

Abstract
A blood test that predicts the extent of amyloid plaques in the brain and risk of Alzheimer’s disease would have important benefits for the early identification of higher risk of dementia and Alzheimer’s disease and the evaluation of new preventative therapies. The goal of this study was to determine whether plasma levels of amyloid-β1–42, 1–40 and the amyloid-β1–42/1–40 ratio among participants in the Pittsburgh centre of the Ginkgo Evaluation of Memory Study were related to the extent of brain fibrillar amyloid plaques measured in 2009 using Pittsburgh compound-B PET imaging, hippocampal volume, cortical thickness in the temporal lobe and white matter lesions. There were 194 participants who had Pittsburgh compound-B measurements in 2009 with the mean age of 85 years; 96% were white and 60% men. Pittsburgh compound-B positivity was defined as a standardized uptake value ratio of ≥1.57. Amyloid-β in blood was measured using a sandwich enzyme-linked immunosorbent assay developed by Eli Lilly and modified at the University of Vermont. All participants were nondemented as of 2008 at the time of study close out. The study sample included 160 with blood samples drawn in 2000–02 and 133 from 2009 and also had brain amyloid measured in 2009. All blood samples were analysed at the same time in 2009. Plasma amyloid-β1–42 was inversely related to the percent Pittsburgh compound-B positive (standardized uptake value ratio ≥1.57), β −0.04, P = 0.005. Practically all participants who were apolipoprotein-E4 positive at older ages were also Pittsburgh compound-B positive for fibrillar amyloid. Among apolipoprotein-E4-negative participants, quartiles of amyloid-β1–42 were inversely related to Pittsburgh compound-B positivity. In multiple regression models, plasma amyloid-β1–42 measured in 2000–02 or 2009 were significantly and inversely related to Pittsburgh compound-B positivity as was the amyloid-β1–42/1–40 ratio. There was a 4-fold increase in the odds ratio for the presence of Pittsburgh compound-B positivity in the brain in 2009 for the first quartile of amyloid-β1–42 as compared with the fourth quartile in the multiple logistic model. This is one of the first longitudinal studies to evaluate the relationship between amyloid-β1–42 in the blood and the extent of brain amyloid deposition measured by PET imaging using Pittsburgh compound-B. Our findings showed that remote and recent low plasma amyloid-β1–42 levels were inversely associated with brain amyloid deposition in cognitively normal individuals. However, changes in plasma amyloid-β1–42 over time (8 years) were small and not related to the amount of Pittsburgh compound-B.
Keywords: Alzheimer’s disease, amyloidosis, longitudinal study, blood–brain barrier, blood amyloid-β1–42
We have found an inverse relationship between plasma levels of amyloid-β1–42 in blood collected in 2000–02 (n = 160) and for same individuals in 2009 (n = 133) and amount of fibrillar amyloid measured by Pittsburgh compound-B in 2009. There was about a 4-fold higher risk of fibrillar amyloid for the first versus fourth quartile of amyloid-β1–42 in blood.
Graphical Abstract
Graphical Abstract.
Introduction
We have reported in the follow-up in the Pittsburgh field centre of Ginkgo Evaluation of Memory Study (GEMS) from 2009 to 2015 that the presence of fibrillar amyloid deposition, as determined by Pittsburgh compound-B (PiB) status was a significant predictor of the risk of dementia, especially when combined with measures of neurodegeneration, such as hippocampal (HIP) volume and brain vascular disease, i.e. white matter lesions (WML; Lopez et al., 2018).
Previous reports suggested that increased plasma amyloid-β1–42 and amyloid-β1–40 in nondemented participants were predictors of Alzheimer’s disease (Mayeux et al., 1999). However, three recent large longitudinal studies reported that lower amyloid-β1–42 or the amyloid-β1–2/1–40 ratio were predictors of the risk of dementia. Also, a recent meta-analysis suggested that the amyloid-β1–42/1–40 ratio was the better predictor of dementia than either amyloid-β1–42 or amyloid-β1–40 alone (Song et al., 2011; Chouraki et al., 2015; Wolters, 2018; Lopez et al., 2019). We recently reported that blood levels of amyloid-β1–42 and amyloid-β1–42/1–40 ratio measured in 2000–02 were inversely related to risk of dementia to 2008 for participants who were cognitively normal (CN) at 2000–02 but not for those with mild cognitive impairment (MCI) in GEMS (Lopez et al., 2019).
The purpose of this study was to examine the association between baseline (2000–02; n = 160) and follow-up (2009; n = 133) plasma amyloid-β1–40 and amyloid-β1–42 levels and amyloid deposition in the brain measured with PiB in 2009 in a group of well-characterized, nondemented participants from the Pittsburgh GEMS.
Materials and methods
The Ginkgo Evaluation of Memory Study
A full description of recruitment and screening procedures of GEMS has been reported previously (DeKosky et al., 2008; Snitz et al., 2009). Briefly, volunteers were recruited from September 2000 to May 2002, using voter registration and other purchased mailing lists, from four US communities with academic medical centres. The current study included only Pittsburgh participants in the GEMS who had measurements of brain amyloid using PiB in 2009, 1 year after the termination of GEMS. PiB was not measured in the other three field centres.
In the Pittsburgh cohort, there were 966 original participants and 132 (13.6%) were demented by 2008: 115 participants were deceased without dementia and there were 66 dropouts (Supplementary Fig. 1).
At the end of the trial, the Pittsburgh field centre recruited 194 participants for repeat cognitive testing, magnetic resonance imaging (MRI), and amyloid imaging using PET for PiB. Eleven of the participants were excluded due to technical problems, three had technical problems with PiB imaging and eight had technical problems with MRI scans. One participant dropped out shortly after testing, leaving 182 eligible for analysis. Participants were selected based on their willingness to be contacted for additional follow-up studies and to have a brain MRI scan and PET imaging. Exclusion criteria were contraindications for an MRI scan or PET imaging for amyloid, had no evidence of dementia or treatment for dementia since the termination of the trial in 2008–09 (Supplementary Fig. 1). The participants completed an abbreviated neuropsychological battery, the 10-question Center for Epidemiologic Studies Depression Scale, a timed walk, an inventory of the subjects’ prescriptions and over-the-counter medications. A total of 160 of the 183 (87%) participants with blood samples for measures of amyloid-β1–42 in blood and PET amyloid imaging for fibrillar amyloid from blood samples drawn in 2000–02 and 133 participants with blood samples drawn in 2009 were eligible and included in this analysis (Fig. 1). All blood samples were analysed at the same time in 2009.
Figure 1.

Description of Pittsburgh GEMS cohort.
PET scans
The PET data were acquired in 2008–09 on a Siemens/CTI ECAT HR+ scanner as previously described in detail. Approximately 40 min following the bolus intravenous injection of 15 positive/−1.5 mCi of [11C]PIB (administered over 10–20 s), subjects were placed in the PET scanner and positioned so that the entire brain is in the field of view (Mathis et al., 2013). The [11C]PIB PET scan was acquired in dynamic, 3D imaging mode for 20 min (4 × 5 min frames) beginning 50 min after the injection of [11C]PIB. After the emission data were acquired, post-injection transmission scans were acquired for attenuation correction, using 68Ge/68Ga rods (Mathis et al., 2013). Data were corrected for photon attenuation, scatter and radioactive decay. The final reconstructed PET image resolution was ∼6 mm (transverse and axial) based on in-house point source measurements.
