

Abstract
Incoherent neutron scattering (INS) is one of the useful experimental methods for studying protein dynamics at the pico-nanosecond timescale. At this timescale, protein dynamics is highly coupled with hydration, which is observed as protein dynamical transition (PDT). INS is very sensitive to hydrogen atomic dynamics because of the large incoherent scattering cross section of hydrogen atom, and thus, the INS of a hydrated protein provides overall dynamic information about the protein, including hydration water. Separation of hydration water dynamics is essential for understanding hydration-related protein dynamics. H2O/D2O exchange is an effective method in the context of INS experiments for observing the dynamics of protein and hydration water separately. Neutron scattering is directly related to the van Hove space-time correlation function, which can be calculated quantitatively by performing molecular dynamics (MD) simulations. Diffusion and hydrogen bond dynamics of hydration water can be analyzed by performing MD simulation. MD simulation is useful for analyzing the dynamic coupling mechanism in hydration-related protein dynamics from the viewpoint of interpreting INS data because PDT is induced by hydration. In the present work, we demonstrate the methodological advantages of the H2O/D2O exchange technique in INS and the compatibility of INS and MD simulation as tools for studying protein dynamics and hydration water.
Keywords: Quasi-elastic neutron scattering, H2O, D2O exchange, Protein dynamics, Protein hydration
Significance.
Dynamical coupling between protein and hydration water is one of the essential subjects of protein biophysics. In this paper, we report incoherent neutron scattering with H2O/D2O exchange enables to derive hydration water dynamics. All atom MD simulations reveal hydrogen bond dynamics specific to hydration water. These results are comparable with each other and give us deep insight for hydration water dynamics.
Proteins, biologically functional molecules, work in an aqueous environment at ambient temperature. Protein structure fluctuates thermally, and its dynamics is strongly influenced by hydration [1–3]. In fact, a theoretical study demonstrated that the low-frequency collective modes in proteins are strongly coupled to hydration. The hydration-coupled modes contribute dominantly to the motional amplitude of proteins [4]. Except for stochastically rare conformational changes from the micro to the milli second timescales, protein structural dynamics under thermal equilibration is characterized at the pico-to-nanosecond timescale and on order of Angstrom units in the spatial scale [5]. Incoherent neutron scattering (INS) facilitates analysis of protein dynamics in these time and space regions [6,7]. Vibrations, relaxation, and diffusion dynamics are characterized using INS [8]. Molecular vibrations appear as inelastic scattering over a wide energy range, and the inelastic contribution can be approximated using the Debye-Waller factor. The relaxation and diffusion dynamics appear as quasi-elastic neutron scattering (QENS), which accounts for a broad part of the spectrum around elastic scattering. One of the hydration-related protein dynamics observed by INS is protein dynamical transition (PDT) [1,5,9]. This is observed as an abrupt increase in mean-square displacements (MSDs) at approximately 240 K with increasing temperature. PDT is enhanced significantly above the threshold hydration level by percolation transition of hydration water and is inhibited at lower hydration levels [3,10].
The neutron scattering function is expressed by the dynamic structural factor, S(Q,ω), which is a Fourier transformation of van Hove’s correlation function in real space and time, G(r,t) [11]. The neutron scattering function is separated into coherent and incoherent scatterings. INS probes the self-correlation of a single particle, which is useful for characterizing molecule dynamics, while the molecular structure is determined by the analysis of coherent neutron scattering. In Table 1, the neutron cross sections of the main constituent atoms in proteins are listed. The cross sections of incoherent and coherent scattering depend on the element species. Because X-rays are scattered by electrons, scattering cross section is almost proportional to atomic number. This is not the case for neutrons. Particularly noteworthy in Table 1 are large incoherent cross section of hydrogen and its significant isotope effect. The cross section of hydrogen is larger than that of deuterium by one order of magnitude. Typically, half of the constituent atoms of proteins is hydrogen atoms. In INS experiments, protein dynamics are observed through scattering from the hydrogen atoms in protein. Therefore, deuteration isotope labeling can be used for observing site-specific dynamics [12,13]. For studying protein dynamics in the hydrated state by INS, D2O is used for hydration during sample preparation to minimize the scattering due to hydration water. Moreover, H2O/D2O exchange is rather effective for analyzing the protein and hydration water separately [10,14,15].
