

Abstract
Group 3 late embryogenesis abundant (G3LEA) proteins, which act as a well-characterized desiccation protectant in anhydrobiotic organisms, are structurally disordered in solution, but they acquire a predominantly α-helical structure during drying. Thus, G3LEA proteins are now accepted as intrinsically disordered proteins (IDPs). Their functional regions involve characteristic 11-mer repeating motifs. In the present study, to elucidate the origin of the IDP property of G3LEA proteins, we applied replica exchange molecular dynamics (REMD) simulation to a model peptide composed of two tandem repeats of an 11-mer motif and its counterpart peptide whose amino acid sequence was randomized with the same amino acid composition as that of the 11-mer motif. REMD simulations were performed for a single α-helical chain of each peptide and its double-bundled strand in a wide water content ranging from 5 to 78.3 wt%. In the latter case, we tested different types of arrangement: 1) the dipole moments of the two helices were parallel or anti-parallel and 2) due to the amphiphilic nature of the α-helix of the 11-mer motif, two types of the side-to-side contact were tested: hydrophilic-hydrophilic facing or hydrophobic-hydrophobic facing. Here, we revealed that the single chain alone exhibits no IDP-like properties, even if it involves the 11-mer motif, and the hydrophilic interaction of the two chains leads to the formation of a left-handed α-helical coiled coil in the dry state. These results support the cytoskeleton hypothesis that has been proposed as a mechanism by which G3LEA proteins work as a desiccation protectant.
Keywords: late embryogenesis abundant proteins, desiccation protectant, α-helical coiled coil
Significance.
Group 3 late embryogenesis abundant (G3LEA) proteins are disordered in solution, but they structuralize into an α-helical structure during drying. To elucidate the origin of this unusual property, we performed replica exchange MD simulations for a peptide composed of two tandem repeat of an 11-mer motif characteristic to G3LEA proteins. It was revealed that its single chain alone exhibits no structuralization, but its two-helix bundle forms a left-handed α-helical coiled coil in the dry state. These results support the cytoskeleton hypothesis that has been proposed as a mechanism by which G3LEA proteins work as a desiccation protectant in anhydrobiotic organisms.
The traditional view of the structure–function paradigm is that a protein’s function is closely related to a uniquely rigid three-dimensional (3D) structure, which is determined by the protein’s primary amino acid sequence. However, it has been accepted that a number of proteins do not adopt a unique 3D structure in solution; instead, they adopt random-coil-like conformations [1–3]. Such proteins undergo a disorder-to-order transition as a requirement for biological function, and they are referred to as intrinsically disordered proteins (IDPs) [1–3].
The late embryogenesis abundant (LEA) proteins, initially identified in cotton seeds at late stages of embryo development [4,5], are now accepted as IDPs [6–9]. In many anhydrobiotic organisms, LEA proteins are likely to be produced for tolerance against drought stresses [10–13]. LEA proteins can be classified into several groups according to the gene expression pattern or amino acid sequence [10–13]. Among them, group 3 LEA (G3LEA) proteins have been characterized well. G3LEA proteins are disordered in solution, but they acquire a secondary, predominantly α-helical structure during drying [6–9]. This property is highly unusual compared with those of usual globular proteins, the dehydration of which most often causes the loss of structure rather than the formation of an ordered structure.
The primary structures of G3LEA proteins consist of several tandem repeats of 11-mer motifs that are loosely conserved between different anhydrobiotes [10–13]. In a previous study, we explored the consensus sequences of the 11-mer motifs for several native G3LEA proteins that originate from anhydrobiotes among insects (Polypedilum vanderplanki), nematodes, and plants [14]. The resulting 11-mer consensus sequence for each organism was shown to contain three Lys, and three acidic residues (Glu and Asp), and these oppositely charged residues were well mixed in the linear sequence. Thus, the 11-mer motifs are categorized into strong polyampholyte in the IDP classification [15]. Our previous study indicated that the peptides that have two or four tandem repeats of the 11-mer consensus motif are disordered in the solution, but form an α-helical coiled coil in the dry state [14]. In addition, we have demonstrated that, similar to native G3LEA proteins, these peptides have the ability to protect biological materials from desiccation stress: for example, a 22-mer peptide composed of two tandem repeats of an 11-mer motif (AKDGTKEKAGE) can protect liposomes and proteins against desiccation stress [16–21]. Hereafter, we call this peptide PvLEA-22, because its 11-mer motif was derived from the G3LEA proteins of Polypedilum vanderplanki [22]. Taken together, for a better understanding of the structure-function relationship of G3LEA proteins, it is of great importance to investigate the disorder-order transition mechanism of the 11-mer motif regions at the atomistic level.
