

Abstract
The metabolic serine hydrolase family, is arguably, one of the largest functional enzyme class in mammals, including humans, comprising of 1 – 2% of the total proteome. This enzyme family uses a conserved nucleophilic serine residue in the active site to perform diverse hydrolytic reactions, and consists of proteases, lipases, esterases, amidases and transacylases as prototypical members of this family. In humans, this enzyme family consists of >250 members, of which, approximately 40% members remain unannotated, both in terms of their endogenous substrates and the biological pathways that they regulate. The enzyme ABHD14B, an outlying member of this family, is also known as CCG1/TAFII250-interacting factor B, as it was found associated with the transcription initiation factor TFIID. The crystal structure of human ABHD14B has been solved for over a decade, however its endogenous substrates remain elusive. In this paper, we annotate ABHD14B as a lysine deacetylase (KDAC), showing this enzyme’s ability in transferring an acetyl group from a post-translationally acetylated-lysine to coenzyme A (CoA), to yield acetyl-CoA, while re-generating the free amine of protein lysine residues. We validate these findings by in vitro biochemical assays using recombinantly purified human ABHD14B, in conjunction with cellular studies in a mammalian cell line by knocking down ABHD14B and by modeling substrates into the enzyme active site. Finally, we report, the development and characterization of a much needed, exquisitely selective ABHD14B antibody, using which, we map the cellular and tissue distribution of ABHD14B, and prospect metabolic pathways that this enzyme might biologically regulate.
Introduction
The advent of genome sequencing technology has resulted in an explosion in the number of available protein sequences1. Concomitant with this exponential rise in the number of protein sequences, the propagation of functional annotation errors or lack of annotation for enzymes in particular, has become more prominent throughout databases that rely heavily on high-throughput computational predictions of protein (enzyme) function without much experimental support2, 3. This in turn, has presented modern biochemists with challenges in studying enzyme mechanisms, and understanding their endogenous functions in the post-genomic era4. Realizing this problem, over the past decade, several large-scale consortia (e.g. The Enzyme Function Initiative5, 6, The Lipid Maps Lipidomics Gateway7, 8) have been set up, where researchers with different expertise have worked together to develop new tools and/or platforms to assign function (or substrates) to enzymes of unknown or misannotated function. Complementary to the high-throughput approaches developed by these consortia, are emerging biochemical technologies9, 10, that have been leveraged to study specific enzyme superfamilies to address the same problem.
One such approach, is the functional chemical proteomic strategy termed activity based protein profiling (ABPP), first described by Cravatt and co-workers, to study enzymes from the serine hydrolase family11. Most members of this enzyme family possess the famed catalytic triad (e.g. Ser-His-Asp)12, and without any exceptions to date, follow a consensus two step catalytic mechanism13, 14. In the first step of the reaction mechanism, a highly conserved nucleophilic serine residue in the enzyme active site, attacks an electron deficient carbon (or phosphorus) center at ester, thioester or amide functionalities, to form a covalent acyl-enzyme intermediate, and a conserved base (generally the histidine residue of the catalytic triad) in the enzyme active site protonates the leaving group. In the second step, a water molecule is activated by the now deprotonated base in the enzyme active site to hydrolytically cleave the acyl-enzyme intermediate, and in doing so, regenerates the enzyme for another round of catalysis (Scheme 1)12, 13.
Scheme 1. The conserved catalytic mechanism of the metabolic serine hydrolase family of enzymes.

In humans, the serine hydrolase family to date, consists of >250 enzymes, split almost equally into the serine proteases (trypsin/chymotrypsin/subtilisin family) and the metabolic serine hydrolases, with the latter constituting lipases, esterases, amidases, peptidases, glycan hydrolases, acyl-CoA hydrolases, and transacylases as prototypical members13. Given their central roles in metabolism and biological signaling, deregulation of these metabolic serine hydrolase enzymes results in several human pathophysiological conditions like cancers, neurodegenerative diseases, (neuro)inflammation, diabetes, obesity and metabolic syndrome13. Yet, for most of these enzymes implicated in the aforementioned human diseases, the biochemical basis for the disease progression and pathophysiology remains largely unknown. Over the past two decades, Cravatt and co-workers have further expanded the ABPP technology15–21, and have tailored several elegant platforms to ascertain function and biochemically characterize several members of the metabolic serine hydrolase family. Yet, despite their heroic efforts, approximately 40% of the metabolic serine hydrolases still lack annotation, in terms of their endogenous substrates and the biological pathways that they influence13, 22.
An example of a metabolic serine hydrolase enzyme with unknown function is the α/β-hydrolase domain fold containing protein # 14B (ABHD14B), an outlying member of this enzyme family13. In search of interacting protein partners of the conserved histone acetyl-transferase (HAT) domain of the largest subunit for the TFIID transcription factor CCG1/TAFII250 (mutations to this protein cause cell cycle arrest in G1 phase, hence the name CCG1), a yeast two-hybrid screen identified ABHD14B as a putative target, thus leading to ABHD14B having the moniker CCG1/TAFII250-interacting factor B (CIB)23. Following up on this discovery, human ABHD14B was subsequently expressed recombinantly, purified from E. coli, shown to have weak hydrolase activity for a surrogate substrate, and the three-dimensional structure of this enzyme (PDB: 1IMJ) has been elucidated over a decade ago23. The three-dimensional structure revealed that human ABHD14B possessed the nucleophilic serine residue (S111) as part of a non-canonical sxSxs motif (where, x = any amino acid, S = S111), along with famed catalytic triad, and was therefore classified as a member to the metabolic serine hydrolase family. Also, it was postulated in the same study that ABHD14B plays a role in transcriptional activation, given its interactions with important transcriptional factors. However, despite these preliminary biochemical and structural studies, the endogenous substrates and the biological pathways that ABHD14B governs in vivo remain cryptic. In this paper, we report different biochemical assays and immunochemical tools, with complementary cellular and structural modelling studies, towards annotating ABHD14B as a novel class of lysine deacetylases, where this enzyme transfers an acetyl-group from a post-translationally modified protein acetyl-lysine residue to a molecule of CoA to produce acetyl-CoA. This functional annotation of ABHD14B expands the repertoire of known activities within the metabolic serine hydrolase family, and adds another enzyme family, capable of deacetylating protein lysine residues24 along with the well-studied sirtuins25–28 and histone deacetylase (HDAC) enzymes29, 30.
Materials and Methods
Materials
All chemicals, buffers and reagents described in this paper were purchased from Sigma Aldrich unless specified otherwise. All mammalian cell culture media and fetal bovine serum (FBS) were purchased from HiMedia unless otherwise mentioned. The anti-ABHD14B primary antibody was generated in rabbit using wild type (WT) human ABHD14B purified from E. coli as antigen at Bioklone Biotech Private Limited, India (www.bioklone.com). The primary anti-GAPDH antibody (cat: ab8245) and horse radish peroxidase (HRP) conjugated secondary anti-mouse antibody (cat: ab6789) were purchased from Abcam, while the HRP-conjugated anti-rabbit secondary antibody (cat: 31460) was purchased from Thermo Fisher Scientific. The primary anti-acetylated-lysine antibody used in this study was a kind gift from Dr. Krishanpal Karmodiya, IISER Pune31. The acetyl-Histone 3 (Lys9/14) peptide (Merck, Millipore, cat: 12-360), and Histones from calf thymus (Roche, cat: 10223565001) used in this study were kind gifts from Dr. Ullas Kolthur, TIFR Mumbai.
Cloning, Expression and Purification of Human ABHD14B
The WT human abhd14b gene was synthesized as a codon-optimized construct for expression in E. coli (GenScript, USA) and subsequently cloned into the pET-30b(+) vector (Millipore) between the HindIII and NdeI restriction sites, after removing the 6X-His-tag from the plasmid. The S111A ABHD14B mutant was generated using standard Quik-Change site directed mutagenesis protocols as per the manufacturer’s instructions (New England Biolabs). The plasmid bearing the desired gene (WT human ABHD14B or S111A human ABHD14B) was transformed into BL21(DE3) E. coli competent cells, following which, a single colony was grown in 5 mL of Luria-Bertani (LB) media containing kanamycin (final concentration 50 μg/mL) for 8 hours at 37 °C with constant shaking. This overnight primary culture was used to inoculate 1 L of the same medium, and the cells were allowed to grow at 37 °C with constant shaking until the OD600 reached ~0.6, at which point the protein expression was induced in the cell culture by the addition of 500 μM isopropyl β-D-1-thiogalactopyranoside (IPTG). Thereafter, the cells were grown at 30 °C for 16 hours, harvested by centrifugation (6,000g for 20 mins) and the cell pellets were stored at -80 °C till further use. The protein overexpression was confirmed by SDS-PAGE analysis. The stored cell pellet was thawed on ice and re-suspended in 45 mL of purification buffer (10 mM Tris, 10 mM 2-mercaptoethanol at pH 7.9). The cell suspension was sonicated at 4 °C using a Vibra Cell VCX 130 probe sonicator (Sonics) at 60% pulse amplitude, for 30 min with cycles of 5 sec ON and 10 sec OFF. The resulting lysate was centrifuged at 20,000g for 45 min at 4 °C to pellet the cellular debris and insoluble proteins from our protein of interest (WT human ABHD14B or S111A human ABHD14B) in the supernatant (soluble) fraction. To this resulting supernatant, protamine sulfate (0.05% w/v) was added dropwise at 4 °C, over 30 min, and this suspension was centrifuged at 20,000g for 45 min at 4 °C to separate nucleic acids. Post-centrifugation, the supernatant was collected, and ammonium sulfate (40% saturation) was added slowly over 30 min with continuous stirring at 4 °C. This mixture was centrifuged at 20,000g for 20 min at 4 °C and the resulting pellet was discarded. To the remaining supernatant, ammonium sulfate at 60% and 70% saturation were added slowly over 30 min, the mixtures centrifuged as described earlier, and the resulting supernatant fractions were discarded. The pellets were then resuspended in purification buffer to a final volume of 10 mL, and dialyzed in 1 L of purification buffer with 4 buffer changes every 2 hours initially, and the final buffer change was kept overnight (~ 14 hours) to dialyze out any residual ammonium sulfate from the previous purification step. Post-dialysis, the protein fraction (10 mL) was applied to an anion exchange column (GE Healthcare HiTrap CaptoQ, 5 mL), pre-equilibrated with purification buffer, and the protein of interest was eluted with a gradient of NaCl (25 – 250 mM) in purification buffer. Both WT and S111A ABHD14B were found to elute at 100 mM NaCl concentration as ascertained by SDS-PAGE analysis. The fractions containing the protein of interest were pooled and concentrated using a 10-kDa molecular weight cut off filter (Millipore) to a final volume of 6 mL. This concentrated protein fraction (6 mL) was loaded onto a preparative High Load 26/60 Superdex 200 gel filtration column (GE Healthcare) previously equilibrated with the purification buffer containing 100 mM NaCl, and 3 mL fractions were collected from this gel filtration column.The fractions were assessed for purity by SDS-PAGE analysis, and those containing WT or S111A ABHD14B were pooled, concentrated using a 10-kDa molecular weight cut off filter (Millipore) to a final concentration of ~50 μg/μL, flash frozen in liquid nitrogen as 10 μL aliquots, and stored at - 80°C till further use.
