

During prostate carcinogenesis, molecular changes occur in prostate epithelial cells such that their Androgen Receptor (AR) protein expression increases significantly (i.e. >5 fold). This elevated AR expression perverts AR-dependent transcriptional programming from a growth suppressor to an oncogenic stimulator of malignant growth (1-3). Due to this malignancy acquired addiction to AR transcriptional programming, androgen deprivation therapy (ADT), which lowers serum testosterone (T) from 600 to <100 ng/dL, not only inhibits prostate cancer cell proliferation, but also results in transcriptional reprogramming that induces cancer cell death (4). Unfortunately, metastatic prostate cancer patients eventually develop resistance to primary castration therapy in addition to secondary hormone-based therapies such as abiraterone and/or enzalutamide. The most common mechanism for the development of resistance is via an adaptive “auto-regulation” that increases AR protein expression by >30-fold (5). This increase results in sufficient AR accumulation in the nuclei of castration-resistant prostate cancer (CRPC) cells to induce by mass action the AR-dependent transcriptional programming needed to both inhibit their death and stimulate their proliferation despite castrate levels of androgen.
While this adaptive increase in AR provides a growth advantage under ADT conditions, it creates the potential for an “induced synthetic lethality” in these malignant cells. The basis for this induced synthetic lethality is derived from the fact that the half-life of the AR protein is more than doubled when fully occupied by androgen (5). Thus, when high AR-expressing CRPC cells are cycled from low (i.e., <100 ng/dL) to supraphysiologic (>700 ng/dL) androgen (SPT), the resultant fully ligand-occupied AR protein is stabilized, driving its accumulation in the nucleus to such a high level that it now “paradoxically” suppresses proliferation and induces cell death (5-10). Several complementary mechanisms for the paradoxical effect of SPT have been described. Using both in vitro cell lines and in vivo xenograft models Isaacs et al. demonstrated that AR is a DNA licensing factor that plays a critical role in DNA replication and must be degraded as a cell goes through the cell cycle (11). These studies also documented that treatment with SPT saturates ligand binding to the AR, maximizing the amount of ligand-bound AR in the nucleus and stabilizing it against degradation, thus inhibiting DNA relicensing, which results in cell death in the subsequent cycle (12,13). In independent studies, Haffner et al. documented that treatment of CRPC cells with SPT increases AR nuclear binding to DNA, generating transient double-strand DNA breaks in these cells through the recruitment of AR and topoisomerase IIβ to androgen response elements leading to death of these cells (14,15). An additional rationale for the use of SPT is the observation that there are differences in the AR transcriptome when CRPC cell lines with high levels of AR are exposed to either high or low androgen concentrations (16,17). Under high androgen conditions, AR represses several genes, including AR itself in addition to those involved in androgen synthesis, DNA synthesis, cell proliferation and survival (e.g., c-MYC, BCL2, NFκB). Therefore, SPT can lead castration-resistant cells to transition from a more oncogenic transcriptome associated with castrate T levels to a high androgen transcriptome that does not support cancer proliferation and survival (16,17).
Indeed, the recent publication of Lam et al. documents that treatment with SPT durably suppresses the in vivo growth of 4 out of 13 AR-expressing patient-derived CRPC xenografts (PDXs) due to a multifaceted growth suppression and impaired DNA damage response transcriptional program (18). In the Lam et al. study, CRPC PDXs were initially established in castrated hosts and then supplemented with exogenous testosterone to rapidly raise and maintain the serum testosterone from <100 ng/dL to a supraphysiologic value of >700 ng/dL (18). In contrast to the SPT maintenance approach of Lam et al. to induce lethality in CRPC cells, our group previously proposed that additional therapeutic advantage can be gained by cycling between short periods (i.e., 2 weeks) of SPT back to a castrate level of T (5,19). This cycling optimally inhibits growth and induces cell death in high AR expressing CRPC cells; a strategy we termed serial bipolar androgen therapy (sBAT). The rationale for this sBAT approach is that during such rapid cycling, any subset of CRPC cells that “auto-regulate” to decrease transcription of AR fast enough to lower AR protein levels, thus preventing both transcriptional reprogramming to a growth suppressive transcriptome and not inhibiting DNA replication licensing or fragmentation, can survive such SPT exposure. However, if these surviving now low AR-expressing cells are subsequently rapidly cycled back to castrate T, they must rapidly re-upregulate AR transcription to elevate AR protein to a high enough level to maintain the oncogenic transcriptome needed to survive and grow in the now low androgen environment. Such AR auto-upregulation, however, re-sensitizes them to another round of SPT. By serially cycling systemic androgen rapidly between the bipolar extremes of SPT and castrate levels, CRPC cells are placed in a highly stressful and energy demanding compromised state of constant transcriptional reprogramming, which in preclinical patient-derived human CRPC xenograft models inhibits their survival and growth. See Figure 1 for an example of such a therapeutic response.