Table 1.
Coherent, incoherent, and total neutron cross sections of a few elements.
| Elements | σcoh(10−24 cm2) | σinc(10−24 cm2) | σtot(10−24 cm2) |
|---|---|---|---|
| H | 1.76 | 80.27 | 82.03 |
| D | 5.59 | 2.05 | 7.64 |
| C | 5.55 | 0.001 | 5.55 |
| N | 11.01 | 0.50 | 11.51 |
| O | 4.23 | 0.00 | 4.23 |
Molecular dynamics (MD) simulation is a powerful method for studying protein dynamics [16]. From MD results, van Hove’s correlation function, which is directly related to the neutron scattering function, can be calculated [8]. Neutron scattering spectra calculated from MD simulation of crystalline protein is comparable quantitatively to the experimental data obtained using hydrated powder protein samples [17,18]. Moreover, MD simulation is an effective tool for analyzing the dynamics of hydration water, such as diffusion and hydrogen-bonding dynamics [19]. The analysis provides a reasonable interpretation of the neutron spectrum of hydrated proteins [20]. Combined analysis of INS with MD is effective for elucidating the dynamic coupling between protein and hydration water.
In the present work, we analyze the dynamics of hydration water in hydration-induced PDT of staphylococcal nuclease (SNase) by performing an INS experiment with H2O/D2O exchange of hydration water and MD simulation.
Materials and Methods
Sample preparation
SNase was expressed in Escherichia coli and purified by urea extraction and with SP-Sepharose Fast Flow column chromatography. The purified protein was dialyzed against D2O and lyophilized. The resultant powder was dissolved in D2O and lyophilized. The procedure was repeated more than three times to ensure complete substitution with D2O. The obtained lyophilized protein was used as the dehydrated sample (Fig. 1(A)), and its hydration level was estimated as to be 0.11 g D2O/g protein by thermogravimetry [3]. Hydrated SNase was prepared by equilibration of the dehydrated sample with a saturated KCl, NaCl, or NaBr solution of D2O. The corresponding hydration levels were 0.44, 0.36, and 0.30 g D2O/g protein, respectively. The water content of the dehydrated SNase was almost identical to the number of water molecules in its crystal structure (1EY0.pdb). The hydration level of 0.44 g D2O/g protein corresponds roughly to a single hydration layer [3].
Figure 1.

(A) Lyophilized protein power sample in aluminum neutron cell and (B) snapshot from MD simulations of hydrated crystalline model system containing four SNase molecules at h=0.40 g H2O/g protein.
Neutron scattering experiment
Neutron scattering experiments were performed with the triple axis spectrometer LTAS in the JRR-3M reactor, which has an energy resolution of ~106 μeV, in Tokai, Japan. The q-range covered was 0.4–2.0 Å−1. Neutron scattering measurements were performed at intervals of 10 K between 80 K and 300 K. First, the temperature was reduced to 80 K. Then, the measurements were performed at the target temperature. The obtained data were corrected by empty cell background subtraction. Data analysis was performed without multiple scattering correction because the sample transmission value was relatively large, approximately 90%.
The dynamics of hydration water can be examined based on the difference in INS between H2O- and D2O-hydrated proteins, as follows [10,14,15],
| (1) |
The concept is depicted in Figure 2.
Figure 2.

Concept of H2O/D2O exchange for estimating scattering intensity of hydration water based on difference in scattering intensity between H2O- and D2O-hydrated proteins.
MD simulation of protein crystalline
MD simulations of crystalline SNase were performed at a constant temperature (300 K) and pressure (1 atm), which were maintained following the Berendsen method (relaxation time of 1 ps for both) by using the program AMBER [18]. The crystal structure of SNase (PDB code: 1EY0.pdb) was used as the initial simulation structure. The simulated system was constructed to reproduce the crystal unit cell, and it had a space group symmetry of P41 (Fig. 1(B)). Missing residues 1–5 and 142–149 in 1EY0.pdb were modeled on the basis of the NMR structures of SNase (PDB code: 1JOR.pdb). The simulations in the protein crystal explain quantitatively the neutron scattering functions with lyophilized SNase [17,18]. The simulation system consisted of four protein molecules with either 1,110 or 1,494 water molecules, which correspond to h=0.30 and 0.40 g H2O/g protein, respectively. Water molecules were placed randomly in the empty space in each system. For bulk water, 440 water molecules were set in a rectangular box. The periodic boundary condition was imposed, and the particle mesh Ewald method was used with a cutoff of 10.0 Å. The AMBER ff14SB force field and the TIP3P water model were employed [21]. After energy minimization (2000 steps), 2 ns MDs were performed to equilibrate the systems at 300 K and 1 atm. Successive 4 ns trajectories were obtained for the analyses. For hydrogen bond analysis, pairs of water molecules were selected as being hydrogen-bonded only if their inter-oxygen distance was smaller than 3.5 Å and simultaneously the O–H...O angle was greater than 120°.