Colorimetric Substrate Hydrolysis Assays
All colorimetric assays were performed in a final reaction volume of 250 μL in a Varioskan Flash plate reader (Thermo Fisher Scientific), monitoring the release of para-nitrophenolate from substrates by measuring absorbance at 405 nm in a 96 well plate assay at 37 °C. All assays were performed in 10 mM Tris buffer at pH 8 in biological triplicates to ensure reproducibility. For the structure activity relationship (SAR) assays, 10 μM of WT ABHD14B was incubated with 500 μM of para-Nitrophenyl (pNp) analogues of varying chain lengths, namely pNp-acetate, pNp-butyrate, pNp-octanoate, pNp-decanoate, pNp-dodecanoate and pNp-palmitate (Figure S1), and absorbance at 405 nm was monitored over 30 min, with plate readings at 10 sec intervals. For the enzyme kinetics assays with pNp-acetate, pNp-butyrate and pNp-octanoate, substrate concentrations of 0 – 1000 μM were used with all other assay conditions and parameters remaining the same. The resulting data from this experiment was fit to a Lineweaver-Burk equation to yield enzyme kinetic constants for the respective substrate. To study the effects of CoA and acetyl-CoA on ABHD14B catalyzed hydrolysis reaction, 10 μM WT ABHD14B was pre-incubated with CoA (1 mM) or acetyl CoA (1 mM) at 37 °C in a 96 well plate for 15 min, following which the hydrolysis reaction was initiated by adding 50 μM pNp-acetate to this mixture, with all other assay parameters remaining the same.
Gel-Based ABPP Experiments
For all the gel-based ABPP experiments, WT or S111A ABHD14B was treated with the fluorophosphonate-rhodamine (FP-Rh) activity probe, at 37 °C for 1 hour with constant shaking in a final volume of 50 μL, following which, the reaction was quenched by adding 4X-SDS loading dye, and then boiling at 95 °C for 10 min. For the protein titration experiments, ABHD14B (WT or S111A) concentrations were varied from 0.5 – 10 μM keeping FP-Rh constant at 5 μM. For the activity probe titration experiments, ABHD14B (WT or S111A) concentrations were kept constant at 5 μM, while the FP-Rh concentrations were varied from 0.2 – 5 μM. Post-quenching the ABPP reactions, the samples were loaded and resolved on a 12.5% SDS-PAGE gel, and the enzyme activity was visualized on Syngene Chemi-XRQ gel documentation system. To ensure accurate protein loading in the gel-based ABPP assays, post-enzyme activity visualization, the gels were stained with Coomassie Brilliant Blue R-250 and imaged on the same system.
Thermal Shift Assays
Thermal shift assay was performed on a CFX-Real Time RT-PCR system (BioRad) using the Sypro-Orange dye as per manufacturer recommended protocols (Sigma-Aldrich) in a 96 well plate32. Briefly, 10 μM of WT or S111A ABHD14B was incubated with CoA (10 mM) or acetyl-CoA (10 mM) at 37 °C for 15 mins, following which, 1X Sypro-Orange dye was added to the reaction mixture, to make up the final volume to 25 μl in 10 mM Tris at pH 8. The fluorescence was measured in the FRET channel with an excitation and emission wavelength of 470 and 569 nm respectively, over a temperature range of 45 °C to 70 °C. All thermal shift assays were done in biological triplicates to ensure reproducibility of data. The raw fluorescence data from the thermal shift assays was converted into % Response using the following equation:
where X = fluorescence at any temperature, A = fluorescence at 45 °C, and B = fluorescence at 75 °C. Thus, based on this equation, the % response at 45 °C and 75 °C will always be 0 and 100 respectively.
Mammalian Cell Culture
All mammalian cell lines (HEK293T, HeLa, A549, THP1 and MCF7) described in this paper were purchased from ATCC, and cultured in complete medium [RPMI1640 supplemented with 10% (v/v) FBS, and 1% (v/v) penicillin-streptomycin (Pen-Strep) (MP Biomedicals)] at 37 °C with 5 % (v/v) CO2 unless otherwise mentioned. The cell cultures were stained routinely with 4’,6-diamidino-2-phenylindole (DAPI) and assessed by microscopy to ensure that they were devoid of any mycoplasma contamination using established protocols33. All cell lines were cultured in 15 cm tissue culture dishes (Eppendorf), and upon 80% confluence were harvested by scrapping, washed with Dulbecco’s phosphate buffer saline (DPBS) (3x times), centrifuged at 200g for 5 mins to remove excess DPBS, and stored at -80 °C till further use. For measuring the cellular levels of CoA and acetyl-CoA, these cell pellets were re-suspended in 150 μL of extraction buffer (1:1 methanol and water with 5% glacial acetic acid) by vortexing, and incubated on ice for 10 min. This was followed by the addition of 1.5 μL of 5 M ammonium formate, vigorous vortexing and a further 5 min incubation on ice. Finally, the extracts were centrifuged at 16,000g for 5 min at 4 °C, the supernatant was transferred in 1 mL glass vials, dried down under vacuum and stored at -40 °C until the LC-MS analysis34.
LC-MS Method to Measure CoA and Acetyl-CoA
The dried extracts were re-suspended in 35 μL of solvent A (2% acetonitrile in water with 100 mM ammonium formate) by vortexing and/or water bath sonication. To remove any particulate impurities, the re-suspended extracts were centrifuged at 16,000g for 5 min at 4 °C, and 20 μL of the supernatant was injected on a Phenomenex Gemini® C18 column (50 X 4.6 mm, 5 μm, 110 Å) fitted with a Phenomenex guard column (4 X 3 mm) using an Exion UHPLC system. The temperature of auto-sampler and column oven were set at 8 °C and 42 °C, respectively and the flow rate for the LC-MS run was 0.2 mL/min. A typical LC run consisted of 30 min, and had the following gradient sequence: (i) 0% solvent B (98% acetonitrile in water with 5 mM ammonium formate) for 2 min; (ii) linear increase in solvent B from 0% to 60% for the next 6 min; (iii) linear increase of solvent B from 60% to 90% in the next 1 min; (iv) maintaining 90% solvent B for 10 min; (v) linear drop in solvent B from 90% to 0% in 1 min; and (vi) re-equilibrating column with 0% solvent B for 10 min. The UHPLC was coupled to X500R Quadrupole Time-Of-Flight (QTOF) mass spectrometer (Sciex), which was operated in high resolution mass spectrometry (HRMS) positive ionization mode with the following source parameters: ion source gas 1 at 40 psi, ion source gas 2 at 50 psi, curtain gas at 30 psi, CAD gas at 7 psi, temperature at 500 °C, spray voltage at +5500 V, declustering potential (DP) at 90 V, DP spread 20 V, collision energy 20 V. The m/z ([M+H]+) of CoA and acetyl-CoA are 768.1225 and 810.1331, respectively. All data was collected and analysed using SCIEX OS-Q (version 1.4.0.18067) software as described earlier35. For the ABHD14B catalysed in vitro acetyl-transfer assays with pNp-acetate, the final reaction volume was 100 μL. In this assay, 10 μM pNp-acetate was incubated with CoA (100 μM), and the reaction was initiated by 10 μM of WT or S111A ABHD14B, and allowed to proceed for 5 mins at 37 °C. To reduce the rate of non-enzymatic acetyl-transfer reaction between pNp-acetate and CoA, this assay was done at pH 7.0. For the in vitro acetyltransferase assays with acetylated-histone 3 peptide, the final reaction volume was 50 μL. In this reaction, 2.5 μM of the peptide was incubated with CoA (25 μM), and the reaction was initiated by 2.5 μM of WT or S111A ABHD14B, and allowed to proceed for 5 mins at 37 °C at pH 7.0. To directly compare the relative rates of the acetyltransferase reaction between acetylated-lysine containing peptides, and pNp-acetate, the aforementioned assay was also performed with pNp-acetate (2.5 μM) in 50 μL volume with the same enzyme and CoA concentrations. For the in vitro acetyl-transferase assays with histones from calf thymus, the final reaction volume was 50 μL. In this reaction, 100 ng of the histone preparation was incubated with CoA (25 μM), and the reaction was initiated by 2.5 μM of WT or S111A ABHD14B, and allowed to proceed for 5 mins at 37 °C at pH 7.0. At the end of the experiment, all reactions were filtered through a 3-kDa molecular weight cut off filter (Millipore) by centrifugation to remove the enzyme, and 20 μL of the resulting flow through from these experiments, was subjected to LC-MS analysis described earlier34.