Standard atomistic structure analysis methods such as X-ray diffraction and cryo-electron microscopy would be inapplicable to highly disordered molecules. Instead, computer simulations can directly provide atomistic information not only on the transient structures but also on their dynamics. Li and He applied a conventional molecular dynamics (cMD) simulation to examine desiccation-induced structural alterations in a 66-amino acid fragment of a G3LEA protein from an anhydrobiotic nematode [23]. In their study, its folded structure in vacuo was first determined to be a hairpin-like, double-bundled, α-helical structure by homology modeling. It was found that this structure becomes disordered in response to the addition of water. Recently, a similar cMD simulation study was reported by Navarro-Retamal, et al. [24], who revealed the hydration-induced unfolding of the intrinsically disordered LEA proteins COR15A and COR15B from Arabidopsis thaliana. These simulations were helpful for the mechanistic understanding of the disorder-order transition of G3LEA proteins. However, from a theoretical viewpoint, the use of a cMD simulation is problematic in the case of IDPs. They are characterized by a comparatively shallow free energy landscape with multiple local minima that lack a dominant global minimum. For obtaining a convergent, statistically relevant picture of the structural ensemble, i.e., an accurate representation of the Boltzmann-weighted distribution of conformations, the use of enhanced sampling methods is required.
In the present study, we adopted the replica exchange molecular dynamics (REMD) method for enhanced conformational sampling [25] and explored the most stable conformations of PvLEA-22 at different water content levels, from 5 to 78.3 wt%. Then, we prepared several different initial structures, including only a single α-helical chain of the peptide or its double-bundled strand. In the latter case, we tested different types of chain arrangements: 1) the dipole moments were parallel or anti-parallel, and 2) due to the amphiphilic nature of the α-helix of the 11-mer motif, two types of the side-to-side contact were tested: hydrophilic-hydrophilic facing or hydrophobic-hydrophobic facing. On the basis of these results, we revealed that the single chain alone exhibited no IDP-like property, and the interaction of at least two chains via the hydrophilic faces was indispensable for structuralization into an α-helix in the dry state. The present results support the cytoskeleton hypothesis that has been proposed as a mechanism by which the G3LEA protein work as a desiccation protectant.
Methods
Simulation systems
To examine the water content dependence of the peptide structures, we prepared eight simulation systems differing in water content (Table 1), where the simulation box size was 9 nm×9 nm×9 nm under the periodic boundary conditions. As described below, one or two peptide molecules were put in the box.
Table 1.
Eight REMD simulation systems differing in the total number of water molecules, water content (wt%) and the number of replicas in the REMD simulation
| number of H2O molecules | H2O content (wt%) | replicas |
|---|---|---|
| 894 | 78.3 | 20 |
| 471 | 65.5 | 18 |
| 271 | 52.2 | 14 |
| 147 | 37.2 | 12 |
| 72 | 22.5 | 8 |
| 40 | 13.9 | 8 |
| 20 | 7.5 | 8 |
| 13 | 5.0 | 8 |
According to our previous FTIR study, PvLEA-22 structuralizes from disorder into an α-helical coiled coil upon dehydration, and this change is fully reversible: the folded peptide returns to the disordered state upon hydration [14]. Therefore, it is reasonable to start the simulation with the folded structure of the peptide in an extremely dehydrated state and monitor the peptide unfolding in solution of a given water content until equilibrium is reached. This protocol is identical with that described in a previous study by Li and He [23], who performed a cMD simulation, not REMD, for each solution with a given water content.
The 11-mer repeating motifs of PvLEA-22 are expected to have an amphiphilic character due to a hydrophobic stripe formed by the apolar residues at the positions 1, 2, 5, and 9 and a wider hydrophilic stripe formed by the polar residues at positions 3, 6, 7, 8, and 11 [14]. Thus, two types of the side-to-side contact are possible when two α-helical chains of the peptide are parallelly arranged: hydrophilic-hydrophilic facing or hydrophobic-hydrophobic facing. Furthermore, two types of dipole moment orientation are possible: parallel or anti-parallel. Taken together, we prepared the following four types of chain arrangements in the initial state: 1) the hydrophilic-hydrophilic facing of two α-helix chains and the anti-parallel arrangement of their dipole moments (Fig. 1a), 2) the same dipole arrangement as in 1), except the hydrophobic-hydrophobic facing (Fig. 1b), 3) the hydrophilic-hydrophilic facing of two chains and the parallel arrangement of their dipole moments and, and 4) the same dipole arrangement as in 3), except the hydrophobic-hydrophobic facing. In each model, the two peptide chains were separated with a minimal Cα-Cα distance of 3.8 Å.
Figure 1.
