

Abstract
Cystic fibrosis is a chronic multisystemic disease originating from functional alterations in CFTR (cystic fibrosis transmembrane conductance regulator) protein. To date, more than 300 pathogenic variants have been described in the literature. However, the diagnosis of CF, which was thought to become easier after the CFTR gene was identified, became more complicated due to the enormous amount of variations. In this study, we present a patient whose clinical findings were consistent with cystic fibrosis (CF) and showed a homozygous missense change that is not previously reported in the CFTR gene as pathogenic. In the next-generation sequencing analysis, homozygous c.4096A > T single-nucleotide exchange (I1366F [p.Ile1366Phe], missense) was shown in both alleles of the patient' CFTR gene. According to our database analysis, this variant has not yet been previously reported (VarSome, ClinVar, MutationTaster, Ensembl, dbSNP, PubMed). We do consider the change as pathogenic since the patient's findings were compatible with CF and the data analysis was in favor of pathogenicity. The most recent consensus report published in 2017 emphasized the importance of CFTR gene analysis, and this study emphasizes the difficulties of associating CFTR gene variations with a clinical picture and constitutes a new data on the genotype–phenotype correlation of CFTR variants. Also, considering the frequency of CF (according to World Health Organization data, every 1 out of 2,000–3,000 infants is born with CF in European Union countries and every 1 out of 3,500 in the United States) as well as the increasing rate of molecular studies performed on CF patients worldwide, reporting novel variation has an additional value.
Keywords: CFTR (cystic fibrosis transmembrane conductance regulator) , novel, pathogenic variant, cystic fibrosis, next-generation sequencing
Introduction
Cystic fibrosis (CF) is a chronic multisystemic disease originating from functional alterations in CFTR (cystic fibrosis transmembrane conductance regulator) protein. The CFTR protein is a chlorine channel and is located on the apical membranes of epithelial cells. Dysfunction in the lungs causes dehydration of surface fluid, mucous stasis, and eventually airway inflammation complicated by recurrent infections, resulting in progressive airway obstruction. Course of the disease is degenerative; interaction of CFTR with other ion channels and cellular pathways lead to inflammation as well as mucous obstruction due to impaired mucociliary clearance, bronchiectasis, recurrent infections, and pH changes (due to bicarbonate secretion imbalance), all contributing to the chronic degenerative process. Estimated incidence of CF in our country is 1:10,000. The majority of patients with CF present with a classical clinical picture (malabsorption and respiratory findings). However, 5% of the patients have no descriptive clinical findings and/or sweat test results. CFTR gene analysis is of great importance both in the diagnosis of the latter group and in the definitive diagnosis of patients with a classic presentation of the disease. CFTR gene is located on the long arm of chromosome 7 (7q31.2) and spans an area of ∼250 kb. A total of 28 exons have been identified. Since this gene has been described, more than 2,000 CFTR variants and more than 300 pathogenic variants have been reported in CFTR gene databases 1 2 However, the diagnosis of CF, which was thought to become easier after CFTR gene was identified, became more complicated due to the enormous amount of variations. 3 In this report, we present a patient whose clinical findings were consistent with CF and showed a homozygous missense change that is not previously reported in the CFTR gene as pathogenic, aiming to further enhance our knowledge about CFTR variants and their genotype–phenotype relationship.
Case Report
A 3.5-month-old male child presented with initial symptoms of inability to feed, difficulty in breathing during eating, and rapid breathing at 3.5 months. The patient is now 1 year old. During his follow-up, he was cachectic, had dyspnea attacks, had productive cough, and was complicated by recurrent respiratory infections, and diagnosed with Serratia Marcescens pneumonia twice and Staphylococcus Hominis pneumonia once on microbiological analysis from sputum samples. During those respiratory tract infections, blood culture revealed bacteremia of those agents too. He had fatty stool and intermittent diarrhea, and a stool testing for steatorrhea was positive. His weight was 3,500 g (below 3rd percentile [<3p]), height was 60 cm (<3p), and head circumference was 36 cm (<3p) at 4 months of age. There were no features in his prenatal history, and no relative was detected in his pedigree with symptoms and findings alike. A next-generation sequence (NGS) analysis of his CFTR gene was performed.
Materials and Methods
A whole blood sample was taken from the patient. After the DNA was isolated, DNA sequence analysis was performed with NextFlex Cystic Fibrosis Amplicon Panel (PerkinElmer Inc.), which targets and sequences 28 exons of the CFTR locus, containing primer pairs and reagents needed to amplify these coding regions of the CFTR gene with DNA isolated from fresh or frozen material and prepare libraries for Illumina sequencing that is able to sequence all 28 exons of the gene simultaneously on Illumina MiniSeq Platform.