Western Blotting Experiments
Adult male C57BL/6 mice were anesthetized with isoflurane and euthanized by cervical dislocation, following which, brain, heart, liver, lungs, kidney and spleen of these mice were harvested. The tissues were washed (3x times) with cold sterile DPBS, re-suspended in cold sterile DPBS and homogenized using a tissue homogenizer (Bullet Blender 24, Next Advance) using one scoop of glass beads (0.5 mm diameter; Next Advance) for brain or zirconium beads (0.5 mm diameter; Next Advance) for all other organs at a speed setting of 8 for 3 min at 4 °C. The lysate was then centrifuged at 1000g for 5 min at 4 °C to pellet the tissue debris. For cells harvested from aforementioned mammalian cell culture section, the cell pellets were re-suspended in 1 mL of cold sterile DPBS, and lysed by sonication using an earlier reported protocol36. Thereafter, all the lysates (tissue or cell) were separated into soluble and membrane proteomic fractions by ultracentrifugation at 100,000g for 1 h at 4 °C using an earlier reported protocol36, and the soluble fraction was used for western blot analysis. The protein concentration of the soluble fractions was estimated using Pierce BCA Protein Assay kit (Thermo Fisher Scientific). 40 μg of tissue soluble proteome or 70 μg of cellular soluble proteome were loaded and resolved on a 12.5% SDS-PAGE gel and subsequently transferred onto a nitrocellulose membrane at 80 V for 2 h at 4 °C. The efficiency of the transfer was assessed by Ponceau staining of the membrane using standard protocols36. The membrane was then blocked with 5% (w/v) milk powder in PBST (1X phosphate buffer saline (PBS) with 0.1% (v/v) Tween-20) for 1 h at 25 °C, and probed with a primary antibody (dilution 1:1000 to 10,000) overnight (12 – 14 h) at 4 °C. The membrane was then washed with PBST (3x times), and probed with an appropriate HRP-conjugated secondary antibody (dilution 1:10,000) for 1 h at 25 °C, and any unreactive secondary antibody was washed off using PBST (3x times). Finally, the western blots were visualized by SuperSignal West Pico PLUS Chemiluminescent substrate (Thermofisher Scientific) and imaged on a Syngene Chemi-XRQ gel documentation system. The ImageJ software (NIH, USA) was used for any densitometric (quantitative) analysis of the western blot images37–39.
Cellular Immunofluorescence Assays
The mammalian HEK293T cells cultured in complete media were plated on coverslips, washed with 1X PBS, fixed with 4% (w/v) paraformaldehyde in 1X PBS for 15 min and permeabilized with 0.5% (w/v) Triton X-100 in 1X PBS for 15 min. The fixed cells were subsequently treated with blocking buffer (5% (w/v) BSA in 1X PBS with 0.1% (v/v) Tween-20) for 30 min, followed by sequential probing with anti-ABHD14B antibody (1:10,000 dilution) in blocking buffer for 2 h and anti-rabbit IgG-Alexa Fluor-488 (1:1000 dilution) along with Phalloidin-594 (1:500 dilution) in blocking buffer for 1 h. For visualization of the nucleus, the fixed cells were incubated with DAPI (50 ng/mL in 2X SSC (saline-sodium citrate) buffer) for 2 min. The entire procedure was carried out in a humid chamber at 25 °C, and after each step, the cells were washed with 1X PBS. The stained cells were imaged using a Zeiss confocal microscope at the IISER Pune – Microscopy Centre, and the data was analysed using the ImageJ software (NIH, USA)37–39.
Generation of ABHD14B Knockdown Cell Lines
Three human abhd14b lentiviral shRNA knockdown constructs and a non-targeting (control) construct were purchased from TransOMIC, USA. These constructs have puromycin resistance and ZsGreen fluorescent reporter coding genes. The abhd14b targeting 5’ sequences are KD_1 → ACATCTGCACTGACAAAATCAA, KD_2 → ACCCATGGGTCAGACCAGCTTA, and KD_3 → CGAAGACTCCAGCTCTGATTGA. The lentivirus cDNAs were generated by co-transfecting HEK293T grown in complete media, with shRNA constructs, plasmids psPAX2 and pMD2.G (1:1:0.5) using the transfection reagent PEI 40,000 using established protocols40. 2 days post-transfection, the lentiviral particles were filtered through a 0.45 μm filter and used to transduce HEK293T cells along with 4 μg/mL polybrene. 1 day post-transduction, the lentiviral containing media was replaced with fresh complete media containing puromycin (5 μg/mL), and the cells bearing the desired constructs were thereafter selected on puromycin. The complete media + puromycin (5 μg/mL) was changed every 2 days and cell selection was checked for, by the amount of green fluorescence in cells. The puromycin selection strategy was continued until all the non-green fluorescent cells were removed (> 99%) from the cell population. Once this was achieved, the cells were harvested and lysed in DPBS by sonication, and the whole cell lysates was used to confirm knockdown of ABHD14B by western blot analysis using protocols described earlier36. All the knockdown HEK293T cell lines were revived and maintained in complete media containing 1 μg/mL puromycin, to ensure that desired constructs are not competed by any other cell populations.
Identification of a Substrate Binding Channel in ABHD14B
The tunnel analysis was conducted using the CAVER v1.0 online server41, 42. In brief, the structure of the monomer of human ABHD14B (PDB: 1IMJ)23 was loaded onto the server, and the catalytic triad sequence (S111, D162, H188) was manually input to provide a starting point for the analysis. Using the default server settings, two tunnels were found, one of which was a subset of the other (essentially forming one, unified tunnel) (Figure 8A). In parallel, tunnels were calculated using the MOLEonline server43, 44 and yielded a near identical result (Figure 8A). The tunnel-defining residues (entrance, exit, and bottleneck) were visualized using PyMol and found to be basically identical for Caver and MOLEonline. An electrostatic calculation using the APBS plugin to PyMol45–47 was also performed on the native monomer of human ABHD14B (PDB: 1IMJ)23. First, the required PQR files were generated using the PARSE force-field and these PQR files were subsequently used to calculate the electrostatic surface diagrams45–47 displayed in Figure 8B. Software packages for analysis and visualization of ABHD14B were compiled by SBGrid48.
Figure 8. A putative active site tunnel in human ABHD14B.

(A) The tunnels calculated by CAVER (teal) and MOLEonline (yellow) are essentially identical albeit slightly different in length using the available structure of human ABHD14B (PDB: 1IMJ)23. The entrance of the tunnel is near the catalytic triad (S111, H188, D162). Right panel is the same view rotated by 90°. All of the residues that line the tunnel are shown as sticks. The bottleneck, or narrowest point of the tunnel is S111 of the catalytic triad. (B) An electrostatic calculation of the structures shown in panel A. The entrance of the tunnel, which houses the catalytic triad of residues has an overall positive charge, which likely assists in the catalysis. (C) The length of the phosphopantetheine arm of CoA is consistent with the length of the calculated tunnel in ABHD14B. At the exit of the tunnel (yellow) are several candidate residues for interacting with the nucleotide 5’-pyrophosphate moiety of CoA. This view of the tunnel is equivalent to that shown in panel A (right side) and has labels for each of the residues that line the tunnel.
Data plotting
All graphs and plots presented in this paper were made using the GraphPad Prism 7 (version 7.0b) software for MacOS-X. Unless otherwise mentioned all data in bar format is presented as mean ± standard deviation for three or more biological replicates from independent experiments.
Results
Purification and Biochemical Properties of Human ABHD14B
The WT enzyme and the active site directed S111A mutant of human ABHD14B were successfully expressed and purified from E. coli in high purity (> 99%) (Figure S2) and apparent homogeneity as assessed by analytical gel filtration and thermal shift assays (Figure S3). Our purification scheme typically afforded 10 – 15 mg ABHD14B/L culture. To determine, whether the S111A mutant was catalytically inactive, we first resorted to established gel-based ABPP assays11, and found that as a function of increasing enzyme concentration, WT human ABHD14B, but not S111A human ABHD14B, displayed robust dose dependent activity against the FP-Rh activity probe, which was kept constant in this assay (Figure 1A). Next, we found that as a function of increasing the FP-Rh activity probe concentration, and keeping the enzyme concentration constant, that WT human ABHD14B again, showed robust dose dependent activity, while the S111A mutant showed no activity at all in this gel-based ABPP experiment (Figure 1B). To complement these gel-based ABPP assays, we also performed substrate hydrolysis assays, as previous studies have shown that ABHD14B has hydrolase activity against acylated-para-nitrophenol derivatives23. Towards this, we incubated WT and S111A human ABHD14B with pNp-acetate (500 μM), and found that WT human ABHD14B produced para-nitrophenolate from pNp-acetate approximately 10-fold better than S111A human ABHD14B (Figure 1C). We hypothesize that, since pNp-acetate is unlikely to be the “real” substrate for this enzyme, and as pNp-acetate is an activated molecule, its binding to the enzyme, and subsequent interactions with the other conserved residues of the enzyme active site (e.g. catalytic base), might result in its slow hydrolysis even by S111A human ABHD14B. Taken together however, these results suggested that the active site directed S111A mutant of human ABHD14B was severely catalytically compromised, if not catalytically dead.
Figure 1. The S111A mutant of human ABHD14B is catalytically inactive.

(A, B) Gel-based ABPP assays with WT and S111A human ABHD14B, showing robust dose dependent activity of WT ABHD14B, but not S111A ABHD14B, as a function of increasing: (A) enzyme concentration (0.5 – 10 μM), and (B) activity probe (FP-Rh) concentration (0.2 – 5 μM), while keeping the other constant (activity probe concentration (5 μM) in A; enzyme concentration (5 μM) in B). The gel-based ABPP experiments were done three independent times with reproducible results each time. (C) Colorimetric enzymatic assay showing ~ 10-fold more activity of WT human ABHD14B, compared to S111A human ABHD14B, against pNp-acetate (500 μM). The colorimetric assays were done three independent times with reproducible results each time.
