

Abstract
Background
Recent preclinical data suggest that neuroendocrine (NE) subtype of small cell lung cancer (SCLC) has strong therapeutic relevance. NE high tumors are associated with immune desert and NE low tumors are considered to have an immune oasis phenotype. Our aim was to investigate the NE phenotypes of surgically resected SCLC tumors according to inter-tumor heterogeneity.
Methods
Expression analysis for 2,560 genes was performed in 32 surgically resected SCLC patients’ primary tumors and corresponding lymph node (LN) metastases. To analyze tumor heterogeneity, we examined the differences in the gene expression of primary tumors versus LN metastases. We performed cluster analysis and heat map to divide patients into NE high and low subtypes by using the top NE-associated genes described in preclinical studies.
Results
We found 6% (n=154) genes with significant differences and only 13.1% (n=336) of all genes in the panel had a strong correlation between the primary tumor and LN metastases. Cluster analysis clearly distinguished SCLC NE high versus low subtypes both in primary tumor (20 vs. 12, respectively) and LNs (23 vs. 9, respectively). As for inter-tumor heterogeneity, in case of five patients, a change in the NE pattern was observed. Specifically, we found significant downregulation of the NE-associated genes CAV1 (P=0.004), CAV2 (P=0.029) and ANXA3 (P=0.035) in their LN metastases compared to their primary tumor.
Conclusions
Our data confirm the results of preclinical studies and clearly distinguish NE low and high differentiation clusters in SCLC. Moreover, they highlight the gene expression discordance between primary tumors and corresponding LN metastases suggesting that the NE pattern of metastatic LNs might not reflect that of the primary tumor. Altogether, by shedding light on the diversity of SCLC, the current study might help to improve patient selection and treatment in this devastating disease.
Keywords
Small cell lung cancer (SCLC); neuroendocrine tumor; lymph node metastasis; tumor heterogeneity; RNA sequencing
Introduction
The pathological diagnosis of small cell lung cancer (SCLC) is based on simple hematoxylin and eosin (H&E) staining with no significant diagnostic advancements in this recalcitrant cancer. As for SCLC therapy, atezolizumab in combination with chemotherapy was recently shown to moderately increase response rate and survival compared to chemotherapy alone (1). However, lack of clinically useful biomarkers and knowledge on inter-tumor and intra-tumor molecular heterogeneity of SCLC tumors might be a key to the long term failure of therapeutic efficacy.
According to recent preclinical studies, the way we consider SCLC has been significantly changed. SCLC cannot be considered as a single disease entity, it is rather a continuous spectrum of tumor cells with different neuroendocrine (NE) marker expressions. SCLC can be classified into NE high and NE low subtypes that have major differences in morphology, growth properties, genetic alterations, and immunogenicity including numerous intracellular signaling pathways (2,3).
Importantly, very recent data suggest that SCLC can present as either an “immune desert” or an “immune oasis” tumor. Immune desert SCLCs are associated with NE high phenotype and characterized by sheets of tumor cells, little stroma, low numbers of infiltrating immune cells and low or absent programmed death receptor ligand-1 (PD-L1) expression (2). In contrast, in the NE low group, the predominant phenotype is the immune oasis, which is characterized by broad bands of fibrosis and numerous inflammatory cells in the fibrous strands. Therefore, NE low SCLC patients are more likely to respond to immunotherapy and targeted therapies as well (2,4). Recent studies also suggest that molecular heterogeneity and changes in NE pattern and in key relevant pathways might help to identify new targets and therapies (2,4).
In contrast to lung adenocarcinoma, a tumor subtype where significant therapeutic advancements have been made over the past fifteen years by sub-segmenting the disease into different targetable active kinase mutation associated subtypes, SCLC has mostly loss of function mutations that are more challenging to target (5,6). Moreover, the initiating molecular events in SCLC are probably loss of the tumor suppressors TP53 and RB1, disruption of signaling networks such as Notch signaling, and also amplification of MYC family genes (7-12). However, the molecular phenotypes and heterogeneity between SCLC cell lines does not apply to those mutations only, but also may be reflected in other types of genetic alterations (2). In addition, a certain level of heterogeneity in terms of somatic mutations between primary and metastatic tumors was also observed, although the difference was minimal among other factors partly due to the short time interval between tumor formation and development of nodal metastases (13). Moreover, because surgical resection in SCLC is rarely performed, little is known about the heterogeneity between LN metastases vs. primary tumors in SCLC.
Exploring the gene expression profile of matched primary and LN metastatic SCLC tumors might provide unique insights into the complexity of this aggressive type of cancer and might also help in the development of new therapeutic approaches. Herein, the aim of our resected cross-sectional study was to confirm recently published preclinical data in the clinical setting. To this end, we investigated the differences in key relevant pathways between primary and LN metastatic SCLC specimens using comparative gene expression assays that might identify new potential pathways with therapeutic significance.
Methods
Ethics statement
The present study was directed in accordance with the guidelines of the Helsinki Declaration of the World Medical Association. The national level ethics committee (Hungarian Scientific and Research Ethics Committee of the Medical Research Council, ETT-TUKEB-7214-1/2016/EKU) approved the study. The need for individual informed consent for this retrospective study was waived. After clinical information was collected, patient identifiers were removed, and subsequently patients cannot be identified either directly or indirectly.