Figure 1.
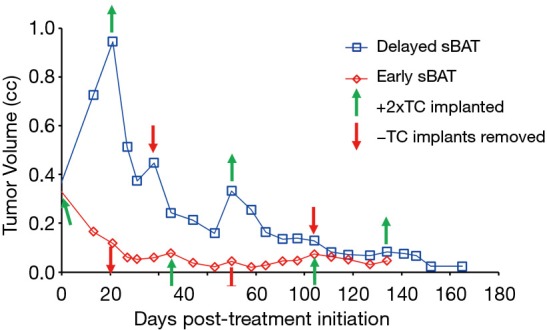
Response of SkCaP-CR PDX in castrated hosts to serial bipolar androgen therapy (sBAT). sBAT cycles initiated when cancers were ~0.3 cc [early sBAT, (red)] vs. ~0.9 cc [delayed sBAT, (blue)]. Testosterone cypionate was given via subcutaneous silastic implants (green up arrow), which elevated serum testosterone levels from <100 ng/dL to 17,300 +/− 900 ng/dL within 48 hours (i.e., SPT). When the silastic implants were removed (red down arrow), serum testosterone decreased to <100 ng/dL within 24 hours (i.e., castrate).
Based on this rationale, sBAT has been translated clinically in metastatic CRPC patients progressing on second line hormone therapies (i.e., abiraterone and/or enzalutamide) through intramuscular administration of 400 mg of T cypionate every 28 days to rapidly raise serum T to supraphysiologic levels >1,500 ng/dL by 2 days post-injection with a decline to near castrate level (~100 ng/dL) by 28 days post-injection, Figure 2. Overall, sBAT has produced minimal side effects and shown clinical responses in ~25% of patients (20-22). These positive results raise the issue of how to increase both the frequency and extent of clinical benefit resulting from the sBAT regimen. The fundamental therapeutic basis of sBAT is the mandatory requirement for rapid, adaptive auto-regulation of AR and its transcriptome by CRPC cells during the rapid extreme changes in androgen concentrations. Therefore, we are presently testing whether the therapeutic benefit of sBAT can be increased by combining it with appropriately timed transcriptional inhibitors targeting different, but complimentary epigenetic regulators to disrupt the adaptive transcriptional reprogramming by CRPC cells needed for their survival and proliferation during rapid extreme changes in androgen level.
Figure 2.
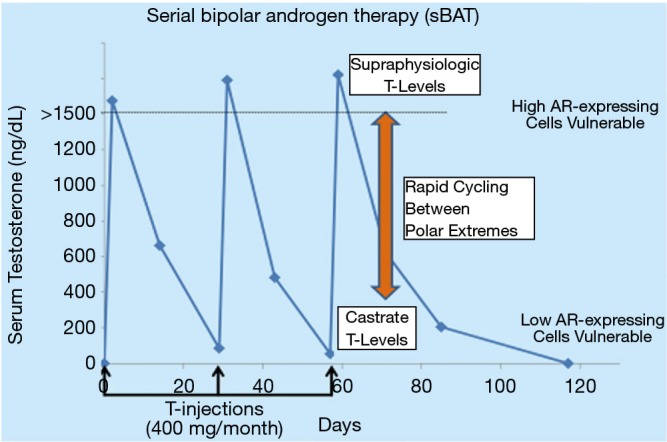
Serum testosterone concentrations during serial bipolar androgen therapy (sBAT) clinical regimen. Testosterone cypionate was provided via intramuscular (IM) injection of 400 mg every 30 days. This results in serial cycling between supraphysiologic and castrate levels of serum testosterone on a monthly schedule.
Acknowledgments
The authors wish to thank M. Rosen, S. Dalrymple, and L. Antony for excellent technical assistance in addition to the clinical research nurses and data managers of the Johns Hopkins Prostate Cancer Research Program who supported this study. We also wish to thank the patients who participated in the clinical studies.
Funding: The preclinical studies are supported by the US Department of Defense, through the Prostate Cancer Research Program under Award No. W81XWH1810349.