Since WT human ABHD14B was able to turnover acylated-para-nitrophenol derivatives, we decided to perform structure activity studies with this class of surrogate substrates, to assess whether this enzyme has any preference for the esterified acyl-group to pNp. In this experiment, we incubated WT human ABHD14B (10 μM) with 500 μM of different pNp-analogues of varying chain lengths, ranging from acetate (two carbon atoms, C2) to palmitate (16 carbon chain, C16), and monitored the release of para-nitrophenolate produced from these surrogate substrates. From these studies, we found a distinct trend, where WT human ABHD14B preferred smaller acylated-esters of pNp as substrates, with pNp-acetate being the best substrate (Figure 2). Further, we found that this enzyme sluggishly turned over pNp-octanoate (C8) but was unable to turnover acylated-esters of pNp having more than 10 carbon atoms (Figure 2). To ascertain this substrate preference profile, we performed enzyme kinetics assays on the three pNp-substrates that WT human ABHD14B was able to turnover. This experiment further confirmed that pNp-acetate was indeed the best substrate for this enzyme (Table 1). Albeit sluggish due to the unnatural nature of the pNp-acetate substrate, these substrate profiling results taken together, suggest that ABHD14B might function as a deacetylase or acetyltransferase in biological settings.
Figure 2. WT human ABHD14B prefers hydrolysis of acetyl-group from a surrogate pNp-substrate.

The colorimetric assays for each substrate were done three independent times with reproducible results each time.
Table 1. Kinetic constants for WT human ABHD14B against different acylated esters of pNp.
| Substrate | kcat (min-1) | Km (mM) | kcat/Km (M-1s-1) |
|---|---|---|---|
| pNp-acetate | 2.2 ± 0.8 | 1.8 ± 0.6 | 20.4 ± 2.5 |
| pNp-butyrate | 0.6 ± 0.2 | 3.1 ± 0.6 | 3.2 ± 0.4 |
| pNp-octanoate | 0.2 ± 0.1 | 3.9 ± 0.7 | 0.9 ± 0.2 |
Characterizing the Acetyl-Transfer Reaction Catalyzed by Human ABHD14B
In findings reported by Padmanabhan et al., describing the three-dimensional structure and preliminary biochemical characterization of ABHD14B23, the authors also speculate that WT human ABHD14B likely binds acetyl-CoA, because of its association to the HAT domains of important transcription factors. Given this literature precedence, we decided to test whether human ABHD14B binds acetyl-CoA and/or CoA using established thermal shift assays32, and chose the S111A mutant for these studies, as we hypothesized that binding interactions (if any) of acetyl-CoA and/or CoA would be longer for a catalytically inactive mutant of an enzyme. From these assays, where S111A human ABHD14B (10 μM) was incubated with acetyl-CoA or CoA (both at 10 mM), we saw a left-ward (lower temperature) shift in the fluorescence profiles (% Response) for both these compounds, with the shift being more prominent for CoA (~ 5 °C) compared to acetyl-CoA (~ 2.5 °C), suggesting that CoA most likely binds to the enzyme tighter than acetyl-CoA (Figure 3A). We also saw similar leftward shift, albeit to a much lesser extent (~ 2 °C), in thermal shift assays after incubating WT human ABHD14B with acetyl-CoA and CoA without a distinct change in binding for acetyl-CoA or CoA (Figure S4). The authors of the same aforementioned paper also speculate that the binding of acetyl-CoA to ABHD14B might result in its hydrolysis23, to yield CoA and acetate, and we also tested this hypothesis, looking for the formation of CoA from acetyl-CoA by LC-MS analysis. However, we did not find any turnover of acetyl-CoA in the presence of WT human ABHD14B, suggesting that this enzyme does not possess any deacetylase (or HAT-type) activity for acetyl-CoA.
Figure 3. ABHD14B performs an acetyl-transfer reaction.

(A) Thermal shift assays showing binding of both CoA and acetyl-CoA to S111A human ABHD14B. The thermal shift assays were done three independent times with reproducible results each time. (B) Colorimetric enzymatic assay with pNp-acetate, showing increase in rate of enzymatic reaction following incubation of ABHD14B with CoA, but not acetyl-CoA. The colorimetric assays were done two independent times with reproducible results each time. (C) Formation of acetyl-CoA by WT human ABHD14B, but not S111A human ABHD14B, when incubated with pNp-acetate and CoA. The LC-MS data represents mean ± standard deviation from six independent experiments.
Next, we wanted to assess whether the binding of either acetyl-CoA or CoA to ABHD14B, has any effect on the pNp-acetate hydrolysis reaction catalysed by WT human ABHD14B. In this assay, WT human ABHD14B (10 μM) was pre-incubated with either acetyl-CoA or CoA (1 mM each), and the reaction was initiated by adding pNp-acetate (50 μM). Acetyl-CoA had no effect on the kinetics of the reaction, however CoA significantly enhanced the rate of this hydrolysis reaction (Figure 3B). We performed another enzyme kinetic assay for WT human ABHD14B against pNp-acetate substrate in the presence of excess CoA (1 mM), and obtained the following kinetic constants: kcat = 9.1 ± 1.4 min-1; Km = 1.7 ± 0.4 mM; kcat/Km = 89 ± 12 M-1s-1, which further showed that the rate of the hydrolysis reaction becomes almost 4.5 times faster (from kcat and kcat/Km values) in the presence of excess CoA (also see Table 1). To note, consistent with the ping-pong mechanism, CoA had little effect on the binding of pNp-acetate to the enzyme. That CoA binds more tightly to ABHD14B and accelerates the enzyme catalysed pNp-acetate hydrolysis reaction, and the inability of this enzyme to hydrolyze acetyl-CoA, suggested that ABHD14B might, contrary to previous speculation23, functions as a deacetylase rather than an acetyltransferase. Finally, to confirm if acetyl-CoA is produced when WT human ABHD14B was incubated with pNp-acetate and CoA, we performed LC-MS analysis34. We found from this experiment, that this was indeed the case, where WT human ABHD14B, but not S111A human ABHD14B, robustly produced acetyl-CoA, when incubated with pNp-acetate and CoA (Figure 3C). Taken together, these results conclusively show that ABHD14B performs an acetyl-transfer reaction, where CoA is the eventual acetyl-group acceptor from an acetylated-substrate, and acetyl-CoA is a product of this enzyme catalysed reaction.
Characterization of an anti-ABHD14B Antibody
Since we were able to obtain WT human ABHD14B with high purity and apparent homogeneity in relatively good yields, we decided to generate an anti-ABHD14B antibody from rabbit, and got this custom synthesized from Bioklone Biotech Private Limited, India. Briefly, the rabbit was immunized with three booster doses of the antigen (1 mg WT human ABHD14B each time), and once appropriate titer levels of the antibody were produced, the polyclonal anti-ABHD14B antibody was purified using standard antibody affinity purification protocols, as per the company’s established procedures. Following immunization and purification from blood, we obtained 1.1 mg of polyclonal anti-ABHD14B antibody from 1 rabbit, and we wanted to validate its compatibility for western blotting and cellular immunofluorescence experiments. Towards this, we first tested this antibody against varying amounts (0.01 – 1 μg) of recombinantly purified WT or S111A human ABHD14B, and found that this polyclonal antibody at a dilution 1:1000, was able to detect even 10 ng of recombinant protein (Figure 4A), giving us confidence that, this polyclonal antibody might in fact be able to detect endogenous levels of human ABHD14B in cell lines by western blotting. To verify this, we cultured 5 human cell lines (HEK293T, A549, HeLa, MCF7 and THP1), and assessed whether ABHD14B was detected in their soluble proteomes by western blotting. We found that in all the 5 human lines, our polyclonal antibody was able to selectively detect endogenous ABHD14B, consistent with available literature49, with almost no cross-reactivity with any other protein in the western blot analysis (Figure 4B). We found from this experiment, that THP1 macrophage cells had the highest levels of ABHD14B, and was therefore initially chosen as the candidate mammalian cell line for studying the cellular function of ABHD14B. However, given the suspension and macrophage nature of this cell line, we were unable to perform cellular immunofluorescence assays (IFA) and/or get significant knockdown of ABHD14B in THP1 cells (described later), and hence had to choose an alternative cell line for performing these studies. We wanted an adherent cell line amenable to cellular IFA experiments and gene knockdown studies, and hence chose HEK293T cells for all subsequent cellular studies described in this paper.
Figure 4. Characterization of the anti-ABHD14B antibody.