Study population
A total of 32 metastatic histologically confirmed SCLC patients with available tumor tissue specimen were included in our study, who underwent surgical resection between 1978 and 2013 at the National Koranyi Institute of Pulmonology. Formalin-fixed, paraffin-embedded (FFPE) tissue samples according to primary and LN metastatic lesions were obtained at the time of lung resection surgery. Clinicopathological data included gender, age at the time of diagnosis, smoking history, operation type, overall survival (OS) and disease-free survival (DFS). Pathological TNM stage according to the Union for International Cancer Control (7th edition) was recorded (14). OS was calculated from the time of surgery until death, or last available follow-up, while DFS until the first documentation of recurrence. Date of the last follow-up included in this analysis was August 2017.
Expression analysis
FFPE tissue samples were macrodissected and sent for molecular analysis. RNA expression analysis of 2,560 cancer-related genes was performed by HTG Molecular Diagnostics, Inc. (“HTG”) using the HTG EdgeSeq Oncology Biomarker (OB) Panel. HTG EdgeSeq is a targeted RNA expression assay that is generated via nuclease protection and consists of hybridization of target RNA to a DNA probe, followed by treatment with a single-strand nuclease. The assay was validated using negative and positive process controls. All samples were run as singletons. Standardization and normalization of the results were performed as described earlier (15-18).
Treatment
Patients who underwent tumor resection including segmentectomy, lobectomy, and pulmonectomy were treated according to the contemporary guidelines with postoperative systemic chemotherapy with a platinum-etoposide doublet regimen or with a combination of cyclophosphamide, epirubicin, and vincristine (CEV).
Data pre-processing and cluster analysis
Both data pre-processing and cluster analysis was performed with ClustVis web tool, which uses several R packages internally, including ggplot2 for PCA plot and pheatmap (R package version 0.7.7) for plotting heat map (19). For data pre-processing, unit variance scaling method was used dividing the values by standard deviation. To divide SCLC tumor samples into NE subgroups first we selected the top RNA genes (n=25) associated with NE differentiation according to Zhang et al. and Gazdar et al. by comparing their list of genes with our gene panel (2,3). Then an agglomerative hierarchical cluster analysis was performed to identify tumors according to NE high vs. NE low subgroup (20).
Statistical methods and pathway analysis
Results of targeted RNA sequencing were grouped according to the place of origin and NE differentiation of the sample and statistical analyses were performed by Student’s t-test and Mann-Whitney U test. Two-sided p-values less than 0.05 were considered statistically significant. The value of linear correlation coefficient (r) varies from −1 to 1 both values inclusive. Non-correlation (−0.3≤r≤0.3), moderate positive linear correlation (0.3<r≤0.7), strong positive linear correlation (0.7<r≤1). To measure effect size and consequently indicate the standardized difference between the two groups Cohen’s d was calculated, which is an estimate of the difference between two means expressed in standard deviation units. Accordingly, Cohen’s d is the ratio of the difference between two means divided by the standard deviation (21). Small effect size (0.2≤d<0.5), medium effect size (0.5≤d<0.8), large effect size (0.8≤d) (21). All statistical analyses were performed using the PASW Statistics 23.0 package (SPSS Inc., Chicago, IL, USA). Patient survival data was examined by univariate survival analysis via Kaplan-Meier approach. Gene pathways were analyzed with ToppGene Suite and ConsensusPathDB which had the following built-in source databases: Reactome, KEGG BioSystem, HumanCyc, PID, IntAct and BioGRID. Genes in the SCLC SuperPath were identified using the PathCards integrated database (22). PathCard provides information on one SuperPath which represents more human pathways relevant in SCLC. Our gene expression data was compared with other available online databases using CBioPortal for Cancer Genomics as previously described (23,24).
Results
Clinicopathological characteristics and gene expression
Major clinicopathological data are shown in Table S1. A total of 32 patients were included in this study: 22 males and 10 females (age range, 34–78 years; median, 58 years). Median OS was 20.7 months and median DFS was 14.9 months, respectively.
Table S1. Major clinicopathological data of study population.
| Patient ID | OS (months) | DFS (months) | Gender | Smoking history | Age |
|---|---|---|---|---|---|
| 3 | 37.6 | 37.6 | Female | Smoker | 62 |
| 6 | 19.4 | 11 | Female | Ex-smoker | 57 |
| 12 | 30 | 15.1 | Female | Smoker | 64 |
| 21 | N/A | N/A | Female | N/A | 58 |
| 26 | 61.7 | 61.7 | Male | Smoker | 62 |
| 28 | 96.2 | 96.2 | Female | Smoker | 40 |
| 29 | 18.6 | 18.6 | Male | N/A | 55 |
| 33 | 29.5 | 29.5 | Male | Smoker | 63 |
| 36 | 4.9 | 4.9 | Female | N/A | 62 |
| 39 | 29.1 | 11.2 | Male | N/A | 58 |
| 41 | 118.5 | 18.4 | Male | N/A | 50 |
| 104 | N/A | N/A | Male | N/A | 68 |
| 126 | N/A | N/A | Male | N/A | 60 |
| 132 | 23 | N/A | Male | N/A | 66 |
| 134 | 9.3 | 1.7 | Male | Smoker | 59 |
| 148 | 5.2 | 5.2 | Female | Smoker | 48 |
| 171 | N/A | N/A | Male | N/A | 43 |
| 188 | N/A | N/A | Male | N/A | 67 |
| 190 | N/A | N/A | Male | N/A | 55 |
| 204 | N/A | N/A | Male | N/A | 59 |
| 209 | N/A | N/A | Female | N/A | 34 |
| 210 | 3.9 | 3.9 | Male | N/A | 45 |
| 216 | N/A | N/A | Male | N/A | 52 |
| 268 | 17.4 | N/A | Male | N/A | 41 |
| 270 | 16.6 | 16.6 | Male | Ex-smoker | N/A |
| 271 | 28.3 | N/A | Male | N/A | 46 |
| 332 | N/A | N/A | Female | N/A | 54 |
| 346 | 32.8 | 32.8 | Male | Smoker | 57 |
| 352 | N/A | N/A | Male | N/A | 63 |
| 354 | 12.5 | 12.5 | Male | Smoker | 70 |
| 355 | 14.7 | 14.7 | Female | N/A | 78 |
| 363 | 20.7 | 12.9 | Male | N/A | 48 |
OS, overall survival; DFS, disease-free survival.