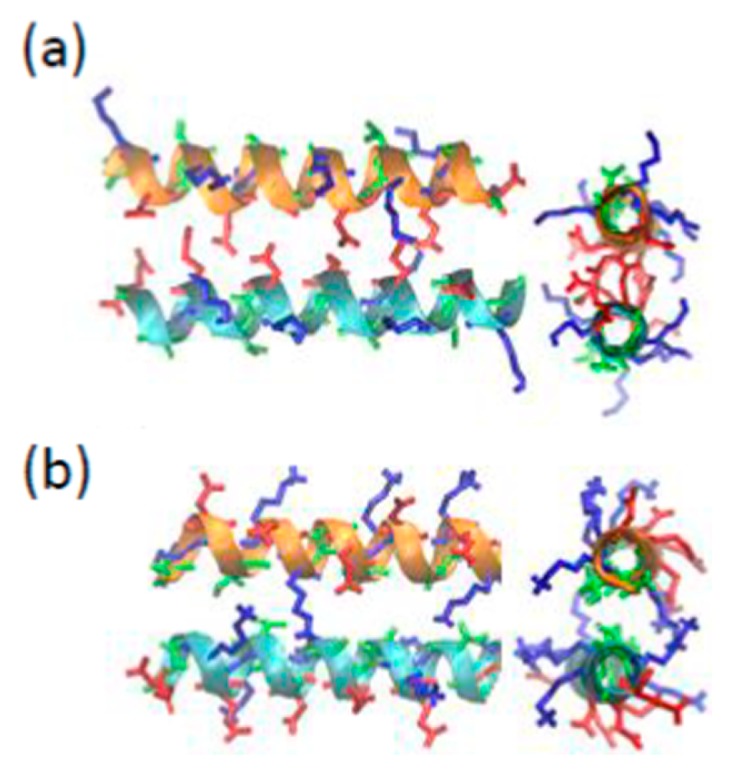
The initial structures for the REMD simulations of PvLEA-22. (a) anti-parallel hydrophilic-facing model, (b) anti-parallel hydrophobic-facing model. In each figure, a side view (left) and a top view along the helix axis (right) are shown. The red, blue and green stick models represent the side chains of acidic (Asp and Glu), basic (Lys) and neutral (Ala and Thr) amino acid residues, respectively.
In advance, we performed the cMD and REMD simulations to examine which chain arrangements are more stable in vacuo (Supplementary Fig. S1). As a result, arrangements 3) and 4) were confirmed to be less stable, probably due to the electrostatic repulsion between the two parallel dipole moments. Therefore, we selected only the 1) and 2) arrangements as the initial structures for the present REMD simulations (Fig. 1): hereafter, these models are called anti-parallel hydrophilic-facing and anti-parallel hydrophobic-facing models, respectively.
For comparison, we prepared a peptide whose amino acid sequence was randomized with the same amino acid composition as that of PvLEA-22 [14]. Hereafter, this peptide will be called the scrambled peptide (AKEKGETDKAGGAKD TEGKEKA) [16–21]. For this peptide, we tested a model in which the dipole moments of two chains were anti-parallelly arranged.
In addition to these two-chain models, we tested a single-chain model where one α-helical chain of PvLEA-22 or the scrambled peptide was put in the simulation box.
The protonation states of Asp, Glu and Lys in the peptides were determined with the assumption of pH=7 in the water content range of 78.3–37.2 wt%, and then the total net charge of the system was zero in all the systems studied. On the other hand, in the water content range of 22.5–5.0 wt%, these dissociative residues were neutralized to avoid their excessive electrostatic interactions due to the dehydrated environment.
Computational details of REMD simulation
The exact choice of temperature is crucial for the optimal performance of the REMD simulation. The temperatures have to be selected in a way that gives the same exchange probability between all adjacent pairs over the entire temperature range. Patriksson and Spoel proposed an algorithm that generates a set of temperatures to obtain a desired exchange probability [26]. In the present study, we determined a set of temperatures according to this algorithm. Then, the exchange probability and the temperature range were selected to be 0.3 and 300 K–400 K, respectively. The resulting number of replicas are listed in Table 1. The total simulation time of each REMD simulation was 140 ns, and the REMD exchanges were attempted every 2 ps. The 10–140 ns trajectory data were subjected to data analyses, including the dictionary of secondary structure of proteins (DSSP) and principal component analysis (PCA).
All simulations in this study were performed using the MD simulation package Gromacs 5.0.4 with the GROMOS 54A7 force field [27] and the SPC water model [28]. The simulations were performed in the isothermal–isochoric (NVT) ensemble with a Nosé–Hoover thermostat [29,30]. During the MD simulations, the covalent bonds were constrained by the LINCS algorithm [31,32], and the SPC water molecules were constrained with the SETTLE algorithm [33]. The van der Waals interactions were used with a cutoff distance of 4 nm. The electrostatic interactions were calculated using the simple cut-off method with a large cut-off distance of 4 nm. The simulation time step was set to 2 fs.
Results
Secondary structure analysis
The results of the DSSP analysis for the anti-parallel hydrophilic-facing and anti-parallel hydrophobic-facing models of PvLEA-22 are summarized in Tables 2 and 3, respectively. In the anti-parallel hydrophilic-facing model, the secondary structure is dominantly a random coil in high water content solutions of 78.3 and 65.5 wt%, and the sum of the coil, bend and turn contents reaches approximately 70%. With the decreasing water content, the percentage of the α-helix increases, and it eventually exceeds that of the random coil at a water content approximately 50 wt%. A similar tendency was also observed for the anti-parallel hydrophobic-facing model, although the inversion between the α-helix and coil contents occurred at a lower water content of approximately 30 wt%.
Table 2.