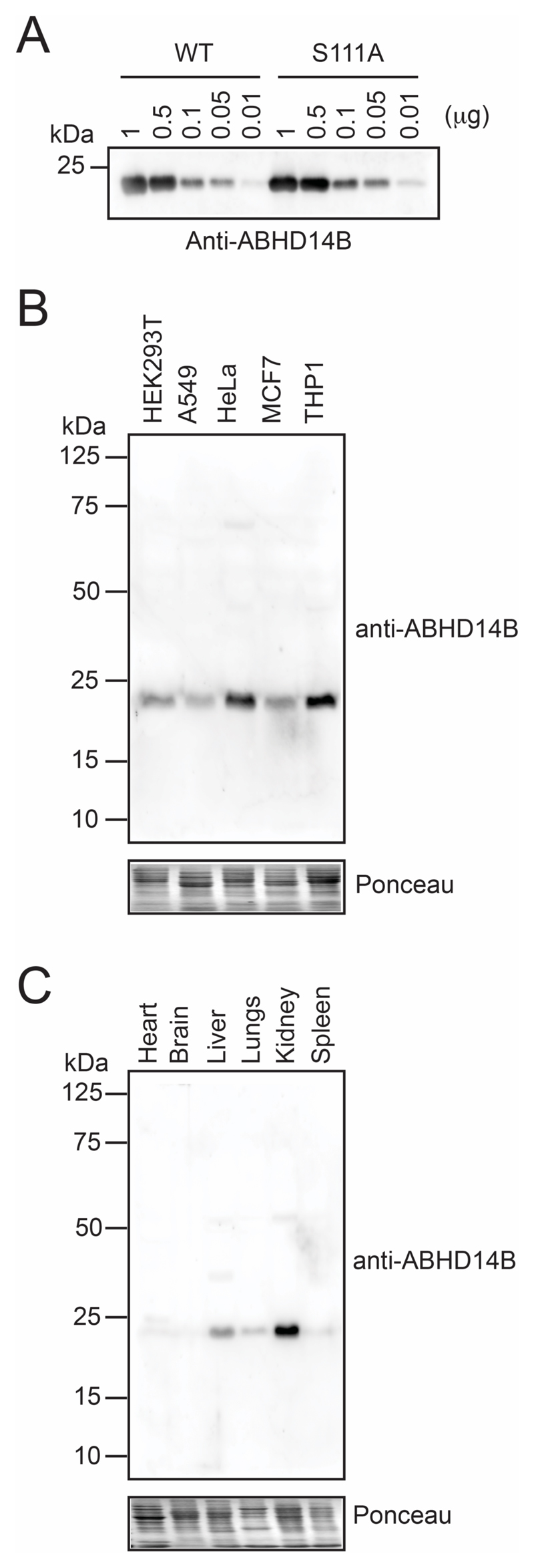
Western blot analysis of a rabbit polyclonal anti-ABHD14B antibody tested against (A) varying amounts (0.01 – 1 μg) of recombinantly purified WT and S111A human ABHD14B; (B) soluble proteomes of different human cell lines; and (C) soluble proteomes of different mouse tissues. The western blots reported in (B) and (C), show selective detection of endogenous mammalian ABHD14B in cells and tissues respectively. All western blot experiments were done three independent times with reproducible results each time.
The human and mouse ABHD14B have ~ 90 % sequence identity, and we wanted to determine whether the ABHD14B antibody produced by us had any cross-species reactivity. To test this, we harvested different tissues (heart, brain, liver, lungs, kidney and spleen) from C57Bl/6 wild type mice, and assessed their soluble proteomes for ABHD14B protein levels by western blot analysis. We found from this immunoblotting experiment, that our polyclonal antibody can robustly and selectively detect endogenous mouse ABHD14B across different tissues expressing ABHD14B with almost no cross-reactivity with other proteins in these mouse tissues (Figure 4C). Further, the protein levels of ABHD14B detected by us across the different mouse tissues, especially the highest expression of ABHD14B in kidney and liver, are consistent with data reported in a large-scale gene expression database (http://biogps.org)50, 51 for ABHD14B (Figure S5), giving us further confidence in the quality of our antibody.
Finally, to assess whether the antibody generated by us was compatible for cellular IFA experiments, we performed an IFA experiment in the human HEK293T cells to determine this (Figure 5). We found from this cellular IFA experiment, that cellular fluorescent signal for ABHD14B (green channel) was only visible in the presence of our polyclonal anti-ABHD14B antibody, and the control (secondary antibody only) samples showed negligible signal for ABHD14B, validating that our anti-ABHD14B antibody was indeed compatible for cellular IFA experiments. Further, consistent with previous cellular localization studies from a yeast-hybrid screen23 and available literature49, we found that ABHD14B was present in both the nucleus and cytosol in HEK293T cells. Given the ubiquitous cellular localization of ABHD14B, we speculated that the putative substrate(s) for ABHD14B is/are likely present throughout the cells, and this enzyme likely serves a general enzymatic reaction.
Figure 5. Cellular localization of ABHD14B in HEK293T cells.

Cellular IFA in HEK293T cells, show that ABHD14B is present in both the nucleus and cytosol, and the cellular fluorescence for ABHD14B (in green channel) is seen only in the presence of the ABHD14B antibody. The cellular IFA were done five independent times with reproducible results each time.
Effects of Genetically Knocking Down ABHD14B in HEK293T Cells
To understand the physiological function of ABHD14B, we decided to genetically knockdown ABHD14B in human HEK293T cells using the established RNA interference technology. We found that compared to a non-targeting (scrambled) “control” line, the cell lines generated with shRNA-targeting ABHD14B (KD_1, KD_2 and KD_3) showed good knockdown of ABHD14B in HEK293T cells (Figure 6A). Since the lines KD_2 and KD_3 showed greater than 90% knockdown of ABHD14B (and KD_1 failed to make this cut off), they were chosen for subsequent studies (Figure 6A). First, we found that the knocking down of ABHD14B expression in cells, did not affect cell growth, and we did not observe any obvious phenotypic defects in the knockdown (KD_2 and KD_3) cell lines compared to the control cell line. Since, previous biochemical studies showed that ABHD14B transfer an acetyl-group to CoA, we decided to measure the cellular levels of CoA and acetyl-CoA in the ABHD14B knockdown cell lines using LC-MS analysis34. We found that following ABHD14B knockdown, the cellular levels (relative to the control cell line) of CoA, the substrate of the ABHD14B catalyzed reaction remained unchanged (Figure 6B). Interestingly however, the cellular levels (relative to the control cell line) of acetyl-CoA, the product of the ABHD14B catalyzed reaction have significantly reduced (~ 50%) following the knockdown of ABHD14B in HEK293T cells (Figure 6B). Consistent with the biochemical assays, these cellular measurements of CoA and acetyl-CoA further suggest that in physiological settings, ABHD14B most likely regulates an acetyl-transferase (deacetylase) reaction, and in doing so, controls the cellular biosynthetic flux of acetyl-CoA.
Figure 6. Effects of ABHD14B genetic knockdown in human HEK293T cells.

(A) Western blot analysis confirming the knockdown of ABHD14B in the KD_1, KD_2 and KD_3 cell lines, relative to the non-targeting control cell line made from HEK293T cells. This western blot experiment was done three independent times with reproducible results each time. (B) The cellular levels of CoA and acetyl-CoA in the KD_2 and KD_3 cell lines, relative to the non-targeting control cell line measured by LC-MS analysis. The LC-MS data represents mean ± standard deviation from six independent experiments. (C) Western blot analysis, showing increased protein lysine acetylation following ABHD14B knockdown in HEK293T cells. This western blot experiment was done three independent times with reproducible results each time.
We established the role for CoA and acetyl-CoA in the ABHD14B catalyzed acetyltransferase reaction, but the biological origins of the acetyl-group remained unknown. In cells, conserved and functionally relevant protein lysine residues are often acetylated by enzymes using acetyl-CoA (e.g. HAT enzymes)52, 53, and these post-translationally protein acetylated-lysine residues serve many functions, important among which are the regulation of gene transcription and controlling cellular metabolism and proliferation54–56. Given its association with important transcription factors, we hypothesized that ABHD14B might be performing a deacetylase-type reaction, transferring the acetyl-group from a post-translationally acetylated-lysine protein residue of a protein to CoA, and making acetyl-CoA in the process. To test this hypothesis, we assessed the lysine acetylation profile of cell lysates prepared from ABHD14B knockdown cell lines (KD_2 and KD_3) and compared them to lysates prepared from the control cell line, by western blot analysis using an anti-acetylated lysine antibody31. Consistent with our hypothesis, we found that ABHD14B knockdown significantly increased the concentration of acetylated-lysine residues in cell lysates (Figure 6C), suggesting that in physiological settings, an acetylated-lysine of a protein is in fact the other substrate of ABHD14B, and the origin of the acetyl-group of that eventually forms acetyl-CoA. That ABHD14B knockdown increases cellular concentrations of acetylated-lysine residues of proteins, and concomitantly decreases the levels of acetyl-CoA, shows a clear substrate-product relationship in physiological settings for the ABHD14B-catalyzed lysine deacetylase reaction.
Characterizing the ABHD14B catalyzed acetyltransferase reaction against acetylated-lysine containing substrates
Thus far, we had only tested the acetyl-transfer reaction catalyzed by ABHD14B using pNp-acetate as the acetyl-donor (Figure 3). However, in view of the results from genetically disrupting this enzyme in human HEK293T cells, where we find increased protein lysine-acetylation (Figure 6C), we wanted to validate if ABHD14B can in fact use acetylated-lysine containing peptides or proteins as substrates for the acetyl-transferase reaction. To test if this was indeed the case, we first chose the commercially available di-acetylated histone H3 21 amino acid peptide with the following sequence: ARTKQTAR(KAC)STGG(KAC)APRKQLC, where KAC is the acetylated lysine residue. We incubated purified WT and S111A human ABHD14B with enzyme equimolar concentrations of the aforementioned peptide in the presence of excess CoA, and assessed the formation of acetyl-CoA in this assay by LC-MS analysis. We found that WT human ABHD14B, but not S111A human ABHD14B, was indeed able to make acetyl-CoA from this acetylated peptide (Figure 7A). To compare the relative production of acetyl-CoA from different acetyl-donors, we also ran that same assay with enzyme equimolar concentrations of pNp-acetate (instead of the acetyated-lysine peptide), and found that the formation of acetyl-CoA was ~ 3.5-fold more when the acetylated-lysine peptide was used as the acetyl donor (Figure 7A). This result suggests that this enzyme in fact prefers acetylated-lysine proteogenic substrates over pNp-acetate. Since the peptide substrate had two acetylated-lysine residues, we wanted to determine, how many acetyl-groups was ABHD14B able to transfer to CoA, and in the same LC-MS experiment looked for masses of the di-acetylated ([M+H]+ = 2370.315), mono-acetylated ([M+H]+ = 2328.304), and non-acetylated ([M+H]+ = 2286.294). We found from the LC-MS analysis, that WT human ABHD14B was in fact able to transfer both acetyl groups from the peptide to CoA forming acetyl-CoA, and yielding a non-acetylated peptide from the di-acetylated starting peptide in the process (Figure 7B). These results taken together show first, that acetylated-lysine containing peptides are better substrates for ABHD14B compared to pNp-acetate, and at first approximation, ABHD14B can perform acetyl-transfer reactions on acetylated-lysine containing peptides, presumably without much sequence consensus.