Concerning the genes from the SCLC SuperPath (n=77), a significantly higher expression of apoptosis regulator gene BCL2 (P=0.047; 95% CI of the difference, 0.007 to 1.222) and a significantly lower expression of Laminin Subunit Gamma 3 gene (LAMC3); (P=0.044; 95% CI of the difference, −1.42 to −0.02) was found in the primary tumor in male vs. female patients.
Heterogeneity in gene expression between primary tumor versus LN metastasis
The expression data of 2,560 cancer-related genes were used to compare primary lesions and LN metastases. Volcano plot shown in Figure 1 represents the gene expression heterogeneity between primary tumors and LN metastases.
Figure 1.

Volcano plot displaying discordance in RNA gene expression between primary tumor and lymph node (LN) metastases. The y-axis corresponds to the significance level [−log10(P value)], while the x-axis shows the log2 fold-change (FC) value of relative gene expression. RNA genes above the red line showed significant expressional differences (P<0.05). Positive fold-change (log2 FC > 0) indicates upregulated genes in LN metastases compared to the primary tumor; negative fold-change (log2 FC < 0) indicates downregulated genes in LN. Red dots indicate genes included in the SCLC SuperPath and a significant difference between primary tumor and lymph node (LN) metastases in RNA gene expression was observed (BAK1, IKBKG, PIK3CD, and LAMC3). Blue lines indicate log2 FC −0.5 and 0.5.
A significant difference was found in the gene expression of 154 genes, including four already reported relevant genes in the SCLC SuperPath (Figure 1).
According to KEGG BioSystem database, in our study the highest proportion of genes with significant inter-tumor heterogeneity between the primary and LN metastatic lesions was observed in the cytokine-cytokine receptor interaction pathway (Figure 2A), that has a role mostly in inflammatory host defenses, cell growth, differentiation, cell death, angiogenesis, development and repair processes aimed at the restoration of homeostasis (n=23). Based on Cohen’s d the effect size in the relative gene expression of these 154 genes between the primary tumor and LN metastases was considered as medium in case of 139 (d≥0.5) and large in case of 15 genes (d≥0.8). Furthermore, only 13.1% (n=336) of all genes in the entire panel had strong correlation (r value >0.7) between the primary tumor and the LNs. Transcription factors had higher percentage of correlation compared to genes in importance in extracellular and cell adhesion receptors and signaling (Figure 2B).
Figure 2.

Inter-tumor heterogeneity between the primary and lymph node (LN) metastatic lesions in RNA gene expression. (A) Pie chart summarizing intracellular pathways according to KEGG BioSystem with significant inter-tumor differences in RNA gene expression detected (n=154). Highest proportion of genes belonged to the cytokine-cytokine receptor interaction pathway (11.1%), that are associated with crucial aspects of inflammation, tumor immunology and differentiation. (B) Strong correlation (0.7≤r<1) and non-correlation (−0.3≤r<0.3) in relative RNA gene expression according to the main molecular cellular components (encoding products). Asterisk, significant (P<0.05) differences in relative gene expression (genes in importance in transcription factors, receptors and signaling, extracellular and cell adhesion).
The top 25 genes with significant difference in gene expression are shown in Table 1. The majority of these top genes was downregulated (n=20) in LN metastases and have a wide range of functions including proliferation, growth, survival, vascular development and angiogenesis but also water homeostasis and protein hydrolyzation. Meanwhile, upregulated top genes (n=5) have a major role in cell adhesion, lymphoid tissue development and inflammatory response.
Table 1. The list of the top 25 genes with expressional differences in primary tumor versus lymph node (LN) metastasis.
