

Cancer cell population exhibits great cellular heterogeneity, and single-cell morphology encodes metastatic potential.
Abstract
A central goal of precision medicine is to predict disease outcomes and design treatments based on multidimensional information from afflicted cells and tissues. Cell morphology is an emergent readout of the molecular underpinnings of a cell’s functions and, thus, can be used as a method to define the functional state of an individual cell. We measured 216 features derived from cell and nucleus morphology for more than 30,000 breast cancer cells. We find that single cell–derived clones (SCCs) established from the same parental cells exhibit distinct and heritable morphological traits associated with genomic (ploidy) and transcriptomic phenotypes. Using unsupervised clustering analysis, we find that the morphological classes of SCCs predict distinct tumorigenic and metastatic potentials in vivo using multiple mouse models of breast cancer. These findings lay the groundwork for using quantitative morpho-profiling in vitro as a potentially convenient and economical method for phenotyping function in cancer in vivo.
INTRODUCTION
Much effort is being made to explore the predictive power of genomic alterations in the detection and prognosis of diseases (1–4). However, a high degree of genomic instability in advanced cancers with metastatic disease endows these genomes with a myriad of abnormalities affecting the expression and function of tens of thousands of genes (5, 6). Recent studies show that individual (clonal) cells can display a broad landscape of properties, such as different gene expression patterns (7) and invasive behaviors (8), further increasing the challenge of deciphering the molecular basis of metastasis in cancer. A potential solution to this problem is to use a surrogate readout of a combinatorial set of genomic alterations that lead to similar outcomes. Previous studies using RNAi screens have shown that cell morphology (CM) can be an informative readout that is highly associated with molecular underpinnings (9, 10). Furthermore, recent studies indicate that the morphological status of cells can be linked to fundamental physiological properties of cells, such as cell cycle progression (11), cell-matrix adhesion properties, responsiveness to drug (8–10), aging (12), gene expression patterns (7), and invasiveness potential (8). To this end, CM in a defined environment is an emergent, yet relatively easily measurable, outcome resulting from the coupling between a cell’s biochemistry and its biophysics that are ultimately encoded by the cell genome.
RESULTS
CM is a highly heritable trait at the single-cell level
We used a long-term, time-lapse recording of MDA-MB-231 human breast cancer cells growing in vitro, which readily suggested a high degree of cellular heterogeneity, including large variations in cell motility, cell size, and CM (movie S1). To determine whether the phenotypic traits presented by individual cells were stochastic or persistent, we used an ultralow-density growth assay to assess the morphology of individual cells in colonies. Cells were morphologically similar to other cells in the same colony but distinct from cells in other colonies. The morphological traits of an individual cell persisted over extended periods of time (>1 month in culture). This observation suggested that morphological traits of individual parental cells were passed on to their progeny either by inheritance or by sharing a similar local microenvironment (Fig. 1A). A similar phenomenon was observed with six cancer cell lines derived from primary pancreatic tumors and metastases (fig. S1).
Fig. 1. Cell polymorphism—cell morphology is a highly heritable trait at the single-cell level.
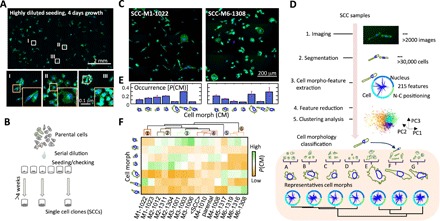
(A) Nuclei (blue) and F-actin organization (green) of MDA-MB-231 breast cancer cells after growth for 4 days from a sparse initial seeding density, showing how cells formed several spatially and morphologically distinct clusters. Representative high-resolution images of different clonal cells highlighted as I, II, and III are shown at the bottom. (B) Schematic plot showing the serial dilution procedure used to establish single-cell clones (SCCs) from a parental cell population. (C) Nuclei (blue) and F-actin organization (green) in cells of two established SCCs, SCC-M1-1022 and SCC-M6-1308, displaying distinct morpho-types. (D) Flow diagram illustrating the process used to quantify cell morphology (CM) through an unsupervised machine learning approach. A classifier model was built on the basis of all 14 SCCs and the parental MDA-MB-231 cells through principal component analysis and k-means clustering analysis. The morphology of all measured cells was classified into one of seven cell morph classes. Representative CM for each cell morph class (A to G) is shown at the bottom. (E) The fraction of cells in each cell morph class was used to quantitatively represent morpho-types of SCCs. Cell morph class fraction profiles for SCC-M1-1022 and SCC-M6-1308 are shown in the histograms. (F) Unsupervised hierarchical clustering of the SCCs based on their morpho-types (i.e., fraction of cells in cell morph classes A to G). The names of established SCCs were further marked as M1 to M6 based on six distinct cell morpho-type clusters revealed in the dendrogram.
To further investigate the clonal architecture of CM, we generated single-cell clones (SCCs) obtained through the expansion of individual parental MDA-MB-231 breast cancer cells. Cells in each SCC displayed a distinct morphology (Fig. 1, B and C, and fig. S2) (7). To quantitatively describe the morphological spectrum of SCCs, we measured the morphology of cells in 14 SCCs and the parental cell line using a previously developed high-throughput microscopy and analysis system (11, 13–18). Briefly, cells and their nuclei were fluorescently stained and imaged using widefield fluorescence microscopy. For each well, a ~6 mm by 6 mm field of view was imaged and reconstructed from 81 (9 by 9) image tiles collected with a 10× objective. The morphology of cells was then automatically measured using a custom software (see details in Materials and Methods).