Ethical Statement: The authors are accountable for all aspects of the work ensuring that questions related to the accuracy or integrity of all parts of the work are appropriately investigated and resolved. All animal procedures were approved by the Johns Hopkins University School of Medicine Institutional Animal Care and Use Committee.
Provenance: This is an invited article commissioned by the Section Editor, Dr. Peng Zhang, MD, PhD (Department of Neurology, Zhongnan Hospital of Wuhan University, Wuhan, China).
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab 2003;88:2972-82. 10.1210/jc.2002-022038 [DOI] [PubMed] [Google Scholar]
- 2.Antony L, van der Schoor F, Dalrymple SL, et al. Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/β-catenin/TCF-4 complex inhibition of c-MYC transcription. Prostate 2014;74:1118-31. 10.1002/pros.22828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci 2014;10:627-42. 10.7150/ijbs.8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res 2001;61:5038-44. [PubMed] [Google Scholar]
- 5.Isaacs JT, D'Antonio JM, Chen S, et al. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate 2012;72:1491-505. 10.1002/pros.22504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonnenschein C, Olea N, Fasanen ME, et al. Negative controls of cell proliferation: Human prostate cancer cells and androgens. Cancer Res 1989;49:3474-81. [PubMed] [Google Scholar]
- 7.Wolf DA, Schulz P, Fittler F. Synthetic androgens suppress the transformed phenotype in the human prostate carcinoma cell line LNCaP. Br J Cancer 1991;64:47-53. 10.1038/bjc.1991.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langeler EG, van Uffelen CJ, Blankenstein MA, et al. Effect of culture conditions on androgen sensitivity of the human prostatic cancer cell line LNCaP. Prostate 1993;23:213-23. 10.1002/pros.2990230304 [DOI] [PubMed] [Google Scholar]
- 9.Kokontis J, Takakura K, Hay N, et al. Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res 1994;54;1566-73. [PubMed] [Google Scholar]
- 10.Joly-Pharaboz MO, Ruffion A, Roch A, et al. Inhibition of growth and induction of apoptosis by androgens of a variant of LNCaP cell line. J Steroid Biochem Mol Biol 2000;73:237-49. 10.1016/S0960-0760(00)00076-5 [DOI] [PubMed] [Google Scholar]
- 11.Litvinov IV, Vander Griend DJ, Antony L, et al. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc Natl Acad Sci U S A 2006;103:15085-90. 10.1073/pnas.0603057103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vander Griend DJ, Litvinov IV, Isaacs JT. Stabilizing androgen receptor in mitosis inhibits prostate cancer proliferation. Cell Cycle 2007;6:647-51. 10.4161/cc.6.6.4028 [DOI] [PubMed] [Google Scholar]
- 13.D'Antonio JM, Vander Griend DJ, Isaacs JT. DNA licensing as a novel androgen receptor mediated therapeutic target for prostate cancer. Endocr Relat Cancer 2009;16:325-32. 10.1677/ERC-08-0205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haffner MC, Aryee MJ, Toubaji A, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet 2010;42:668-75. 10.1038/ng.613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haffner MC, De Marzo AM, Meeker AK, et al. Transcription-induced DNA double strand breaks: Both oncogenic force and potential therapeutic target? Clin Cancer Res 2011;17:3858-64. 10.1158/1078-0432.CCR-10-2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai C, He HH, Chen S, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 2011;20:457-71. 10.1016/j.ccr.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao S, Gao Y, He HH, et al. Androgen receptor tumor suppressor function is mediated by recruitment of retinoblastoma protein. Cell Reports 2016;17:966-76. 10.1016/j.celrep.2016.09.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lam HM, Nguyen HM, Labrecque MP, et al. Durable Response of Enzalutamide-resistant Prostate Cancer to Supraphysiological Testosterone Is Associated with a Multifaceted Growth Suppression and Impaired DNA Damage Response Transcriptomic Program in Patient-derived Xenografts. Eur Urol 2019. [Epub ahead of print]. 10.1016/j.eururo.2019.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denmeade SR, Isaacs JT. Bipolar androgen therapy: The rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. Prostate 2010;70:1600-7. 10.1002/pros.21196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schweizer MT, Antonarakis ES, Wang H, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci Transl Med 2015;7:269ra2. 10.1126/scitranslmed.3010563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teply BA, Wang H, Luber B, et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol 2018;19:76-86. 10.1016/S1470-2045(17)30906-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee P, Schweizer MT, Lucas JM, et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J Clin Invest 2019;130:4245-60. 10.1172/JCI127613 [DOI] [PMC free article] [PubMed] [Google Scholar]