Results and Discussion
Selective observation of protein and hydration water dynamics by H2O/D2O exchange
Figure 3(A) shows the INS profiles of the D2O- and H2O-hydrated proteins, and the hydration water. The protein dynamics can be observed with the D2O-hydrated sample, where the INS due to hydration water (D2O) is minimized. As shown in Figure 2, the INS due to the hydration water is obtained by the difference in the scattering intensities of an H2O-hydrated sample and a D2O-hydrated sample according to equation (1).
Figure 3.

(A) Neutron scattering profiles at Q=1.1 Å−1 of H2O- and D2O-hydrated proteins at h=0.44 g D2O/g protein and 300 K, and that of hydration water. INS data of hydration water are obtained by subtracting D2O-hydrated protein from H2O-hydrated protein, according to equation (1). The thin line indicates the instrumental resolution function. Quasi-elastic neutron scattering (QENS) of hydration water (B) and bulk water (C) at 300 K. Closed circle and the solid lines denote the observed QENS data and the curve fitted using equation (2), respectively. The thin and dotted lines indicate Lorentzian and constant background, respectively. The under-panel shows the difference between the observed and the fitted QENS intensities.
Figures 3(B) and (C) show the INS due to hydration water at h=0.44 and that due to bulk liquid water (H2O) at 300 K, respectively, where the strong broad QENS spectrum is observed. The scattering function can be expressed approximately as follows [22]:
| (2) |
where A(Q) is the Debye–Waller factor; EISF is an elastic incoherent structural factor; L(Q,ω) is a Lorentzian function with half width at half maximum (HWHM), Γ(Q); and B(ω) is the constant background due to inelastic scattering. The scattering profile can be fitted reasonably by equation (2) at all Q values. EISF decreased to be zero substantially in the present case. This is reasonable for water molecules because EISF is related to the probability of finding an atom at the same position at t=0 and ∞. Therefore, the elastic term was not included in the subsequent data fitting. Figure 4 shows the variation of Γ(Q) with Q2 for the hydration water and the bulk liquid water. The Γ(Q) of bulk water increased with Q from the origin. This result indicates that translational diffusion was dominant over localized motions such as rotation. We analyzed the translational diffusion dynamics due to jump-diffusion model by using the following equation [22]:
Figure 4.

(A) Half-width at half-maximum of Lorentzian function in fitting equation (2) for bulk water and hydration water against Q2. The solid curves correspond to the data fit with the jump-diffusion models. Closed circle, open circle, closed triangle, and open triangle denote hydration water with h=0.44 at 300 K, hydration water with h=0.30 at 300 K, bulk water at 280 K, and bulk water at 300 K, respectively. (B) Enlarged view of (A).
| (3) |
where Dtrans and τ are the diffusion constant and residence time, respectively, and the obtained values are listed in Table 2. Dtrans and τ of bulk liquid water are consistent with the values obtained in a previous work [23]. In case of the hydration water, Γ(Q) values are significantly smaller than that of the bulk liquid water. This suggests that the diffusional dynamics of hydration water in protein is suppressed owing to the interaction with protein surface.
Table 2.