Results
In the NGS analysis, homozygous c.4096A > T single nucleotide exchange (I1366F [p.Ile1366Phe], missense) was shown in both alleles of the patient's CFTR gene ( Figs. 1 and 2 ). According to our database analysis, this variant has not yet been previously reported (VarSome [ https://varsome.com/variant/hg19/rs770345073 ], 4 ClinVar, MutationTaster, Ensembl, dbSNP, PubMed). The DANN score (deleterious annotation of genetic variants using neural networks) was calculated as 0.9882 through in silico data analysis from the VarSome database. 5 Pathogenic protein product of the patient, UniProt protein CFTR_HUMAN domain “ABC transporter 2,” has 76 classified variants, of which 68 are pathogenic (88.2%, greater than 66.7%); this is a result that strengthens the possibility that the variant is pathogenic. Computational analysis from DANN, GERP (Genomic Evolutionary Rate Profiling), dbNSFP (Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs), FATHMM (Functional Analysis through Hidden Markov Models), LRT (likelihood ratio test), MetaLR (likelihood ratio meta-analysis), MetaSVM (meta-analytic support vector machine), MutationAssessor, MutationTaster, and PROVEAN databases showed 0 estimated benign results versus 9 estimated pathogenic results. There is no validated genotype–phenotype association information on the dbSNP database, and reference sequence (rs770345073) seems to be validated only by frequency. We do consider the change as pathogenic since the patient's findings were compatible with CF and the data analysis was in favor of pathogenicity.
Fig. 1.
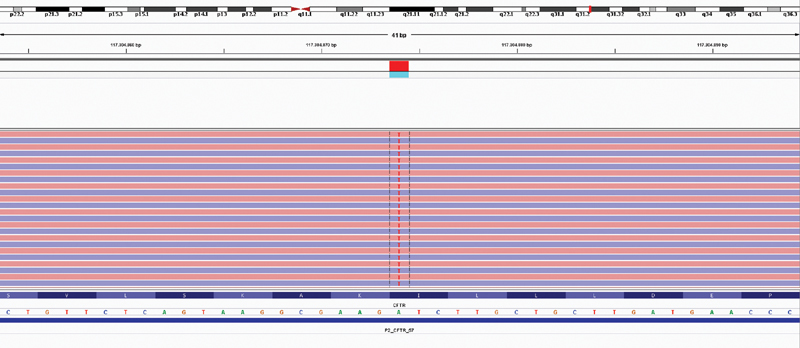
Results of CFTR sequence analysis (Illumina MiniSeq, NextFlex Cystic Fibrosis Amplicon Panel) of the patient. The A > T change present in the patient (red “T” at every line corresponds to one strand each, meaning this change is present in every strand read by the platform) is shown.
Fig. 2.
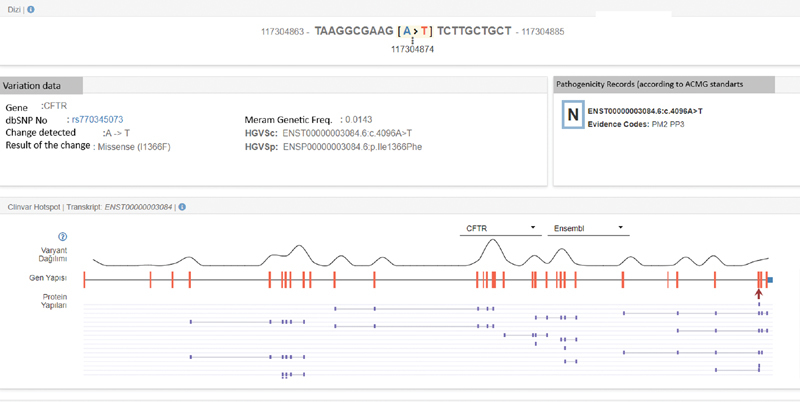
Results of CFTR sequence analysis (Illumina MiniSeq, NextFlex Cystic Fibrosis Amplicon Panel) of the patient. Variant's position within the exon, describing the ENST number and dbSNP code is shown.
Discussion
CFTR gene analysis is becoming increasingly important, and defining the absolute relationship between genotype and phenotype is a complex matter. There are only a few articles on the detection of CFTR gene by NGS. 6 Difficulties in assessing genotype–phenotype relationship make it important to report new changes in clinically compatible patients and their accessibility to genetic counselors. 7 Moreover, large-scale population genetics studies in our country are insufficient, and this makes reporting the previously unreported variations more important.