MRI scans
MRI scanning was performed on a GE Signa 1.5 T scanner prior to the PET imaging procedure with a standard head coil, 19 including fluid-attenuated inversion recovery, and has been previously described in detail (Lopez et al., 2018). Total intracranial volume was computed using the Brain Extraction Tool. HIP volumes were calculated using the automated labelling pathway, an atlas-based segmentation technique using a fully deformable registration approach to measure predefined regions of interest (ROIs), and anatomical ROIs were from the automated anatomical labelling atlas. HIP ROIs were defined on the reference brain (Montreal Neurological Institute) and transformed to fit each individual’s anatomical image; trilinear interpolation was used. Volumes were calculated as the number of cubic millimetre voxels. The HIP volumes were evaluated as the proportion of the intracranial volume. A participant was considered to have small hippocampi (HIP positive) when either the right or left hippocampus was <25th percentile of that in individuals who remained CN throughout follow-up. A fuzzy connectedness algorithm was used to segment the WML from each individual’s T2-weighted, fluid-attenuated inversion recovery images. The volume of WML is presented as the proportion of the intracranial volume, and volumes >75th percentile of those in normal participants were considered abnormal, or WML positive. These classifications were done before the data analysis (Lopez et al., 2018).
Cortical thickness was defined as the average of four regions: entorhinal cortex, fusiform gyrus, middle temporal gyrus and inferior temporal gyrus with the composite MRI-based cortical thickness were determined using the automated surface-based algorithm implemented in the FreeSurfer software package (v5.3, Laboratory for Computational Neuroimaging, Athinoula A. Martinos Center for Biomedical Imaging, Harvard University, Cambridge, MA, USA; Fischl and Dale, 2000; Fischl, 2012; Jack et al., 2015).
Data analysis of PET
The co-registration of the MRI and PiB PET image has been described (Rosario et al., 2008). Briefly, each subject’s native PET and MRI data were co-registered using Statistical Parametric Mapping (SPM8) software. The individual’s co-registered MRI scan was then spatially normalized to an MCI MRI template using default and best parameter settings for mid-life/elderly subjects. ROIs were hand-drawn on the template that was a high-resolution MRI of a single elderly MCI subject (79 years of age, with mild atrophy and ventricular enlargement) that was also used for Alzheimer’s Disease Neuroimaging PiB PET data analysis (Rissman et al., 2012). The ROIs included six cortical areas (i.e. anterior cingulate gyrus, anterior ventral striatum, frontal cortex, lateral temporal cortex, parietal cortex, precuneus cortex). PiB retention was measured using the standardized uptake value (SUV) ratio (SUVR) that is the regional summed uptake over the 50–70 min scan (or SUV: scaled to injected dose, body mass) that is then normalized by the SUV value of the cerebellum reference region. The SUVR values (mean and standard deviation) from each of six cortical brain sub-regions and a global cortical SUVR level (average of the six regions) were calculated (Mathis et al., 2013). PiB positivity was defined by a previously described iterative-outlier approach (Aizenstein et al., 2008). A subject was defined as positive if the partial volume corrected global cortical SUVR was >1.57 (Mathis et al., 2013). The PET imaging measures fibrillar amyloid.
Laboratory methods
All subjects had blood tests at baseline (2000–02), including complete blood count, metabolites, including liver enzymes, creatinine and cystatin-C to evaluate kidney function, electrolytes, thyroid-stimulating hormone and vitamin B12 levels (DeKosky et al., 2008). The amyloid-β proteins were measured using a sandwich enzyme-linked immunosorbent assay initially developed by Eli Lilly and further implemented at the University of Vermont’s Laboratory for Clinical Biochemistry Research. The antibodies, stock standard proteins and heat-inactivated rat plasma were provided by Eli Lilly. Capture antibodies were 2G3 for the amyloid-β1–40 assay and 21F12 for the amyloid-β1–42 assay. The plasma samples were denatured using guanidine hydrochloride (GuHCL) (GuHCL; 1.67 M GuHCL for amyloid-β1–40 samples, 0.83 M GuHCL for amyloid-β1–42 samples) with protease inhibitors for 15 min at room temperature. Samples were then diluted to 0.5 M GuHCL with Phosphate-buffered saline and used in the assay. Standard stock RS0546 was diluted to range from 250 to 3.9 pg/ml for the amyloid-β1–40 assay. Standard stock RS0548 was diluted to range from 125 to 0.49 pg/ml for the amyloid-β1–42 assay. Biotinylated 3D6 was the detection antibody for both assays. Streptavidin-conjugated horseradish peroxidase provided enzyme activity for detection using 3,3ʹ,5,5ʹ-tetramethylbenzidine as a substrate. Inter-assay coefficients of variation ranged from 3.1 to 7.9% for the amyloid-β1–40 and 12 to 20% for the amyloid-β1–42 (Shah et al., 2012).
There were 856 Pittsburgh subjects that had amyloid-β1–42 measured at baseline in 2000–02, and 580 (68%) were alive and not demented by 2008–09. There were 160 participants who had both PiB measured in 2009 (Fig. 1) and amyloid-β1–42 measured in blood drawn in 2000–02 and 133 participants who had both a PiB measurement in 2009 and amyloid-β1–42 evaluated in the blood in 2009. All assays were analysed in 2009 at the same time for both 2000–02 and 2009 samples. The laboratory was blinded to the diagnosis. Pearson correlation between the two measurements at 2000–02 and 2009 of amyloid-β1–42 was 0.63, and that of amyloid-β1–40 was 0.53 (P = 0.0001 for both). There was only a small change in amyloid-β1–42 over time (0.84 ± 14.9 pg/ml, median 0.70, 25th percentile −4.4) and in amyloid-β1–40 (median 7.6 ± 7.1, 25th percentile −0.38 pg/ml). Therefore, there were very small within-individual differences over 8 years from 2000–02 to 2009 in amyloid-β1–42.
The mean blood levels of amyloid-β1–42 were similar in Pittsburgh and for the total GEMS cohort, 15.7 pg/ml for all participants as compared with 15.2 pg/ml in the Pittsburgh cohort in 2000–02.
The distribution of amyloid-β1–42 is shown in Fig. 2 and follows a log normal distribution. There was only one outlier ≥3 SD from mean. The risk of dementia was directly related to PiB levels with the increase in risk at a PiB SUVR of ∼1.6 (Lopez et al., 2019). Mean and median amyloid-β1–42 blood levels were also very similar for blood samples drawn in 2000–02 and 2009 (11.9 and 11.3 pg/ml).
Figure 2.

Distribution of amyloid-β1–42 (pg/ml) as measured in 2000–02 and 2009 in the Pittsburgh GEMS cohort. (A) Frequency distribution of log amyloid-β1–42 in blood in 2000–02. (B) frequency distribution of log amyloid-β1–42 in blood in 2009.
Statistical analysis
Plasma amyloid levels, WML, creatinine, alanine aminotransferase, aspartate aminotransferase, gamma-glutamyl transferase, vitamin B12 and thyroid-stimulating hormone were log-transformed. Plasma amyloid levels were also analysed in quartile groups where the cut points of the quartiles were listed in the tables. All multi-group comparisons were carried out using two-sample t-tests, analysis of variance and Wilcoxon rank sum test for continuous outcomes. For categorical outcomes, contingency table methods with chi-square tests were used. For chi-square tests where the expected count was <5, exact tests of significance levels were computed. Correlation and regression coefficients were computed between amyloid deposition in selected brain regions and plasma amyloid levels in 2000 and 2009 while controlling for age. Censored values of the amyloid markers were replaced by the limit of detection for each of the markers. This approach was chosen over a Tobit model since the censoring rate was relatively small. To compare the coefficients in Table 1, a new dataset was created by appending the data used in Model a to the data used in Model b and a dummy variable indicating whether the data were for Model a or for Model b was also created. The significant of the interaction term between the dummy variable and the predicting variable (e.g. PiB score) would suggest that the regression coefficients in Model a and Model b were different. Logistic regression model was performed to assess the relation between brain amyloid deposition (positive vs. negative) and plasma amyloid levels while controlling forage, years of education, number of blocks walked per week, hypertension, apolipoprotein-E4 (APOE4), Ginkgo intervention (yes/no) and creatinine. Analyses were performed with SAS 9.4 (SAS Institute, Cary, NC, USA). There were 18 participants with MCI at baseline 2000–02 included in this study. The definition of MCI was having impairments at or below the 10th percentile of the Cardiovascular Health Study normative data stratified by age and education on at least 2 of the 10 selected neuropsychological test scores from five cognitive domains and a Clinical Dementia Rating global score of 0.5 (Lopez et al., 2019).