It has been previously shown that using a limited number of representative cell shapes is an effective strategy to explore complex CM datasets (9, 10). Here, we found that the morphology of cells in SCCs was categorized into seven CM classes (denoted A to G), which were themselves derived from a clustering analysis based on morphological features describing all >30,000 cells analyzed (Fig. 1D and Materials and Methods). This analysis provides visual and quantitative representations of CM across SCCs by assessing the distributions of these seven CM classes (e.g., Fig. 1E). These CM classes are associated with distinct properties of traditional CM parameters such as size, shape factor, and aspect ratio of cells and nuclei (fig. S3A).
On the basis of unsupervised hierarchical clustering of the CM distributions of the 14 SCCs, we classified the SCCs into six distinct morpho-types (M1 to M6) (Fig. 1F). All SCCs showed a certain degree of morphological heterogeneity as measured by Shannon’s entropy of the morpho-types. The parental cells had a substantially higher level of morphological heterogeneity compared with SCCs (fig. S3, B and C). The global CM distribution obtained by ensemble-averaging the CM distributions of the 14 SCCs (denoted here <SCC>) was approximately the same as the CM distribution of the parental cells (Fig. 1F). Furthermore, SCCs displayed morphologies similar to colonies in the ultralow-density growth assay (Fig. 1, A and C).
Together, these results suggest that the parental MDA-MB-231 breast cancer cell population is composed of distinct, coexisting classes of cells with heritable morphological traits that persist over long time scales.
Morphological phenotypes in vitro and differential tumor progression in vivo
Individual MDA-MB-231 breast cancer cells can show distinct behavior in vivo, including differential ability to disseminate from the primary tumor and differential organ dissemination (8). To determine whether different cell morphologies of SCCs derived from MDA-MB-231 breast cancer cells corresponded to distinct outcomes in vivo, SCCs were injected into the mammary fat pad of mice, which were monitored for 50 days. We found that SCCs displayed a broad range of tumorigenicity (as measured by the weight and size of the primary tumors) and metastatic potential (as measured by human DNA content in the lungs) (Fig. 2A and fig. S4). For instance, the subclone SCC-M5-1317 formed tumors that were 50% larger than tumors produced by the parental MDA-MB-231 cells, but had a 99% decrease in its ability to form metastasis compared with parental cells (fig. S4). In contrast, SCC-M6-1308 and SCC-M6-1319 formed tumors of sizes similar to those produced by parental cells, but displayed 5 to 10 times more effective metastasis than parental cells. We also identified a group of SCCs (e.g., SCC-M2-1012) that formed small tumors that did not metastasize. Implanted SCCs also produced substantially different numbers of circulating tumor cells (CTCs) in the blood (Fig. 2B). Analysis of histological sections of the mouse lungs showed multiple metastatic lesions in mice bearing tumors formed by SCC-M6-1308 and SCC-M6-1319, but no metastatic lesions for SCC-M5-1317 and SCC-M2-1012 (Fig. 2C and fig. S4). Short tandem repeat (STR) analysis showed that SCCs with distinct metastatic potential (SCC-M1-1022, SCC-M6-1308, and parental cells) had the exact same STR profiles, confirming their common ancestral origin.
Fig. 2. Morphological phenotypes in vitro and differential tumor progression in vivo.
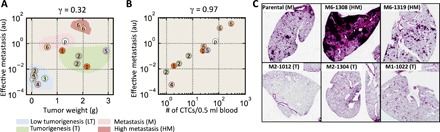
(A and B) Scatter plot showing both tumor size and the extent of lung metastasis resulting from the injection of 14 SCCs and parental MDA-MB-231 cells into the mammary pad of SCID mice. At least four mice were tested for each SCC (A). The number within each circle represents the morpho-type class of the corresponding SCC. On the basis of tumorigenicity and metastatic burden in the lung, these SCCs were further classified into four groups: low tumorigenicity (LT), tumorigenic (T), metastatic (M), and hypermetastatic (HM). The Pearson’s correlation coefficient between the effective metastasis and tumor weight among all SCCs is 0.32. The number of circulating tumor cells (CTCs) is highly correlated with lung metastasis, with a correlation coefficient of 0.96 (B). (C) Histological sections of mice lung show that clear metastatic lesions are present for SCC-M6-1308, SCC-M6-1319, and parental cells, but not in other SCCs, including SCC-M2-1012, SCC-M2-1304, and SCC-M2-1022. au, arbitrary units.
We classified the SCCs into four grades of aggressiveness based on their tumorigenicity and metastatic potentials: (i) low tumorigenicity (LT), (ii) tumorigenic (T), (iii) metastatic (M), and (iv) hypermetastatic (HM) (summary of information about SCCs is given in table S1). We found only a weak correlation between tumor size and lung metastasis (Pearson’s correlation coefficient γ = 0.32) (Fig. 2A). This is consistent with the fact that SCCs that were highly tumorigenic could be either metastatic or not metastatic. In contrast, the number of CTCs per volume of blood was highly correlated with lung metastasis (γ = 0.97) (Fig. 2B), but poorly correlated with tumor size (fig. S4). The high correlation between the number of CTCs and metastatic burden in the lungs indicates that SCCs that can extravasate to blood vessels have higher potential for lung metastasis. Nevertheless, that these CTCs could come from lung metastases as well cannot be ruled out (19). We note that SCCs enriched with cells displaying a spindle-like morphology (i.e., cell shape with a high aspect ratio) are not the ones showing a high metastatic potential (fig. S4H).
Together, these results indicate that SCCs with the same morpho-types displayed similar in vivo outcomes, including tumorigenicity, tumor cells in circulation, and metastatic potential (Fig. 2A).