Several consensus reports have been published for the diagnosis of CF since 1998. 8 9 10 11 The most recent consensus report published in 2017 emphasized the importance of CFTR gene analysis. 12 Since its approval by the U.S. Food and Drug Administration in 2013, NGS has been used to understand bacterial and nonhuman eukaryotic genomes to analyze transcriptome signals of the cells, determine the epigenetic chromosomal patterns, and analyze gene variations, giving successful results. 13 The percentage of diagnoses and the rates of new variant detection are remarkably high in existing studies. 14 15 16 17 18 In the comprehensive study of Grosu et al with two different CFTR kits, the success rate of the tests was found to be between 99.7 and 100.0% compared with Sanger sequencing, which was accepted as the reference method. 14
Apparently, this variant (c.4096A > T, I1366F) was mentioned in two previous studies. In the study of Sheth et al, 19 which included primary sclerosing cholangitis (PSC), primary biliary cirrhosis, and inflammatory bowel disease patients, I1366F change was detected in one patient with PSC. No data were asserted about whether this particular change is disease-causing for PSC or CF. In another study, by Gallegos-Orozco et al, 20 aiming to further describe the relationship between CFTR gene and PSC, one PSC patient was found to be a carrier of I1366F variant. Authors addressed the change as a variant with unknown functional and phenotypic effect.
The clinical data of our case are overlapping with the characteristics of CF. It is well known that CF patients demonstrate defective cellular immunity against gram-negative bacteria. There are several studies linking Serratia Marcescens to CF either by demonstrating diminished lymphocyte response 21 or by observing the increased frequency of Serratia pneumonia in CF patients. 22 23 Increased Serratia Marcescens bacteremia was also demonstrated in CF patients. 24 Adding to that, sequence variation that we have seen in our NGS-based assay was found in both alleles, was in a homozygous state, was missense, and had a very high DANN score, and proteomic/genomic data were correlated with pathogenicity since it creates significant changes in CFTR protein structure. Data from assay platform show that this is the first detection of this variant with this assay; out of 5,383 patients studied with this assay in different centers, this particular variant was never detected, both heterogeneously and homogenously. Therefore, we report this change as a novel pathogenic variant. The main limitation of this report is that this variant is shown in only one patient; hence, it is quite hard to infer a definitive result. Though it contributes to the path that leads to more concise practice, clinical data on this variant needs further accumulation.
This study emphasizes the difficulties of associating CFTR gene variations with a clinical picture and constitutes new data about the genotype–phenotype correlation of CFTR variants. Also, considering the frequency of CF (according to World Health Organization data, every 1 out of 2000–3000 infants is born with CF in European Union countries and every 1 out of 3,500 in the United States 25 ) as well as the increasing rate of molecular studies performed on CF patients worldwide, reporting novel variation has an additional value.
Footnotes
Conflict of Interest None declared.
References
- 1.Clinical and Functional Translation of CFTR.https://cftr2.org/. Accessed September 29, 2019
- 2.Cystic fibrosis mutation database.Hospital for Sick Children, Toronto www.genet.sickkids.on.ca. Accessed September 29, 2018
- 3.Poulou M, Fylaktou I, Fotoulaki M, Kanavakis E, Tzetis M. Cystic fibrosis genetic counseling difficulties due to the identification of novel mutations in the CFTR gene. J Cyst Fibros. 2012;11(04):344–348. doi: 10.1016/j.jcf.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Kopanos C, Tsiolkas V, Kouris A et al. VarSome: the human genomic variant search engine. Oxford Bioinformatics. 2019;35(11):1978–1980. doi: 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31(05):761–763. doi: 10.1093/bioinformatics/btu703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes E E, Stevens C F, Saavedra-Matiz C A et al. Clinical sensitivity of cystic fibrosis mutation panels in a diverse population. Hum Mutat. 2016;37(02):201–208. doi: 10.1002/humu.22927. [DOI] [PubMed] [Google Scholar]