Table 1.
Linear regression coefficients of amyloid-β1–42 and measures of brain MRI (WMH, cortical thickness, HIP volume and PiB score) in the Pittsburgh GEMS cohort
| Model | Independent variable | Estimate | SE | P-value | 95% CI | Standardized coefficient * |
|---|---|---|---|---|---|---|
| Amyloid-β1–42 measured in 2000–02 | ||||||
| 1a | HIP volume | 1.19 | 1.94 | 0.54 | −2.61 to 4.99 | 0.06 |
| 2a | Cortical thickness | 0.21 | 0.15 | 0.15 | −0.07 to 0.50 | 0.13 |
| 3a | PiB score | −0.40 | 0.14 | 0.00 | −0.67 to −0.13 | −0.25 |
| 4a | WMH | −8.15 | 11.58 | 0.48 | −30.85 to 14.54 | −0.07 |
| Amyloid-β1–42 measured in 2009 | ||||||
| 1b | HIP volume | −0.55 | 1.90 | 0.77 | −4.27 to 3.19 | −0.03 |
| 2b | Cortical thickness | 0.24 | 0.14 | 0.10 | −0.04 to 0.52 | 0.15 |
| 3b | PiB score | −0.26 | 0.14 | 0.06 | −0.53 to 0.01 | −0.17 |
| 4b | WMH | −11.57 | 11.33 | 0.31 | −33.77 to 10.64 | −0.10 |
Each independent variable was in separate regression model. WMH = white matter hyperintensities.
Standardized coefficient to 1 SD of independent and dependent variables. Model 1a vs 1b, P = 0.533; Model 2a vs 2b, P = 0.903; Model 3a vs 3b, P = 0.476; Model 4a vs 4b, P = 0.823.
Data availability
The data of the current study are available from the corresponding author upon reasonable request.
Results
Neuroimaging study
Measurement of PiB was available in the Pittsburgh cohort from 194 (27%) GEMS participants and not for 525 participants, 1 year after GEMS close out (Supplementary Table 1 and Supplementary Fig. 1). Participants without PiB measurements had significant greater history at the baseline of heart attack, higher white blood cell count, lower haematocrit and higher aspartate aminotransferase, a liver enzyme. There were no differences in baseline amyloid-β1–40, amyloid-β1–42, amyloid-β1–42/1–40 ratio or prevalence of APOE4 between those who did or did not have PiB measurements (Supplementary Table 1).
The prevalence of PiB+ (SUVR ≥1.57) in the 182 eligible participants was higher for those who were classified as having MCI in 2009, n = 38, 26 (68%) PiB+ positive as compared with 75 (52%) PiB positive among 144 classified as CN. The prevalence of PiB+ was much higher among APOE4 carriers, 29 of 34 (85%), as compared with 62 of 134 (46%) among-APOE4 noncarriers. PiB+ was also associated in 2009 with higher levels of systolic blood pressure and higher pulse wave velocity as measured in 2009. There were, however, no differences in PiB+ in relationship to average age at the time of the scans within the very narrow age range, gender, education and 3MSE scores within CN and MCI participants.
In previous analysis from the total GEMS sample (n = 1824), baseline (2000–02) amyloid-β1–42 was associated with poorer kidney function, higher creatinine levels, history of hypertension and diabetes, age, higher in women but not related to APOE4, education, body mass index or history of CHD (Lopez et al., 2019). In the current study, diabetes and creatinine were significantly related to amyloid-β1–42 in 2000–02. None of the baseline risk factors in 2000–02 were significantly related to blood amyloid-β1–42 levels measured in 2009 (Supplementary Table 2).
Pittsburgh compound-B standardized uptake value ratio and blood amyloid
The analysis included 160 participants with blood drawn in 2000–02, of whom 142 participants diagnosed as CN and 18 participants diagnosed as MCI in 2000–02, and 133 participants with blood drawn in 2009, of whom 107 participants diagnosed as CN and 16 participants diagnosed as MCI in 2000–02.
There was an inverse association between blood amyloid-β1–42 and PiB SUVR, from both plasma samples from 2000–02 and 2009. For example, in 2000–02, the mean PiB SUVR was 1.93, median 1.91, for participants with plasma amyloid in first quartile of blood amyloid-β1–42 and a mean PiB SUVR of 1.63, median 1.49, in the fourth quartile (Table 2). However, there was very substantial overlap between PiB SUVR and plasma amyloid-β1–42 at any level of amyloid-β1–42 (Fig. 3). There was no association between amyloid-β1–40 and PiB (not shown).
Table 2.
Variables by quartile of plasma amyloid-β1–42 at 2000–02 and 2009 in the Pittsburgh GEMS cohort
| Plasma amyloid-β1–42 | HIP volume |
Cortical thickness (mm) |
PiB score |
WMH |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Mean | SD | Median | n | Mean | SD | Median | n | Mean | SD | Median | n | Mean | SD | Median | |
| At 2000–02 | ||||||||||||||||
| 1Q: ≤6.23 | 39 | 0.255 | 0.028 | 0.249 | 39 | 3.00 | 0.33 | 3.05 | 39 | 1.93 | 0.49 | 1.91 | 39 | 0.009 | 0.005 | 0.009 |
| 2Q: 6.24–9.35 | 41 | 0.253 | 0.028 | 0.251 | 41 | 3.07 | 0.38 | 3.06 | 41 | 1.69 | 0.43 | 1.53 | 41 | 0.010 | 0.006 | 0.008 |
| 3Q: 9.36–13.92 | 41 | 0.254 | 0.039 | 0.252 | 41 | 2.99 | 0.52 | 3.11 | 41 | 1.75 | 0.46 | 1.59 | 41 | 0.007 | 0.005 | 0.006 |
| 4Q: >13.92 | 38 | 0.257 | 0.035 | 0.261 | 37 | 3.20 | 0.33 | 3.20 | 38 | 1.63 | 0.43 | 1.49 | 38 | 0.009 | 0.004 | 0.009 |
| P = 0.932, age-adjusted P = 0.924 | P = 0.092, age-adjusted P = 0.097 | P = 0.028, age-adjusted P = 0.023 | P = 0.013, age-adjusted P = 0.007 | |||||||||||||
| At 2009 | ||||||||||||||||
| 1Q: ≤5.81 | 31 | 0.254 | 0.036 | 0.244 | 31 | 3.00 | 0.43 | 3.06 | 31 | 1.75 | 0.45 | 1.68 | 31 | 0.010 | 0.005 | 0.009 |
| 2Q: 5.82–9.34 | 34 | 0.254 | 0.025 | 0.249 | 34 | 3.05 | 0.33 | 3.08 | 34 | 1.80 | 0.42 | 1.86 | 34 | 0.009 | 0.007 | 0.007 |
| 3Q: 9.35–14.01 | 34 | 0.254 | 0.032 | 0.253 | 34 | 3.09 | 0.43 | 3.11 | 34 | 1.58 | 0.43 | 1.47 | 34 | 0.010 | 0.006 | 0.008 |
| 4Q: >14.01 | 34 | 0.253 | 0.035 | 0.256 | 33 | 3.21 | 0.41 | 3.28 | 34 | 1.62 | 0.42 | 1.49 | 34 | 0.008 | 0.004 | 0.008 |
| P = 1.000, age-adjusted P = 0.998 | P = 0.198, age-adjusted P = 0.199 | P = 0.099, age-adjusted P = 0.080 | P = 0.396, age-adjusted P = 0.461 | |||||||||||||
WMH comparisons were based on log-transformed values. Q = quartile; WMH = white matter hyperintensities.
