

Abstract
Diatoms emerged in the Mesozoic period and presently constitute one of the main primary producers in the world’s ocean and are of a major economic importance. In the current study, using whole genome sequencing of ten accessions of the model diatom Phaeodactylum tricornutum, sampled at broad geospatial and temporal scales, we draw a comprehensive landscape of the genomic diversity within the species. We describe strong genetic subdivisions of the accessions into four genetic clades (A–D) with constituent populations of each clade possessing a conserved genetic and functional makeup, likely a consequence of the limited dispersal of P. tricornutum in the open ocean. We further suggest dominance of asexual reproduction across all the populations, as implied by high linkage disequilibrium. Finally, we show limited yet compelling signatures of genetic and functional convergence inducing changes in the selection pressure on many genes and metabolic pathways. We propose these findings to have significant implications for understanding the genetic structure of diatom populations in nature and provide a framework to assess the genomic underpinnings of their ecological success and impact on aquatic ecosystems where they play a major role. Our work provides valuable resources for functional genomics and for exploiting the biotechnological potential of this model diatom species.
Subject terms: Comparative genomics, Biodiversity
Introduction
Diatoms are unicellular predominantly diploid and photosynthetic eukaryotes. They belong to a big group of heterokonts, constituent of chromalveolate (SAR group), which are believed to be derived from serial endosymbiosis combining genes from green and red algae predecessors and further diversified via horizontal gene transfer from a wide range of prokaryotes [1–3]. Ehrenberg [4] first discovered diatoms in the 19th century in dust samples collected by Charles Darwin in the Azores. According to the earliest fossil records, they are believed to be in existence since at least 190 million years [5]. In nature, most diatoms likely live in obligate relationships with bacteria [6] but many, like Phaeodactylum tricornutum, can be propagated in axenic conditions. In spite of its low abundance in the open ocean [7], P. tricornutum is extensively used as a model to study and characterize diatom metabolism, and to understand diatom evolution [1, 8–12].
P. tricornutum is a coastal diatom found under highly unstable environments like estuaries and rock pools. Although it has never been reported to undergo sexual reproduction, factors such as sensitivity to many nonspecific abiotic components and the general lack of knowledge on sexual reproduction in this species [13–15] limit our ability to constrain the sexual cycle of these organisms. Since the discovery of P. tricornutum by Bohlin in 1897 and the characterization of different morphologies, denoted fusiform, triradiate, oval, round and cruciform, ten isolates from nine different geographic locations (sea shores, estuaries, rock pools, and tidal creeks) around the world, from sub-polar to tropical latitudes, have been accessioned (Fig. S1), well described in [16]. These accessions have been collected within the time frame of approximately one century, from 1908 (Plymouth isolate, Pt2/3) to 2000 (Dalian isolate, Pt10) (Fig. S1) [16]. All the isolates have been maintained either axenically or with native bacterial populations in different stock centers and have been cryopreserved after isolation. Previous studies have reported distinct functional behaviors of different accessions as adaptive responses to various environmental cues [17–20], but very little is known about their genetic diversity. However, based on sequence similarity of the ITS2 region within the 28S rDNA repeat sequence, the accessions can be divided into four genotypes (Genotype A: Pt1, Pt2, Pt3 and Pt9; Genotype B: Pt4; Genotype C: Pt5 and Pt10; Genotype D: Pt6, Pt7, and Pt8), with genotypes B and C being the most distant [16]. P. tricornutum is among the few diatom species with a whole genome sequence available to the community [21], and the only diatom for which extensive state-of-the-art functional and molecular tools have been developed over the past few decades [22–35]. These resources have advanced P. tricornutum as a model diatom species and provided a firm platform for future genome wide structural and functional studies.
The accumulated effects of diverse evolutionary forces such as recombination, mutation, and selection have been found to dictate the structure and diversity of genomes in a wide range of species [36–39]. The existence of genomic diversity within a species reflects its potential to adapt to a changing environment. Exploring the genomic diversity within a species not only provides information about its evolution, it also offers opportunities to understand the role of various biotic and abiotic interactions in structuring a genome [40]. Such studies in diatoms are rare and estimates of genetic diversity within diatom populations are mostly inferred using microsatellite-based genotyping approaches [41–43]. Although these techniques have revealed a wealth of information about diatom evolution, their dispersal and reproductive physiology [40], additional insights can be obtained using state-of-the-art whole genome comparative analysis techniques [43]. Deciphering the standing genomic variation of P. tricornutum across different accession populations, sampled at broad geospatial scale, is an important first step to assess the role of various evolutionary forces in regulating the adaptive capacities of diatoms in general (e.g. [44]). To understand the underlying genomic diversity within different accessions of P. tricornutum and to establish the functional implications of such diversity, we performed deep whole genome sequencing of the ten most studied accessions, referred to as Pt1–Pt10 [16, 19, 45]. We present a genome-wide diversity map of geographically distant P. tricornutum accessions, describing a stable genetic structure in the environment. This work further provides the community with whole genome sequences of the accessions, which will be a valuable genetic resource for functional studies of accession-specific ecological traits in the future.
Results
Reference-assisted assembly reveals low nucleotide diversity across multiple accessions of P. tricornutum
We sequenced the whole genomes of ten accessions of P. tricornutum using Illumina HiSeq 2000, and performed a reference-based assembly using the genome sequence of the reference strain Pt1 8.6 [1]. Across all accessions, the percentage of sequence reads mapped on the reference genome ranged between ~65% and ~80% (Table 1), with an alignment depth ranging between 26× and 162×, covering 92–98% of the reference genome (Table 1). Many regions on the reference genome that are observed as being unmapped by reads from individual ecotypes are annotated as rich in transposable elements (TEs) (Fig. S2). At >90% identity, the repeated proportion of unmapped reads varies between ~38% (Pt1) and 75% (Pt4).
Table 1.