Water content dependence of the secondary structure distributions (given in %) for the anti-parallel hydrophilic-facing model
| H2O content (wt%) | α-Helix | β-sheet | β-bridge | turn | bend | coil |
|---|---|---|---|---|---|---|
| 78.3 | 23 | 3 | 2 | 12 | 20 | 39 |
| 65.5 | 27 | 1 | 1 | 11 | 22 | 38 |
| 52.2 | 37 | 0 | 0 | 8 | 18 | 37 |
| 37.2 | 45 | 4 | 3 | 6 | 11 | 31 |
| 22.5 | 57 | 0 | 1 | 5 | 9 | 28 |
| 13.9 | 60 | 0 | 0 | 9 | 7 | 24 |
| 7.5 | 62 | 0 | 2 | 7 | 6 | 24 |
| 5.0 | 61 | 0 | 0 | 7 | 7 | 25 |
Table 3.
Water content dependence of the secondary structure distributions (given in %) for the anti-parallel hydrophobic-facing model
| H2O content (wt%) | α-helix | β-sheet | β-bridge | turn | bend | coil |
|---|---|---|---|---|---|---|
| 78.3 | 22 | 4 | 2 | 21 | 9 | 38 |
| 65.5 | 31 | 1 | 1 | 10 | 10 | 39 |
| 52.2 | 29 | 0 | 1 | 10 | 18 | 40 |
| 37.2 | 31 | 6 | 4 | 11 | 11 | 34 |
| 22.5 | 49 | 1 | 3 | 9 | 9 | 27 |
| 13.9 | 48 | 2 | 3 | 9 | 10 | 48 |
| 7.5 | 48 | 1 | 1 | 12 | 8 | 33 |
| 5.0 | 42 | 1 | 3 | 21 | 8 | 23 |
Figure 2a shows the water content dependence of the α-helix rate in the anti-parallel hydrophilic-facing and anti-parallel hydrophobic-facing models, whose results are plotted in blue and red, respectively. The α-helix rate of the former model increases in an accelerated fashion with a loss of water and reaches larger than 60% in the extremely dry solutions of 7.5 and 5.0 wt%. Similarly, the α-helix rate of the latter model also increases with the loss of water, although it reaches a plateau below 20 wt%. In contrast, in the case of the double-bundled strand of the scrambled peptide (green plots in Fig. 2b), the α-helix rate is always low (<20%) over the whole examined water content range.
Figure 2.

Water content dependence of the helix rates of PvLEA-22. (a) The anti-parallel hydrophilic-facing (blue) and hydrophobic-facing (red) models. The results for the single-chain model are represented in orange. (b) The antiparallel double helix model (green) and single-chain model (violet) for the scrambled peptide.
Interestingly, in the single-chain model of PvLEA-22 (orange plots in Fig. 2a), no apparent increase of the α-helix rate was observed in low water content solutions; its α-helix rate was kept below 20%, except for in the two water content cases, where the rates were 65.5 and 52.2 wt%. The origin of such irregular behavior approximately 60 wt% will be explained later. Similarly, in the single-chain model of the scrambled peptide, the α-helix rate was also low (<20%) over the whole examined water content range (violet plots in Fig. 2b).
Tertiary structure analysis
To examine the three-dimensional structure distribution of the peptide chains for each water content, the trajectory data at 300 K were subjected to PCA, and the sampled structures were projected on the plane that was defined by the first and second principal axes (PC1 and PC2). Several examples of such clustering analysis for PvLEA-22 are shown in Figures 3a-c, which correspond to the results for the anti-parallel hydrophilic-facing model. As expected, at a high water content of 78.3 wt% (Fig. 3a), the structures are widely distributed over the PC1-PC2 plane: there are many clusters with shallow minima (yellow and orange regions). With decreasing water content, the number of clusters tends to decrease, while the minimum of each cluster becomes deep. In Figures 3b and c, we picked up relatively deep minima as Basins 1 and 2, which correspond to the clusters with a distribution probability of >30% and 15–30%, respectively. In the case of an extremely low water content of 7.5 wt%, most of the sampled structures are located in Basin 1.
Figure 3.

Free energy maps were obtained from the PCA analysis for the anti-parallel hydrophilic-facing model. (a), (b) and (c) represent the results for the water content levels of 78.3, 65.5 and 7.5 wt%, respectively. The energy values (kJ/mol) are indicated by the color code.
The representative structure with the highest appearance probability at each water content is given in Figure 4. In the higher water content solutions of 78.3 and 65.5 wt%, the peptides are almost disordered. With the decreasing water content, the ratio of α-helical fragments increases, and eventually, an anti-parallel double-bundled structure becomes predominant in extremely low water content, which is consistent with the results of the DSPP analysis. As shown in Figure 5, the number of the interchain hydrogen bonds steeply increases with the decreasing water content (decrease of the peptide-water hydrogen bonds). This implies that the formation of the interchain hydrogen bonds compensates for the energy loss due to the decrease of hydrogen bonds with the surrounding water molecules and simultaneously stabilizes the anti-parallel helix arrangement.
Figure 4.

Representative structure of the anti-parallel hydrophilic-facing model in each water content level (given in wt%).
Figure 5.

Water content dependence of the number of interchain hydrogen bonds (blue) and of the peptide-water hydrogen bonds (red) in the anti-parallel hydrophilic-facing model.