Figure 3.
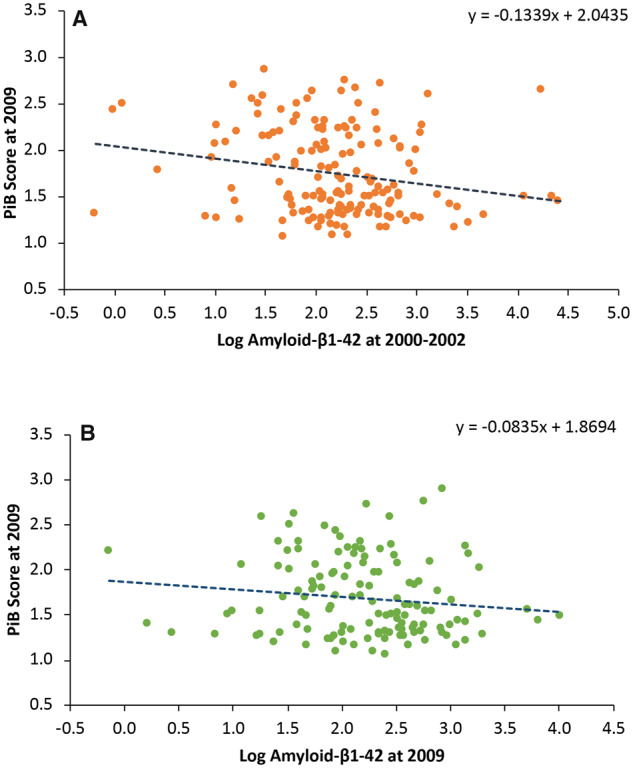
PiB score at 2009 and log amyloid-β1–42 at 2000–02 and 2009 in the Pittsburgh GEMS cohort. (A) Relationship between log amyloid-β1–42 in blood in 2000–02 and PiB score at 2009. (B) Relationship between log amyloid-β1–42 in blood in 2009 and PiB score in 2009.
Age-adjusted regression coefficients with amyloid-β1–42 levels as the dependent variables were significant for PiB SUVR with amyloid-β1–42 measured in blood from 2000 to 2002, β = −0.40, SE 0.14 (P = 0.005), and amyloid-β1–42 measured in blood samples drawn in 2009, β = −0.26, SE 0.14 (P = 0.06). The difference between the two regression coefficients was not significant (Table 1). The association between amyloid-β1–42 and cortical thickness was not significant for 2000–02 blood measurements while there was a significant yet not linear association of amyloid-β1–42 in 2000–02 and white matter abnormalities but not for blood amyloid-β1–42 measured in 2009. (Table 2). There was no significant association between blood amyloid-β1–42 and HIP volume at either time period (Table 2).
Plasma amyloid-β1–42 was divided into quartiles, and the percentage of PiB+ by each quartile was evaluated. Blood amyloid-β1–42 both at baseline 2000–02 and at follow-up 2009 was significantly and inversely related to the percentage of PiB+. The results were similar when the analysis was restricted to CN participants at baseline and at follow-up. Blood levels of amyloid-β1–40 both at baseline in 2000–02 and at follow-up in 2009 were not related to the percentage PiB+ (data not shown). Among the 18 participants with MCI in 2000–02, 5 of the 11 (45%) participants in the first quartile of amyloid-β1–42 were PiB+ versus 3 of the 7 (43%) PiB+ in the other three quartiles, while for 2009 blood samples, 3 of the 5 (60%) participants in the first quartile were PiB+ compared with 4 of the 11 (36%) participants in the other three quartiles based on very small sample sizes.
The amyloid-β1–42/1–40 ratio in blood as measured in 2000–02 was not significantly related to PiB amyloid levels (test statistics, P = 0.622 for linear trend), but ratio from bloods measured at 2009 was significantly related to PiB+ (test statistics, P = 0.039 for linear trend; Supplementary Table 3). These results were similar when the analysis was restricted to those who were CN at 2000–02 or at 2009.
Apolipoprotein-E4 and amyloid-β1–42 in blood and brain amyloid
Most of the participants who were APOE4 carriers were also PiB+ at 2009 (Table 3). Among APOE4 carriers, there was no relationship between amyloid-β1–42 at either baseline or follow-up and PiB+ but only four PiB− at 2000–02 and 2009, severely limiting any statistical analysis. Among APOE4 noncarriers, age-adjusted prevalence of PiB+ was inversely related to the amyloid-β1–42 at both baseline (not significant) and follow-up (Table 3).
Table 3.
PiB status by APOE4 and quartiles of blood amyloid-β1–42 at baseline (2000–02) and follow-up (2009) in the Pittsburgh GEMS cohort
| Quartile | APOE4− |
APOE4+ |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| n | # PiB+ | % PiB+ | Age-adjusted P-value | n | # PiB+ | % PiB+ | Age-adjusted P-value | ||
| Amyloid-β1–42 baseline | 1 | 27 | 16 | 59.3 | 11 | 10 | 90.9 | ||
| 2 | 30 | 11 | 36.7 | 7 | 6 | 85.7 | |||
| 3 | 28 | 14 | 50.0 | 9 | 8 | 88.9 | |||
| 4 | 33 | 10 | 30.3 | 0.072 | 4 | 3 | 75.0 | 0.844 | |
| Amyloid-β1–42 follow-up | 1 | 26 | 13 | 50.0 | 7 | 7 | 100.0 | ||
| 2 | 25 | 16 | 64.0 | 6 | 5 | 83.3 | |||
| 3 | 28 | 9 | 32.1 | 3 | 1 | 33.3 | |||
| 4 | 23 | 6 | 26.1 | 0.026 | 7 | 6 | 85.7 | 0.450 | |
Quartile cut points—amyloid-β1–42 baseline: 1Q: ≤6.36, 2Q: 6.37–9.38, 3Q: 9.39–13.86, 4Q: >13.86; amyloid-β1–42 follow-up: 1Q: ≤6.13, 2Q: 6.14–9.54, 3Q: 9.55–14.01, 4Q: >14.01. Q = quartile.
Brain amyloid by specific brain regions
PiB was evaluated in six areas (anterior cingulate gyrus, anterior ventral striatum, frontal cortex, lateral temporal cortex, parietal cortex and precuneus cortex), and amyloid-β1–42 levels at 2000–02 and 2009 were evaluated in relationship to the extent of PiB retention in each of these regions. The regression coefficient for amyloid-β1–42 and amyloid at specific sites was negative and significant at all six sites based on the measurement of blood amyloid-β1–42 in 2000–02. The strongest associations were within the anterior cingulate gyrus and frontal cortex regions. Similarly, the regression coefficients based on 2009 bloods were negative, smaller in magnitude than those based on 2000–02 bloods and no longer significant but not significantly different from the 2000–02 regression coefficients (Table 4).
Table 4.