Reference-assisted mapping statistics
| Library name | Accession number/axenicity | Origin | Year of isolation | Mapped read pairs | % mapped read pairs | Alignment depth (X) | Genome coverage (%) |
|---|---|---|---|---|---|---|---|
| Pt1 | CCMP2561 (axenic) | Blackpool, UK | 1956 | 3,642,044 | 79.41 | 26.5 | 98.0 |
| Pt2 | CCMP2557 (xenic) | Plymouth, UK | Prior to 1910 | 6,016,241 | 78.23 | 43.8 | 98.0 |
| Pt3 | CCMP2558 (axenic) | Plymouth, UK | 1930s | 6,373,591 | 65.62 | 46.4 | 98.3 |
| Pt4 | CCMP2559 (xenic) | Finland | 1951 | 15,583,665 | 67.31 | 113.5 | 94.0 |
| Pt5 | CCMP630 (axenic) | West Dennis, MA, USA | 1972 | 5,346,009 | 75.50 | 38.9 | 93.2 |
| Pt6 | CCMP631 (xenic) | MA, USA | 1956 | 3,922,830 | 64.50 | 28.5 | 94.1 |
| Pt7 | CCMP1327 (axenic) | Long Island, NY, USA | 1952 | 4,937,516 | 67.30 | 35.9 | 94.9 |
| Pt8 | CCMP2560 (xenic) | Vancouver, Canada | 1987 | 22,235,170 | 78.36 | 162.1 | 94.4 |
| Pt9 | CCMP633 (axenic) | Guam, Micronesia | 1981 | 7,551,099 | 74.68 | 55.2 | 97.5 |
| Pt10 | CCMP2928 (axenic) | Dalian, China | 2000 | 5,436,057 | 72.59 | 39.6 | 92.1 |
The table summarizes the origin and year of sampling of each accession of P. tricornutum along with the number of total reads mapped on the reference. Average depth (X = average number of reads aligned on each base covered across the entire genome) was estimated using the number of mapped read pairs and the horizontal coverage (aka. coverage breadth) across the whole genome
Following the assembly, we performed variant calling using Genome Analysis Toolkit [46] and discovered 462,514 (depth ≥ 4×) single nucleotide polymorphisms (SNPs) including ~25% singleton sites, 573 insertions (of varying lengths from 1 to 312 bp) and 1801 deletions (of lengths from 1 to 400 bp) (Fig. 1a), across all the accessions. The spectrum of SNPs across all the accessions further reveals a higher rate of transitions (Ts) over transversions (Tv) (Ts/Tv = 1.6). In total, compared with the reference alleles from Pt1 8.6, six possible types of single nucleotide changes could be distinguished, among which G:C -> A:T and A:T -> G:C accounted for more than ∼60% of the observed mutations (Fig. S3A). Further, most SNPs and INDELs (insertions and deletions) are shared between different accessions, except for Pt4, which possesses the highest proportion of specific SNPs (~35%) and INDELs (~75%) (Fig. 1b). Interestingly, we found that most of the SNPs are heterozygous, and the proportion of heterozygous variants across all the accessions varies between ~45% (in Pt5 and Pt10) to ~98% (in Pt1, Pt2, and Pt3) (Fig. 1c). Most of the variant alleles in the accessions with high proportions of heterozygous variants were further found to be significantly deviated from Hardy–Weinberg equilibrium (HWE) (chi-square test, P-value < 0.05) (Fig. 1c), possibly linked to prolonged asexual reproduction [47]. Surprisingly, despite significant differences in the proportion of heterozygote variant alleles between the accessions, which ranges between 45 and 98%, the average pairwise synonymous nucleotide diversity (πS) estimated from genes with callable sites across all the accessions is 0.007 per synonymous site. This indicates that any two homologous sequences taken at random across different populations will on average differ by only ∼0.7% on synonymous positions. The nonsynonymous pairwise diversity (πN) over the same genes is 0.003, consistent with an excess of nonsynonymous mutations being deleterious. An average nonsynonymous (N) to synonymous (S) variant ratio (πN/πS) was estimated to be ~0.43, which is higher than in the Ostreococcus tauri, πN / πS = 0.2 [48]. Since πN/πS is negatively correlated with the effective population size, which is the number of distinct clones in unicellular organisms [49], this could reflect either a lower census population size in P. tricornutum, and/or a lower rate of sexual reproduction. Linkage disequilibrium (LD) analysis using only homozygous SNP sites revealed, on average, high LD (>0.7) over pairs of variations, genome wide (Fig. S3B). Further, based on the difference in the allelic frequencies of the SNPs, the pairwise Fst between the populations ranges from ∼0.005 (between Pt1 and Pt3) to ∼0.4 (between Pt4 and Pt10) (Fig. 1d). Considering Fst as a measure of genetic differentiation or structuring between the populations, the ten P. tricornutum accessions can be clustered into 4 genetic groups/clades with Pt1, Pt2, Pt3, and Pt9 in clade A; Pt4 in clade B; Pt5, Pt10 in clade C; and Pt6, Pt7, Pt8 in clade D, reflecting low intra-group Fst (∼0.02) and high inter-group Fst (0.2–0.4) (Fig. 1d).
Fig. 1.

Genetic diversity between P. tricornutum accessions. a The bar plot represents total number of discovered SNPs, with the proportion of heterozygous SNPs (dark blue) and homozygous SNPs (light blue), insertions (orange) and deletions (yellow) in each accession compared with the reference genome. b The stack bar plot represents the proportion of total vs specific polymorphic variant sites, including SNPs, insertions and deletions (from left to right, respectively) across all the accessions. c The world map indicates proportion of heterozygous (dark violet) and homozygous SNPs (violet) in each accession, represented as pie charts. The outer ring represents the proportion of variant alleles being significantly deviated from HWE (deep red). d The heat-map shows the genetic differentiation or association between all possible pairs of accessions. The colors indicate Fst values, which range from 0.02 to 0.4, with a color gradient from yellow to green, respectively. Values closer to 0 signify close genetic makeup and values closer to one indicate strong genetic structuring between the populations
Four genetic clades of P. tricornutum
With the exception of Pt4, where we found the maximum number of variant alleles to be accession-specific, most of the variant alleles are shared between at least two accessions, indicating close genetic relatedness (Fig. 1b). Therefore, in order to cluster the accessions based on the genome structure shared among them, we used Bayesian clustering approach by applying Markov Chain Monte Carlo (MCMC) estimations, programmed within the ADMIXTURE software [50]. Based on the allelic composition of the ten genomes, six genomic clusters (K = 6) can be formed, which is distributed within each accession genome in different proportions (represented by pie-chart colors) (Fig. 2a). Further, depending on the pattern (both qualitative and quantitative) of distributed genomic clusters across different accession genomes, the ten accessions revealed four genetic clusters with Pt1, Pt2, Pt3, and Pt9 in one, Pt4 in a second, Pt5, Pt10 in a third, and Pt6, Pt7, Pt8 in a fourth cluster (Fig. 2b). These clusters (Fig. 2b) are in broad agreement with Fst-based genetic clades (Fig. 1d), phylogenetic clusters inferred using ribosomal marker genes (18S (Fig. S4A), and ITS2 (Fig. S4B)), as also reported previously [16], and at whole genome scale (this study) as inferred by a phylogenetic tree generated using maximum likelihood algorithm based on all (Fig. 2c) and only homozygous polymorphic sites (SNVs and INDELS) (Fig. S4C).
Fig. 2.

Clustering of P. tricornutum accessions. a Principal component analysis (PCA) showing the distribution of the ten accessions based on their shared genome structure, revealing four genetic clusters referred to as clades A, B, C, and D. b Pie charts showing the genetic makeup of the genomes of each accession. Based on the allelic composition of the ten genomes, six genomic clusters (K = 6) are formed, which are distributed across individual accession genome in different proportions (represented by six different colors in the pie-chart). c Phylogenetic association of the accessions based on 468,188 genome-wide polymorphic sites (including SNP and INDELS) using a maximum likelihood approach. The numbers on the branches indicate the bootstrap values. Pie charts adjacent to each node of the whole genome tree correspond to the proportion of SNPs and INDELs over all functional features of the genome; GENEs (blue), TEs (yellow), IGRs (Intergenic Regions, represented in gray)
Further sequential assessment of the 18S and ITS2 rDNA gene sequences across different clades indicated the presence of multiple variations, including both heterozygous and homozygous variant alleles (Fig. S4D, E). Because the ribosomal DNA region including 18S and ITS2 is highly repetitive, which is on average ~4 times more than non-ribosomal genes (Fig. 3a), these differences can be understood as intra-genomic variations within the genome. However, taxonomists and ecologists use differences within 18S gene sequences as a measure of species assignation and to estimate species delineation [7]. This latter practice has been previously shown to be very conservative as no differences in the 18S gene were found between reproductively isolated species [51]. Alternatively, the possibility of sub-populations or cryptic populations cannot be ignored, as previously reported in planktonic foraminifers [52] and coccolithophores [53].
Fig. 3.

Large structural variations within accessions. a The heat-map displays the fold change (FC) of read depth between each reference gene and median of read depth of all the reference genes, within each accession. Using Z-score as a measure of normalized read depth, log2 fold change (FC) is calculated as a ratio of Z-score per gene to the average normalized read depth of all the genes per accession. A blue to red color gradient in the heat-map represents low to high log2FC. From all the accessions only those genes are plotted where log2FC is more than 2 in at least one of the accessions and are considered to exhibit copy number variation (CNV). b The bar plots represent the total and specific numbers of genes, denoted on the Y-axis, that exhibit a loss or multiple copies (CNV) within one or more accessions. c The bar plots represent the number of total- and accession-specific TEs exhibiting CNVs across one or more accessions. d With similar principle esthetics as a of this figure, the heat-map shows the patterns of log2FC only across all the accessions of those TEs exhibiting CNV in at least one of the ten accessions studied
We examined the possible presence of sub-populations on 18S gene heterozygosity in some of the accessions. We confirmed the expression of all the heterozygous alleles within the 18S rDNA gene using whole genome and total-RNA sequencing of a monoclonal culture (propagated from a single cell) from Pt8 (constituent of Clade D) and Pt3 (constituent of Clade A) population, referred to as Pt8Tc and Pt3Ov (Fig. S4D), respectively. This experiment indicates that the cultures (Pt1–Pt10) are a single population with no or undetectable heterogeneity.