- 7.Terlizzi V, Castaldo G, Salvatore D et al. Genotype-phenotype correlation and functional studies in patients with cystic fibrosis bearing CFTR complex alleles. J Med Genet. 2017;54(04):224–235. doi: 10.1136/jmedgenet-2016-103985. [DOI] [PubMed] [Google Scholar]
- 8.Rosenstein B J, Cutting G R; Cystic Fibrosis Foundation Consensus Panel.The diagnosis of cystic fibrosis: a consensus statement J Pediatr 199813204589–595. [DOI] [PubMed] [Google Scholar]
- 9.De Boeck K, Wilschanski M, Castellani C et al. Cystic fibrosis: terminology and diagnostic algorithms. Thorax. 2006;61(07):627–635. doi: 10.1136/thx.2005.043539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Boeck K, Derichs N, Fajac I et al. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros. 2011;10 02:S53–S66. doi: 10.1016/S1569-1993(11)60009-X. [DOI] [PubMed] [Google Scholar]
- 11.Smyth A R, Bell S C, Bojcin S et al. European cystic fibrosis society standards of care: best practice guidelines. J Cyst Fibros. 2014;13 01:S23–S42. doi: 10.1016/j.jcf.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Farrell P M, White T B, Ren C Let al. Diagnosis of cystic fibrosis: consensus guidelines from the cystic fibrosis foundation J Pediatr 2017181SS4–S15., 15.e1 [DOI] [PubMed] [Google Scholar]
- 13.Bahassi M, Stambrook P J. Next-generation sequencing technologies: breaking the sound barrier of human genetics. Mutagenesis. 2014;29(05):303–310. doi: 10.1093/mutage/geu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grosu D S, Hague L, Chelliserry M et al. Clinical investigational studies for validation of a next-generation sequencing in vitro diagnostic device for cystic fibrosis testing. Expert Rev Mol Diagn. 2014;14(05):605–622. doi: 10.1586/14737159.2014.916618. [DOI] [PubMed] [Google Scholar]
- 15.Baker M W, Atkins A E, Cordovado S K, Hendrix M, Earley M C, Farrell P M. Improving newborn screening for cystic fibrosis using next-generation sequencing technology: a technical feasibility study. Genet Med. 2016;18(03):231–238. doi: 10.1038/gim.2014.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trujillano D, Ramos M D, González J et al. Next generation diagnostics of cystic fibrosis and CFTR-related disorders by targeted multiplex high-coverage resequencing of CFTR. J Med Genet. 2013;50(07):455–462. doi: 10.1136/jmedgenet-2013-101602. [DOI] [PubMed] [Google Scholar]
- 17.Loukas Y L, Thodi G, Molou E, Georgiou V, Dotsikas Y, Schulpis K H. Clinical diagnostic Next-Generation sequencing: the case of CFTR carrier screening. Scand J Clin Lab Invest. 2015;75(05):374–381. doi: 10.3109/00365513.2015.1031689. [DOI] [PubMed] [Google Scholar]
- 18.Essawi O, Farraj M, De Leeneer K et al. Next generation sequencing to determine the cystic fibrosis mutation spectrum in Palestinian population. Dis Markers. 2015;2015:458653. doi: 10.1155/2015/458653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheth S, Shea J C, Bishop M D et al. Increased prevalence of CFTR mutations and variants and decreased chloride secretion in primary sclerosing cholangitis. Hum Genet. 2003;113(03):286–292. doi: 10.1007/s00439-003-0963-z. [DOI] [PubMed] [Google Scholar]
- 20.Gallegos-Orozco J F, Wang N et al. Lack of association of common cystic fibrosis transmembrane conductance regulator gene mutations with primary sclerosing cholangitis. Am J Gastroenterol. 2005;100(04):874–878. doi: 10.1111/j.1572-0241.2005.41072.x. [DOI] [PubMed] [Google Scholar]
- 21.Sorensen R U, Stern R C, Chase P, Polmar S H. Defective cellular immunity to gram-negative bacteria in cystic fibrosis patients. Infect Immun. 1979;23(02):398–402. doi: 10.1128/iai.23.2.398-402.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrer Marcelles A, Bellver Moreira P, Cobos Barroso N, Liñán Cortés S, Codina Grau G, Fernández Pérez F. Cystic fibrosis: a microbiological study over an 8-year period [in Spanish] Arch Bronconeumol. 1995;31(10):494–500. [PubMed] [Google Scholar]
- 23.Blau H, Linnane B, Carzino R et al. Induced sputum compared to bronchoalveolar lavage in young, non-expectorating cystic fibrosis children. J Cyst Fibros. 2014;13(01):106–110. doi: 10.1016/j.jcf.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 24.McCarthy M M, Rourk M H, Spock A. Bacteremia in patients with cystic fibrosis. Clin Pediatr (Phila) 1980;19(11):746–748. doi: 10.1177/000992288001901106. [DOI] [PubMed] [Google Scholar]
- 25.World Health Organization.https://www.who.int/genomics/public/geneticdiseases/en/index2.html#CF. Accessed September 20, 2018