The morphological diversity of SCC correlates with distinct gene expression patterns
We next determined whether distinct morpho-types and corresponding tumorigenicity and metastatic potential were associated with distinct gene expression patterns. Transcriptomic microarray analysis showed that the gene expression profiles of SCCs at approximately the same passage number were strongly associated with specific SCC morpho-types (Fig. 3A). SCCs belonging to the same morpho-type were located in close proximity of each other in the gene expression space, spanned by the first two principal components PC1 and PC2. Similar results were obtained using an unsupervised hierarchy clustering analysis based on expression data from genes that displayed the highest expression variations—measured by standard deviations—among SCCs (200 gene probes with 144 unique genes; see full list in table S2).
Fig. 3. Morphological diversity of SCCs is driven by distinct gene expression patterns.
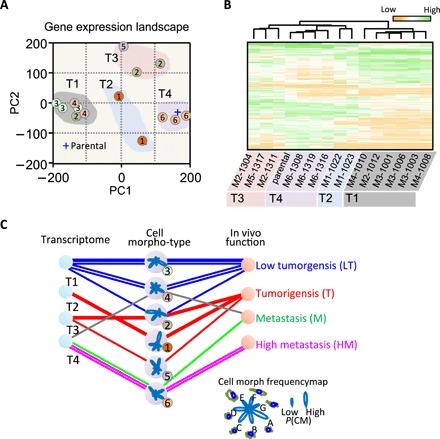
(A) The first and second principal components obtained from the principal component analysis of gene expression data were used to show the landscape of whole genome expression profile of the SCCs. The number within each circle represents the morpho-type class of each SCC. SCCs with the same morpho-type classes in general clustered together. (B) Unsupervised hierarchy clustering analysis using differentially expressed genes among these SCCs (see detailed list of genes in table S2) shows four distinct gene expression classes (G1 to G4). SCCs within the same morpho-type class are classified within the same gene expression class with the exception of SCC-M2-1012. SCCs within G1 and G3 gene expression classes exhibit multiple morpho-type classes. (C) Diagram showing mutual relations between morpho-type, gene expression class, and outcomes in vivo for different SCCs. Polar-petal plots were used to visualize fraction profiles of cell morph classes for the six different morpho-types. The length of a petal indicates the fraction size for the corresponding CM class.
Overall, we found that there were four distinct gene expression subtypes among SCCs and the parental breast cancer cells (Fig. 3B). In the list of 200 gene candidates that were potentially responsible for the cell polymorphism, the SPANX family (SPANXB2 and SPANXE)—cancer-testis antigens that are often highly expressed in tumor cells—featured the most variation, with approximately 1000-fold difference between SCCs with the lowest and highest levels of expression. A recent study has shown that the expression of members of the SPANX gene family promotes breast cancer invasion (20). Several genes in this list have been previously associated with patient survival and cancer metastasis, including CDH11 (21), KISS1 (22), MAGEA3 (12, 23), MAGEC1 (24), TNFSF10 (25), CXCR4 (26), and GDF15 (18).
Mutual correlations between morpho-types, gene expression classes, and aggressiveness further confirmed that gene expression profiles (Fig. 3) and aggressiveness in vivo (Fig. 2, A to C) are reflected by the morphology of SCCs (Fig. 3C). This analysis shows that cells of LT are particularly small (enriched in cell morphs A and C); enrichment in elongated cells (cell morph F) was found only in groups of tumorigenic—but not metastatic—SCCs. The shapes of cells that were exclusively metastatic exhibited enriched cell morphs E and G: Their morphology tended to be rounder and larger (enriched in cell morph E) (Fig. 3C).
Distinct gene expression profiles of SCCs reveal prognostic genes
We further evaluated the genes that were differentially expressed among functionally distinct SCCs. A total of 218 genes (table S3) were either significantly down-regulated or significantly up-regulated [>5-fold and P value from one-way analysis of variance (ANOVA) <0.05] when comparing SCCs of different tumorigenicity and metastatic potential (Fig. 4A). Among these 218 genes, 189 genes (87%) were associated with the comparison of LT and M′ tumors, in contrast to 38 genes that were associated with the comparison of T and M′ tumors (Fig. 4A). This indicates that at the transcriptomic level, SCCs of LT were more different from metastatic SCCs (M′) than tumorigenic SCCs. Of 38 genes that were differentially regulated between T and M′, 28 (74%) also could differentiate LT from T tumors, suggesting that tumorigenic (T) SCCs represent an intermediate transcriptomic state between LT SCCs and M′ SCCs.
Fig. 4. Distinct gene expression profiles of SCCs reveal prognostic genes.