In Figure 6, the structure in the water content of 22.5 wt% is illustrated along two sight lines that are different from that in Figure 4. Obviously, the two chains are twisted to each other, resulting in a coiled coil-like structure (see discussion). Such a twisted structure was maintained down to the lowest water content level. To understand how the coiled coil structure is stabilized, we examined the orientations of the side chains of the dissociative residues (Glu and Asp: red, Lys: blue). As shown in Supplementary Figure S2, where the water molecules are deleted, the side chains of Lys residues are oriented to those of Glu and Asp, which are located at the helix-helix interface. Consequently, the hydrogen bonds are formed between Lys and Glu/Asp residues, which would be a main driving force for the formation of coiled coil.
Figure 6.

Representative structure of the anti-parallel hydrophilic-facing model at a water content level of 22.5 wt%. The red and blue stick models represent the side chains of acidic (Asp and Glu) and basic (Lys) amino acid residues, respectively.
Similar PCA analyses were performed for the anti-parallel hydrophobic-facing model, and representative structures were picked up for each water content level. The results are shown in Supplementary Figure S3. Similar to Figure 4, the anti-parallel double-bundled α-helical structure was stabilized with the decreasing water content. Interestingly, during the REMD simulation, the way of lateral contact of the two helices spontaneously changed from the hydrophobic-facing (initial structure) to hydrophilic-facing arrangement (Supplementary Fig. S4). Such a drastic structural change is thought to be driven by the formation of the interchain hydrogen bonds in place of the peptide-water hydrogen bonds (Supplementary Fig. S5).
In the scrambled peptide, which cannot form an amphiphilic helix, the double-bundled α-helical structure in the initial state was disrupted, and the two chains were aggregated in the low water content solutions (Supplementary Fig. S6). Similarly, the structure of the single-chain model was, as a whole, disordered over the whole water content range, except at 52.2 and 65.5 wt% (Supplementary Fig. S7). In a medium water content range approximately 60 wt%, the peptide chain tends to fold back at the central Gly residue to form an intramolecular anti-parallel double-bundled α-helical structure, which is consistent with the maximal peak of the α-helix rate approximately 50–60 wt% found in Figure 2 (orange plots).
Discussion
Anhydrobiotic organisms can survive severe drought state in which they show no detectable metabolism but retain the ability to revive after rehydration. In the case of the larva of P. vanderplanki, for example, the water content in the anhydrobiotic state is <5 wt% [34]. Usually, such an extremely dehydrated state should cause the structural disruption of cell and intracellular components, such as proteins and membranes. To avoid this, anhydrobiotic organisms accumulate desiccation protectants, such as disaccharide trehalose and LEA proteins in cells [35]. The mechanism by which trehalose works as a protectant has been extensively investigated [36–38]. However, the functional mechanism of LEA proteins remains less clear than those of trehalose. In this study, we focused on LEA proteins, especially G3LEA proteins.
In general, globular proteins are stabilized by a hydrophobic core. However, such a core is not a distinct feature of G3LEA proteins, because hydrophobic and hydrophilic amino acids are involved in a similar ratio. Accordingly, different from usual globular proteins, G3LEA proteins are disordered in water but structuralize to form an α-helix in the dry state, as demonstrated by the FTIR observation of the amide I band [6,8]. This unique character is thought to be responsible for the biological function of a desiccation protectant. However, the mechanism by which such structuralization is induced by desiccation has not been clarified. According to our previous studies, the repeat region of the 11-mer motif characteristic to G3LEA proteins is the core functional region [16–21]. Thus, we here focus on a peptide that is composed of the two tandem repeats of the 11-mer motif, PvLEA-22, and extensively explored the structures of single- and two-chain systems of PvLEA-22 over a wide range of water content levels from 5 to 78.3 wt%.
In the single-chain model, the α-helix rate of PvLEA-22 was not increased upon dehydration (Fig. 2a (orange), Supplementary Fig. S7). In addition, PCA analysis indicated that the structure is distributed widely over the PC1-PC2 plane (Supplementary Fig. S8), which means that there is no predominant chain conformation. In the present REMD simulations, the maximal water content tested was 78.3 wt%. To examine the behavior of the peptide in higher water content states, we carried out cMD simulations for the system in which a single α-helical chain of PvLEA-22 is dissolved in bulk water in the initial state. The root mean square displacement (RMSD) data of three runs all indicated that the peptide conformation was disrupted within approximately 20 ns (Supplementary Figs. S9a and b). Thus, the single chain of PvLEA-22 is intrinsically disordered irrespective of the water content. In this regard, the 11-mer motif region is different from single α-helical (SAH) domains that have had much attention recently because they form isolated α-helices [39].
The α-helical conformation of PvLEA-22 was also unstable in the two-chain models when water content was increased up to 78.3 wt% (Figs. 3a and 4) and the peptide was dissolve in bulk water (Supplementary Figs. S9c-e). However, the double-bundled α-helical strand became predominant in the extremely dry state (Fig. 2a, red and blue). This structure is likely stabilized by the formation of interchain hydrogen bonds through which the hydrophilic surfaces of the two amphiphilic chains come into contact with each other (Fig. 6, Supplementary Figs. S2 and S4). In contrast, in the scrambled peptide, whose α-helix does not have the amphiphilic character, two chains were aggregated (Supplementary Fig. S6). These results are consistent with our previous FTIR study, which indicated that the band positions of the amide I spectrum of dried PvLEA-22 agreed well with those of typical coiled-coil proteins, and the scrambled peptide was disordered in both aqueous and dry states [14].