Dynamic parameters of liquid bulk water and protein hydration water at two hydration levels, h=0.30 and 0.44 g D2O/g protein, obtained by quasi-elastic neutron scattering.
| Sample | D*10−5 [cm2/s] | τ [ps] |
|---|---|---|
| h=0.44, 300 K | 0.799±0.073 | 39.75±0.22 |
| h=0.30, 300 K | 0.190±0.131 | 121.62±40.57 |
| Bulk water, 280 K | 2.225±0.032 | 3.01±0.08 |
| Bulk water, 300 K | 3.372±0.042 | 1.44±0.10 |
Figures 5(A) and (B) shows the MSDs of protein and its hydration water, respectively, at different hydration levels as a function of temperature, as obtained in the INS experiments. As for the protein dynamics in Figure 5(A), the MSDs increase monotonously with temperature, and the MSDs of the samples with h=0.36 and 0.44 deviate from those of the samples with h=0.30 above 240 K, which indicates PDT. Figure 5(B) shows that the MSDs of hydration water, which are almost comparable to the MSDs of the proteins below the PDT temperature at all hydration levels. At h=0.30, the MSDs of the protein and the hydration water are similar across the entire temperature range. At h=0.36 and 0.44, the MSDs of the hydration water are greater than those of the proteins above the PDT temperature, and the increment of MSD is greater for h=0.44 than it is for h=0.33. These results indicate that the dynamics of the protein and its hydration water are tightly coupled to each other, and PDT can be controlled by hydration water dynamics. There is a threshold hydration level, h=0.36, for the onset of PDT [10,24]. Dynamics of the hydration water is activated by the percolation transition, and consequently PDT is facilitated by the activated hydration water dynamics via hydrogen bond between them. The percolation of hydration water is the origin of the onset of PDT [10]. The present results are consistent with the previous findings [10,24].
Figure 5.

MSD of protein (A) and hydration water (B) at three hydration levels obtained by INS experiments. Closed circle, open circle, and cross denote h=0.44, 0.36, and 0.30, respectively. The thin line in (A) indicate the regression line below 240 K for h=0.44.
Diffusion and Hydrogen bond dynamics of hydration water by MD simulation
In MD simulation with a crystalline protein system, larger scale motions of protein are suppressed due to the direct contact among neighboring proteins (crowded environment). Due to this effect, the results from MD simulation in this system can reproduce neutron scattering data quantitatively, because samples in INS is powder state, where the direct interaction exists. Furthermore, in principle, incoherent neutron scattering is able to observe the single atomic dynamics. The amplitude of the fluctuation is an order of a few Å. This is much smaller than the periodic box length. Thus, the MD with the periodic box condition is relevant to compare with the observed protein dynamics. The hydration-dependent protein dynamics in MD simulation are compatible with neutron scattering experiments [17,18]. To examine the hydration-level-dependent diffusion and the hydrogen bond dynamics of the hydration water, MD simulations of an all-atom model were performed with hydrated crystalline SNase. From the MD simulation of two hydration levels, h=0.30 and 0.40, at 300 K, the MSDs of hydration water and bulk water were estimated. Hydrogen bond dynamics were analyzed as well. The selected two hydration levels were below and above the hydration threshold for PDT onset [10].
The obtained time-dependent MSDs of hydration water and bulk water are shown in Figure 6. Life time of hydrogen bond between hydration water and protein is pico-second time scale [19]. The hydration water molecules sometimes move to the different hydration sites or the outer hydration layers, and then do not always stay in the same hydration site in this time scale. They are not always oscillating in the harmonic potential. Thus, the dynamics of hydration water can be described as Brownian motion rather than harmonic motion. The Brownian processes in the short-time and longtime are considered as ballistic motions and diffusion motions, respectively [25]. The MSDs of both hydration water and bulk water increase rapidly until 0.2 ps, which can be ascribed to ballistic motions. The ballistic motions of hydration water are indistinguishable from those of liquid water, suggesting that these motions are not affected by the interaction with protein. Over longer timescales, the MSDs of the hydration water are significantly smaller than those of the bulk water. This reduction in the amplitudes of water dynamics can possibly be ascribed to the interaction of water with protein, such as a hydrogen-bond or van der Waals contact and so on. The diffusion constants estimated at 10 ps of bulk water and hydration water at h=0.40 and h=0.30 are 2.7×10−5 cm2/s, 1.32×10−5 cm2/s, and 0.15×10−5 cm2/s, respectively. These values are reasonably comparable to the diffusion constants obtained in INS experiments (see Table 2). Under thermal equilibrium, the hydrogen bonds of water molecules fluctuate temporally. To examine the dynamics of the hydrogen bonds associated with the water molecules, a hydrogen bond population operator, h, was introduced [26]. Its value was unity when a given tagged pair of water molecules was hydrogen-bonded and was zero otherwise. The set of values h for all pairs of water molecules specifies the bond organization at a given instant. The average number of hydrogen bonds in an equilibrium of N water molecules is
Figure 6.