| Symbol | Gene name | Mean expression | P value | Cohen’s d | 95% CI of the difference | |
|---|---|---|---|---|---|---|
| Primary tumor | LN metastasis | |||||
| AQP4 | Aquaporin 4 | 7.29 | 5.53 | <0.0001 | 1.106 | 0.96 to 2.54 |
| CFTR | Cystic fibrosis transmembrane conductance regulator | 6.08 | 3.95 | <0.0001 | 1.402 | 1.37 to 2.86 |
| COL6A6 | Collagen type VI alpha 6 chain | 6.26 | 4.34 | <0.0001 | 1.031 | 0.98 to 2.84 |
| CYP2B6 | Cytochrome P450 family 2 subfamily B member 6 | 7.27 | 6.12 | <0.0001 | 1.001 | 0.57 to 1.72 |
| FIGF | C-Fos Induced Growth Factor | 4.45 | 1.67 | <0.0001 | 1.279 | 1.69 to 3.86 |
| PGC | Progastricsin | 5.13 | 1.73 | <0.0001 | 1.367 | 2.15 to 4.62 |
| ROS1 | ROS proto-oncogene 1, receptor tyrosine kinase | 6.48 | 2.94 | <0.0001 | 1.563 | 2.4 to 4.66 |
| TMPRSS2 | Transmembrane serine protease 2 | 6.50 | 4.10 | <0.0001 | 1.195 | 1.39 to 3.40 |
| ZNF385B | Zinc finger protein 385B | 7.28 | 6.15 | <0.0001 | 0.932 | 0.52 to 1.73 |
| ANGPT1 | Angiopoietin 1 | 7.92 | 6.67 | 0.001 | 0.910 | 0.56 to 1.92 |
| IGFBP7 | Insulin like growth factor binding protein 7 | 10.75 | 11.38 | 0.001 | 0.884 | −0.99 to −0.27 |
| RSPO2 | R-spondin 2 | 5.34 | 3.67 | 0.001 | 0.899 | 0.73 to 2.59 |
| AGER | Advanced glycosylation end-product specific receptor | 8.01 | 6.96 | 0.002 | 0.807 | 0.40 to 1.70 |
| GML | Glycosylphosphatidylinositol anchored molecule like | 2.05 | 0.59 | 0.002 | 0.807 | 0.55 to 2.36 |
| *LAMC3 | Laminin subunit gamma 3 | 6.56 | 5.77 | 0.002 | 0.815 | 0.30 to 1.27 |
| CEACAM3 | Carcinoembryonic antigen related cell adhesion molecule 3 | 6.35 | 5.48 | 0.003 | 0.785 | 0.31 to 1.41 |
| LCN2 | Lipocalin 2 | 5.84 | 3.60 | 0.003 | 0.782 | 0.80 to 3.66 |
| LTB | Lymphotoxin beta | 7.45 | 8.45 | 0.003 | 0.760 | −1.65 to −0.34 |
| POU5F1 | POU class 5 homeobox 1 | 8.14 | 7.40 | 0.003 | 0.786 | 0.27 to 1.22 |
| CAV1 | Caveolin 1 | 8.83 | 8.08 | 0.004 | 0.738 | 0.24 to 1.25 |
| CCL21 | C-C motif chemokine ligand 21 | 8.09 | 9.48 | 0.004 | 0.753 | −2.32 to −0.47 |
| CCR7 | C-C motif chemokine receptor 7 | 5.66 | 6.67 | 0.004 | 0.738 | −1.69 to −0.32 |
| FCER2 | Fc fragment of IgE receptor II | 3.68 | 5.02 | 0.004 | 0.754 | −2.21 to −0.45 |
| CCL8 | C-C motif chemokine ligand 8 | 5.72 | 4.66 | 0.005 | 0.726 | 0.32 to 1.77 |
| MUC1 | Mucin 1, cell surface associated | 10.38 | 9.49 | 0.005 | 0.730 | 0.28 to 1.50 |
*, gene belonging to SCLC SuperPath. The majority of genes (n=20) was downregulated in LN metastases and have a wide range of functions including proliferation, growth, survival, vascular development and angiogenesis but also water homeostasis and protein hydrolyzation. Upregulated genes (n=5) have a major role in cell adhesion, lymphoid tissue development and inflammatory response.
NE pattern of primary tumor versus LN metastasis
In our gene panel, we identified NE high (n=6) and NE low (n=20) genes among the top genes previously reported (25 and 25, respectively) as shown in Figure 3. The NE associated genes had a wide range of functions including substrate attachment, cell migration, invasion and metastasis, inward rectifier potassium channel inhibitor activity, phospholipase A2 inhibitor activity, G protein signaling and epithelial cell differentiation. Gene expression heat map including all primary and LN metastatic tumor samples according to NE associated genes are shown in Figure 3. Based on these genes, hierarchical cluster analysis clearly identified SCLC NE subtypes according to primary tumors (NE high vs. low, 20 vs. 12, respectively) and LNs (NE high vs. low, 23 vs. 9, respectively). Moreover, based on previously reported cell line gene expression, in our study NE associated key genes REST and MYC expression was significantly (P<0.001) lower while DLL3 was significantly higher (P<0.001) in NE high subtypes. According to the tumor suppressor genes, relative expression of TP53 was significantly higher in the NE high (vs. NE low, P=0.009; 95% CI of the difference, 0.321 to 2.082) subgroup and RB1 was significantly higher in the NE low subgroup (vs. NE high, P=0.015; 95% CI of the difference, −1.549 to −0.176). Interestingly, in five patients we observed a change in NE pattern in primary vs. LN metastatic samples: four patients had NE low specific gene expression in their primary tumor but NE high specific expression in their LN metastases, while only one patient’s primary tumor was identified in the NE high subtype but changed pattern to NE low subtype according to LN metastasis. In fact, due to this heterogeneity between primary and LN metastases regarding the overall diagnosis related to NE differentiation of a patient’s tumor there was a higher number of NE high (72% vs. 62.5%) and a lower number of NE low (28% vs. 37.5%) patients theoretically diagnosed when using the LN specimens compared to primary tumors (Figure 4A). As shown in Figure 4B, the correlation between primary and LN samples regarding NE pattern was categorized as moderate (r=0.664), having a match rate of 84.38%.
Figure 3.

Cluster and simultaneous heat map analysis identifies NE high (vs. NE low) subgroups based on top NE associated genes in our gene panel. Each row represents an RNA gene and each column a single sample. Cluster analysis clearly identified main cluster A and B that represents NE low and NE high subgroups, respectively. Of note, irrespective of tissue origin both primary and lymph node (LN) metastatic samples were included. The Arabic numeral from sample name corresponds to sample ID, while the Roman number refers to the tissue sample origin (I—primary tumor, III—LN metastasis). Red dots indicate samples that changed NE pattern during metastatic spread means that the matched pair was not categorized in the same NE subgroup (samples from patients #6, 21, 26, 33, and 270).