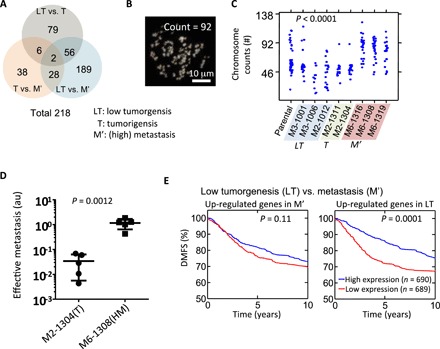
(A) Venn diagram showing the number of genes that are found to be significantly different (>5-fold and P value from one-way ANOVA <0.05) between three different in vivo grades of aggressiveness for SCCs (i.e., LT versus T, T versus M′, and LT versus M′). M′ includes both M and HM. (B and C) Representative image showing 4′,6-diamidino-2-phenylindole (DAPI)–stained spreading chromosome of SCC-M6-1308 (B). Chromosome number counted using the metaphase spreading assay for parental cells (n = 44), and cells from SCC-M3-1001 (n = 24), SCC-M3-1006 (n = 11), SCC-M2-1012 (n = 22), SCC-M2-1311 (n = 18), SCC-M2-1304 (n = 18), SCC-M6-1316 (n = 26), SCC-M6-1308 (n = 31), and SCC-M6-1319 (n = 22). One-way ANOVA test shows there is a significant difference, with a P < 0.0001 (C). (D) Score for effective metastasis to the lung in the tail-vein injection mouse model (n = 5) shows significant difference (P = 0.0012 by Student t test) between tumorigenic clone SCC-M2-1304 (mean lung effective metastasis score, 0.034) and metastatic clone SCC-M6-1308 (1.159). (E) Differentially expressed genes between LT SCC versus M′ SCC were used to investigate their prognostic power. A cohort of 1379 tumors from patients with breast cancer was used to test the predictive potential of identified gene sets. Patients were separated into two groups based on the average expression level of these identified genes, and the Kaplan-Meier survival curves for the two groups of patients were plotted. For the genes that were up-regulated in the M′ SCCs, no significant prognostic effect was found. However, the results show that patients with higher expression levels of metastasis suppressor genes (i.e., up-regulated genes in LT) have a significantly longer survival time than those with low expression (P = 0.0001). P value is evaluated using log-rank test.
To explore the relation between morpho-types and gene expression of SCCs, we cross-compared the list of 218 genes with the list of 883 genes functionally annotated as “regulation of cell shape” (GO:0000902) by gene ontology (27–29). We found that 22 genes co-occurred in both lists, which corresponds to a P value of 2.42 × 10−4 (table S4). Since our findings show that morpho-types of SCCs are highly associated with their functions in vivo, this result strongly suggests that the morphological heterogeneity of SCCs is a result of differential expressed genes, such as interleukin-6 (IL-6), IL-7R, etc., which may play a role in tumor progression (30).
We further investigated the potential mechanisms by which morpho-type M6 may encode metastatic potential in vivo. M6 is characterized by cells displaying large nuclei, which may be indicative of an increase in ploidy (31, 32). To test this hypothesis, we analyzed the degree of ploidy of SCCs. Measurement of the distribution of the number of chromosomes for each SCC, using the metaphase-spread assay, showed that SCCs with high metastatic potential (M′ SCC) displayed a substantial higher average number of chromosomes (77 to 86) than LT SCCs and T SCCs (50 to 59) and parental MDA-MB-231 cells (~59) (Fig. 4C). This is consistent with experimental and clinical evidence that suggests that tetraploidization is a frequent genomic abnormality associated with enhanced metastasis, possibly due to a high rate of aneuploidy production in subsequent cell divisions and/or better tolerance of aneuploidy due to higher basal ploidy (33–36). All SCCs exhibited wide distributions in chromosome numbers (Fig. 4B), suggesting that high chromosome instability is inherent in all cells derived from the parental breast cancer cell population.
Another morphological characteristic of the morpho-type M6 is the highly symmetric shape of the cells compared with the much more spindle-like morphology of the other morpho-types on two-dimensional (2D) glass surfaces. As cell shape is often a readout of the production of cortical cytoskeletal forces (17), we compared the motility of different SCCs in 3D matrices of controlled collagen I content (37, 38). For all motility models tested, including cells on 2D substrates of controlled stiffness and cells embedded in 3D collagen matrices (39, 40), we did not find a correlation between motility and metastatic potential (fig. S5). The proliferation rate of SCCs in culture showed that the highly metastatic SCC clones and parental MDA-MB-231 cells had the highest population growth rates among SCCs (fig. S5B).
Direct injection of tumorigenic and metastatic SCCs into blood vessels through the tail vein showed that metastatic SCCs (SCC-M6-1308) could more effectively extravasate and colonize the lung, suggesting that the metastatic potential of the SCCs is determined by their lung-seeding capacity (Fig. 4D and fig. S6).
Last, we compared the distant metastasis–free survival (DMFS) of patients with breast cancer, which was stratified by expression levels using a cohort of 1379 tumors obtained from patients with breast cancer (GOBO database, Gene expression–based outcome for breast cancer) (41). Of the 218 genes identified by our SCC classification, we found 155 genes in this cohort (see summary in table S3). Kaplan-Meier survival analysis showed that patients with tumors showing a higher level of expression of tumor-suppressor genes—the genes that were up-regulated when comparing LT and M—had a significantly improved DMFS (P = 0.0001) than patients with tumors showing low expression of these genes (Fig. 4D). Consistent with this result, tumors of patients with a high expression of genes that were up-regulated in T or LT tumors in comparison to M′ or T tumors had significantly longer DMFS (fig. S7), confirming the role of cancer cell polymorphism in tumor evolution and progression.
DISCUSSION
Together, our analysis of SSCs derived from a parental breast cancer cell line demonstrates clonally persistent morphological heterogeneity. These SCCs show a wide range of distinct tumorigenic and metastatic potentials in vivo. The progression and outcomes of SSC-derived cancers in mice are associated with distinct patterns of gene expression. The same genes that are differentially regulated when comparing metastatic to nonmetastatic SCCs are of prognostic value to assess metastasis-free patient survival. These results support our hypothesis that CM is a holistic readout (in physics, CM would be called an “emergent” property) of the complex genomic and gene expression changes in cancer cells. The morphological features that predict metastatic potential are associated with increased ploidy. High basal ploidy provides better tolerance of diverse aneuploid karyotypes, which produce the phenotypic variation driving adaption of metastatic tumors to novel microenvironments.