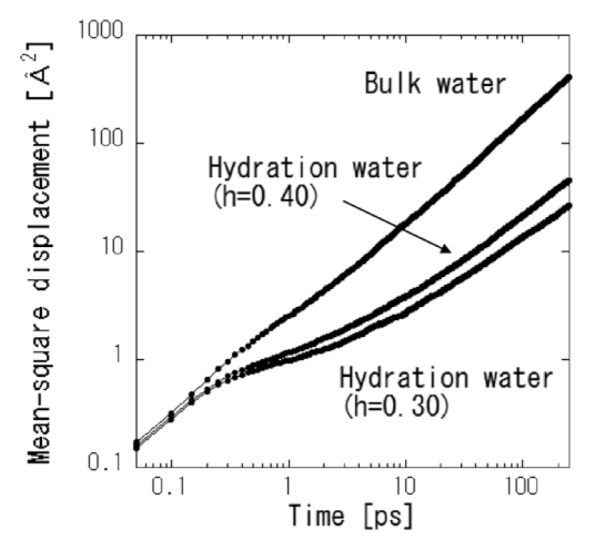
Time-dependent MSDs of hydration water at two hydration levels and bulk water at 300 K obtained by MD simulations, averaged over all water molecules in the system at time zero.
where 〈h〉 denotes the time average of h. The fluctuation is characterized by the correlation function, c(t). The correlation function of c(t) is the probability that two arbitrary water molecules that are hydrogen-bonded at time zero are bonded at time t as well, regardless of whether the bond is broken at an intermediate time [19]. Figure 7 shows the c(t) of hydration water and bulk water. The initial decay at the sub-pico second scale can be ascribed to the ballistic motions observed in the time-dependent MSDs shown in Figure 6. Beyond the initial period of the ballistic motions, the decay of c(t) indicates rearrangement of the hydration water cluster or network [19]. The function of the higher hydration level decays faster than that of the lower hydration level. The two hydration levels are above and below the percolation transition of hydration water [10]. The decay of c(t) originates from the extension of the hydration water cluster or network by percolation. Decay of the c(t) of the bulk water was significantly faster than that of the hydration water. This indicated the deceleration of water dynamics by hydration, and this result is consistent with the variation of the diffusion constant. The decay of c(t) cannot be described by a simple exponential function, suggesting the heterogeneity of hydrogen bond dynamics. The Kohlrausch-Williams-Watts (KWW) function is often used for the heterogeneous relaxation system [27], as following,
Figure 7.
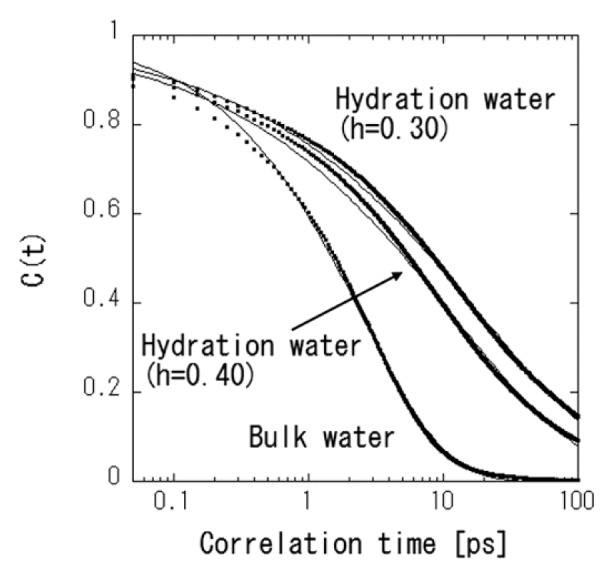
Time evolution of water-water hydrogen bond correlation functions for hydration water in protein at two hydration levels and bulk water at 300 K. The thin curves indicate the data fitting by KWW function.