Figure 7. ABHD14B performs a lysine deacetylase reaction on peptide and protein substrates.

(A) Formation of acetyl-CoA by WT human ABHD14B, but not S111A human ABHD14B, when incubated with enzyme equimolar concentration of the di-acetylated (Lys9/14) histone H3 peptide (1 – 20) and excess CoA (25 μM). The control samples for this assay is the entire mixture without enzyme. (B) Formation of non-acetylated peptide from the starting di-acetylated (Lys9/14) histone H3 peptide (1 – 20) (2.5 μM) by WT human ABHD14B (2.5 μM) in the presence of excess CoA (25 μM). The S111A human ABHD14B (2.5 μM) has no activity against the di-acetylated peptide substrate. (C) Formation of acetyl-CoA by WT human ABHD14B (2.5 μM), but not S111A human ABHD14B (2.5 μM), when incubated with histone preparations from calf thymus (100 ng) and excess CoA (25 μM). The control samples for this assay is the entire mixture without enzyme. All LC-MS data represented in (A), (B), and (C) is the mean ± standard deviation from three independent experiments.
Having shown that WT human ABHD14B can transfer an acetyl-group from an acetylated-lysine containing peptide to CoA, we wanted to determine if this enzyme can also perform a similar lysine deacetylase reaction using acetylated-lysine containing protein substrates, and we chose histone preparations from calf thymus test this premise, as previous studies have shown that these proteins are acetylated57. In this assay, we incubated WT or S111A human ABHD14B with histone preparations from calf thymus in the presence of excess CoA, and assessed whether acetyl-CoA was formed at the end of the reaction by LC-MS analysis. We found from this LC-MS assay, that WT human ABHD14B, was indeed able to produce acetyl-CoA from histone preparations obtained from calf thymus (that are presumably acetylated)57, while the catalytically inactive S111A human ABHD14B mutant did not produce an acetyl-CoA of significance, and had no enzyme control levels of acetyl-CoA in this assay (Figure 7C). Taken together, these results suggest that this enzyme can indeed perform a lysine deacetylase reaction on both peptide and protein substrates containing post-translationally modified acetylated-lysine, and corroborates the findings from the from the genetic knockdown of ABHD14B in HEK293T cells shown in Figure 6.
A putative substrate binding site identified in ABHD14B
To address how substrates could approach the catalytic triad of residues in ABHD14B, we used two computational tools to search for tunnels in the protein41–44. Both the computational programs calculated similar tunnels that were slightly different in length, but superimpose very well with one another (Figure 8A). These tunnels run from the surface of the protein near the active site and emerge on the opposite side. The combined length of the calculated tunnels is approximately 20 Å, which is appropriate for binding the phosphopantetheine arm of CoA58 and positioning the thiol moiety near the catalytic serine residue (S111) for accepting the acetyl-group. It is important to note that the tunnel entrance shows an overall positive charge (Figure 8B) that is likely a key factor in the binding of a sulfate ion in the enzyme active site from the crystallography experiment in its entrance, but could also stabilize formation of the tetrahedral intermediate during the catalytic reaction cycle (Scheme 1). Importantly, the presence of a tunnel that enters and exits form the surface of the protein serves to explain how this protein could mediate the participation of two substrates (e.g. an acetylated-lysine protein residue and CoA) in the reaction. It is also important to note that at the tunnel exit, there are several protein residues (W198, H99, H55, and R56) that could be used to facilitate binding of the pyrophosphate moiety of CoA (Figure 8C).
Discussion
The general transcription initiation factor TFIID is a multiprotein complex mainly comprised of the TATA-box binding protein (TBP)59 and the TBP-associated factors (TAFs)60, 61, and is shown to bind the TATA-box region of a promoter, and initiate transcription by forming the core of the pre-initiation complex along with other transcriptional factors, and RNA polymerase II62–65. The TFIID has been shown to activate cellular transcription by binding to both naked DNA and chromatin66–68, and has been the subject of several landmark studies pertaining to regulation of eukaryotic transcription over the past three decades. The largest unit of the TFIID complex is the protein CCG1/TAFII250, that gets its name as a fusion of two acronyms: (i) CCG1 = cell cycle arrest in G1 phase69, and (ii) TAFII250 = TAF protein with a molecular weight of 250 kDa70. Several studies characterizing the biochemical function of the CCG1/TAFII250 protein have now shown that this large protein is central to eukaryotic transcription and has three major structural domains that perform specific functions. These include: (i) the N-terminal and C-terminal kinase domains71; (ii) two bromodomains that are involved in ubiquitinylation of histones and chromatin proteins72, 73; and (iii) HAT domain responsible for incorporating epigenetic acetyl-marks on histones74. It has further been shown that deletion or amino acid mutations to the HAT domain of the CCG1/TAFII250 protein specifically results in cells being arrested in G1 phase of cell cycle, and this eventually results in cellular apoptosis69, 74. Given its central role in activating the eukaryotic transcription process, the CCG1/TAFII250 protein also interacts with other structural proteins, enzymes and cofactors during the formation of the pre-initiation complex, and the identification of these partners has been the focus of several research groups. A pull-down study aiming to identify proteins that CCG1/TAFII250 interacts with resulted in the identification of ABHD14B, given this interaction, ABHD14B was also termed CIB23. The same study went on to recombinantly purify this enzyme, show its ability to perform hydrolytic reactions, and determine its three-dimensional structure over a decade ago23. Yet, the functional annotation of this enzyme has remained elusive since then, and this study to the best of our knowledge, remains the only biochemical characterization of this cryptic enzyme.
In this paper, we aimed to annotate function to this orphan enzyme from the serine hydrolase family. We show here that ABHD14B catalyzes an unprecedented lysine deacetylase (KDAC) reaction, transferring an acetyl-group from a post-translationally acetylated lysine residue of a protein to CoA, and in the process, makes acetyl-CoA, and regenerates the amine of the lysine residue of proteins (Figure 9), and provide several lines of compelling evidence towards this annotation. First, we show by substrate hydrolysis assays using surrogate acylated-pNp-analogs, that, ABHD14B has a very tight substrate SAR, strongly prefers pNp-acetate as a substrate, and that, as the chain length of the acylated group of pNp increases, the enzymatic hydrolysis efficiency of ABHD14B concomitantly decreases (Figure 2, Table 1). Second, we show that ABHD14B binds CoA, that this binding of CoA significantly increases the pNp-acetate hydrolysis, and this enzyme can in fact transfer the acetyl group from pNp-acetate to CoA to form acetyl-CoA in the process (Figure 3). Third, we show that genetically knocking down ABHD14B in human HEK293T cells, results in increased levels of protein lysine acetylation, and decreased cellular concentrations of acetyl-CoA, thereby confirming in cell physiological settings, that ABHD14B indeed functions as a novel lysine deacetylase (Figure 6). Fourth, and most importantly, we validate these cellular findings by complementary in vitro acetyltransferase assays using acetylated-lysine containing peptide and protein substrates, and show that ABHD14B can in fact produce acetyl-CoA from these substrates (Figure 7). Interestingly, we also find in these assays that in comparison to the “surrogate” pNp-acetate substrate, the “native” acetylated-lysine containing peptide and protein substrates, produce more acetyl-CoA for similar substrate concentrations, suggesting that the latter are preferred substrates for ABHD14B (Figure 7). Finally, we have identified an active site tunnel in the WT human ABHD14B, that extends from the surface of the protein near the catalytic triad to the far end of the protein, suggesting that indeed this enzyme’s active site can accommodate two different substrates and more specifically is comparable in length to the phosphopantetheine arm of CoA, that eventually accepts the acetyl group (Figure 8).
Figure 9. The lysine deacetylase (KDAC) reaction catalyzed by ABHD14B.

The genetic knockdown of ABHD14B in human HEK293T cells, consistent with other studies reported in this paper, results in increased protein lysine-acetylation, and decreased cellular acetyl-CoA levels, as shown by the red arrows in this figure.
Why is the ABHD14B catalyzed lysine deacetylase (KDAC)24 novel? There are currently two known enzyme families that can perform a lysine deacetylase reaction: the sirtuins27, 28 and the HDACs29, 30. Both of these enzyme classes have distinct enzymatic mechanisms for deacetylating lysines. The sirtuins, for instance, use a molecule of NAD+ to perform the deacetylase reaction, and in the process, generate acetyl-ADP-ribose, the eventual fate of the acetyl group from the post-translationally acetylated lysine25. In contrast, the HDACs, have conserved acidic (Asp) and basic (His) residues in the enzyme active site that act in tandem to activate a water molecule, and perform a cofactor-less hydrolysis reaction29. The fate of the acetyl group in the enzymatic reaction catalyzed by HDACs, is the formation of a free acetate molecule that is eventually recruited back into metabolism29. Here, by functionally characterizing ABHD14B, we present a third mechanism for deacetylating protein lysine residues, using the consensus serine hydrolase mechanism13, 14 (Scheme 1), where in the first step the acetyl group from lysine in transferred to the nucleophilic serine residue of ABHD14B to form an acetyl-enzyme covalent intermediate. In the second step, a molecule of CoA binds to the enzyme (instead of water), and the acetyl group from the acetyl-enzyme covalent intermediate is transferred to the free thiol of the phosphopantetheine arm of CoA to yield acetyl-CoA. The unique and distinguishing features of the ABHD14B catalyzed reaction in comparison to sirtuins and HDACs, is that, the reaction catalyzed by the former involves the formation of a covalent acetyl-enzyme intermediate, and the eventual fate of the acetyl-group is the formation of acetyl-CoA.