Next, concerning the observed polymorphisms within the 18S ribosomal marker gene, we investigated whether the four clades can be considered as different species. We looked for the existence of compensatory base changes (CBCs) within secondary structures of the ITS2 gene between all pairs of accessions. The presence of CBCs within ITS2 has been recently suggested to account for reproductive isolation in multiple plant species [54] and between diatom species [55, 56]. By comparing the ITS2 secondary structure from all the accessions, we did not find any CBCs between any given pair of accessions (Fig. S5). As a control, we compared the ITS2 secondary structure of all the P. tricornutum accessions with the ITS2 sequences of other diatom species (Cyclotella meneghiniana, Pseudo-nitzschia delicatissima, Pseudo-nitzschia multiseries, Fragilariopsis cylindrus) that have significant degrees of evolutionary divergence as depicted previously using multiple molecular marker genes [21, 57], and found multiple CBCs in them (Fig. S5).
Close genetic relatedness depicted by large structural genomic variations among accessions
Next, using a normalized measure of read depth (see Experimental procedures), we found that 259 and 590 genes, representing ~2% and ~5% of the total gene content, respectively, have been lost or exhibit copy number variation (CNV), across the ten accessions (Fig. 3a, b) (File S1). Multiple randomly chosen loci were also validated by PCR for their loss from certain accessions compared with the reference strain Pt1 8.6 (Fig. S6). Compared with the reference, ~70% of the genes that are either lost or show CNV are shared among multiple accessions with an exception of Pt10, which displays the maximum number of lost genes and accession-specific genes exhibiting CNV (Fig. 3b). In addition, we detected 207 TEs (~6% of the total annotated TEs) (File S2) showing CNVs across one or more accessions (Fig. 3c, d), 80% of which are shared among two or more accessions, with Pt10 again possessing the maximum number of accession-specific TEs exhibiting CNVs (Fig. 3c). Not surprisingly, across all the accessions, class I-type TEs, which undergo transposition via a copy-and-paste mechanism, show more variation in the estimated number of copies than class II-type TEs (Figs. 3d and S7) that are transposed by a cut-and-paste mechanism. Euclidean distance estimated between accessions, based on the variation in the number of copies of different genes and TEs displaying CNVs, followed by hierarchical clustering, depicted three genetic clusters: Pt1, Pt2, Pt3, Pt9 in cluster1; Pt5, Pt10 in cluster 2, and Pt4, Pt6, Pt7, Pt8 in cluster 3 (Fig. 3a, d). These clusters are in broad agreement with the ones described by Fst and indicate the closer genetic makeup between accessions within a cluster than between the clusters. Further, biological processes can only be traced for ~40% of the genes exhibiting accession-specific CNVs. Among all the enriched biological processes (chi-square test, P < 0.01) (File S1), a gene associated to nitrate assimilation (Phatr3_EG02286) is observed to have higher copy number specifically in Pt4. Likewise, each accession can be characterized by specific genetic features, represented by ~0.3% to ~28% accession-specific CNVs (Fig. 3b), possibly linked to the explicit functional behavior of some accessions in response to various environmental cues, as reported previously [17–19].
Evolutionary adaptation in P. tricornutum clades
Species are under continuous pressure to adapt to a changing environment over time. We therefore wanted to understand the functional consequences of the genetic diversity between the accessions. Localization of the polymorphic sites over genomic features (genes, TEs, and intergenic regions) revealed highest rate of variation within genes (Fig. 2c), specifically on exons, and was consistent across all the studied accessions. We further identified genes within different phylogenetic clades experiencing different selection pressure based on lowest and highest πN/πS ratios. Across all the accessions, 241 genes displaying πN/πS > 1 and a higher frequency of nonsynonymous to synonymous polymorphism, as expected under balancing selection (BS) [58] (File S3). Furthermore, across all the accessions, 128 genes exhibit a signature of relaxed selection (RS) with accumulation of NS polymorphisms, among which 47% are specific to one or other clades (Fig. 4a). In addition, many genes (902) were found to have loss-of-function (LoF hereafter) variant alleles (Fig. 4a), including frame-shift mutations and mutations leading to theoretical start/stop codon loss and/or gain.
Fig. 4.

Evolutionary and functional consequences of polymorphisms. a The bar plot represents total and specific numbers of genes that are subject to relaxed selection, or experiencing loss-of-function (LoF) mutations. For each category, the accessions are plotted as stack plots with total and specific numbers of genes. Numbers of genes in each category are indicated. b The box plot represents the number of gene families affected by loss-of-function (LoF) mutations and suggests a bias of such mutations on the genes belonging to large gene families. The Y-axis represents, as log scale, the number of genes in the gene families vs those that are not affected by LoF mutations
Based on the presence of functional domains (Pfam domains), all P. tricornutum annotated genes [59] were grouped into 3020 gene families. These families can be as large as the reverse transcriptase gene family, which is highly abundant in marine plankton [60], representing 149 candidate genes having reverse transcriptase domains, or as small as families that constitute single gene candidates. Across all the accessions, we observed that most genes experiencing LoF mutations belong to large gene families (Fig. 4b). This is consistent with a previous observation of the existence of functional redundancy in gene families as a balancing mechanism for null mutations in yeast [61]. Therefore, to estimate an unbiased effect of any evolutionary pressure (e.g., LoF allele mutations) on different gene families, we calculated a ratio, termed the effect ratio (EfR, see Experimental procedures), which normalizes that if any gene family has enough candidates to buffer the effect on some genes influencing evolutionary pressure, it will be considered as being less affected compared with those for which all or most of the constituents are under selection pressure. From this analysis, each genetic clade displayed a specific set of gene families as being under selection (Fig. 5). Functional enrichment of constrained genes revealed enrichment of (1) AAA family proteins that often perform chaperone like functions that assist in the assembly or disassembly of proteins complexes, protein transport and degradation as well as other functions such as replication, recombination, repair and transcription [62], (2) tetratricopeptide-like repeats known for their role in a variety of biological processes, such as cell cycle regulation, organelle targeting and protein import, vesicle fusion and biomineralization [63]. A redox class of enzymes are common to both groups of genes and a significant proportion of unknown function proteins is found in the group of genes under BS (File S3).
Fig. 5.

Relaxed selection within each genetic clade. Based on the EfR metric, the network displays highly affected gene families experiencing balancing selection. Gene families associated with MetH gene in all the accessions are indicated within the blue circles. The red circles group individual accessions as clades
Finally, considering all pairwise correlated gene families exhibiting similar selection signals, measured using EfR among the ten accessions, we used hierarchical clustering to examine the functional closeness of accessions with one another. Consistent with the population structure, accessions within individual clades are more closely related than the accessions belonging to other clades (Fig. S8A, B), suggesting variation in functional relatedness between different proposed phylogenetic clades.
Selection of MetH-facilitated methionine biosynthesis over MetE
Apart from the clade-specific genes that are under high selection pressure as depicted by high rate of N/S divergence when compared with the reference, a group of gene families associated with methionine biosynthesis (MetH, Phatr3_J23399) was also observed to accumulate nonsynonymous polymorphisms in all the accessions. In P. tricornutum, MetE (cobalamin-independent methionine synthase) and MetH (cobalamin-dependent methionine synthase) are known to catalyze conversion of homocysteine to methionine in the presence of symbiotic bacteria and vitamin B12 in the growth media, respectively. Previous reports have suggested that growing axenic cultures in conditions of high cobalamin (vitamin B12) availability results in repression of MetE, leading to its LoF and high expression of the MetH gene in P. tricornutum and Chlamydomonas reinhardtii [64–66]. In accordance with these results, we observed a high expression of MetH in axenically grown laboratory cultures (Fig. 6a) compared with its expression in cells cultured with their natural co-habitant bacteria. However, we were not able to trace any significant signature for the loss of MetE gene although its expression is significantly lower in axenic cobalamin-containing cultures (Fig. 6b). Similar observations were obtained for CBA1 and SHMT genes (Fig. 6c, d), which under cobalamin scarcity enhance cobalamin acquisition and manage reduced methionine synthase activity, respectively [65].
