliation.24,25 It is likely that varied patterns of expression in multiple genes secondary to the underlying CHD7 mutation, including those contributory to cerebellar cellular migration, are responsible for the cerebellar dysgenesis in CHARGE syndrome.
CONCLUSIONS
We present the novel imaging finding of characteristic cerebellar neuronal heterotopias in patients with CHARGE syndrome, commonly associated with disordered foliation and abnormal inferior cerebellar morphology, thereby expanding the phenotype of cerebellar dysgenesis in this syndrome. The heterotopias are symmetrically located in the subcortical white matter of the bilateral inferior cerebellar hemispheres and were common in our cohort. Recognition of these findings should prompt consideration of CHARGE syndrome in the undiagnosed patient, and they can serve as supportive neuroimaging markers for the condition in patients for whom the diagnosis is being considered.
ABBREVIATION:
- CHARGE
Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, and Ear abnormalities and deafness
Footnotes
Disclosures: Joe Rutledge—UNRELATED: Travel/Accommodations/Meeting Expenses Unrelated to Activities Listed: College of American Pathologists, Comments: reimbursement of travel to committee meetings (3 times a year).
References
- 1. Vissers LE, van Ravenswaaij CM, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004;36:955–57 10.1038/ng1407 [DOI] [PubMed] [Google Scholar]
- 2. Blake KD, Davenport SL, Hall BD, et al. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila) 1998;37:159–73 10.1177/000992289803700302 [DOI] [PubMed] [Google Scholar]
- 3. Amiel J, Attieé-Bitach T, Marianowski R, et al. Temporal bone anomaly proposed as a major criteria for diagnosis of CHARGE syndrome. Am J Med Genet 2001;99:124–27 10.1002/1096-8628(20010301)99:2<124::aid-ajmg1114>3.0.co;2-9 [DOI] [PubMed] [Google Scholar]
- 4. Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A 2005;133A:306–08 [DOI] [PubMed] [Google Scholar]
- 5. Yu T, Meiners LC, Danielsen K, et al. Deregulated FGF and homeotic gene expression underlies cerebellar vermis hypoplasia in CHARGE syndrome. Elife 2013;2:e01305 10.1002/ajmg.a.30559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sohn YB, Ko JM, Shin CH, et al. Cerebellar vermis hypoplasia in CHARGE syndrome: clinical and molecular characterization of 18 unrelated Korean patients. J Hum Genet 2016;61:235–39 10.1038/jhg.2015.135 [DOI] [PubMed] [Google Scholar]
- 7. Fujita K, Aida N, Asakura Y, et al. Abnormal basioccipital development in CHARGE syndrome. AJNR Am J Neuroradiol 2009;30:629–34 10.3174/ajnr.A1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Natung T, Goyal A, Handique A, et al. Symmetrical chorioretinal colobomata with craniovertebral junction anomalies in CHARGE syndrome: a case report with review of literature. J Clin Imaging Sci 2014;4:5 10.4103/2156-7514.126046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoch MJ, Patel SH, Jethanamest D, et al. Head and neck MRI findings in CHARGE syndrome. AJNR Am J Neuroradiol 2017;38:2357–63 10.3174/ajnr.A5297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mahdi E, Whitehead MT.. Coronal clival cleft in CHARGE syndrome. Neuroradiol J 2017;30:574–77 10.1177/1971400916678248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mahdi ES, Whitehead MT.. Clival malformations in CHARGE syndrome. AJNR Am J Neuroradiol 2018;39:1153–56 10.3174/ajnr.A5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Geus CM, Bergman JEH, van Ravenswaaij-Arts CM, et al. Imaging of clival hypoplasia in CHARGE syndrome and hypothesis for development: a case-control study. AJNR Am J Neuroradiol 2018;39:1938–42 10.3174/ajnr.A5810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lalani SR, Hefner MA, Belmont JW, et al. CHARGE syndrome In: Adam MP, Pagon RA, Ardinger HH, et al., eds. GeneReviews. Seattle: University of Washington; 2012 [Google Scholar]
- 14. Rorke LB, Fogelson MH, Riggs HE.. Cerebellar heterotopia in infancy. Dev Med Child Neurol 1968;10:644–50 10.1111/j.1469-8749.1968.tb02951.x [DOI] [PubMed] [Google Scholar]
- 15. Legendre M, Gonzales M, Goudefroye G, et al. Antenatal spectrum of CHARGE syndrome in 40 fetuses with CHD7 mutations. J Med Genet 2012;49:698–707 10.1136/jmedgenet-2012-100926 [DOI] [PubMed] [Google Scholar]
- 16. Lin AE, Chin AJ, Devine W, et al. The pattern of cardiovascular malformation in the CHARGE association. Am J Dis Child 1987;141:1010–13 10.1001/archpedi.1987.04460090087034 [DOI] [PubMed] [Google Scholar]
- 17. Patel S, Barkovich AJ.. Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol 2002;23:1074–87 [PMC free article] [PubMed] [Google Scholar]
- 18. Barkovich AJ, Millen KJ, Dobyns WB.. A developmental and genetic classification for midbrain-hindbrain malformations. Brain 2009;132:3199–230 10.1093/brain/awp247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salvati A, Dicuonzo F, Carella A, et al. A case of isolated focal cerebellar heterotopia in a patient affected by HHT: MR and MR spectroscopy findings. J Neurol Neurophysiol 2010;01:107 10.4172/2155-9562.1000107 [DOI] [Google Scholar]
- 20. Brun A. Zur Kenntnis der Bildungsfehler des Kleinhirns; Epikritische Bemerkungen zur Entwicklungspathologie, Morphologie und Klinik der umschriebenen Entwicklung-shemmungen des Neozerebellums. Schweiz Arch Neurol Psych 1917;1:48–105 [Google Scholar]
- 21. Demaerel P. Abnormalities of cerebellar foliation and fissuration: classification, neurogenetics and clinicoradiological correlations. Neuroradiology 2002;44:639–46 10.1007/s00234-002-0783-1 [DOI] [PubMed] [Google Scholar]
- 22. Millen KJ, Millonig JH, Wingate RJ, et al. Neurogenetics of the cerebellar system. J Child Neurol 1999;14:574–82 10.1177/088307389901400905 [DOI] [PubMed] [Google Scholar]
- 23. Sudarov A, Joyner AL.. Cerebellum morphogenesis: the foliation pattern is orchestrated by multi-cellular anchoring centers. Neural Dev 2007;2:26 10.1186/1749-8104-2-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuwamura M, Shirota A, Yamate J, et al. Analysis of aberrant neuronal migrations in the hereditary cerebellar vermis defect (CVD) rat using bromodeoxyuridine immunohistochemistry. Acta Neuropathol 1998;95:143–48 10.1007/s004010050778 [DOI] [PubMed] [Google Scholar]
- 25. Kuramoto T, Kuwamura M, Serikawa T.. Rat neurological mutations cerebellar vermis defect and hobble are caused by mutations in the netrin-1 receptor gene Unc5h3. Brain Res Mol Brain Res 2004;122:103–08 10.1016/j.molbrainres.2003.12.003 [DOI] [PubMed] [Google Scholar]