Figure 4.
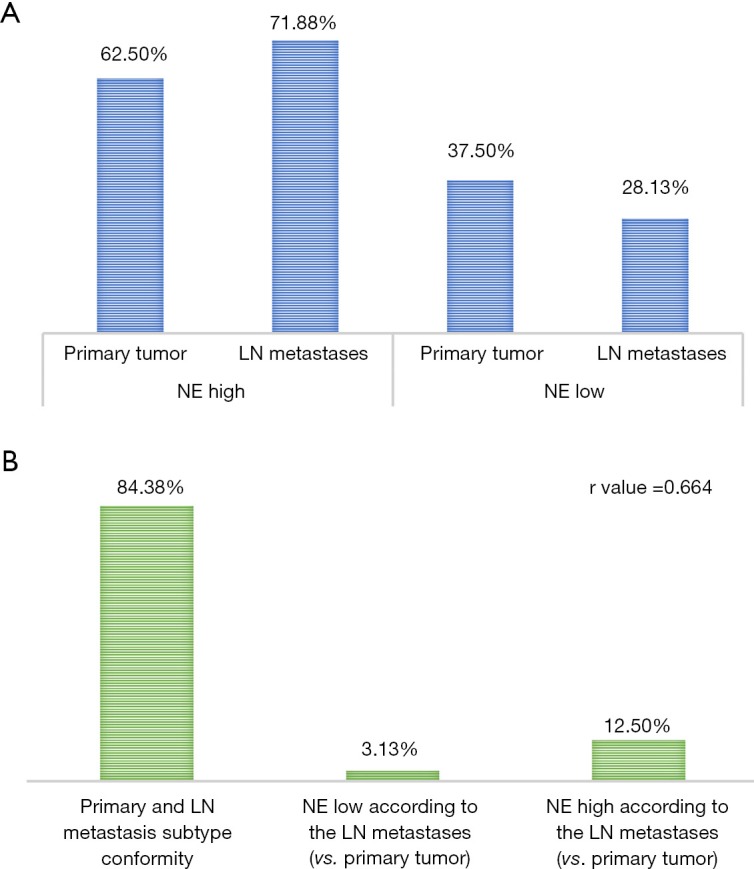
Differences in patients tumor neuroendocrine (NE) subtype classification according to primary tumor or lymph node (LN) metastasis. (A) Percentage of patients in NE high vs NE low subgroup according to tissue origin (primary tumor, 62.50% vs. 37.50%; LN metastasis, 71.88% vs. 28.13%, respectively). (B) Conformity of NE pattern in LN metastatic samples and the primary tumor (r value: 0.664; 84.38% conformity; 12.50% NE high subtype and 3.13% NE low subtype according to LN metastases vs. primary tumors).
NE related gene expression including the pooled expression of all primary tumors vs. LN metastases is shown in Figure 5. A non-significant but clearly detectable heterogeneity irrespective of specific NE pattern (including all NE associated genes) was found in gene expression signature between primary tumor and LN, and a significant downregulation of genes CAV1, CAV2, and ANXA3 in LN metastases that has a potential role in cancer development, proliferation and apoptosis.
Figure 5.

Heat map analysis of the top genes in our gene panel associated with neuroendocrine (NE) differentiation in primary tumor versus lymph node (LN) metastases. All genes appeared to be differently expressed, with a significant difference in Caveolin 1 (CAV1) (P=0.004; 95% CI of the difference, 0.24 to 1.25), Caveolin 2 (CAV2) (P=0.029; 95% CI of the difference, 0.06 to 1.21), and Annexin A3 (ANXA3) (P=0.035; 95% CI of the difference, 0.06 to 1.63). CAV1 and CAV2 are antagonists for the regulation of several essential cellular processes such as endothelial proliferation, endocytosis, infection, inflammatory response, cellular growth control and apoptosis, while ANXA3 plays a role in the regulation of cellular growth and in signal transduction pathways. All three latter mentioned genes were significantly down-regulated in LN metastases.
Figure S1A shows the OS of patients according to primary tumor NE pattern. There were no significant OS differences according to NE high vs. NE low subgroups (median OS, 20.7 vs. 19.4 months; P=0.56). The median OS was non-significantly higher among patients with tumors that change NE pattern from low to high vs. patients constantly classified to NE high subgroup when considered LN metastases additionally to the primary tumors (median OS, 29.1 vs. 20.7 months; P=0.77; Figure S1B).
Discussion
Recent studies show that SCLC can no longer be considered as a single disease entity, and a clearer understanding of pathogenesis and in depth NE features of SCLC might help lay the framework for treatment including immunotherapy administration (12,25-30). Therefore, the aim of our study was to confirm the preclinical data on NE low vs. NE high tumor features in the clinical setting in resected-stage, histologically diagnosed SCLC patients. Our further aim was to explore inter-tumor heterogeneity by comparing gene expression and key relevant intracellular pathways of tumor samples obtained from matched primary tumor and mediastinal LN metastases.
Our main finding from this study highlights NE low vs. NE high tumor subtypes in SCLC.
First, we analyzed the associations of gene expression and clinicopathological parameters. Importantly, in primary tumors, relative expression of the key relevant gene BCL2, which recently was demonstrated to be potentially targetable with venetoclax was significantly higher in primary tumors in males (31).