Fig. 6.

Selection of MetH-facilitated methionine biosynthesis over MetE. The bar plots represent relative expression of a MetH, b MetE, c CBA1, and d SHMT genes in four (Pt2, Pt3, Pt4, and Pt8) of the ten accessions with the presence of vitamin B12 in axenic cultures (light gray), and with natural bacteria and no vitamin B12 in the growing media (black)
Discussion
Using whole genome sequence analysis of accessions sampled across multiple geographic locations around the world (Fig. S1), the aim of this study was to describe the global genetic and functional diversity of the model diatom P. tricornutum. By defining a comprehensive landscape of natural variations across multiple accessions, we could investigate genetic structure between P. tricornutum populations, and a summary of our results is presented in Fig. 7. To do so, we first performed reference-based assembly and found consistently high genome coverage (>90%) mapped by sequencing reads from respective accessions, where some accessions have more coverage (>98%, Pt1, Pt2, Pt3, and Pt9) than others (Table 1). This difference is independent of the size of the sequencing library, as it does not correlate with the genome coverage (Table 1), and a portion of unmapped reads is likely a consequence of the incomplete reference genome, which contains several gaps [1]. In addition, given the redundant nature of unmapped reads together with the fact that the unmapped reference genome is annotated as being rich in TEs (Fig. S2), a major portion of unmapped reads likely account for large structural variability within the genomes of individual accessions. This explanation is most clear in Pt10, which is shown to have the largest number of gene losses (Fig. 3b) and the highest number of accession-specific TEs with high copy numbers (Fig. 3c) and covers at least (92%) of the reference genome (Table 1). This suggests the role of TEs in creating substantial genetic diversity as also shown in many species of plants and animals [67, 68].
Fig. 7.
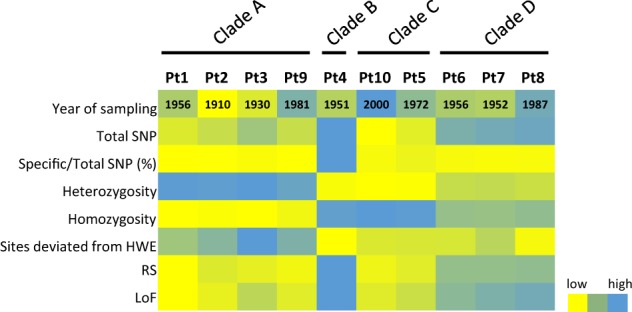
Population structure of the ecotypes. The color gradient from yellow to blue indicates low to high numerical values across each ecotype (indicated on the top X-axis of a) within different functional categories indicated on the Y-axis. These functional categories include (from top to bottom), Year of sampling = year in which the respective ecotype was sampled, Total SNP = absolute number of SNPs found in each ecotype, Specific/Total SNP (%) = percentage of ecotype specific SNPs, Heterozygosity = number of heterozygous SNPs from a set of total SNPs within each ecotype, Homozygosity = number of homozygous SNPs from a set of total SNPs within respective ecotype, sites deviated from HWE = number of SNP
Next, based on patterns of variations discovered over the whole genomes and on the molecular marker genes (18S and ITS2) of all the accessions, and by using various clustering algorithms (see “Results”), the ten accessions could be grouped into four genetic clades. Clade A clusters Pt1, Pt2, Pt3, Pt9; clade B includes Pt4; clade C clusters Pt5, Pt10; and clade D clusters Pt6, Pt7, Pt8. Most of the structural variants discovered, both small (SNPs and INDELS) and large (CNV and Gene Loss), are shared among populations within a clade rather than between clades. This suggests high intra-clade relatedness over a variety of structural, functional and possibly ecological traits.
P. tricornutum is a coastal species with limited dispersal potential, which is consistent with the reports of its absence in the open ocean from Tara Oceans data [7]. Consequently, the Fixation index (Fst) between different genetic clades is very high (0.2–0.4) (Fig. 1d), confirming the existence of accessions subdivisions into four genetic clades. As also expected for an organism with limited dispersal potential, the accessions show partial geographical structuring (Fig. 2a, b), as Pt5, Pt9, and Pt10 clusters with accessions not sampled from proximal locations. These dispersals to different localities may be fostered by ocean currents [42], human activities like rafting, ballasting [69, 70], and migration of birds [71–73]. In addition, the fact that the subdivisions do not correlate with the sampling time (Figs. 7 and S1), which spans approximately a century, suggests long and stable genetic populations, which is in line with reports from other diatom species [41, 42]. This suggests as reported in these studies that environmental conditions have a more important impact in structuring the populations than dispersal potential and generation time.
Although there exist partial genetic structuring within the accessions, the average nucleotide diversity (π), estimated across all the accessions, is remarkably low (0.2%) compared with the diversity estimates in other unicellular eukaryotes [36, 48, 74–76] but in line with previous estimations in marine phytoplanktonic eukaryotes [77]. Given the observation that there exist a large proportion of heterozygous variant alleles (Fig. 1c), the high Fst between the clades, and the low nucleotide diversity across the accessions, we propose that allele frequency plays a significant role in the genetic differentiation of the clades. The difference in allele frequencies is possibly linked to adaptive selection. This phenomenon has recently been studied in diatoms where allele-specific expression of numerous loci has been demonstrated to be a significant source of adaptive evolution in the cold-adapted diatom species F. cylindrus [78]. Furthermore, high proportions of heterozygous variant alleles in some clades (clade A, 98%, Fig. 7) compared with others (clade B, 45%, Fig. 7) suggests a high selection pressure in the clade B accession Pt4. High Fst, and yet low nucleotide diversity across all the accessions, suggests some degree of genetic and functional convergence among the accessions. This can be explained as a consequence of laboratory culture mediated domestication where some genes accumulate nonsynonymous mutations which might result from the relaxation of selective constraints in an artificial environment.
It is also worth considering that genetic homogenization or functional convergence across the meta-population can also be a consequence of continuous gene flow between the accessions. However, in the case of P. tricornutum, gene flow seems limited as highly differentiated populations show partial geographical structure, except Pt9 of clade A, Pt5 of clade C and Pt8 of clade D (Fig. 2a, b). This is consistent with earlier findings in Emilinia huxleyi and Thalassiosira pseudonana in which genomes of isolates from different geographical locations clustered together in the phylogeny [79, 80]. In addition, P. tricornutum is not known to reproduce sexually, although various components (genes) of the meiosis pathway are conserved in P. tricornutum as well as in other diatom species known to undergo sexual reproduction [81]. Furthermore, the absence of contemporary base changes (CBC) within ITS2 secondary structure between all the accessions compared with the presence of many CBCs between P. tricornutum accessions and other diatom species suggests that the accessions may be able to exchange genetic material sexually. However, because P. tricornutum is a coastal diatom with only limited dispersal capacity, which is further supported by its apparent absence in the open ocean [7], the possibility of gene flow within different populations is likely to be limited at best.
Next, high linkage disequilibrium (>0.7) observed across all the accessions (Fig. S3B) can be explained by prolonged asexual reproduction [82], a common behavior among diatoms [80]. Asexual reproduction results in higher proportions of divergent alleles within loci with less genetic variation among individuals, and a significant deviation from HWE [82]. Therefore, it is likely that clade B with one isolate, Pt4 also undergoes sexual reproduction reasonably often compared with clade A accessions, as Pt4 possesses the smallest number of heterozygous variant alleles, most of which follow HWE (Figs. 1c and 7). To our surprise, despite high variability in the levels of heterozygosity between different accessions (Fig. 7), the mutational spectrum, compared with the reference, and across all the accessions consisted of high G:C -> A:T and A:T -> G:C transitions (Fig. S3A). Deamination of cytosines dominantly dictates C to T transitions in both plants and animals [83, 84], and CpG methylation potential of the genome is greatly influenced by heterozygous SNPs in CpG dinucleotides [85]. Previous studies have demonstrated low DNA methylation in P. tricornutum, using Pt1 8.6, a monoclonal isolate accessioned from a Pt1 single cell as a reference [32, 86]. Because there exist significant differences in the proportion of heterozygote variant alleles between the accessions (45–98%), testing for DNA methylation patterns across different accessions may provide an interesting opportunity to dissect cross-talk between loss of heterozygosity and DNA methylation in the selection of certain traits [87].