Regression coefficients of amyloid-β1–42 at 2000–02 and 2009 as predictors of amyloid SUVR in six brain regions in the Pittsburgh GEMS cohort
|
Regression coefficient for log amyloid-β1–42 at 2000–02 |
Regression coefficient for log amyloid-β1–42 at 2009 |
|||||
|---|---|---|---|---|---|---|
| Estimate | SE | P-value | Estimate | SE | P-value | |
| PiB score in ACG region | −0.18 | 0.06 | 0.006 | −0.13 | 0.07 | 0.066 |
| PiB score in AVS region | −0.12 | 0.05 | 0.012 | −0.09 | 0.05 | 0.079 |
| PiB score in FRC region | −0.18 | 0.06 | 0.002 | −0.10 | 0.06 | 0.124 |
| PiB score in LTC region | −0.09 | 0.04 | 0.042 | −0.04 | 0.05 | 0.433 |
| PiB score in PAR region | −0.12 | 0.05 | 0.016 | −0.07 | 0.05 | 0.210 |
| PiB score in PRC region | −0.13 | 0.06 | 0.019 | −0.08 | 0.06 | 0.233 |
ACG = anterior cyngulate gyrus; AVS = anterior ventral striatum; FRC = frontal cortex; LTC = lateral temporal cortex; PAR = parietal cortex; PRC = precuneus cortex.
There was no significant relationship between the change in amyloid-β1–42 or amyloid-β1–40 between 2000–02 and 2009 and percentage PiB+ among those who were CN at 2000–02 or at 2009 (Supplementary Table 4).
The logistic regression model included blood amyloid-β1–42 as a discrete variable, i.e. quartiles of amyloid-β1–42 with the highest quartile as comparator. The lowest quartile <6.23 pg/ml in 2000–02 was associated with an apparent 4-fold (P = 0.015) increased risk of amyloid positivity (Table 5). Similarly, for amyloid-β1–42 measured at 2009, the odds ratio (OR) was 2.78 (0.84–9.17, P = 0.09). The second quartile was associated with a higher OR than the first quartile. The CIs for these estimates were very wide because of the small sample size in each quartile. In the logistic regression that included the log of amyloid-β1–42 as a continuous variable, the OR for amyloid-β1–42 for blood samples drawn in 2000–02 was 0.56 (0.52–0.99, P = 0.05) and, for 2009 blood samples, the regression coefficient was 0.48 (0.26–0.06, P = 0.023).
Table 5.
Logistic regression model of predictors of PiB+ (SUVR ≥1.57), log plasma amyloid-β1–42 as measured at 2000–02 and 2009 separately in the Pittsburgh GEMS cohort
| Outcome = PiB+ at 2009, N = 143 (76 PiB+) |
Outcome = PiB+ at 2009, N = 120 (61 PiB+) |
||||
|---|---|---|---|---|---|
| OR (95% CI) | P-value | OR (95% CI) | P-value | ||
| Age, per year | 1.12 (0.97, 1.28) | 0.123 | Age, per year | 1.16 (0.99, 1.36) | 0.072 |
| Years of education, per year | 1.03 (0.89, 1.19) | 0.680 | Years of education, per year | 1.02 (0.86, 1.20) | 0.829 |
| Number id="991" of blocks walked, ≤58/>58 | 1.28 (0.54, 3.08) | 0.575 | Number id="994" of blocks walked, ≤58/>58 | 0.93 (0.34, 2.56) | 0.883 |
| Hypertension, yes/no | 1.23 (0.54, 2.82) | 0.618 | Hypertension, yes/no | 2.08 (0.79, 5.48) | 0.137 |
| APOE4, yes/no | 8.84 (2.72, 28.73) | 0.000 | APOE4, yes/no | 7.01 (1.97, 24.96) | 0.003 |
| Ginkgo biloba intervention, yes/no | 1.36 (0.64, 2.90) | 0.428 | Ginkgo biloba intervention, yes/no | 1.28 (0.55, 2.97) | 0.571 |
| Log creatinine | 1.81 (0.27, 12.19) | 0.540 | Log creatinine | 0.92 (0.13, 6.72) | 0.936 |
| Amyloid-β1–42 at 2000 | Amyloid-β1–42 at 2009 | ||||
| ≤6.23 | 3.99 (1.30, 12.19) | 0.015 | ≤5.81 | 2.78 (0.84, 9.17) | 0.093 |
| 6.24–9.35 | 1.24 (0.43, 3.57) | 0.693 | 5.82–9.34 | 5.99 (1.76, 20.37) | 0.004 |
| 9.36–13.92 | 1.87 (0.65, 5.40) | 0.246 | 9.35–14.01 | 1.05 (0.32, 3.43) | 0.933 |
| N | # PiB positive | N | # PiB positive | ||
| Amyloid-β1–42 at 2000 | Amyloid-β1–42 at 2009 | ||||
| 1Q | 35 | 25 | 1Q | 29 | 16 |
| 2Q | 37 | 17 | 2Q | 30 | 23 |
| 3Q | 37 | 21 | 3Q | 31 | 10 |
| 4Q | 34 | 13 | 4Q | 31 | 12 |
Reference group for amyloid-β1–42 at 2000: >13.92. Reference group for amyloid-β1–42 at 2009: >14.01. Quartile cut points—amyloid-β1–42 baseline: 1Q: ≤6.36, 2Q: 6.37–9.38, 3Q: 9.39–13.86, 4Q: >13.86; amyloid-β1–42 follow-up: 1Q: ≤6.13, 2Q: 6.14–9.54, 3Q: 9.55–14.01, 4Q: >14.01. Q = quartile.
Approximately 80–85% of APOE4 carriers were PiB+. These analyses were therefore repeated for APOE4 noncarriers only (44% PiB+). The hazards ratio (HR) for the first quartile of amyloid-β1–42 measured in 2000–02 was 4.7 (95% confidence interval (CI) 1.39–16.0, P = 0.01), and that of amyloid-β1–42 measured in 2009 was 2.51 (95% CI 0.67–9.37, P = 0.17). Again, the second quartile was associated with the highest risk, 8.8 (2.2–35.11, P = 0.002). A further sensitivity analysis was restricted to both participants who were CN at 2009 and APOE4 noncarriers (n = 89, 38 PiB+ in 2000–02 blood samples, and n = 71, 29 PiB+ for analysis of 2009 blood samples). The HR for the first quartile remained very high, 3.47 (95% CI 0.86–14.0), for the first quartile of 2000–02 blood analysis of amyloid-β1–42 and 6.7 (95% CI 1.05–41.2, P = 0.04) for the first quartile of 2009 blood analysis (not shown).
Discussion
Our findings showed that remote (2000–02) and recent (2009) low plasma amyloid-β1–42 levels were significantly inversely associated with 2009 brain fibrillar amyloid deposition in CN individuals. However, changes in plasma amyloid-β1–42 over time between 2000–02 and 2009 were small and not related to the amount of 2009 PiB retention. Unfortunately, the lack of brain amyloid (PiB) at 2000–02 precluded us to determine whether decreased amyloid-β1–42 levels preceded amyloid deposition in this study, although it is probable that some PiB-positive participants in 2009 were also PiB positive at 2000–02, at the time of initial blood samples. It is possible that larger decreases in amyloid-β1–42 over time in between 2000–02 and 2009 were associated with the risk of dementia over the time period and therefore excluded from the analysis. No repeat blood samples were available for participants who were incident dementia. The absence of a baseline brain amyloid measurement therefore limits the evaluation of change in plasma amyloid-β1–42 and change in brain amyloid.
If we assumed that individuals in the lowest quartile of amyloid-β1–42 were ‘at high risk’ for brain amyloid, then 25 of the 76 PiB+ (sensitivity 33%) would be identified and 57 of the 67 (specificity 85%) individuals would be identified as PiB− (Table 5). This blood amyloid-β1–42 measure would not be a good diagnostic test as compared with CSF measures of amyloid-β1–42 or some of the newer, much more expensive blood tests because of this low sensitivity in a population of older individuals with the high prevalence of PiB+ [85 of 160 (53%)] (Table 6). In contrast, it could be used as a very low cost screening method to identify individuals at the higher risk of amyloid positivity as well as the risk of dementia, as occurs when higher serum cholesterol levels are indicative of risk of clinical CHD (Verberk et al., 2018; Lopez et al., 2019).
Table 6.