Next, we analyzed the site-specific inter-tumor heterogeneity, including all genes available in our panel. The most significant and relevant increase in primary tumor gene expression was detected in the cytokine-cytokine receptor interaction pathway which is one of the immune-related signaling pathways and has a crucial role in inflammatory host defense, cell growth and differentiation (32-34). This may hold implications in potential future targets and developments of immunotherapy in SCLC (35). Furthermore, we found a relatively high number of genes (n=154) that show significant differences in gene expression with medium and large effect size based on Cohen’s d, along with a small percentage of genes with high correlation (13.1%), which might indicate a non-homogeneous nature of the tumor mass at different anatomical locations within a patient.
Our findings were compared with known cancer research datasets, such as The Cancer Genome Atlas (TCGA), and with previously reported studies. Unfortunately, unlike in lung adenocarcinoma and squamous cell carcinoma, in SCLC no RNA-Seq data was available in TCGA (36). Nevertheless, using the cBioPortal for Cancer Genomics we compared our RNA-genes (n=154) that were differently expressed in primary tumor and LN metastases with data only available on somatic gene mutation in primary SCLC. According to cBioPortal, in a pooled cohort of 210 patients/samples, from our 154 queried genes molecular alterations were present in 100 genes. Furthermore, our results are in line with a preclinical experimental research that used two genetically engineered mouse models to examine tumor heterogeneity via RNA-seq on fluorescent-activated cell sorting (FACS)-isolated cancer cells from both models and revealed widespread changes in gene-expression programs during metastatic progression (37). Meanwhile, our results are in contrast to a study performed on stage 4 distant organ autopsy samples including only 2 SCLC patients, which showed n=359 mutations in 354 genes and n=271 mutations in 247 genes, but a 95% concordance in mutation profile in primary and distant organ metastatic lesions using whole-exome sequencing (WES) (13). A possible explanation to the discordance might be related to the different materials and methods used. Gene expression assay in our study might be more sensitive and therefore more valuable in differentiating tumor phenotypes based on gene expression. Inter-tumor heterogeneity was also reported in terms of overall frequencies of allelic imbalance and actionable driver genes (TP53, PIK3CA, and ERBB2) in breast cancer and in gene expression in non-small cell lung cancer (38-41). Furthermore, in previous studies, the coexistence of several subclones and a variable level of intra-tumor heterogeneity within the primary tumors were also described in other histological types of lung cancer (42-44). Accordingly, gene profiling of a single tumor-site biopsy might not be sufficient for diagnostic purposes and LN metastatic site biopsy samples might have future relevance due to potentially relevant divergences in gene expression.
Next, we performed the cluster analysis using the top genes reported in earlier studies that are associated with NE differentiation (2,3). We confirmed that SCLC tumor samples regardless of tumor localization express NE associated genes. To our knowledge, this is the first study, indicating that NE high and low subsets of SCLC tumors clearly separate in a clinical setting.
Of note, differences in NE pattern were also observed in primary tumor and LN metastases suggesting a possible inter-tumor heterogeneity. A significant downregulation of NE associated genes CAV1, CAV2 and ANXA3 was found in LN metastases. Although CAV1 is involved in cancer development, proliferation and apoptosis, its exact role is still controversial. Preclinical data suggest that higher expression of CAV1 can inhibit apoptosis in SCLC and might be associated with poor response to chemotherapy and radiation therapy (45). Due to inter-tumor heterogeneity, four patients with NE low and one patient with NE high primary tumors presented with opposing NE phenotype according to their matched LN metastases. This fact further supports the additional relevance of LN biopsies and can also have therapeutic implications. Latest preclinical studies suggest that NE low subtype is radioresistant and chemoresistant compared with the NE high subtype, but is more likely to respond to immunotherapy due to its immune oasis phenotype (2,3,46). Consequently, during treatment decisions, knowledge on NE differentiation regarding both the primary tumor and LN metastasis might provide additional information and help clinicians since LN metastatic lesion alone might not reflect the NE phenotype of the original tumor pattern.
In our study the lower expression of NE associated key genes REST and MYC, and the higher expression of DLL3 in NE high subtype are in accordance with the preclinical findings of Gazdar et al., and confirms the accuracy of the cluster analysis performed (2). Characterizing the MYC and DLL3 expression according to the NE pattern may also lay the framework for developing future targeted therapies based on NE subtypes. In fact, a recent preclinical study performed on human and mouse cell lines suggest that SCLC with high MYC expression is vulnerable to Aurora kinase inhibition (8). Furthermore, Aurora kinase inhibition combined with chemotherapy suppresses tumor progression and might increases survival. Meanwhile, in another preclinical study, a DLL3-targeted antibody-drug conjugate induced durable tumor regression in vivo across multiple patient-derived xenograft models (11).
Finally, we investigated the prognostic role of NE subtypes. We found no major differences in OS between the two NE subgroups or constantly NE high vs. NE pattern changing tumors during metastatic spread. Our findings are in contrast to a previous study which found that NE IHC expressing tumors are associated with better prognosis compared to basaloid (BA) phenotypes (47). However, the later study analyzed only routine IHC staining compared to our study on NE low vs. high associated gene analysis. Furthermore, there were no NE subtype specific treatment administered that could potentially alter the prognosis and we included limited number of patients who underwent surgical resection over a long time period.
This study has limitations. First of all, this is a retrospective cross-sectional study with limited clinicopathological data available. The patient population is unique regarding resected sample size but is small even in the light of the fact that matched tumor samples are usually not available in case of SCLC. Prognostic data in our study might be influenced by the intraoperative techniques and differences in administration of oncotherapy over a long time period. Gene expression was examined via targeted expression assay, consequently, results were aligned to the probe sequences and there was no de facto human genome version used for alignment. It is also important to note that the gene expression of LN metastases might be influenced by the lymphoid tissues pattern and by the presence of lymphatics-associated genes.