We anticipate that incorporating single-cell analysis of intratumoral heterogeneity could further improve diagnosis and prognosis for individual patients and that quantitative cell phenotyping analysis in vitro could offer an effective and economical method to decipher complex cellular heterogeneity in tumors to identify lethal cancer cell subtypes for diagnostic and therapeutic purposes.
MATERIALS AND METHODS
Cell lines and culture
The parental breast cancer cell line MDA-MB-231 (42) and derived SCCs were maintained in high-glucose (4.5 mg/ml) Dulbecco’s minimum essential medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were maintained at 37°C in a 5% CO2, 95% air incubator.
Establishment of SCCs
A suspension of parental MDA-MB-231 cells was diluted using culture medium to a cell density of approximately 1 cell/0.1 μl. A droplet of 0.1 μl of cell suspension was placed in each well of a 96-well plate by pipetting followed by microscopy inspection to examine the number of cells in the deposited droplet. For wells containing a single cell, 200 μl of culture medium was subsequently added to allow for cell growth into SCCs. The culture medium was then replaced regularly every 3 to 4 days, and SCCs were subsequently transferred to 24-well plates, 6-well plates, and 10-cm petri dishes after they became confluent. SCCs were then frozen down and thawed for further experiments.
Orthotopic implantation and metastasis assays
Studies using 7- to 10-week-old female severe combined immunodeficient (SCID) mice [National Cancer Institute (NCI)] were performed according to protocols approved by the Johns Hopkins University Animal Care and Use Committee. Briefly, 2 × 106 cells were resuspended in 1:1 ratio of phosphate-buffered saline (PBS) to Matrigel (BD Biosciences) and injected into the second left mammary fat pad. Tumor growth was monitored by caliper measurements. Tumor volume (cubic millimeter) was calculated as length by width by depth by 0.52. After indicated times, mice were sacrificed, and the lungs were perfused with PBS. The left lung was inflated by injecting with low–melting point agarose. Uninflated lungs were used for human genomic DNA extraction. Lungs were digested with lysis buffer and proteinase K at 55°C overnight, and genomic DNA was isolated by phenol/chloroform extraction and isopropanol precipitation. Genomic DNA (200 ng) was used for quantitative polymerase chain reaction (qPCR) to quantify human HK2 and mouse 18S transcripts.
To count CTCs, 500 μl of blood from each mouse was collected. Red blood cells were lysed using ammonium chloride solution (Stem Cell Technologies, catalog no. 07800). RNA from the remaining cells in the blood was extracted (Life Technologies, catalog no. 15596-026) and reverse transcribed to complementary DNA (cDNA; Bio-Rad iScript Reverse Transcriptase, catalog no. 170-8840). The cDNA was then used for qPCR to quantify human-only 18S rRNA and mouse and human 18S rRNA. In each sample, we measured normalized human 18S gene expression by 2−Csample where Csample = Chu − average C(hu and mu 18S). The calibration curve between measured 18S gene expression and the number of MDA-MB-231 cells was obtained by spiking controlled numbers of MDA-MB-231 cells in naïve mouse blood samples.
For the tail-vein injection model, MDA-MB-231 subclones were harvested by trypsinization, resuspended at 107 cells/ml in PBS, and injected (1 × 106 cells) intravenously into SCID mice. After 2 weeks, lungs were perfused with PBS. One lung was inflated for formalin fixation and paraffin embedding. The other lung was used to isolate genomic DNA for qPCR analysis with human-specific HK2 primers.
Immunostaining and fluorescence microscopy
Approximately 12,000 cells were plated in each well of a 24-well glass bottom plate (MatTek, MA), corresponding to approximately 20% surface coverage to ensure single-cell dispersion. After 16 hours of incubation, cells were fixed with 3.7% paraformaldehyde for 12 min at room temperature. Cells were then permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) for 10 min; nonspecific binding was blocked with PBS supplemented with 1% albumin from bovine serum for 40 min. Nuclear DNA was stained with Hoechst 33342 (Sigma-Aldrich) at 1:50 dilution; F-actin was stained with phalloidin Alexa Fluor 488 (Invitrogen) at a 1:40 dilution. Fluorescently labeled cell samples were visualized with a Nikon digital sight DS-Qi1MC camera mounted on a Nikon TE300 epifluorescence microscope (Nikon Melville, NY) and equipped with a motorized stage and motorized excitation and emission filters (Prior Scientific, Rockland, MA) controlled by NIS-Elements (Nikon). For each sample, 81 (9-by-9 square grid) fields of view from a low-magnification lens (10× Plan Fluor lens; numerical aperture, 0.3; Nikon) were used, which covered a contiguous area of 6.03 mm by 4.73 mm (28.5 mm2). The fluorescence channels for Hoechst 33342 and Alexa Fluor 488 were recorded to obtain the necessary morphometric information about the nucleus and cellular body of each individual cell within the scanning region.