The four genetic clades are further supported by functional specialization of grouped populations (Fig. 5), nicely illustrated with Pt4 in clade B. Pt4 shows a low nonphotochemical quenching capacity [18], which was proposed to be an adaptive trait to low light conditions. Specifically, this accession has been proposed to establish an upregulation of a peculiar light harvesting protein LHCX4 in extended dark conditions [18, 20]. In line with these observations, a gene involved in nitrate assimilation (Phatr3_EG02286) in Pt4 shows high copy numbers, suggesting an altered mode of nutrient acquisition. Nitrate assimilation was shown to be regulated extensively under low light or dark conditions to overcome nitrate limitation of growth in Thalassiosira weissflogii [88]. Pt4 is likely adapted to the low light and highly seasonal environment that characterizes the high latitudes where it was found, which may well affect its nitrate assimilation capacity [89, 90]. Additional functions emerging from clade C (Pt5 and Pt10) include vacuolar sorting and vesicle-mediated transport gene families, which could be an indication of altered intracellular trafficking [91].
In conclusion, the study presents pan-genomic diversity of the model diatom P. tricornutum. This is the first study within diatoms that provides a comprehensive landscape of diversity at whole genome sequence level and brings new insights to our understanding of diatom functional ecology and evolution. Given our observation that P. triconrutum accessions possess high numbers of heterozygous alleles, it would be interesting to think of possible selective functional preferences of one allele over the other under different environmental conditions or during the life/cell cycle. In the future, such studies could be crucial for deciphering the mechanisms underpinning allele divergence and selection within diatoms. Likewise, more than answers, our study delivers more questions, which should help address the genetic basis of diatom success in diverse ocean ecosystems. Finally, this study provides the community with genomic sequences of P. tricornutum accessions that can be useful for functional studies.
Experimental procedures
Sample preparation, sequencing, and mapping
Ten different accessions of P. tricornutum were obtained from the culture collections of the Provasoli-Guillard National Center for Culture of Marine Phytoplankton (CCMP, Pt1 = CCMP632, Pt5 = CCMP630, Pt6 = CCMP631, Pt7 = CCMP1327, Pt9 = CCMP633), the Culture Collection of Algae and Protozoa (CCAP, Pt2 = CCAP 1052/1A, Pt3 = CCAP 1052/1B, Pt4 = CCAP 1052/6), the Canadian Center for the Culture of Microorganisms (CCCM, Pt8 = NEPCC 640), and the Microalgae Culture Collection of Qingdao University (MACC, Pt10 = MACC B228). All of the accessions were grown axenically in batch cultures with a photon fluency rate of 75 μmol photons m−2 s−1 provided by cool-white fluorescent tubes in a 12:12 light: dark (L:D) photoperiod at 20 °C. Exponentially growing cells were harvested and total DNA was extracted with the cetyltrimethylammonium bromide method [92]. At least 6 μg of genomic DNA from each accession was used to construct a sequencing library following the manufacturer’s instructions (Illumina Inc.). Paired-end sequencing libraries with a read size of 100 bp and an insert size of approximately 400 bp were sequenced on an Illumina HiSeq 2000 sequencer at Berry Genomics Company (China). The corresponding data can be accessed using bioSample accessions: SAMN08369620, SAMN08369621, SAMN08369622, SAMN08369623, SAMN08369624, SAMN08369625, SAMN08369626, SAMN08369627, SAMN08369628, SAMN08369629, SAMN12551644 (Pt3Ov genomic), SAMN12551645 (Pt3Ov Transcriptomic), and SAMN12551646 (Pt8Tc genomic). Low quality read pairs were discarded using FASTQC with a read quality (Phred score) cutoff of 30. Using the genome assembly published in 2008 as reference [1], we performed reference-assisted assembly of all the accessions. We used BOWTIE (-n 2 –X 400) for mapping the high quality NGS reads to the reference genome followed by the processing and filtering of the alignments using SAMTOOLS and BEDTOOLS. Detailed methods are provided in File S4.
Discovery of small polymorphisms and large structural variants
GATK [46], configured for diploid genomes, was used for variant calling, which included SNPs, small insertions (of varying lengths from 1 to 312 bp) and deletions (of lengths from 1 to 400 bp). The genotyping mode was kept default (genotyping mode = DISCOVERY), Emission confidence threshold (-stand_emit_conf) was kept 10 and calling confidence threshold (-stand_call_conf) was kept at 30. The minimum number of reads per base, to be called as a high quality SNV, was kept to 4 (read depth ≥ 4×). Following this filtration step, the number of sites in the protein coding genes covered for all ten accessions, and therefore callable to estimate the genome wide synonymous and nonsynonymous polymorphism, added up 11.0 Mbp. The average pairwise synonymous and nonsynonymous diversity πS and πN [93] were estimated for all genes using in-house R script from [93] equation 22 for each gene and the complete callable coding sequences (available from the authors upon request).
Next, considering Z-score as a normalized measure of read-depth, gene and TE candidates showing multiple copies (representing CNV) or apparently being lost (representing gene loss) were determined. For TE CNV analysis, TEs that are more than 100 bp lengths were considered. We measured the fold change (Fc) by dividing normalized read depth per genomic feature (Z-score per gene or TE) by average of normalized read depth of all the genes/TEs (average Z-score), per sample. Genes or TEs with log2 scaled fold change > = 2 were reported and considered to exist in more than one copy in the genome. Genes where the reads from individual accession sequencing library failed to map on the reference genome were considered as potentially lost within that accession and reported. Detailed method is provided in File S4. Later, some randomly chosen loci were picked and validated for the loss in the accessions compared with the reference genome by PCR analysis.
Validation of gene loss and quantitative PCR analysis
In order to validate gene loss, DNA was extracted from all the accessions as described previously [22] and PCR was performed with the primers listed in Table S1. PCR products were loaded in 1% agarose gel and after migration gels were exposed to UV light and photographs were taken using a gel documentation apparatus to visualize the presence and absence of amplified fragment. To assess gene expression, RNA was extracted as described in [23] from three biological replicates of accessions grown axenically in artificial sea water (ASW) [94] supplemented with vitamins as well as in the presence of their endemic bacteria in ASW without vitamins. qPCR was performed as described previously [23]. Briefly, cDNA was synthetized from 1 µg RNA using High-Capacity cDNA Reverse Transcription Kit (catalog number 4368813) from Fischer scientific and according to manufacturer instructions. 1 µl of cDNA was used in the QPCR reaction with the LightCycler® 480 SYBR Green I Master (catalogue number 04707516001) from Roche and according to the manufacturer’s instructions. Two reference genes were used, Tata Box binding Protein and Ribosomal Protein Small subunit 30S for normalization [23].
P. tricornutum population structure
Haplotype analysis
First, to cluster the accessions as haplogroups, ITS2 gene (chr13: 42150–43145) and 18S gene (chr13: 43553–45338) were used. Polymorphic sites across all the accessions within ITS2 and 18S genes were called and used to generate their corresponding accession-specific sequences, which were then aligned using CLUSTALW. The same approach was employed to perform haplotype analysis at the whole genome scale. Later, a maximum likelihood algorithm was used to generate the 18S, ITS2 and, whole genome tree with bootstrap values of 1,000. We used MEGA7 [95] to align and deduce the phylogenetic trees.