Conclusions
To conclude, our study highlights the gene discordance between primary tumors and corresponding LN metastases in SCLC. Differences in gene expression are suggestive for a relatively high mutational rate in tumor cells and thus for a potential higher chance of developing drug resistance-inducing mutations. Furthermore, we confirm the results of preclinical studies and we identified NE low vs. NE high gene expressing tumors including inter-tumor heterogeneity between the primary and LN metastatic lesions in term of NE pattern as well. Importantly among other genes discussed, CAV1 and BCL2 as a potential regulator of apoptosis, and also DLL3 and MYC might be a promising consideration in future SCLC therapy. Additional profiling of LN metastatic samples, besides primary tumor biopsy, performed through minimally invasive techniques such as endobronchial ultrasound guided biopsy can provide prognostic and predictive information for treating SCLC patients. Further prospective studies are needed to define the impact of inter-tumor heterogeneity and the predictive relevance of NE subtypes in the treatment of SCLC including immunotherapy administration.
Figure S1.

Kaplan-Meier analysis of neuroendocrine (NE) subgroups. (A) Overall survival of the patients belonging to each NE subgroup according to the primary tumor; (B) survival curve of the patients belonging constantly to NE high subgroup vs. survival curve of the patients belonging to NE low subgroup based on the primary tumor and to NE high subgroup based on the LN metastasis. No significant differences were found in overall survival.
Acknowledgments
The authors thank the patients and clinical teams. Furthermore, we thank Dr. Glen J. Weiss for critical review of this manuscript. We thank Leslie Rozeboom for her assistance in processing tissue samples.
Funding: Z Lohinai was supported by the 2018 LCFA-BMS/IASLC Young Investigator Scholarship Award and the ESMO Translational Research Fellowship. Z Lohinai, B Dome and J Moldvay acknowledge funding from the Hungarian National Research, Development and Innovation Office (OTKA #124652 and OTKA #129664, Z Lohinai; KH130356, B Dome; NAP2-2017-1.2.1-NKP-0002, J Moldvay; K129065, B Dome, J Moldvay). B Dome and V Laszlo were also supported by the Austrian Science Fund (FWF I 3522, V Laszlo; FWF I 3977, B Dome). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The present study was directed in accordance with the guidelines of the Helsinki Declaration of the World Medical Association. The national level ethics committee (Hungarian Scientific and Research Ethics Committee of the Medical Research Council, ETT-TUKEB-7214-1/2016/EKU) approved the study. The need for individual informed consent for this retrospective study was waived.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Horn L, Mansfield AS, Szczęsna A, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 2018;379:2220-9. 10.1056/NEJMoa1809064 [DOI] [PubMed] [Google Scholar]
- 2.Gazdar AF. MS32.04: Molecular Phenotypes of SCLC. In Proceedings from the International Association for the Study of Lung Cancer - 19th World Conference on Lung Cancer, September 23-26, 2018; Toronto, Canada. J Thor Oncol 2018;13:S309 10.1016/j.jtho.2018.08.211 [DOI] [Google Scholar]
- 3.Zhang W, Girard L, Zhang YA, et al. Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl Lung Cancer Res 2018;7:32-49. 10.21037/tlcr.2018.02.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saito M, Shiraishi K, Goto A, et al. Development of targeted therapy and immunotherapy for treatment of small cell lung cancer. Jpn J Clin Oncol 2018;48:603-8. 10.1093/jjco/hyy068 [DOI] [PubMed] [Google Scholar]
- 5.Chan BA, Hughes BGM. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res 2015;4:36-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koinis F, Kotsakis A, Georgoulias V. Small cell lung cancer (SCLC): no treatment advances in recent years. Transl Lung Cancer Res 2016;5:39-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peifer M, Fernandez-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012;44:1104-10. 10.1038/ng.2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mollaoglu G, Guthrie MR, Böhm S, et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017;31:270-85. 10.1016/j.ccell.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer 2017;17:725-37. 10.1038/nrc.2017.87 [DOI] [PubMed] [Google Scholar]
- 10.Suda K, Rozeboom L, Yu H, et al. Potential effect of spliceosome inhibition in small cell lung cancer irrespective of the MYC status. PLoS One 2017;12:e0172209. 10.1371/journal.pone.0172209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saunders LR, Bankovich AJ, Anderson WC, et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med 2015;7:302ra136. 10.1126/scitranslmed.aac9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardnell RJ, Li L, Sen T, et al. Protein expression of TTF1 and cMYC define distinct molecular subgroups of small cell lung cancer with unique vulnerabilities to aurora kinase inhibition, DLL3 targeting, and other targeted therapies. Oncotarget 2017;8:73419-32. 10.18632/oncotarget.20621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saber A, Hiltermann TJN, Kok K, et al. Mutation patterns in small cell and non-small cell lung cancer patients suggest a different level of heterogeneity between primary and metastatic tumors. Carcinogenesis 2017;38:144-51. [DOI] [PubMed] [Google Scholar]
- 14.Mirsadraee S, Oswal D, Alizadeh Y, et al. The 7th lung cancer TNM classification and staging system: Review of the changes and implications. World J Radiol 2012;4:128-34. 10.4329/wjr.v4.i4.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seyednasrollah F, Laiho A, Elo LL. Comparison of software packages for detecting differential expression in RNA-seq studies. Brief Bioinform 2015;16:59-70. 10.1093/bib/bbt086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rapaport F, Khanin R, Liang Y, et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol 2013;14:R95. 10.1186/gb-2013-14-9-r95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suda K, Murakami I, Yu H, et al. CD44 facilitates epithelial to mesenchymal transition phenotypic change at acquisition of resistance to EGFR kinase inhibitors in lung cancer. Mol Cancer Ther 2018;17:2257-65. 10.1158/1535-7163.MCT-17-1279 [DOI] [PubMed] [Google Scholar]