Projecting ahead, the functional annotation of ABHD14B, presents several new questions. First and foremost, since the deacetylation mechanism catalyzed by ABHD14B is novel, having three-dimensional structures with putative acetylated lysine substrate (or their analogs), and CoA and/or acetyl-CoA bound to the enzyme, will provide new insights into the structural basis for this catalytic reaction. Second, we report a selective anti-ABHD14B antibody, and show that this enzyme is present throughout the cell (nucleus and cytosol) (Figure 5), and is mostly expressed in metabolically rich tissues (kidney, liver) (Figure 4). This leads us to speculate that the lysine deacetylase reaction catalyzed by ABHD14B is likely to affect the metabolic status of cells. Towards understanding this metabolism, advanced mass spectrometry based metabolomics and physiological (e.g. cellular respiration, mitochondrial function, lipid flux etc.) studies are warranted to map the different biological pathways that are under the influence of ABHD14B in mammalian cells and tissues. Third, we show that disruption of ABHD14B in mammalian cells results in increased lysine acetylation of cellular proteins, suggesting that these are in fact the substrates of ABHD14B. Towards mapping these, the initial identification of proteins that interact or tightly bind to ABHD14B, can be done by established immunoprecipitation (IP) experiments in conjunction with mass spectrometry-based proteomics using the antibody we report here (Figure 4). Additionally, established mass spectrometry-based proteomics platforms can also be leveraged to map the “global acetylome profile” in mammalian cells following ABHD14B disruption, to cover substrates that might be missed by IP experiments75–77. Fourth, publicly available databases49, and genome-wide association studies78–81, have found that deregulated (over)expression of ABHD14B is linked to progression of aggressive cancers in humans. Thus, it would be interesting to study the role of this enzyme in these cancers, and understand the metabolic pathways that are regulated by ABHD14B in this physiological context. Lastly, there are no specific inhibitors, or knockout mice described in literature for ABHD14B, and having such pharmacological tools and genetic animal models respectively, would greatly benefit researchers studying the biology of this metabolically exciting enzyme moving forward.
Supplementary Material
Acknowledgements
We thank the IISER Pune–Leica microscopy facility for providing access and technical expertise to microscopes needed for the immunofluorescence experiments, and the National Facility for Gene Function in Health and Disease, IISER Pune for providing the mice tissues for the immunoblotting experiments. Dr. Krishanpal Karmodiya, IISER Pune is thanked for providing the anti-acetyl-lysine antibody used in this study. Dr. Ullas Kolthur, TIFR Mumbai is thanked for providing the acetylated-lysine containing peptide and protein substrates used in this study.
Funding Sources.
This work was supported by a Wellcome Trust DBT India Alliance Intermediate Fellowship (IA/I/15/2/502058 to S.S.K.), a DST-FIST Infrastructure Development Grant (to IISER Pune Biology Department), and the Searle Scholars Program (to J.B.R.). The Council for Scientific and Industrial Research (CSIR) is thanked for a graduate student fellowship to A.R.
Footnotes
Accession Codes.
The Uniprot IDs for the human and mouse ABHD14B are Q96IU4 and Q8VCR7 respectively. The PDB code for the WT human ABHD14B is 1IMJ23.
Author Contributions.
A.R. and S.S.K. conceived the project, designed the experiments, and wrote the manuscript with inputs from all authors. A.R. and K.V. performed all the biochemical assays, cellular and immunoblotting experiments. J.M. and J.B.R. performed the computational analyses of the structure.
The authors declare no competing financial interests.
All animal studies reported in this manuscript have received formal approval from the Indian Institute of Science Education and Research, Pune – Institutional Animal Ethics Committee (IISER-P IAEC) constituted as per the guidelines provided by the Committee for the Purpose of Control and Supervision of Experiments in Animals (CPCSEA), Government of India.
References
- [1].Galperin MY, Koonin EV. 'Conserved hypothetical' proteins: prioritization of targets for experimental study. Nucleic Acids Res. 2004;32:5452–5463. doi: 10.1093/nar/gkh885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Brown SD, Babbitt PC. Inference of Functional Properties from Large-scale Analysis of Enzyme Superfamilies. Journal of Biological Chemistry. 2012;287:35–42. doi: 10.1074/jbc.R111.283408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation Error in Public Databases: Misannotation of Molecular Function in Enzyme Superfamilies. Plos Comput Biol. 2009;5 doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Saghatelian A, Cravatt BF. Assignment of protein function in the postgenomic era. Nat Chem Biol. 2005;1:130–142. doi: 10.1038/nchembio0805-130. [DOI] [PubMed] [Google Scholar]
- [5].Gerlt JA, Allen KN, Almo SC, Armstrong RN, Babbitt PC, Cronan JE, Dunaway-Mariano D, Imker HJ, Jacobson MP, Minor W, Poulter CD, et al. The Enzyme Function Initiative. Biochemistry. 2011;50:9950–9962. doi: 10.1021/bi201312u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, Whalen KL. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim Biophys Acta. 2015;1854:1019–1037. doi: 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schmelzer K, Fahy E, Subramaniam S, Dennis EA. The lipid maps initiative in lipidomics. Methods Enzymol. 2007;432:171–183. doi: 10.1016/S0076-6879(07)32007-7. [DOI] [PubMed] [Google Scholar]
- [8].Cotter D, Maer A, Guda C, Saunders B, Subramaniam S. LMPD: LIPID MAPS proteome database. Nucleic Acids Res. 2006;34:D507–510. doi: 10.1093/nar/gkj122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Saghatelian A, Cravatt BF. Global strategies to integrate the proteome and metabolome. Curr Opin Chem Biol. 2005;9:62–68. doi: 10.1016/j.cbpa.2004.12.004. [DOI] [PubMed] [Google Scholar]
- [10].Saghatelian A, Cravatt BF. Discovery metabolite profiling--forging functional connections between the proteome and metabolome. Life Sci. 2005;77:1759–1766. doi: 10.1016/j.lfs.2005.05.019. [DOI] [PubMed] [Google Scholar]
- [11].Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends Biochem Sci. 1998;23:347–352. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- [13].Long JZ, Cravatt BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev. 2011;111:6022–6063. doi: 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Simon GM, Cravatt BF. Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J Biol Chem. 2010;285:11051–11055. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Adam GC, Sorensen EJ, Cravatt BF. Chemical strategies for functional proteomics. Mol Cell Proteomics. 2002;1:781–790. doi: 10.1074/mcp.r200006-mcp200. [DOI] [PubMed] [Google Scholar]
- [16].Adam GC, Sorensen EJ, Cravatt BF. Proteomic profiling of mechanistically distinct enzyme classes using a common chemotype. Nat Biotechnol. 2002;20:805–809. doi: 10.1038/nbt714. [DOI] [PubMed] [Google Scholar]
- [17].Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- [18].Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR, 3rd, Jeffrey SS, Cravatt BF. A streamlined platform for high-content functional proteomics of primary human specimens. Nat Methods. 2005;2:691–697. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- [19].Li W, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J Am Chem Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- [20].Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- [21].Niphakis MJ, Cravatt BF. Enzyme inhibitor discovery by activity-based protein profiling. Annu Rev Biochem. 2014;83:341–377. doi: 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- [22].Bachovchin DA, Cravatt BF. The pharmacological landscape and therapeutic potential of the serine hydrolases. Nat Rev Drug Discov. 2012;11:52–68. doi: 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Padmanabhan B, Kuzuhara T, Adachi N, Horikoshi M. The crystal structure of CCG1/TAF(II)250-interacting factor B (CIB) J Biol Chem. 2004;279:9615–9624. doi: 10.1074/jbc.M312165200. [DOI] [PubMed] [Google Scholar]
- [24].Van Dyke MW. Lysine deacetylase (KDAC) regulatory pathways: an alternative approach to selective modulation. ChemMedChem. 2014;9:511–522. doi: 10.1002/cmdc.201300444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bheda P, Jing H, Wolberger C, Lin HN. The Substrate Specificity of Sirtuins. Annual Review of Biochemistry. 2016;85:405–429. doi: 10.1146/annurev-biochem-060815-014537. 85. [DOI] [PubMed] [Google Scholar]
- [26].Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Bio. 2012;31:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Guarente L. Sirtuins in aging and disease. Cold Spring Harb Symp Quant Biol. 2007;72:483–488. doi: 10.1101/sqb.2007.72.024. [DOI] [PubMed] [Google Scholar]