CBC analysis
CBC analysis was done by generating the secondary structure of ITS2 sequences, using RNAfold [96], across all P. tricornutum accessions and other diatom species. The other species include one centric diatom species C. meneghiniana (AY906805.1), and three pennate diatoms P. delicatissima (EU478789.1), P. multiseries (DQ062664.1), F. cylindrus (EF660056.1). The centroid secondary structures of ITS2 gene with lowest minimum free energy were used for CBC analysis. We used 4SALE [97] for estimating the presence of CBCs between the secondary structure of ITS2 gene across all the species.
Population genetics
Further, we measured various population genetic functions to estimate the effect of evolutionary pressure in shaping the diversity and resemblance between different accession populations. Within individual accessions, by using approximate allelic depths of reference/alternate alleles, we calculated the alleles that are deviated from HWE. We used chi-square estimation to evaluate alleles observed to deviate significantly (P-value < 0.05) from the expected proportion as per p2 (homozygous) + 2pq (heterozygous) + q2 (homozygous) = 1 and should be 0.25% + 0.50% + 0.25%. Alleles were considered heterozygous if the proportion of ref/alt allele is between 20 and 80%. The proportion of ref/alt allele was calculated by dividing the number of reads supporting ref/alt base change by total number of reads mapped at the position. We evaluated average R2 as a function to measure the linkage disequilibrium with increasing distance (1, 5, 10, 20, 30, 40, and 50) between any given pair of mutant alleles across all the accessions using expectation-maximization algorithm deployed in the VCFtools. Although no recombination was observed within the accessions, attempts were made to look for recombination signals using LDhat [98] and RAT [99]. Genetic differentiation or variability between the accessions was further assessed using the mathematical function of Fixation index (Fst), as described by Weir and Cockerham [100].
Genetic clustering
Genetic clustering of the accessions was done using Bayesian clustering approach by applying MCMC estimation programmed within ADMIXTURE (version linux-1.3.0) [50]. Accessory tools like PLINK (version 1.07-x86_64) [101] and VCFtools (version 0.1.13) [102] were used to format the VCF files to ADMIXTURE accepted formats. In the absence of data from individuals of each accession/sample, we assumed the behavior of each individual in a sample to be coherent. Conclusively, instead of estimating the genetic structure within an accession, we compared it across all the accessions. We first estimated the possible clusters of genomes, (K), across all the accessions, by using cross-validation error (CV error) function of ADMIXTURE [103]. We chose the value of K with lowest CV error (see extended methods, File S4). Finally, we used ADMIXTURE with 200 bootstraps, to estimate the genome clusters within individual accessions by considering the possible number of genomes derived via CV-error function.
Functional characterization of polymorphisms
snpEff [104] and KaKs [105] calculator were used to annotate the functional nature of the polymorphisms. Along with the nonsynonymous, synonymous, LoF alleles, transition to transversion ratio and mutational spectrum of the single nucleotide polymorphisms were also measured. πN/πS ratios were calculated for 5232 protein coding genes containing more than ten SNPs. Ten percent of genes with lower πN/πS were considered as under strong purifying selection on amino-acid composition (File S3). Genes with πN/πS > 1, and average frequency on nonsynonymous polymorphism higher than the average frequency of synonymous polymorphism were considered as candidate genes under BS on amino-acid composition (File S3). Various in-house scripts were also used at different levels for analysis and for plotting graphs. Data visualization and graphical analysis were performed principally using ClicO [106], CYTOSCAPE [107], IGV [108] and R (https://www.r-project.org/about.html). Based on the presence of functional domains all the Phatr3 genes [59] were grouped into 3020 gene families. Subsequently, the constituents of each gene family were checked for being either affected by LoF mutations or under relaxed selective constraints. To estimate an unbiased effect of any evolutionary pressure (LoF allele or BS mutations) on different gene families, induced because of high functional redundancies in the gene families, a normalized ratio named as EfR, was calculated. Precisely, the EfR normalizes the fact that if any gene family have enough candidates to buffer the effect on some genes influencing evolutionary pressures, it will be considered as less affected compared with the situation where all or most of the constituents are under selection pressure. The ratio was estimated as shown below and gene families with EfR larger than 1 were considered as being significantly affected.
In addition, significantly enriched (chi-square test, P-value < 0.05) biological processes associated within genes experiencing LoF mutations, purifying selection, BS, or showing CNV, or being lost (GnL), were estimated by calculating observed to expected ratio of their percent occurrence within the given functional set (BS, LoF, and CNV) and their occurrence in the complete annotated Phatr3 (http://protists.ensembl.org/Phaeodactylum_tricornutum/Info/Index) biological process catalog. Later, considering gene family EfR as a function to measure the association rate, we deduced Pearson pairwise correlations between different accessions. The correlation matrix describes that if many equally affected gene families are shared between any given pair of accessions, they will have higher correlation compared with others. Finally, hierarchical clustering using Pearson pairwise correlation matrix assessed the association between the accessions.
Supplementary information
Acknowledgements
HH acknowledges support from National Natural Science Foundation of China (grant no. 91751117). GW acknowledges the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDA17010502). CB acknowledges funding from the ERC Advanced Award ‘Diatomite’, the LouisD Foundation of the Institut de France, the Gordon and Betty Moore Foundation, and the French Government ‘Investissements d’Avenir’ programs MEMO LIFE (ANR-10-LABX-54), PSL* Research University (ANR-1253 11-IDEX-0001-02), and OCEANOMICS (ANR-11-BTBR-0008). CB also thanks the Radcliffe Institute of Advanced Study at Harvard University for a scholar’s fellowship during the 2016–2017 academic year. LT acknowledges funds from the CNRS, MEMO LIFE (ANR-10-LABX-54) and the region of Pays de la Loire (ConnecTalent EPIALG project). AR was supported by an International PhD fellowship from MEMO LIFE (ANR-10-LABX-54).
Author contributions
LT, HH, and CB conceived the study. LT, AR, and GP designed the study. GW, PV, AFDC, CC and LT did the experiments. AR, FRJV, AV, and GP developed and performed the bioinformatics analysis. AR, GP, FRJV, and LT interpreted the results. AR and LT wrote the paper with input from all the authors. LT supervised the study. LT, CB, HH and GP coordinated the study.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hanhua Hu, Phone: +86-27-68780078, Email: hanhuahu@ihb.ac.cn.
Leila Tirichine, Phone: +33-276645058, Email: tirichine-l@univ-nantes.fr.
Supplementary information
The online version of this article (10.1038/s41396-019-0528-3) contains supplementary material, which is available to authorized users.