The present REMD result that the anti-parallel double-bundled α-helical strand is the most stable structure in the dry state of PvLEA-22 strongly rationalizes the previous simulation studies for other LEA proteins/peptides. In a previous study by Li and He [23], the water content dependence of the structure of a 66-amino acid fragment, including four repeating motifs of 11 amino acids of a G3LEA protein from an anhydrobiotic nematode, was investigated using a cMD simulation with a production run of 5 ns. Then, the initial structure was constructed by homology modeling and was assigned to a hairpin-like, double-bundled, α-helical 3D conformation. The simulations indicated that this initial structure was almost maintained in the water deficit state, while the protein was largely unstructured in an aqueous solution. Although the short (5 ns) cMD simulations explored only a limited region of the conformational space, the obtained conclusion is thought to be no problem in light of our present REMD result. Recently, Navarro-Retamal, et al. conducted cMD simulations for the full length LEA proteins COR15A and COR15B from Arabidopsis thaliana [24]. Their simulations for the dry state also started from homology modeling structures in which two α-helices with an amphipathic character with opposing polar and apolar faces oriented along the longitudinal protein axes face in different directions, and the two α-helices are connected by a loop. The simulations indicated that such initial structures were rapidly disrupted in water, which is again consistent with the present REMD results.
After the REMD simulation, the anti-parallel α-helical chains in the initial structure (Fig. 1) converged to a coiled coil structure, as shown in Figure 6. Coiled coils are a common structural feature in proteins [40]. The sequence of a typical coiled coil is characterized by a series of contiguous heptad repeats or hendecad repeats. In the former, apolar residues occur predominantly in the 1st and 4th positions, resulting in the formation of a left-handed apolar stripe along the surface of the right-handed α-helix. In the latter, apolar residues occupy in the 1st, 4th, 5th and 8th positions, resulting in the formation of a right-handed apolar stripe. Usually, it is thought that coiled-coils are formed through the interaction of apolar stripes to be buried at the center of the molecule. As a result, heptad-based and hendecad-based helices dimerize to form the left-handed and right-handed coiled coil, respectively. The 11-mer motif of LEA proteins forms a right-handed apolar stripe [41]. Thus, in an early study by Dure [41], the LEA proteins were predicted to form a right-handed coiled coil. However, the coiled coil formation in the water deficit state does not necessarily follow the above conventional rule because the hydrophobic interaction may not be a main driving force in the structural formation of two protein/peptide chains. Indeed, our REMD simulation indicated that the hydrophobic-facing structure spontaneously changed to the hydrophilic facing one, as demonstrated in Supplementary Figure S4. The resulting coiled coil is the left-handed one, as may be guessed from Figure 6. This was confirmed from the observation of a model in which the double-strand model of Figure 6 was repeated two times along the helix axis (see Supplementary Fig. S10).
According to our experimental study [14], PvLEA-22 forms highly stable glass at room temperature in the dry state: the glass transition temperature Tg was observed at 102°C. Although the scrambled peptide also vitrifies, its Tg (84°C) was significantly lowered than that of PvLEA-22. It is thought that this difference originates from the difference in microstructure between these glassy matrixes. Namely, the α-helical coiled coil should be mechanically more stable than the random coil, leading to the formation of more stable (higher Tg) glass in PvLEA-22 than in the scrambled peptide. To support this, we examined whether the coiled coil of PvLEA-22 is stable up to high temperatures around its Tg. In Supplementary Figure S11 is shown the result of the PCA analysis for the REMD trajectory at 400 K and a representative structure in the case of water content of 7.5 wt%. These data clearly indicate that the coiled coil of PvLEA-22 is stable at such a high temperature. In fact, the secondary structure analysis indicated that the average α-helical rate over the 400 K trajectory was 58%, while that of the scrambled peptide was 17%.
In nature, P. vanderplanki larva can live in dried mud on rocks whose surface temperature reaches 60°C (333 K) [34]. PvLEA-22 is a model of G3LEA proteins in P. vanderplanki. Based on the above results for the stability of this peptide, the coiled coil of the G3LEA proteins is expected to be stable under the inhabitation conditions of this anhydrobiotic organism.
When α-helical coiled coils are assembled to form filaments, they work as a strong structural skeleton. If such filamentation occurs in dried cells, the resulting filaments would work as the cytoskeleton (cf. steels in buildings) to avoid the desiccation-induced disruption of the cells. This is the cytoskeleton hypothesis that has been proposed as one of the mechanisms by which G3LEA proteins function as desiccation protectants [10]. The present REMD study supports this hypothesis.