Quartiles of blood amyloid-β1–42 at baseline (2000–02) and follow-up (2009) and percent PiB positive (SUVR ≥1.57) at 2009 in the Pittsburgh GEMS cohort
| Quartile | Total cohort |
CN only at 2000–02 |
|||||
|---|---|---|---|---|---|---|---|
| n | % PiB+ | Trend P-value | n | % PiB+ | Trend P-value | ||
| Amyloid-β1–42 baseline | 1 | 40 | 70.00 | 0.013 | 29 | 79.31 | 0.013 |
| 2 | 40 | 47.50 | 38 | 44.74 | |||
| 3 | 40 | 57.50 | 38 | 57.89 | |||
| 4 | 40 | 37.50 | 37 | 40.54 | |||
| Amyloid-β1–42 follow-up | 1 | 34 | 61.76 | 0.016 | 29 | 62.07 | 0.029 |
| 2 | 33 | 66.67 | 28 | 67.86 | |||
| 3 | 32 | 34.38 | 29 | 37.93 | |||
| 4 | 34 | 41.18 | 31 | 41.94 | |||
In the current study, the laboratory variability within individuals was low based on split samples as described. There was little difference in levels of amyloid-β1–42 over time in same individuals, within-individual variability, and no evidence of decline in levels due to blood storage. Therefore, the reproducibility of the measurement technique, both laboratory and within-individual variability, for amyloid-β1–42 is good. The accuracy, i.e. levels of amyloid-β1–42 as reported by our laboratory technique in comparison with the actual true levels of amyloid-β1–42 in the blood, requires a new ‘gold standard’, such as mass spectrometry or other newer techniques, and further evaluation in different populations.
The literature of the study of a blood test that can diagnose or predict incident Alzheimer’s disease has been growing over the past years. There is a need to have an accurate peripheral biomarker that can reduce the costs of diagnosis and research. A blood test, i.e. amyloid-β1–42 or amyloid-β1–42/1–40 ratio in blood, could identify individuals at risk during asymptomatic period prior to the development of Alzheimer’s disease (Hampel et al., 2018). Zetterberg recently reviewed the blood-based biomarkers for the Alzheimer’s disease. The brain biomarker molecules are present in much smaller quantities in blood than CSF, creating substantial challenges in laboratory analysis (Zetterberg, 2019).
Previous studies reported a weak correlation between CSF amyloid-β1–42 levels and plasma amyloid levels (Galasko et al., 1998; Mehta et al., 2001; Fagan et al., 2009), while CSF amyloid-β1–42 levels are strongly correlated with cerebral amyloid deposition (Fagan et al., 2006). Lower plasma amyloid-β1–42 and especially the amyloid-β1–42/1–40 ratio were inversely correlated with amyloid deposition in patients with Alzheimer’s disease in several studies (Lui et al., 2010; Burnham et al., 2014; Kaneko et al., 2014; Swaminathan et al., 2014; Tzen et al., 2014). A recent study evaluated plasma tau and amyloid-β1–42 in blood among individuals with prevalent dementia, MCI and CN and found that both high tau and low amyloid-β1–42 were associated with higher tau and amyloid in the brain (Park et al., 2019).
Palmqvist reported a fully automated method of measuring plasma amyloid-β and tau as well as other blood biomarkers. The CSF amyloid-β1–42/1–40 ratio was used as an indicator of brain amyloid status. Plasma amyloid-β1–42 was reduced in CN subjects as compared with participants with MCI and Alzheimer’s disease. Participants with probable amyloid in the brain based on the CSF examination had lower levels of amyloid-β1–42 and amyloid-β1–42/1–40 ratio. The amyloid-β1–42 levels in the blood were lowest in those with Alzheimer’s disease (Palmqvist et al., 2019).
Park and colleagues further evaluated the effect of chemically treating the plasma amyloid-β and brain amyloid as measured by PiB in 358 individuals. Lower levels of modified amyloid-β1–42 were associated with amyloid deposition in the brain (Park et al., 2017). Measures of misfolded proteins related to amyloid-β1–40 and amyloid-β1–42 in the blood using immuno-infrared techniques that measure the secondary structural changes in amyloid-β peptides have successfully identified patients with MCI with brain amyloid from CN without brain amyloid with very high sensitivity and specificity (Nabers et al., 2019).
Another study evaluated plasma amyloid-β1–42/1–40 ratio and other biomarkers and cerebral amyloidosis among 276 participants aged 70–85 years with subjective memory complaints. Seventy-three participants were amyloid-β-PET positive (SUVR 1.07, SD 0.27), and 203 participants were amyloid-β-PET negative (SUVR 0.608, SD 0.05). Plasma levels of amyloid-β1–42 were significantly lower in those classified as positive versus negative (15.1 versus 18.4 pg/ml, respectively). The sensitivity of amyloid-β to identify brain amyloid was very high (75%), and specificity was 77%. The study also noted that other blood biomarkers, such as tau and neurofilament light chain, did not impact the results (Vergallo et al., 2019).
We do not know the initial trigger of Alzheimer’s disease pathology and the origin of amyloid-β. It is possible that there is an increased deposition of amyloid in the brain, primarily amyloid-β1–42, in older individuals caused by decreased clearance of amyloid from the CNS into the blood and hence lower blood amyloid-β1–42 levels, while in younger individuals with genetic forms of Alzheimer’s disease, both increased production and decreased clearance likely contribute to greater brain amyloid deposition (Holtzman, 2004; Mawuenyega et al., 2010; Rissman et al., 2012). Experimental studies using parabiosis techniques and labelling in mice have shown that amyloid-β in the brain can be cleared through the peripheral circulation and that such clearance, i.e. in parabiosis experiments, results in a decrease in Alzheimer’s disease pathology (Xiang et al., 2015).
Differences in the association between amyloid deposition in the brain in older individuals and plasma amyloid may stem from at least three factors. First, it is possible that at least some of the plasma amyloid-β1–42 and amyloid-β1–40 are being produced in the periphery and not in the brain (Rosenberg et al., 1997). Second, there are various factors that can modulate plasma amyloid levels, including age, renal function and liver disease, both excretion and metabolism of amyloid in the blood. In recent studies, the association between plasma amyloid levels was attenuated when renal function, MRI infarcts or evidence of hypertensive vascular disease or diabetes were included in the models (Arvanitakis et al., 2002; Lopez et al., 2008; Gronewold et al., 2016; Hilal et al., 2017; Wang et al., 2017). Third, as noted, there are different methods to measure plasma amyloid that provide varying quantification of levels of these amyloid-β peptides (Lambert et al., 2009; Figurski et al., 2012; Perez-Grijalba et al., 2016; Nakamura et al., 2018). Some of the new techniques, such as mass spectrometry, provide higher sensitivity of levels of amyloid-β1–42 in the blood to predict, and amyloid in the blood may become a diagnostic test for brain amyloid (Nakamura et al., 2018). These mass spectrometry tests are currently more expensive than the methods used in the current study, and unlikely at present to be a screening but rather a potential diagnostic test much like current CSF examinations for PiB+.