References
- 1.Bowler C, Allen AE, Badger JH, Grimwood J, Jabbari K, Kuo A, et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature. 2008;456:239–44. doi: 10.1038/nature07410. [DOI] [PubMed] [Google Scholar]
- 2.Moustafa A, Beszteri B, Maier UG, Bowler C, Valentin K, Bhattacharya D. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science. 2009;324:1724–6. doi: 10.1126/science.1172983. [DOI] [PubMed] [Google Scholar]
- 3.Dorrell RG, Gile G, McCallum G, Meheust R, Bapteste EP, Klinger CM, et al. Chimeric origins of ochrophytes and haptophytes revealed through an ancient plastid proteome. Elife. 2017;6:1–45. [DOI] [PMC free article] [PubMed]
- 4.C.G. E: Die Infusionsthierchen als vollkommene Organismen. Ein Blick in das tiefere organische Leben der Natur. Leipzig: Leopold Voss; 1838. [PubMed]
- 5.Armbrust EV. The life of diatoms in the world’s oceans. Nature. 2009;459:185–92. doi: 10.1038/nature08057. [DOI] [PubMed] [Google Scholar]
- 6.Amin SA, Parker MS, Armbrust EV. Interactions between diatoms and bacteria. Microbiol Mol Biol Rev. 2012;76:667–84. doi: 10.1128/MMBR.00007-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malviya S, Scalco E, Audic S, Vincent F, Veluchamy A, Poulain J, et al. Insights into global diatom distribution and diversity in the world’s ocean. Proc Natl Acad Sci USA. 2016;133:1516–25. [DOI] [PMC free article] [PubMed]
- 8.Allen AE, Dupont CL, Obornik M, Horak A, Nunes-Nesi A, McCrow JP, et al. Evolution and metabolic significance of the urea cycle in photosynthetic diatoms. Nature. 2011;473:203–7. doi: 10.1038/nature10074. [DOI] [PubMed] [Google Scholar]
- 9.Huysman MJ, Fortunato AE, Matthijs M, Costa BS, Vanderhaeghen R, Van den Daele H, et al. AUREOCHROME1a-mediated induction of the diatom-specific cyclin dsCYC2 controls the onset of cell division in diatoms (Phaeodactylum tricornutum) Plant Cell. 2013;25:215–28. doi: 10.1105/tpc.112.106377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrissey J, Sutak R, Paz-Yepes J, Tanaka A, Moustafa A, Veluchamy A, et al. A novel protein, ubiquitous in marine phytoplankton, concentrates iron at the cell surface and facilitates uptake. Curr Biol. 2015;25:364–71. doi: 10.1016/j.cub.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka T, Maeda Y, Veluchamy A, Tanaka M, Abida H, Marechal E, et al. Oil accumulation by the oleaginous diatom Fistulifera solaris as revealed by the genome and transcriptome. Plant Cell. 2015;27:162–76. doi: 10.1105/tpc.114.135194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fortunato AE, Jaubert M, Enomoto G, Bouly JP, Raniello R, Thaler M, et al. Diatom phytochromes reveal the existence of far-red-light-based sensing in the ocean. Plant Cell. 2016;28:616–28. doi: 10.1105/tpc.15.00928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Godhe A, Kremp A, Montresor M. Genetic and microscopic evidence for sexual reproduction in the centric diatom Skeletonema marinoi. Protist. 2014;165:401–16. doi: 10.1016/j.protis.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Moore ER, Bullington BS, Weisberg AJ, Jiang Y, Chang J, Halsey KH. Morphological and transcriptomic evidence for ammonium induction of sexual reproduction in Thalassiosira pseudonana and other centric diatoms. PLoS One. 2017;12:e0181098. doi: 10.1371/journal.pone.0181098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mouget JL, Gastineau R, Davidovich O, Gaudin P, Davidovich NA. Light is a key factor in triggering sexual reproduction in the pennate diatom Haslea ostrearia. FEMS Microbiol Ecol. 2009;69:194–201. doi: 10.1111/j.1574-6941.2009.00700.x. [DOI] [PubMed] [Google Scholar]
- 16.De Martino A, Meichenin A, Shi J, Pan KH, Bowler C. Genetic and phenotypic characterization of Phaeodactylum tricornutum (Bacillariophyceae) accessions. J Phycol. 2007;43:992–1009. [Google Scholar]
- 17.Stanley MS, Callow JA. Whole cell adhesion strength of morphotypes and isolates of Phaeodactylum tricornutum (Bacillariophyceae) Eur J Phycol. 2007;42:191–7. [Google Scholar]
- 18.Bailleul B, Rogato A, de Martino A, Coesel S, Cardol P, Bowler C, et al. An atypical member of the light-harvesting complex stress-related protein family modulates diatom responses to light. Proc Natl Acad Sci USA. 2010;107:18214–9. doi: 10.1073/pnas.1007703107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abida H, Dolch LJ, Mei C, Villanova V, Conte M, Block MA, et al. Membrane glycerolipid remodeling triggered by nitrogen and phosphorus starvation in Phaeodactylum tricornutum. Plant Physiol. 2015;167:118–36. doi: 10.1104/pp.114.252395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taddei L, Stella GR, Rogato A, Bailleul B, Fortunato AE, Annunziata R, et al. Multisignal control of expression of the LHCX protein family in the marine diatom Phaeodactylum tricornutum. J Exp Bot. 2016;67:3939–51. doi: 10.1093/jxb/erw198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tirichine L, Rastogi A, Bowler C. Recent progress in diatom genomics and epigenomics. Curr Opin Plant Biol. 2017;36:46–55. doi: 10.1016/j.pbi.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Falciatore A, Casotti R, Leblanc C, Abrescia C, Bowler C. Transformation of nonselectable reporter genes in marine diatoms. Mar Biotechnol. 1999;1:239–51. doi: 10.1007/pl00011773. [DOI] [PubMed] [Google Scholar]
- 23.Siaut M, Heijde M, Mangogna M, Montsant A, Coesel S, Allen A, et al. Molecular toolbox for studying diatom biology in Phaeodactylum tricornutum. Gene. 2007;406:23–35. doi: 10.1016/j.gene.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 24.De Riso V, Raniello R, Maumus F, Rogato A, Bowler C, Falciatore A. Gene silencing in the marine diatom Phaeodactylum tricornutum. Nucleic Acids Res. 2009;37:e96. doi: 10.1093/nar/gkp448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huysman MJ, Martens C, Vandepoele K, Gillard J, Rayko E, Heijde M, et al. Genome-wide analysis of the diatom cell cycle unveils a novel type of cyclins involved in environmental signaling. Genome Biol. 2010;11:R17. doi: 10.1186/gb-2010-11-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maheswari U, Jabbari K, Petit JL, Porcel BM, Allen AE, Cadoret JP, et al. Digital expression profiling of novel diatom transcripts provides insight into their biological functions. Genome Biol. 2010;11:R85. doi: 10.1186/gb-2010-11-8-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maheswari U, Mock T, Armbrust EV, Bowler C. Update of the diatom EST database: a new tool for digital transcriptomics. Nucleic Acids Res. 2009;37:D1001–1005. doi: 10.1093/nar/gkn905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaur S, Spillane C. Reduction in carotenoid levels in the marine diatom Phaeodactylum tricornutum by artificial microRNAs targeted against the endogenous phytoene synthase gene. Mar Biotechnol. 2015;17:1–7. doi: 10.1007/s10126-014-9593-9. [DOI] [PubMed] [Google Scholar]
- 29.Diner RE, Bielinski VA, Dupont CL, Allen AE, Weyman PD. Refinement of the diatom episome maintenance sequence and improvement of conjugation-based DNA delivery methods. Front Bioeng Biotechnol. 2016;4:65. doi: 10.3389/fbioe.2016.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nymark M, Sharma AK, Sparstad T, Bones AM, Winge P. A CRISPR/Cas9system adapted for gene editing in marine algae. Sci Rep. 2016;6:24951. doi: 10.1038/srep24951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rastogi A, Murik O, Bowler C, Tirichine L. PhytoCRISP-Ex: a web-based and stand-alone application to find specific target sequences for CRISPR/CAS editing. BMC Bioinf. 2016;17:261. doi: 10.1186/s12859-016-1143-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veluchamy A, Lin X, Maumus F, Rivarola M, Bhavsar J, Creasy T, et al: Insights into the role of DNA methylation in diatoms by genome-wide profiling in Phaeodactylum tricornutum. Nat Commun. 2013;4:1–9. [DOI] [PubMed]
- 33.Veluchamy A, Rastogi A, Lin X, Lombard B, Murik O, Thomas Y, et al. An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol. 2015;16:102. doi: 10.1186/s13059-015-0671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daboussi F, Leduc S, Marechal A, Dubois G, Guyot V, Perez-Michaut C, et al. Genome engineering empowers the diatom Phaeodactylum tricornutum for biotechnology. Nat Commun. 2014;5:3831. doi: 10.1038/ncomms4831. [DOI] [PubMed] [Google Scholar]