Conclusion
The present REMD simulations allowed the extensive structure exploration of PvLEA-22 over a wide water content range from 5 to 78.3 wt%. One of the important findings is that the single chain of PvLEA-22 alone exhibits no IDP-like property, and instead, the interaction of two α-helical chains via the hydrophilic faces enables the structuralization of a mechanically stronger coiled coil in the extremely dehydrated state, in which the water content is approximately 5 wt%, which is close to that in the dried larva of P. vanderplanki. This scenario is likely true for longer peptides with many tandem repeats of 11-mer motifs and native G3LEA proteins, which could form an intramolecular α-helical coiled coil by folding back their chains, as was already examined in the previous cMD simulation studies [23,24]. The coiled coil is stable enough to be maintained under stressful inhabitation conditions like high temperature, as confirmed for the coiled coil of PvLEA-22. Therefore, it is likely that G3LEA proteins work as a cytoskeleton in dried cells to enable anhydrobiotic organisms to survive drought environments.
Supplementary Material
Acknowledgment
This work was supported by JSPS KAKENHI, Grant Number JP15H02378.
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest.
Author Contributions
T. N., Y. T. and S. M. performed the simulations and analyses.
T. F. and M. S. directed the entire project and cowrote the manuscript.
References
- 1.Uversky V. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol. 2011;43:1090–1103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci. 2012;37:509–516. doi: 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta. 2013;1834:932–951. doi: 10.1016/j.bbapap.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Dure L, III, Greenway SC, Galau GA. Developmental biochemistry of cottonseed embryogenesis and germination: changing messenger ribonucleic acid populations as shown by in vitro and in vivo protein synthesis. Biochemistry. 1981;20:4162–4168. doi: 10.1021/bi00517a033. [DOI] [PubMed] [Google Scholar]
- 5.Grzelczak ZF, Sattolo MH, Hanley-Bowdoin LK, Kennedy TD, Lane BG. Synthesis and turnover of proteins and mRNA in germinating wheat embryos. Can J Biochem. 1982;60:389–397. doi: 10.1139/o82-046. [DOI] [PubMed] [Google Scholar]
- 6.Goyal K, Tisi L, Basran A, Browne J, Burnell A, Zurdo JA, et al. Transition from natively unfolded to folded state induced by desiccation in an anhydrobiotic nematode protein. J Biol Chem. 2003;278:12977–12984. doi: 10.1074/jbc.M212007200. [DOI] [PubMed] [Google Scholar]
- 7.Tolleter D, Jaquinod M, Mangavel C, Passirani C, Saulnier P, Manon S, et al. Structure and function of a mitochondrial late embryogenesis abundant protein are revealed by desiccation. Plant Cell. 2007;19:1580–1589. doi: 10.1105/tpc.107.050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolkers WF, McReady S, Brandt WF, Lindsey GG, Hoekstra FA. Isolation and characterization of a D-7 LEA protein from pollen that stabilizes glasses in vitro. Biochim Biophys Acta. 2001;1544:196–206. doi: 10.1016/s0167-4838(00)00220-x. [DOI] [PubMed] [Google Scholar]
- 9.Hatanaka R, Hagiwara-Komoda Y, Furuki T, Kanamori Y, Fujita M, Cornette R, et al. An abundant LEA protein in the anhydrobiotic midge, PvLEA4, act as a molecular shield by limiting growth of aggregating protein particles. Insect Biochem Mol Biol. 2013;43:1055–1067. doi: 10.1016/j.ibmb.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Wise MJ, Tunnacliffe A. POPP the question: what do LEA proteins do? Trends Plant Sci. 2004;9:13–17. doi: 10.1016/j.tplants.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 11.Tunnacliffe A, Wise MJ. The continuing conundrum of the LEA proteins. Naturwissenschaften. 2007;94:791–812. doi: 10.1007/s00114-007-0254-y. [DOI] [PubMed] [Google Scholar]
- 12.Tunnacliffe A, Hincha DK, Leprince O, Macherel D. LEA proteins: Versatility of Form and Function. In: Lubzens E, Cerdà J, Clark MS, editors. Dormancy and Resistance in Harsh Environments. Springer-Verlag; Berlin, Heidelberg: 2010. pp. 91–108. [Google Scholar]
- 13.Hand SC, Menze MA, Toner M, Boswell L, Moore D. LEA proteins during water stress: not just for plants anymore. Annu Rev Physiol. 2011;73:115–134. doi: 10.1146/annurev-physiol-012110-142203. [DOI] [PubMed] [Google Scholar]
- 14.Shimizu T, Kanamori Y, Furuki T, Kikawada T, Okuda T, Takahashi T, et al. Desiccation-induced structuralizatoin and glass formation of group 3 late embryogenesis abundant protein model peptides. Biochemistry. 2010;49:1093–1104. doi: 10.1021/bi901745f. [DOI] [PubMed] [Google Scholar]
- 15.van de Lee R, Buljina M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, et al. Classification of intrinsically disorderd regions and proteins. Chem Rev. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuki T, Shimizu T, Kikawada T, Okuda T, Sakurai M. Salt effects on the structural and thermodynamic properties of group 3 late embryogenesis abundant protein model peptides. Biochemistry. 2011;50:7093–7103. doi: 10.1021/bi200719s. [DOI] [PubMed] [Google Scholar]