Analysis of CM
Image processing for quantification of cellular morphological features from fluorescence images was carried out using a custom program developed in MATLAB (Mathworks, MA) (13–15, 43). In brief, we first segmented individual cells and their nuclei. We used five different categories of morphological features with a total number of 215 features to characterize nucleus and cell shapes. These features correspond to classes of morphological features that include basic morphological features, boundary signature, curvature, nucleus-cell positioning, and protrusion (fig. S8). The full list of features is summarized in table S5. In general, basic morphology features are features such as area, perimeter, long axis, short axis, and aspect ratio. Boundary signature of a shape (R) is the distance profile from all boundary coordinates to the centroid points of a shape, and boundary signature features are the statistical profiles of R, such as mean, median, and SD. To obtain curvature features, we first calculated the curvature (k) along the boundary of smoothed cell shapes. The smoothing of shape is processed by convolving the x and y coordinates of the shape with a 1D Gaussian filter, which has unit SD and size of 11 pixels. Statistical descriptors for the curvature along the boundary of shape, such as mean, median, SD, were measured as curvature features. The detailed list of statistical descriptors used can be found in the table S5. The same statistical properties used for boundary signature were extracted to represent the curvature features of a shape. Nucleus-cell positioning profile (R′) is represented by the distance from the nucleus edge to the cell edges in different orientations based on the centroid of the nucleus. The nucleus-cell positioning features are a set of statistical properties of R′. For the quantification of protrusion morphology, we adopted a previous approach (15, 43). In brief, we first determined the morphological skeleton of individual cell contours and identified the main body region of the cells. The protrusions were identified as the skeletal structures that were extended beyond the main body of the cell. The protrusions were further classified into two subtypes: primary and secondary protrusions. The primary protrusions were considered to be the protrusions stemming directly from the cell body, while the secondary protrusions were the ones branching from other protrusions. The length of each protrusion was measured, and the total number of protrusions for individual cells was determined as the summation of primary and secondary protrusions. The total number of protrusions, mean length of protrusions, primary protrusion number, secondary protrusion number, and the ratio of secondary to primary protrusions were used as parts of the CM features. To quantitatively classify cell morpho-types, the morphology feature space of cells was first reduced and was represented by projection scores at 36 eigenvectors that spanned 95% of variations of among all measured cells from the principal component analysis (fig. S3). K-means clustering analysis with cityblock distance function was implemented to identify the seven distinct clusters among CM data of all measured cells.
Metaphase spreading assay
Cells were grown up to 60% confluency after plating. Colcemid (Invitrogen) was added to the cultural medium at a concentration of 100 ng/ml, and cells were incubated at 37°C for 3 to 4 hours. Cells were harvested using trypsin and resuspend in 1 ml of culture medium after spinning down. Five microliters of 37°C prewarmed KCI was added slowly to the cell suspension and incubated at room temperature for 7 to 10 min followed by adding 120 μl of freshly prepared fixative solution (methanol:acetic acid in 3:1 volume ratio). Cells were incubated in 9.5 ml of fixative solution for 10 min after being spun down at 1000 rpm for 8 min and having discarded the supernatant. Cells were then resuspended in 0.3 ml of fixative and dropped on a glass slide before being placed onto slide warmer at 65°C for 20 min followed by treatment with RNAse A (1 mg/ml; 1:100 from Qiagen) and propidium iodide (1 mg/ml stock and 1:1000 final) in 2× SSC for ~45 min at 37°C. Slides were air dried before mounting using mounting medium with DAPI (Vectashield). Chromosome spreads were imaged with 63× oil objective mounted on a Ti-E microscope (Nikon) and analyzed using previously established software (44, 45).
Microarray transcriptional profiling
Total RNA was isolated from MDA-MB-231 cells and its SCCs with the RNeasy Mini kit and analyzed using the Affymetrix GeneChip PrimeView Human Gene Expression Array (Johns Hopkins Deep Sequencing and Microarray Core Facility). Partek Genomic Suite was used to normalize expression data of all extended level probe sets using the following options: GC content prebackground adjustment, robust multi-array average (RMA) background correction, and quantile normalization. Gene expression level was defined as the average expression level of all exons for that gene. One-way ANOVA test was used to obtain P value and fold change (FC) values. The differentially expressed genes were detected for P < 0.05 and |FC| >5 (linear).
Statistics
Data were represented as averages ± SEM unless otherwise specified. One-way ANOVA test was performed to determine significance using MATLAB (MathWorks) unless otherwise specified.
Supplementary Material
Acknowledgments
We thank K. Burns and F. M. Gabhann for critically reading the manuscript. Funding: We acknowledge funding from the National Cancer Institute (U54CA143868 and R01CA174388), the National Institute of Neurological Disorders and Stroke (R21NS087485), the National Institute of General Medical Sciences (R35GM118172), and the American Heart Association (12POST12050638). Author contributions: P.-H.W. and D.W. designed the experiments. P.-H.W., D.M.G., J.M.P., A.N., J.M., T.W.-T.C., and M.-H.L. collected the data. P.-H.W. developed the analytical tools and analyzed the data. P.-H.W. composed the figures and wrote the Supplementary Materials and supplementary figures. P.-H.W. and D.W. wrote the manuscript. D.M.G. and R.L. edited the manuscript. Competing interests: D.W., P.-H.W., and J.M.P. are inventors on a patent related to this work (no. US8934698B2, expiring 12 October 2032). All authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaaw6938/DC1
Fig. S1. Cell polymorphism in cancer cells derived from primary pancreatic tumors and metastases.
Fig. S2. Morphological spectrum of SCCs.
Fig. S3. Quantitative morphology analysis of metastasis of breast cancer cells.
Fig. S4. SCCs derived from the same parental breast cancer cells show divergent invasive behavior in vivo.
Fig. S5. Analysis of SCC with migration and proliferation assay.
Fig. S6. Lung tissue sections in tail vein metastasis model for different SCCs.
Fig. S7. Distinct gene expression profiles of SCCs reveal prognostic genes.
Fig. S8. Morphological features.
Table S1. Summary of SCC information.
Table S2. Genes that are highly differentially expressed among SCP.