| (4) |
where τKWW and β are relaxation time and the distribution of the relaxation function. For stretching exponential, β is between 0 and 1 (β=1 is the normal exponential function.). The data fittings by KWW were reasonable. The calculated τKWW and β are 19.6 ps and 0.43 for h=0.30, 12.1 ps and 0.44 for h=0.40, and 2.53 ps and 0.70 for bulk water, respectively. The obtained values of β indicate that the heterogeneity of relaxation time is broader for hydration water than bulk water. The dynamics of the hydration water might be more complex than bulk water by the interaction of the protein surface. Tarek et al., defined the hydrogen bond relaxation time τR as c(τR)=e−1 [19]. The calculated τR of the bulk liquid water at 300 K and those of the hydration water at h=0.40 and h=0.30 are 2.65, 13.80, and 30.25 ps, respectively. These results are not quantitatively consistent with the residence times from a jump-diffusion model analysis of INS (Table 2). The sequence of the residence times is the same between them. Therefore, the experimentally obtained residence time can be interpreted as the hydrogen bond relaxation time of water molecules. In the future, we will determine the relationship between PDT and hydration water dynamics by MD simulation.
Conclusion
One of the main goals of this study was to demonstrate the effectiveness of H2O/D2O exchange and MD simulation for INS experiment. Information of hydration water dynamics can be described separately from protein dynamics by conducting INS experiments with H2O/D2O exchange. The experimentally obtained dynamic parameters of hydration water and bulk water, such as MSDs, diffusion constants, and residence times, are compatible with the values calculated by MD simulation. The residence times of water molecules obtained using the jump-diffusion model were interpreted as the relaxation time required for the rearrangement of the hydration water cluster or network. Incoherent scattering does not intrinsically include structural information because scattering expresses the self-correlation of atomic motions, while MD simulation can provide all-atom information. INS experiments with H2O/D2O exchange combined with MD simulation should serve as a powerful next-generation tool for studying proteins and hydration water dynamics.
Acknowledgment
We thank Naoto Metoki and Koji Kaneko for their help in the neutron scattering experiments. This work was partly supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan awarded to H. N. (16H04928, 18H05229, 18K18828). MK is grateful to Professor Shuji Akiyama for his financial support by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and technology (MEXT) of Japan as a member of project (17H06165). The Salt Science Research Foundation, No. 1046 and 1748, Shimazu Science Foundation and Toray Science Foundation.
Footnotes
Conflict of Interest
All the authors declare that they have no conflict of interest.
Author Contributions
H. N. conducted all experiments and MD simulations.
H. N. and M. K. directed the entire project and co-wrote the manuscript.
References
- 1.Doster W, Cusack S, Petry W. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature. 1989;337:754–756. doi: 10.1038/337754a0. [DOI] [PubMed] [Google Scholar]
- 2.Tournier AL, Xu JC, Smith JC. Translational hydration water dynamics drives the protein glass transition. Biophys J. 2003;85:1871–1875. doi: 10.1016/S0006-3495(03)74614-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakagawa H, Joti Y, Kitao A, Kataoka M. Hydration Affects Both Harmonic and Anharmonic Nature of Protein Dynamics. Biophys J. 2008;95:2916–2923. doi: 10.1529/biophysj.107.128546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitao A, Hirata F, Go N. The effects of solvent on the conformational and the collective motions of protein: normal mode analysis and molecular dynamics simulations of melittin in water and in vacuum. Chem Phys. 1991;158:447–472. [Google Scholar]
- 5.Henzler-Wildman KA, Lei M, Thai V, Kern SJ, Karplus M, Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 6.Zaccai G. How soft is a protein? A protein dynamics force constant measured by neutron scattering. Science. 2000;288:1604–1607. doi: 10.1126/science.288.5471.1604. [DOI] [PubMed] [Google Scholar]