- 19.Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 2015;43:W566-70. 10.1093/nar/gkv468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilks DS. Chapter 15: Cluster Analysis. Statistical Methods in the Atmospheric Sciences. Oxford and Waltham: Academic Press, 2011:603-16. [Google Scholar]
- 21.Cohen J. A Power Primer. Psychol Bull 1992;112:155-9. 10.1037/0033-2909.112.1.155 [DOI] [PubMed] [Google Scholar]
- 22.Belinky F, Nativ N, Stelzer G, et al. PathCards: multi-source consolidation of human biological pathways. Database (Oxford) 2015. doi: . 10.1093/database/bav006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. doi: . 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov 2012;2:401-4. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McColl K, Wildey G, Sakre N, et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 2017;8:73745-56. 10.18632/oncotarget.20572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allison Stewart C, Tong P, Cardnell RJ, et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017;8:28575-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calbo J, van Montfort E, Proost N, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 2011;19:244-56. 10.1016/j.ccr.2010.12.021 [DOI] [PubMed] [Google Scholar]
- 28.Lim JS, Ibaseta A, Fischer MM, et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017;545:360-4. 10.1038/nature22323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47-53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shue YT, Lim JS, Sage J. Tumor heterogeneity in small cell lung cancer defined and investigated in pre-clinical mouse models. Transl Lung Cancer Res 2018;7:21-31. 10.21037/tlcr.2018.01.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lochmann TL, Floros KV, Naseri M, et al. Venetoclax Is Effective in Small-Cell Lung Cancers with High BCL-2 Expression. Clin Cancer Res 2018;24:360-9. 10.1158/1078-0432.CCR-17-1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong C, Wang X, Xu H, et al. Identification of a cytokine-cytokine receptor interaction gene signature for predicting clinical outcomes in patients with colorectal cancer. Int J Clin Exp Med 2017;10:9009-18. [Google Scholar]
- 33.Li Z, Liu X, Liu J, et al. Transcriptome profiling based on protein-protein interaction networks provides a core set of genes for understanding blood immune response mechanisms against Edwardsiella tarda infection in Japanese flounder (Paralichthys olivaceus). Dev Comp Immunol 2018;78:100-13. 10.1016/j.dci.2017.09.013 [DOI] [PubMed] [Google Scholar]
- 34.Spangler JB, Moraga I, Mendoza JL, et al. Insights into cytokine-receptor interactions from cytokine engineering. Annu Rev Immunol 2015;33:139-67. 10.1146/annurev-immunol-032713-120211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guven-Maiorov E, Acuner-Ozbabacan SE, Keskin O, et al. Structural pathways of cytokines may illuminate their roles in regulation of cancer development and immunotherapy. Cancers 2014;6:663-83. 10.3390/cancers6020663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byers LA, Rudin CM. Small cell lung cancer: where do we go from here? Cancer 2015;121:664-72. 10.1002/cncr.29098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang D, Denny SK, Greenside PG, et al. Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov 2018;8:1316-31. 10.1158/2159-8290.CD-17-0987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Becker TE, Ellsworth RE, Deyarmin B, et al. The genomic heritage of lymph node metastases: implications for clinical management of patients with breast cancer. Ann Surg Oncol 2008;15:1056-63. 10.1245/s10434-008-9815-3 [DOI] [PubMed] [Google Scholar]
- 39.Bertucci F, Finetti P, Guille A, et al. Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget 2016;7:27208-19. 10.18632/oncotarget.8349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu J, Byran J, Wang LL, et al. Genetic differences between primary and metastatic tumors from cross-institutional data. J Clin Oncol 2018;36:e18572 10.1200/JCO.2018.36.15_suppl.e18572 [DOI] [Google Scholar]
- 41.Sherwood J, Dearden S, Ratcliffe M, et al. Mutation status concordance between primary lesions and metastatic sites of advanced non-small-cell lung cancer and the impact of mutation testing methodologies: a literature review. J Exp Clin Cancer Res 2015;34:92. 10.1186/s13046-015-0207-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Fujimoto J, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014;346:256-9. 10.1126/science.1256930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Bruin EC, McGranahan N, Swanton C. Analysis of intratumor heterogeneity unravels lung cancer evolution. Mol Cell Oncol 2015;2:e985549. 10.4161/23723556.2014.985549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swanton C. Cancer evolution: the final frontier of precision medicine? Ann Oncol 2014;25:549-51. 10.1093/annonc/mdu005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X, Xiong H, Guan ZZ, et al. Higher expression of Caveolin-1 inhibits human small cell lung cancer (SCLC) apoptosis in vitro. Cancer Invest 2012;30:453-62. 10.3109/07357907.2012.675384 [DOI] [PubMed] [Google Scholar]
- 46.Ito T, Matsubara D, Tanaka I, et al. Loss of YAP1 defines neuroendocrine differentiation of lung tumors. Cancer Sci 2016;107:1527-38. 10.1111/cas.13013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamanaka W, Motoi N, Ishikawa S, et al. A subset of small cell lung cancer with low neuroendocrine expression and good prognosis: a comparison study of surgical and inoperable cases with biopsy. Hum Pathol 2014;45:1045-56. 10.1016/j.humpath.2014.01.001 [DOI] [PubMed] [Google Scholar]