- [29].Seto E, Yoshida M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Csh Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics. 2012;4 doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Karmodiya K, Anamika K, Muley V, Pradhan SJ, Bhide Y, Galande S. Camello, a novel family of Histone Acetyltransferases that acetylate histone H4 and is essential for zebrafish development. Sci Rep-Uk. 2014;4 doi: 10.1038/srep06076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huynh K, Partch CL. Analysis of protein stability and ligand interactions by thermal shift assay. Curr Protoc Protein Sci. 2015;79 doi: 10.1002/0471140864.ps2809s79. 28 29 21-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kelkar DS, Ravikumar G, Mehendale N, Singh S, Joshi A, Sharma AK, Mhetre A, Rajendran A, Chakrapani H, Kamat SS. A chemical-genetic screen identifies ABHD12 as an oxidized-phosphatidylserine lipase. Nat Chem Biol. 2019;15:169–178. doi: 10.1038/s41589-018-0195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li QL, Zhang SH, Berthiaume JM, Simons B, Zhang GF. Novel approach in LC-MS/MS using MRM to generate a full profile of acyl-CoAs: discovery of acyl-dephospho-CoAs. Journal of Lipid Research. 2014;55:592–602. doi: 10.1194/jlr.D045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pathak D, Mehendale N, Singh S, Mallik R, Kamat SS. Lipidomics Suggests a New Role for Ceramide Synthase in Phagocytosis. ACS Chem Biol. 2018;13:2280–2287. doi: 10.1021/acschembio.8b00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Joshi A, Shaikh M, Singh S, Rajendran A, Mhetre A, Kamat SS. Biochemical characterization of the PHARC-associated serine hydrolase ABHD12 reveals its preference for very-long-chain lipids. Journal of Biological Chemistry. 2018;293:16953–16963. doi: 10.1074/jbc.RA118.005640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics. 2017;18:529. doi: 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Arena ET, Rueden CT, Hiner MC, Wang S, Yuan M, Eliceiri KW. Quantitating the cell: turning images into numbers with ImageJ. Wiley Interdiscip Rev Dev Biol. 2017;6 doi: 10.1002/wdev.260. [DOI] [PubMed] [Google Scholar]
- [39].Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev. 2015;82:518–529. doi: 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kamat SS, Camara K, Parsons WH, Chen DH, Dix MM, Bird TD, Howell AR, Cravatt BF. Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat Chem Biol. 2015;11:164–171. doi: 10.1038/nchembio.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stourac J, Vavra O, Kokkonen P, Filipovic J, Pinto G, Brezovsky J, Damborsky J, Bednar D. Caver Web 1.0: identification of tunnels and channels in proteins and analysis of ligand transport. Nucleic Acids Research. 2019;47:W414–W422. doi: 10.1093/nar/gkz378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vavra O, Filipovic J, Marques S, Plhak J, Bednar D, Brezovsky J, Matyska L, Damborsky J. CaverDock: a novel method for the analysis of the ligand transport processes based on iterative docking. Febs Open Bio. 2018;8:449–449. [Google Scholar]
- [43].Pravda L, Sehnal D, Tousek D, Navratilova V, Bazgier V, Berka K, Varekova RS, Koca J, Otyepka M. MOLEonline: a web-based tool for analyzing channels, tunnels and pores (2018 update) Nucleic Acids Research. 2018;46:W368–W373. doi: 10.1093/nar/gky309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Berka K, Hanak O, Sehnal D, Banas P, Navratilova V, Jaiswal D, Ionescu CM, Varekova RS, Koca J, Otyepka M. MOLEonline 2.0: interactive web-based analysis of biomacromolecular channels. Nucleic Acids Research. 2012;40:W222–W227. doi: 10.1093/nar/gks363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Research. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Research. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen JH, Liles K, Chun MJ, et al. Improvements to the APBS biomolecular solvation software suite. Protein Science. 2018;27:112–128. doi: 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P. Collaboration gets the most out of software. Elife. 2018;2 doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, Backstrom A, et al. A subcellular map of the human proteome. Science. 2017;356 doi: 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- [50].Wu C, Jin X, Tsueng G, Afrasiabi C, Su AI. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 2016;44:D313–316. doi: 10.1093/nar/gkv1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, 3rd, Su AI. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms, and specificities. Biochim Biophys Acta. 2009;1789:58–68. doi: 10.1016/j.bbagrm.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- [54].Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta. 2016;1864:1372–1401. doi: 10.1016/j.bbapap.2016.06.007. [DOI] [PubMed] [Google Scholar]
- [55].Marmorstein R, Zhou MM. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol. 2014;6:a018762. doi: 10.1101/cshperspect.a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Vidall G, Gershey EL, Allfrey VG. Chemical Studies of Histone Acetylation - Distribution of Epsilon-N-Acetyllysine in Calf Thymus Histones. Journal of Biological Chemistry. 1968;243:6361. [PubMed] [Google Scholar]
- [58].Wittenborn EC, Jost M, Wei YF, Stubbe J, Drennan CL. Structure of the Catalytic Domain of the Class I Polyhydroxybutyrate Synthase from Cupriavidus necator. Journal of Biological Chemistry. 2016;291:25264–25277. doi: 10.1074/jbc.M116.756833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Horikoshi M, Wang CK, Fujii H, Cromlish JA, Weil PA, Roeder RG. Cloning and structure of a yeast gene encoding a general transcription initiation factor TFIID that binds to the TATA box. Nature. 1989;341:299–303. doi: 10.1038/341299a0. [DOI] [PubMed] [Google Scholar]
- [60].Takada R, Nakatani Y, Hoffmann A, Kokubo T, Hasegawa S, Roeder RG, Horikoshi M. Identification of human TFIID components and direct interaction between a 250-kDa polypeptide and the TATA box-binding protein (TFIID tau) Proc Natl Acad Sci U S A. 1992;89:11809–11813. doi: 10.1073/pnas.89.24.11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dynlacht BD, Hoey T, Tjian R. Isolation of Coactivators Associated with the Tata-Binding Protein That Mediate Transcriptional Activation. Cell. 1991;66:563–576. doi: 10.1016/0092-8674(81)90019-2. [DOI] [PubMed] [Google Scholar]
- [62].Van Dyke MW, Roeder RG, Sawadogo M. Physical analysis of transcription preinitiation complex assembly on a class II gene promoter. Science. 1988;241:1335–1338. doi: 10.1126/science.3413495. [DOI] [PubMed] [Google Scholar]
- [63].Buratowski S, Hahn S, Guarente L, Sharp PA. Five intermediate complexes in transcription initiation by RNA polymerase II. Cell. 1989;56:549–561. doi: 10.1016/0092-8674(89)90578-3. [DOI] [PubMed] [Google Scholar]
- [64].Hahn S, Buratowski S, Sharp PA, Guarente L. Yeast TATA-binding protein TFIID binds to TATA elements with both consensus and nonconsensus DNA sequences. Proc Natl Acad Sci U S A. 1989;86:5718–5722. doi: 10.1073/pnas.86.15.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hahn S, Buratowski S, Sharp PA, Guarente L. Isolation of the gene encoding the yeast TATA binding protein TFIID: a gene identical to the SPT15 suppressor of Ty element insertions. Cell. 1989;58:1173–1181. doi: 10.1016/0092-8674(89)90515-1. [DOI] [PubMed] [Google Scholar]
- [66].Hai TW, Horikoshi M, Roeder RG, Green MR. Analysis of the role of the transcription factor ATF in the assembly of a functional preinitiation complex. Cell. 1988;54:1043–1051. doi: 10.1016/0092-8674(88)90119-5. [DOI] [PubMed] [Google Scholar]
- [67].Horikoshi M, Hai T, Lin YS, Green MR, Roeder RG. Transcription factor ATF interacts with the TATA factor to facilitate establishment of a preinitiation complex. Cell. 1988;54:1033–1042. doi: 10.1016/0092-8674(88)90118-3. [DOI] [PubMed] [Google Scholar]
- [68].Horikoshi M, Carey MF, Kakidani H, Roeder RG. Mechanism of action of a yeast activator: direct effect of GAL4 derivatives on mammalian TFIID-promoter interactions. Cell. 1988;54:665–669. doi: 10.1016/s0092-8674(88)80011-4. [DOI] [PubMed] [Google Scholar]
- [69].Hisatake K, Hasegawa S, Takada R, Nakatani Y, Horikoshi M, Roeder RG. The p250 subunit of native TATA box-binding factor TFIID is the cell-cycle regulatory protein CCG1. Nature. 1993;362:179–181. doi: 10.1038/362179a0. [DOI] [PubMed] [Google Scholar]
- [70].Ruppert S, Wang EH, Tjian R. Cloning and expression of human TAFII250: a TBP-associated factor implicated in cell-cycle regulation. Nature. 1993;362:175–179. doi: 10.1038/362175a0. [DOI] [PubMed] [Google Scholar]
- [71].Dikstein R, Ruppert S, Tjian R. TAFII250 is a bipartite protein kinase that phosphorylates the base transcription factor RAP74. Cell. 1996;84:781–790. doi: 10.1016/s0092-8674(00)81055-7. [DOI] [PubMed] [Google Scholar]
- [72].Pham AD, Sauer F. Ubiquitin-activating/conjugating activity of TAFII250, a mediator of activation of gene expression in Drosophila. Science. 2000;289:2357–2360. doi: 10.1126/science.289.5488.2357. [DOI] [PubMed] [Google Scholar]
- [73].Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- [74].Mizzen CA, Yang XJ, Kokubo T, Brownell JE, Bannister AJ, Owen-Hughes T, Workman J, Wang L, Berger SL, Kouzarides T, Nakatani Y. The TAF(II)250 subunit of TFIID has histone acetyltransferase activity. Cell. 1996;87:1261–1270. doi: 10.1016/s0092-8674(00)81821-8. [DOI] [PubMed] [Google Scholar]
- [75].Philp A, Rowland T, Perez-Schindler J, Schenk S. Understanding the acetylome: translating targeted proteomics into meaningful physiology. Am J Physiol Cell Physiol. 2014;307:C763–773. doi: 10.1152/ajpcell.00399.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Smith KT, Workman JL. Introducing the acetylome. Nat Biotechnol. 2009;27:917–919. doi: 10.1038/nbt1009-917. [DOI] [PubMed] [Google Scholar]
- [77].Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, Cravatt BF. Global profiling of lysine reactivity and ligandability in the human proteome. Nat Chem. 2017;9:1181–1190. doi: 10.1038/nchem.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Posorski N, Kaemmerer D, Ernst G, Grabowski P, Hoersch D, Hommann M, von Eggeling F. Localization of sporadic neuroendocrine tumors by gene expression analysis of their metastases. Clin Exp Metastas. 2011;28:637–647. doi: 10.1007/s10585-011-9397-5. [DOI] [PubMed] [Google Scholar]
- [79].Korkola JE, Houldsworth J, Chadalavada RSV, Olshen AB, Dobrzynski D, Reuter VE, Bosl GJ, Chaganti RSK. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66:820–827. doi: 10.1158/0008-5472.CAN-05-2445. [DOI] [PubMed] [Google Scholar]
- [80].Andersson A, Ritz C, Lindgren D, Eden P, Lassen C, Heldrup J, Olofsson T, Rade J, Fontes M, Porwit-MacDonald A, Behrendtz M, et al. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia. 2007;21:1198–1203. doi: 10.1038/sj.leu.2404688. [DOI] [PubMed] [Google Scholar]
- [81].Namani A, Cui QQ, Wu YH, Wang HY, Wang XJ, Tang XW. NRF2-regulated metabolic gene signature as a prognostic biomarker in non-small cell lung cancer. Oncotarget. 2017;8:69847–69862. doi: 10.18632/oncotarget.19349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.