The levels of amyloid-β in blood are determined by multiple factors. Most amyloid in the peripheral circulation is bound primarily to albumin, an acute phase protein produced in the liver, and possibly complement in the blood (Zipser et al., 2007; Crane et al., 2018). Consequently, low levels of albumin secondary to multiple factors (e.g. inflammation) could result in a decrease in plasma albumin levels and consequently lower the availability of albumin for the transport of amyloid-β1–42 and amyloid-β1–40 in the blood. There was no significant association of albumin levels in blood with either amyloid-β1–42 or extent of amyloid in the brain in these analyses (data not shown). Amyloid-β1–42 is metabolized in the liver, and high levels of amyloid-β1–42 are found in individuals with severe liver disease (Wang et al., 2017). We did not find any association between liver enzymes and amyloid-β1–42 levels, brain fibrillar amyloid or risk of dementia. We did find and association between plasma amyloid-β1–42 level and kidney function in this cohort. The amyloid-β1–42 is excreted by the kidney, and high levels of creatinine, markers of glomerular function, were associated with higher levels of amyloid-β1–42 in blood (Lopez et al., 2008). The relationship between kidney function and amyloid in plasma may explain, in part, the association between hypertension and diabetes, major risk factors for chronic kidney disease in the elderly, and amyloid deposition in the brain and dementia (Janelidze et al., 2016; van Leijsen et al., 2018). Interestingly, several studies have reported decreases in amyloid-β1–42 levels following renal dialysis and possible change in in cognition (Kitaguchi et al., 2015, 2018). There are also ongoing studies of plasma apheresis to reduce amyloid-β1–42 levels in the blood and potentially decrease amyloid in the brain and improve cognition (Boada et al., 2017).
Conclusions
There is a growing research effort to improve the use of peripheral markers of amyloid deposition in the brain. Clearance of amyloid from the brain to the periphery may be an important determinant of the amount of brain amyloid, especially in the elderly. However, the mechanisms for which peripheral amyloid reflect CNS amyloid are not fully understood. Several investigators have suggested that brain vascular disease may play an important role in decreasing blood–brain barrier function and in the clearance of amyloid (Zlokovic, 2011), including the glymphatic pathways (Donahue and Johanson, 2008; Tarasoff-Conway et al., 2015; Wang et al., 2017; Bowman et al., 2018; Cai et al., 2018).
All future studies of blood biomarkers should include the reproducibility of laboratory methods, biological variability within individuals over time and associations with other methods of measurement of similar variables, i.e. amyloid-β1–42, as well as dependent variables, i.e. brain amyloid, Alzheimer’s disease, Alzheimer’s dementia. If blood amyloid-β1–42 or ratio to other amyloid-β species is a determinant of brain amyloid and dementia, suggested by this and other recent studies, then one of the most important questions is what are the determinants of blood levels?
With continued work, blood amyloid amyloid-β1–42 and other blood markers could be clinically used in in the near future, similarly to blood low density lipoprotein cholesterol or apolipoprotein-B level as a predictor of the extent of atherosclerosis and risk of a heart attack. The availability of blood biomarkers would have a substantial impact on human Alzheimer’s disease research or possibly to preventive therapies.
Funding
This study was supported by grant U01 AT000162 from the National Center for Complementary and Alternative Medicine and the Office of Dietary Supplements and grants P01-AG025204 and P50-AG05133 from the National Institute on Aging.
Competing interests
The authors report no competing interests.
Supplementary Material
Glossary
- APOE4
apolipoprotein-E4
- CN
cognitively normal
- GEMS
Ginkgo Evaluation of Memory Study
- HIP
hippocampal
- MCI
mild cognitive impairment
- MRI
magnetic resonance imaging
- PiB
Pittsburgh compound-B
- ROIs
regions of interest
- SUVR
standardized uptake value ratio
- WML
white matter lesions
References
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 2008; 65: 1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis Z, Lucas JA, Younkin LH, Younkin SG, Graff-Radford NR.. Serum creatinine levels correlate with plasma amyloid beta protein. Alzheimer Dis Assoc Disord 2002; 16: 187–90. [DOI] [PubMed] [Google Scholar]
- Boada M, Anaya F, Ortiz P, Olazarán J, Shua-Haim JR, Obisesan TO, et al. Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid-beta concentrations and cognition outcomes in Alzheimer's disease patients: a multicenter, randomized, controlled clinical trial. J Alzheimers Dis 2017; 56: 129–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman GL, Dayon L, Kirkland R, Wojcik J, Peyratout G, Severin IC, et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimers Dement 2018; 14: 1640–50. [DOI] [PubMed] [Google Scholar]
- Burnham SC, Faux NG, Wilson W, Laws SM, Ames D, Bedo J, et al. A blood-based predictor for neocortical Abeta burden in Alzheimer's disease: results from the AIBL study. Mol Psychiatry 2014; 19: 519–26. [DOI] [PubMed] [Google Scholar]
- Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q.. Role of blood-brain barrier in Alzheimer's disease. J Alzheimers Dis 2018; 63: 1223–34. [DOI] [PubMed] [Google Scholar]
- Chouraki V, Beiser A, Younkin L, Preis SR, Weinstein G, Hansson O, et al. Plasma amyloid-beta and risk of Alzheimer's disease in the Framingham Heart Study. Alzheimers Dement 2015; 11: 249–57.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane A, Brubaker WD, Johansson JU, Trigunaite A, Ceballos J, Bradt B, et al. Peripheral complement interactions with amyloid beta peptide in Alzheimer's disease: 2. Relationship to amyloid beta immunotherapy. Alzheimers Dement 2018; 14: 243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Williamson JD, Fitzpatrick AL, Kronmal RA, Ives DG, Saxton JA, et al. ; Ginkgo Evaluation of Memory Study Investigators. Ginkgo biloba for prevention of dementia: a randomized controlled trial. JAMA 2008; 300: 2253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue JE, Johanson CE.. Apolipoprotein E, amyloid-beta, and blood-brain barrier permeability in Alzheimer disease. J Neuropathol Exp Neurol 2008; 67: 261–70. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee S-Y, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 2006; 59: 512–9. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer's disease. EMBO Mol Med 2009; 1: 371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figurski MJ, Waligórska T, Toledo J, Vanderstichele H, Korecka M, Lee VMY, et al. ; Alzheimer's Disease Neuroimaging Initiative. Improved protocol for measurement of plasma beta-amyloid in longitudinal evaluation of Alzheimer's Disease Neuroimaging Initiative study patients. Alzheimers Dement 2012; 8: 250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B. FreeSurfer. Neuroimage 2012; 62: 774–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Dale AM.. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA 2000; 97: 11050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galasko D, Chang L, Motter R, Clark CM, Kaye J, Knopman D, et al. High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol 1998; 55: 937–45. [DOI] [PubMed] [Google Scholar]
- Gronewold J, Klafki H-W, Baldelli E, Kaltwasser B, Seidel UK, Todica O, et al. Factors Responsible for Plasma beta-Amyloid Accumulation in Chronic Kidney Disease. Mol Neurobiol 2016; 53: 3136–45. [DOI] [PubMed] [Google Scholar]
- Hampel H, O’Bryant SE, Molinuevo JL, Zetterberg H, Masters CL, Lista S, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol 2018; 14: 639–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilal S, Akoudad S, van Duijn CM, Niessen WJ, Verbeek MM, Vanderstichele H, et al. Plasma amyloid-beta levels, cerebral small vessel disease, and cognition: the Rotterdam study. J Alzheimers Dis 2017; 60: 977–87. [DOI] [PubMed] [Google Scholar]