- 7.Diehl M, Doster W, Petry W, Schober H. Water-Coupled Low-Frequency Modes of Myoglobin and Lysozyme Observed by Inelastic Neutron Scattering. Biophys J. 1997;73:2726–2732. doi: 10.1016/S0006-3495(97)78301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith JC. Protein dyanmics: comparison of simulations with inelestic neutron scattering experiments. Q Rev Biophys. 1991;24:227–291. doi: 10.1017/s0033583500003723. [DOI] [PubMed] [Google Scholar]
- 9.Rasmussen BF, Stock AM, Ringe D, Petsko GA. Crystalline ribonuclease A loses function below the dynamical transition at 220 K. Nature. 1992;357:423–424. doi: 10.1038/357423a0. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa H, Kataoka M. Percolation of Hydration Water as a Control of Protein Dynamics. J Phys Soc Jpn. 2010;79:083801. [Google Scholar]
- 11.Hove LV. Correlations in Space and Time and Born Approximation Scattering in Systems of Interacting Particles. Phys Rev. 1954;95:249–262. [Google Scholar]
- 12.Wood K, Lehnert U, Kessler B, Zaccai G, Oesterhelt D. Hydration Dependence of Active Core Fluctuations in Bacteriohodopsin. Biophys J. 2008;95:194–202. doi: 10.1529/biophysj.107.120386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood K, Grudinin S, Kessler B, Weik M, Johnson M, Kneller GR, et al. Dynamical Heterogeneity of Specific Amino Acids in Bacteriorhodopsin. J Mol Biol. 2008;380:581–591. doi: 10.1016/j.jmb.2008.04.077. [DOI] [PubMed] [Google Scholar]
- 14.Wood K, Plazanet M, Gabel F, Kessler B, Oesterhelt D, Tobias DJ, et al. Coupling of protein and hydration-water dynamics in biological membranes. Proc Natl Acad Sci USA. 2007;104:18049–18054. doi: 10.1073/pnas.0706566104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wood K, Frölich A, Paciaroni A, Moulin M, Härtlein M, Zaccai G, et al. Coincidence of Dynamical Transitions in a Soluble Protein and Its Hydration Water: Direct Measurememnts by Neutron Scattering and MD simulations. J Am Chem Soc. 2008;130:4586–4587. doi: 10.1021/ja710526r. [DOI] [PubMed] [Google Scholar]
- 16.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 17.Tarek M, Tobias DJ. Environmental Dependence of the Dynamics of Protein Hydration Water. J Am Chem Soc. 1999;121:9740–9741. [Google Scholar]
- 18.Tarek M, Martyna GJ, Tobias DJ. Amplitudes and Frequencies of Protein Dynamics: Analysis of Discrepancies between Neutron Scattering and Molecular Dynamics Simulations. J Am Chem Soc. 2000;122:10450–10451. [Google Scholar]
- 19.Tarek M, Tobias DJ. Role of Protein-Water Hydrogen Bond Dynamics in the Protein Dynamical Transition. Phys Rev Lett. 2002;88:138101. doi: 10.1103/PhysRevLett.88.138101. [DOI] [PubMed] [Google Scholar]
- 20.Tarek M, Tobias DJ. The Dynamics of Protein Hydration Water: A Quantitative Comparison of Molecular Dynamics Simulations and Neutron-scattering Experiments. Biophys J. 2000;79:3244–3257. doi: 10.1016/S0006-3495(00)76557-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham TE, III, Darden, et al. AMBER 2015. University of California; San Francisco: 2015. [Google Scholar]
- 22.Bee M. Quasielastic Neutron Scattering, Principles and Applications in Solid State Chemistry, Biology and Materials Science. CRC Press; 1988. [Google Scholar]
- 23.Teixeira J, Bellissent-Funel M-C, Chen SH, Dianoux AJ. Experimental determination of the nature of diffusive motions of water molecules at low temperatures. Phys Rev A Gen Phys. 1985;31:1913–1917. doi: 10.1103/physreva.31.1913. [DOI] [PubMed] [Google Scholar]
- 24.Oleinikova A, Smolin N, Brovchenko I. Influence of Water Clustering on the Dynamics of Hydration Water at the Surface of a Lysozyme. Biophys J. 2007;93:2986–3000. doi: 10.1529/biophysj.107.108753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riahi MK, Qattan IA, Hassan J, Homouz D. Identifying short- and ling-time modes of the mean-square displacement: An improved nonlinear fitting approach. AIP Adv. 2019;9:055112. [Google Scholar]
- 26.Luzar A, Chandler D. Hydrogen-bond kinetics in liquid water. Nature. 1996;379:55–57. [Google Scholar]
- 27.Mashita R, Inoue R, Tominaga T, Shibata K, Kishimoto H, Kanaya T. Quasielastic neutron scattering study of microscopic dynamics in polybutadiene reinforced with an unsaturated carboxylate. Soft Matter. 2017;13:7862–7869. doi: 10.1039/c7sm01262d. [DOI] [PubMed] [Google Scholar]