- 17.Furuki T, Shimizu T, Chakrabortee S, Yamakawa K, Hatanaka R, Takahashi T, et al. Effects of group 3 LEA protein model peptides on desiccation-induced protein aggregation. Biochim Biophys Acta. 2012;1824:891–897. doi: 10.1016/j.bbapap.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 18.Furuki T, Sakurai M. Group 3 LEA model peptides protect liposomes during desiccation. Biochim Biophys Acta. 2014;1838:2757–2766. doi: 10.1016/j.bbamem.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Furuki T, Sakurai M. Group 3 LEA model peptides protect enzymes against desiccation stress. Biochim Biophys Acta. 2016;1864:1237–1243. doi: 10.1016/j.bbapap.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Furuki T, Watanabe T, Furuta T, Takano K, Shirakashi R, Sakurai M. The dry preservation of giant vesicles using a group 3 LEA protein model peptide and its molecular mechanism. Bull Chem Soc Jpn. 2016;89:1493–1499. [Google Scholar]
- 21.Furuki T, Niwa T, Taguchi H, Hatanaka R, Kikawada T, Sakurai MA. LEA model peptide protects the function of a red fluorescent protein in the dry state. Biochem Biophys Rep. 2019;17:27–31. doi: 10.1016/j.bbrep.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kikawada T, Nakahara Y, Kanamori Y, Iwata K, Watanabe M, McGee B, et al. Dehydration-induced expression of LEA proteins in an anhydrobiotic chironomid. Biochem Biophys Res Commun. 2006;348:56–61. doi: 10.1016/j.bbrc.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Li D, He X. Desiccation induced structural alterations in a 66-amino acid fragment of an anhydrobiotic nematode late embryogenesis abundant (LEA) protein. Biomacromolecules. 2009;10:1469–1477. doi: 10.1021/bm9002688. [DOI] [PubMed] [Google Scholar]
- 24.Navarro-Retamal C, Bremer A, Alzate-Morales J, Caballero J, Hincha DK, González WA, et al. Molecular dynamics simulations and CD spectroscopy reveal hydration-induced unfolding of the intrinsically disordered LEA proteins COR15A and COR15B from Arabidopsis thaliana. Phys Chem Chem Phys. 2016;18:25806–25816. doi: 10.1039/c6cp02272c. [DOI] [PubMed] [Google Scholar]
- 25.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 26.Patriksson A, van der Spoel D. A temperature predictor for parallel tempering simulations. Phys Chem Chem Phys. 2008;10:2073–2077. doi: 10.1039/b716554d. [DOI] [PubMed] [Google Scholar]
- 27.Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, et al. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 28.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B, editor. Intermolecular Forces. D. Reidel Publishing Co; Dordrecht, The Netherlands: 1981. pp. 331–342. [Google Scholar]
- 29.Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol Phys. 1984;52:255–268. [Google Scholar]
- 30.Hoover WG. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 31.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A Linear Constraint Solver for Molecular Simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 32.Hess-Coelho TAA. Redundant Parallel Spherical Mechanism for Robotic Wrist Applications. J Mech Des. 2007;129:891–895. [Google Scholar]
- 33.Miyamoto S, Kollman PA. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 34.Sakurai M, Furuki T, Akao K, Tanaka D, Nakahara Y, Kikawada T, et al. Vitrification is essential for anhydrobiosis in an African chironomid, Polypedilum vanderplanki. Proc Natl Acad Sci USA. 2008;105:5093–5098. doi: 10.1073/pnas.0706197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furuki T, Sakurai M. Physicochemical Aspects of the Biological Functions of Trehalose and Group 3 LEA Proteins as Desiccation Protectantsin, in Survaival Strategies. In: Inoue M, Sakurai M, Uemura M, editors. Extreme Cold and Desiccation. Springer; Nature, Singapore: 2018. pp. 271–286. [DOI] [PubMed] [Google Scholar]
- 36.Crowe JH, Carpenter JF, Crowe LM. The role of vitrification in anhydrobiosis. Annu Rev Physiol. 1998;60:73–103. doi: 10.1146/annurev.physiol.60.1.73. [DOI] [PubMed] [Google Scholar]
- 37.Clegg JS. Cryptobiosis―a peculiar state of biological organization. Comp Biochem Physiol B, Biochem Mol Biol. 2001;128:613–624. doi: 10.1016/s1096-4959(01)00300-1. [DOI] [PubMed] [Google Scholar]
- 38.Crowe LM. Lessons from nature: the role of sugars in anhydrobiosis. Comp Biochem Physiol, Part A Mol Integr Physiol. 2002;131:505–513. doi: 10.1016/s1095-6433(01)00503-7. [DOI] [PubMed] [Google Scholar]
- 39.Swanson CJ, Sivaramakrishnan S. Harnessing the unique structural properties of isolated α-Helices. J Biol Chem. 2014;289:25460–25467. doi: 10.1074/jbc.R114.583906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parry DAD, Fraser RDB, Squire JM. Fifty years of coiled-coils and α-helical bundles: A close relationship between sequence and structure. J Struct Biol. 2008;163:258–269. doi: 10.1016/j.jsb.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Dure L., III A repeating 11-mer amino acid motif and plant desiccation. Plant J. 1993;3:363–369. doi: 10.1046/j.1365-313x.1993.t01-19-00999.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.