- Holtzman DM. In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology. JMN 2004; 23: 247–54. [DOI] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Weigand SD, Knopman DS, Mielke MM, Vemuri P, et al. Different definitions of neurodegeneration produce similar amyloid/neurodegeneration biomarker group findings. Brain 2015; 138: 3747–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, et al. Plasma beta-amyloid in Alzheimer's disease and vascular disease. Sci Rep 2016; 6: 26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko N, Nakamura A, Washimi Y, Kato T, Sakurai T, Arahata Y, et al. Novel plasma biomarker surrogating cerebral amyloid deposition. Proc Jpn Acad Ser B 2014; 90: 353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaguchi N, Hasegawa M, Ito S, Kawaguchi K, Hiki Y, Nakai S, et al. A prospective study on blood Abeta levels and the cognitive function of patients with hemodialysis: a potential therapeutic strategy for Alzheimer's disease. J Neural Transm 2015; 122: 1593–607. [DOI] [PubMed] [Google Scholar]
- Kitaguchi N, Kato T, Matsunaga S, Hirano K, Iwata K, Kawaguchi K, et al. Removal of blood amyloid-beta with hemodialysis reduced brain amyloid-beta, confirmed by brain imaging: a case report. NDT 2018; Volume 14: 2931–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J-C, Schraen-Maschke S, Richard F, Fievet N, Rouaud O, Berr C, et al. Association of plasma amyloid beta with risk of dementia: the prospective Three-City Study. Neurology 2009; 73: 847–53. [DOI] [PubMed] [Google Scholar]
- Lopez OL, Becker JT, Chang Y, Klunk WE, Mathis C, Price J, et al. Amyloid deposition and brain structure as long-term predictors of MCI, dementia, and mortality. Neurology 2018; 90: e1920–e1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez OL, Chang Y, Ives DG, Snitz BE, Fitzpatrick AL, Carlson MC, Kuller LH.. Blood amyloid levels and risk of dementia in the Ginkgo Evaluation of Memory Study (GEMS): a longitudinal analysis. Alzheimers Dement 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez OL, Kuller LH, Mehta PD, Becker JT, Gach HM, Sweet RA, et al. Plasma amyloid levels and the risk of AD in normal subjects in the Cardiovascular Health Study. Neurology 2008; 70: 1664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JK, Laws SM, Li Q-X, Villemagne VL, Ames D, Brown B, et al. Plasma amyloid-beta as a biomarker in Alzheimer's disease: the AIBL study of aging. J Alzheimers Dis 2010; 20: 1233–42. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Kuller LH, Klunk WE, Snitz BE, Price JC, Weissfeld LA, et al. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Ann Neurol 2013; 73: 751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 2010; 330: 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux R, Tang M-X, Jacobs DM, Manly J, Bell K, Merchant C, et al. Plasma amyloid beta-peptide 1-42 and incipient Alzheimer's disease. Ann Neurol 1999; 46: 412–6. [DOI] [PubMed] [Google Scholar]
- Mehta PD, Pirttila T, Patrick BA, Barshatzky M, Mehta SP.. Amyloid beta protein 1-40 and 1-42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci Lett 2001; 304: 102–6. [DOI] [PubMed] [Google Scholar]
- Nabers A, Hafermann H, Wiltfang J, Gerwert K.. Abeta and tau structure-based biomarkers for a blood- and CSF-based two-step recruitment strategy to identify patients with dementia due to Alzheimer's disease. Alzheimers Dement (Amst) 2019; 11: 257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-beta biomarkers for Alzheimer's disease. Nature 2018; 554: 249–54. [DOI] [PubMed] [Google Scholar]
- Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related beta-amyloid status. JAMA Neurol 2019; 76: 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-C, Han S-H, Cho HJ, Byun MS, Yi D, Choe YM, et al. Chemically treated plasma Abeta is a potential blood-based biomarker for screening cerebral amyloid deposition. Alzheimers Res Ther 2017; 9: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-C, Han S-H, Yi D, Byun MS, Lee JH, Jang S, et al. Plasma tau/amyloid-beta1-42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer's disease. Brain 2019; 142: 771–86. [DOI] [PubMed] [Google Scholar]
- Perez-Grijalba V, Fandos N, Canudas J, Insua D, Casabona D, Lacosta AM, et al. Validation of immunoassay-based tools for the comprehensive quantification of Abeta40 and Abeta42 peptides in plasma. J Alzheimers Dis 2016; 54: 751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, Trojanowski JQ, Shaw LM, Aisen PS.. Longitudinal plasma amyloid beta as a biomarker of Alzheimer's disease. J Neural Transm 2012; 119: 843–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario BL, Ziolko SK, Weissfeld LA, Price JC.. Assessment of parameter settings for SPM5 spatial normalization of structural MRI data: application to type 2 diabetes. Neuroimage 2008; 41: 363–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg RN, Baskin F, Fosmire JA, Risser R, Adams P, Svetlik D, et al. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch Neurol 1997; 54: 139–44. [DOI] [PubMed] [Google Scholar]
- Shah NS, Vidal J-S, Masaki K, Petrovitch H, Ross GW, Tilley C, et al. Midlife blood pressure, plasma beta-amyloid, and the risk for Alzheimer disease: the Honolulu Asia Aging Study. Hypertension 2012; 59: 780–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitz BE, O'Meara ES, Carlson MC, Arnold AM, Ives DG, Rapp SR, et al. ; Ginkgo Evaluation of Memory Study Investigators. Ginkgo biloba for preventing cognitive decline in older adults: a randomized trial. JAMA 2009; 302: 2663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Poljak A, Valenzuela M, Mayeux R, Smythe GA, Sachdev PS.. Meta-analysis of plasma amyloid-beta levels in Alzheimer's disease. J Alzheimers Dis 2011; 26: 365–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S, Risacher SL, Yoder KK, West JD, Shen L, Kim S, et al. ; Alzheimer's Disease Neuroimaging Initiative. Association of plasma and cortical amyloid beta is modulated by APOE epsilon4 status. Alzheimers Dement 2014; 10: e9–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 2015; 11: 457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzen K-Y, Yang S-Y, Chen T-F, Cheng T-W, Horng H-E, Wen H-P, et al. Plasma Abeta but not tau is related to brain PiB retention in early Alzheimer's disease. ACS Chem Neurosci 2014; 5: 830–6. [DOI] [PubMed] [Google Scholar]
- van Leijsen EMC, Kuiperij HB, Kersten I, Bergkamp MI, van Uden IWM, Vanderstichele H, et al. Plasma Abeta (amyloid-beta) levels and severity and progression of small vessel disease. Stroke 2018; 49: 884–90. [DOI] [PubMed] [Google Scholar]
- Verberk IMW, Slot RE, Verfaillie SCJ, Heijst H, Prins ND, van Berckel BNM, et al. Plasma amyloid as prescreener for the earliest Alzheimer pathological changes. Ann Neurol 2018; 84: 648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergallo A, Mégret L, Lista S, Cavedo E, Zetterberg H, Blennow K, et al. Plasma amyloid beta 40/42 ratio predicts cerebral amyloidosis in cognitively normal individuals at risk for Alzheimer's disease. Alzheimers Dement 2019; 15: 764–75. [DOI] [PubMed] [Google Scholar]
- Wang J, Gu BJ, Masters CL, Wang YJ.. A systemic view of Alzheimer disease—insights from amyloid-beta metabolism beyond the brain. Nat Rev Neurol 2017; 13: 703. [DOI] [PubMed] [Google Scholar]
- Wang Y-R, Wang Q-H, Zhang T, Liu Y-H, Yao X-Q, Zeng F, et al. Associations between hepatic functions and plasma amyloid-beta levels—implications for the capacity of liver in peripheral amyloid-beta clearance. Mol Neurobiol 2017; 54: 2338–44. [DOI] [PubMed] [Google Scholar]
- Wolters FJ. 2018. Amyloid in cardiovascular disease In: Wolters FJ, editors. On the origin of dementia. A population perspective on risk and aetiology. Rotterdam, the Netherlands: F.J. Wolters; p. 189–206. [Google Scholar]
- Xiang Y, Bu X-L, Liu Y-H, Zhu C, Shen L-L, Jiao S-S, et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer's disease. Acta Neuropathol 2015; 130: 487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H. Blood-based biomarkers for Alzheimer's disease—an update. J Neurosci Methods 2019; 319: 2–6. [DOI] [PubMed] [Google Scholar]
- Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, et al. Microvascular injury and blood-brain barrier leakage in Alzheimer's disease. Neurobiol Aging 2007; 28: 977–86. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 2011; 12: 723–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data of the current study are available from the corresponding author upon reasonable request.