Table S3. Differentially expressed genes among LT versus T versus M’.
Table S4. List of genes affiliated with regulation of cell shape.
Table S5. Summary of CM features.
Table S6. Whole-genome gene expression data of MDA-MB-231 SCCs.
REFERENCES AND NOTES
- 1.Stratton M. R., Campbell P. J., Futreal P. A., The cancer genome. Nature 458, 719–724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A., Kinzler K. W., Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paez J. G., Jänne P. A., Lee J. C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F. J., Lindeman N., Boggon T. J., Naoki K., Sasaki H., Fujii Y., Eck M. J., Sellers W. R., Johnson B. E., Meyerson M., EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Miki Y., Swensen J., Shattuck-Eidens D., Futreal P., Harshman K., Tavtigian S., Liu Q., Cochran C., Bennett L., Ding W. et al. , A strong candidate for the breast and ovarian-cancer susceptibility gene brca1. Science 266, 66–71 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Negrini S., Gorgoulis V. G., Halazonetis T. D., Genomic instability - an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Potapova T. A., Zhu J., Li R., Aneuploidy and chromosomal instability: A vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metast. Rev. 32, 377–389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen A., Yoshida M., Goodarzi H., Tavazoie S. F., Highly variable cancer subpopulations that exhibit enhanced transcriptome variability and metastatic fitness. Nat. Commun. 7, 11246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minn A. J., Kang Y., Serganova I., Gupta G. P., Giri D. D., Doubrovin M., Ponomarev V., Gerald W. L., Blasberg R., Massagué J., Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Invest. 115, 44–55 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin Z., Sadok A., Sailem H., McCarthy A., Xia X., Li F., Garcia M. A., Evans L., Barr A. R., Perrimon N., Marshall C. J., Wong S. T. C., Bakal C., A screen for morphological complexity identifies regulators of switch-like transitions between discrete cell shapes. Nat. Cell Biol. 15, 860–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakal C., Aach J., Church G., Perrimon N., Quantitative morphological signatures define local signaling networks regulating cell morphology. Science 316, 1753–1756 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Wu P. H., Phillip J. M., Khatau S. B., Chen W. C., Stirman J., Rosseel S., Tschudi K., Van Patten J., Wong M., Gupta S., Baras A. S., Leek J. T., Maitra A., Wirtz D., Evolution of cellular morpho-phenotypes in cancer metastasis. Sci. Rep. 5, 18437 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayyoub M., Scarlata C. M., Hamai A., Pignon P., Valmori D., Expression of MAGE-A3/6 in primary breast cancer is associated with hormone receptor negative status, high histologic grade, and poor survival. J. Immunother. 37, 73–76 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Chen W. C., Wu P. H., Phillip J. M., Khatau S. B., Choi J. M., Dallas M. R., Konstantopoulos K., Sun S. X., Lee J. S. H., Hodzic D., Wirtz D., Functional interplay between the cell cycle and cell phenotypes. Integr. Biol. 5, 523–534 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu P. H., Hung S. H., Ren T. N., Shih I. M., Tseng Y., Cell cycle-dependent alteration in NAC1 nuclear body dynamics and morphology. Phys. Biol. 8, 015005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee M. H., Wu P. H., Gilkes D., Aifuwa I., Wirtz D., Normal mammary epithelial cells promote carcinoma basement membrane invasion by inducing microtubule-rich protrusions. Oncotarget 6, 32634–32645 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambliss A. B., Wu P. H., Chen W. C., Sun S. X., Wirtz D., Simultaneously defining cell phenotypes, cell cycle, and chromatin modifications at single-cell resolution. FASEB J. 27, 2667–2676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillip J. M., Wu P. H., Gilkes D. M., Williams W., McGovern S., Daya J., Chen J., Aifuwa I., Lee J. S. H., Fan R., Walston J., Wirtz D., Biophysical and biomolecular determination of cellular age in humans. Nat. Biomed. Eng. 1, 0093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yong K. M. A., Zeng Y., Vindivich D., Phillip J. M., Wu P. H., Wirtz D., Getzenberg R. H., Morphological effects on expression of growth differentiation factor 15 (GDF15), a marker of metastasis. J. Cell. Physiol. 229, 362–373 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minn A. J., Gupta G. P., Siegel P. M., Bos P. D., Shu W., Giri D. D., Viale A., Olshen A. B., Gerald W. L., Massagué J., Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maine E. A., Westcott J. M., Prechtl A. M., Dang T. T., Whitehurst A. W., Pearson G. W., The cancer-testis antigens SPANX-A/C/D and CTAG2 promote breast cancer invasion. Oncotarget 7, 14708–14726 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pishvaian M. J., Feltes C. M., Thompson P., Bussemakers M. J., Schalken J. A., Byers S. W., Cadherin-11 is expressed in invasive breast cancer cell lines. Cancer Res. 59, 947–952 (1999). [PubMed] [Google Scholar]
- 22.Martin T. A., Watkins G., Jiang W. G., KiSS-1 expression in human breast cancer. Clin. Exp. Metastas. 22, 503–511 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Kim J., Reber H. A., Hines O. J., Kazanjian K. K., Tran A., Ye X., Amersi F. F., Martinez S. R., Dry S. M., Bilchik A. J., Hoon D. S. B., The clinical significance of MAGEA3 expression in pancreatic cancer. Int. J. Cancer 118, 2269–2275 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Kruger S., Ola V., Feller A. C., Fischer D., Friedrich M., Expression of cancer-testis antigen CT7 (MAGE-C1) in breast cancer: An immunohistochemical study with emphasis on prognostic utility. Pathol. Oncol. Res. 13, 91–96 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Kuribayashi K., Krigsfeld G., Wang W., Xu J., Mayes P. A., Dicker D. T., Wu G. S., El-Deiry W. S., TNFSF10 (TRAIL), a p53 target gene that mediates p53-dependent cell death. Cancer Biol. Ther. 7, 2034–2038 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Muller A., Homey B., Soto H., Ge N., Catron D., Buchanan M. E., McClanahan T., Murphy E., Yuan W., Wagner S. N., Barrera J. L., Mohar A., Verástegui E., Zlotnik A., Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G., Gene Ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gene Ontology Consortium , Gene Ontology Consortium: Going forward. Nucleic Acids Res. 43, D1049–D1056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carbon S., Ireland A., Mungall C. J., Shu S. Q., Marshall B., Lewis S.; the AmiGO Hub; the Web Presence Working Group , AmiGO: Online access to ontology and annotation data. Bioinformatics 25, 288–289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jayatilaka H., Tyle P., Chen J. J., Kwak M., Ju J., Kim H. J., Lee J. S. H., Wu P. H., Gilkes D. M., Fan R., Wirtz D., Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat. Commun. 8, 15584 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe T., Tanaka Y., Age-related alterations in the size of human hepatocytes. A study of mononuclear and binucleate cells. Virchows Arch. B. Cell Pathol. Icnl. Mol. Pathol. 39, 9–20 (1982). [DOI] [PubMed] [Google Scholar]
- 32.Conlon I., Raff M., Size control in animal development. Cell 96, 235–244 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Frankfurt O. S., Slocum H. K., Rustum Y. M., Arbuck S. G., Pavelic Z. P., Petrelli N., Huben R. P., Pontes E. J., Greco W. R., Flow cytometric analysis of DNA aneuploidy in primary and metastatic human solid tumors. Cytometry 5, 71–80 (1984). [DOI] [PubMed] [Google Scholar]
- 34.Korenaga D., Okamura T., Saito A., Baba H., Sugimachi K., DNA Ploidy Is Closely linked to tumor invasion, lymph-node metastasis, and prognosis in clinical gastric-cancer. Cancer 62, 309–313 (1988). [DOI] [PubMed] [Google Scholar]
- 35.Navin N., Kendall J., Troge J., Andrews P., Rodgers L., McIndoo J., Cook K., Stepansky A., Levy D., Esposito D., Muthuswamy L., Krasnitz A., McCombie W. R., Hicks J., Wigler M., Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavelka N., Rancati G., Li R., Dr Jekyll and Mr Hyde: Role of aneuploidy in cellular adaptation and cancer. Curr. Opin. Cell Biol. 22, 809–815 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fraley S. I., Feng Y., Krishnamurthy R., Kim D. H., Celedon A., Longmore G. D., Wirtz D., A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat. Cell Biol. 12, 598–604 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fraley S. I., Wu P. H., He L., Feng Y., Krisnamurthy R., Longmore G. D., Wirtz D., Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions. Sci. Rep. 5, 14580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu P. H., Giri A., Sun S. X., Wirtz D., Three-dimensional cell migration does not follow a random walk. Proc. Natl. Acad. Sci. U.S.A. 111, 3949–3954 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu P. H., Giri A., Wirtz D., Statistical analysis of cell migration in 3D using the anisotropic persistent random walk model. Nat. Protoc. 10, 517–527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ringner M., Fredlund E., Hakkinen J., Borg A., Staaf J., GOBO: gene expression-based outcome for breast cancer online. PLOS ONE 6, e17911 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee M. H., Wu P. H., Staunton J. R., Ros R., Longmore G. D., Wirtz D., Mismatch in mechanical and adhesive properties induces pulsating cancer cell migration in epithelial monolayer. Biophys. J. 102, 2731–2741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo Q. Y., Phillip J. M., Majumdar S., Wu P. H., Chen J., Calderón-Colón X., Schein O., Smith B. J., Trexler M. M., Wirtz D., Elisseeff J. H., Modulation of keratocyte phenotype by collagen fibril nanoarchitecture in membranes for corneal repair. Biomaterials 34, 9365–9372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng W., Tsao S. W., Lucas J. N., Leung C. S., Cheung A. L. M., A new method for improving metaphase chromosome spreading. Cytom. Part. A 51A, 46–51 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Potapova T. A., Unruh J. R., Box A. C., Bradford W. D., Seidel C. W., Slaughter B. D., Sivagnanam S., Wu Y., Li R., Karyotyping human and mouse cells using probes from single-sorted chromosomes and open source software. Biotechniques 59, 335–346 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaaw6938/DC1
Fig. S1. Cell polymorphism in cancer cells derived from primary pancreatic tumors and metastases.
Fig. S2. Morphological spectrum of SCCs.
Fig. S3. Quantitative morphology analysis of metastasis of breast cancer cells.
Fig. S4. SCCs derived from the same parental breast cancer cells show divergent invasive behavior in vivo.
Fig. S5. Analysis of SCC with migration and proliferation assay.
Fig. S6. Lung tissue sections in tail vein metastasis model for different SCCs.
Fig. S7. Distinct gene expression profiles of SCCs reveal prognostic genes.
Fig. S8. Morphological features.
Table S1. Summary of SCC information.
Table S2. Genes that are highly differentially expressed among SCP.
Table S3. Differentially expressed genes among LT versus T versus M’.
Table S4. List of genes affiliated with regulation of cell shape.
Table S5. Summary of CM features.
Table S6. Whole-genome gene expression data of MDA-MB-231 SCCs.