- 35.Serif M, Dubois G, Finoux AL, Teste MA, Jallet D, Daboussi F. One-step generation of multiple gene knock-outs in the diatom Phaeodactylum tricornutum by DNA-free genome editing. Nat Commun. 2018;9:3924. doi: 10.1038/s41467-018-06378-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flowers JM, Hazzouri KM, Pham GM, Rosas U, Bahmani T, Khraiwesh B, et al. Whole-genome resequencing reveals extensive natural variation in the model green alga Chlamydomonas reinhardtii. Plant Cell. 2015;27:2353–69. doi: 10.1105/tpc.15.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao J, Schneeberger K, Ossowski S, Gunther T, Bender S, Fitz J, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–63. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- 38.Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, et al. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–41. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lachance J, Tishkoff SA. Population genomics of human adaptation. Annu Rev Ecol Evol Syst. 2013;44:123–43. doi: 10.1146/annurev-ecolsys-110512-135833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Godhe Anna, Rynearson Tatiana. The role of intraspecific variation in the ecological and evolutionary success of diatoms in changing environments. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1728):20160399. doi: 10.1098/rstb.2016.0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harnstrom K, Ellegaard M, Andersen TJ, Godhe A. Hundred years of genetic structure in a sediment revived diatom population. Proc Natl Acad Sci USA. 2011;108:4252–7. doi: 10.1073/pnas.1013528108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whittaker KA, Rynearson TA. Evidence for environmental and ecological selection in a microbe with no geographic limits to gene flow. Proc Natl Acad Sci USA. 2017;114:2651–6. doi: 10.1073/pnas.1612346114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rengefors K, Kremp A, Thorsten BH, Reusch A, Wood M. Genetic diversity and evolution in eukaryotic phytoplankton: revelations from population genetic studies. J Plankton Res. 2017;39:165–79. [Google Scholar]
- 44.Matuszewski S, Hermisson J, Kopp M. Catch me if you can: adaptation from standing genetic variation to a moving phenotypic optimum. Genetics. 2015;200:1255–74. doi: 10.1534/genetics.115.178574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailleul B, Berne N, Murik O, Petroutsos D, Prihoda J, Tanaka A, et al. Energetic coupling between plastids and mitochondria drives CO2 assimilation in diatoms. Nature. 2015;524:366–9. doi: 10.1038/nature14599. [DOI] [PubMed] [Google Scholar]
- 46.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Gang, Rynearson Tatiana A. Genetically distinct populations of a diatom co-exist during the North Atlantic spring bloom. Limnology and Oceanography. 2016;61(6):2165–2179. [Google Scholar]
- 48.Blanc-Mathieu R, Krasovec M, Hebrard M, Yau S, Desgranges E, Martin J, et al. Population genomics of picophytoplankton unveils novel chromosome hypervariability. Sci Adv. 2017;3:e1700239. doi: 10.1126/sciadv.1700239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai IJ, Bensasson D, Burt A, Koufopanou V. Population genomics of the wild yeast Saccharomyces paradoxus: quantifying the life cycle. Proc Natl Acad Sci USA. 2008;105:4957–62. doi: 10.1073/pnas.0707314105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piganeau G, Eyre-Walker A, Jancek S, Grimsley N, Moreau H. How and why DNA barcodes underestimate the diversity of microbial eukaryotes. PLoS One. 2011;6:e16342. doi: 10.1371/journal.pone.0016342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Vargas C, Norris R, Zaninetti L, Gibb SW, Pawlowski J. Molecular evidence of cryptic speciation in planktonic foraminifers and their relation to oceanic provinces. Proc Natl Acad Sci USA. 1999;96:2864–8. doi: 10.1073/pnas.96.6.2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saez AG, Probert I, Geisen M, Quinn P, Young JR, Medlin LK. Pseudo-cryptic speciation in coccolithophores. Proc Natl Acad Sci USA. 2003;100:7163–8. doi: 10.1073/pnas.1132069100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolf M, Chen S, Song J, Ankenbrand M, Muller T. Compensatory base changes in ITS2 secondary structures correlate with the biological species concept despite intragenomic variability in ITS2 sequences—a proof of concept. PLoS One. 2013;8:e66726. doi: 10.1371/journal.pone.0066726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaczmarska Irena, Mather Laura, Luddington Ian A., Muise France, Ehrman James M. Cryptic diversity in a cosmopolitan diatom known as Asterionellopsis glacialis (Fragilariaceae): Implications for ecology, biogeography, and taxonomy. American Journal of Botany. 2014;101(2):267–286. doi: 10.3732/ajb.1300306. [DOI] [PubMed] [Google Scholar]
- 56.Amato A, Kooistra WH, Ghiron JH, Mann DG, Proschold T, Montresor M. Reproductive isolation among sympatric cryptic species in marine diatoms. Protist. 2007;158:193–207. doi: 10.1016/j.protis.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Medlin LK. A timescale for diatom evolution based on four molecular markers: reassessment of ghost lineages and major steps defining diatom evolution. Vie Milieu/Life Environ. 2015;65:219–38.
- 58.Bitarello BD, de Filippo C, Teixeira JC, Schmidt JM, Kleinert P, Meyer D, et al. Signatures of long-term balancing selection in human genomes. Genome Biol Evol. 2018;10:939–55. doi: 10.1093/gbe/evy054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rastogi A, Maheswari U, Dorrell RG, Vieira FRJ, Maumus F, Kustka A, et al. Integrative analysis of large scale transcriptome data draws a comprehensive landscape of Phaeodactylum tricornutum genome and evolutionary origin of diatoms. Sci Rep. 2018;8:4834. doi: 10.1038/s41598-018-23106-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lescot M, Hingamp P, Kojima KK, Villar E, Romac S, Veluchamy A, et al. Reverse transcriptase genes are highly abundant and transcriptionally active in marine plankton assemblages. ISME J. 2016;10:1134–46. doi: 10.1038/ismej.2015.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gu Z, Steinmetz LM, Gu X, Scharfe C, Davis RW, Li WH. Role of duplicate genes in genetic robustness against null mutations. Nature. 2003;421:63–6. doi: 10.1038/nature01198. [DOI] [PubMed] [Google Scholar]
- 62.Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure—diverse function. Genes Cells. 2001;6:575–97. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 63.Zeytuni N, Zarivach R. Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure. 2012;20:397–405. doi: 10.1016/j.str.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 64.Helliwell KE, Wheeler GL, Leptos KC, Goldstein RE, Smith AG. Insights into the evolution of vitamin B12 auxotrophy from sequenced algal genomes. Mol Biol Evol. 2011;28:2921–33. doi: 10.1093/molbev/msr124. [DOI] [PubMed] [Google Scholar]
- 65.Bertrand EM, Allen AE, Dupont CL, Norden-Krichmar TM, Bai J, Valas RE, et al. Influence of cobalamin scarcity on diatom molecular physiology and identification of a cobalamin acquisition protein. Proc Natl Acad Sci USA. 2012;109:E1762–71. doi: 10.1073/pnas.1201731109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Helliwell KE, Collins S, Kazamia E, Purton S, Wheeler GL, Smith AG. Fundamental shift in vitamin B12 eco-physiology of a model alga demonstrated by experimental evolution. ISME J. 2015;9:1446–55. doi: 10.1038/ismej.2014.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quadrana L, Bortolini Silveira A, Mayhew GF, LeBlanc C, Martienssen RA, Jeddeloh JA, et al. the Arabidopsis thaliana mobilome and its impact at the species level. Elife. 2016;5:1–25. [DOI] [PMC free article] [PubMed]
- 68.Bonchev G, Parisod C. Transposable elements and microevolutionary changes in natural populations. Mol Ecol Resour. 2013;13:765–75. doi: 10.1111/1755-0998.12133. [DOI] [PubMed] [Google Scholar]
- 69.Thiel MaG L. The ecology of rafting in the marine environment. II. The rafting organisms and community. Oceanogr Mar Biol: Annu Rev. 2005;43:279–418. [Google Scholar]
- 70.Nikula R, Spencer HG, Waters JM. Passive rafting is a powerful driver of transoceanic gene flow. Biol Lett. 2013;9:20120821. doi: 10.1098/rsbl.2012.0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schlichting HE. The rôle of waterfowl in the dispersal of algae. Trans Am Microsc Soc. 1960;79:160–6. [Google Scholar]
- 72.Proctor VW. Dispersal of desmids by birds. Phycologia. 1966;5:227–32. [Google Scholar]
- 73.Foissner W. Biogeography and dispersal of micro-organisms: a review emphasizing protists. Acta Protozool. 2006;45:111–36. [Google Scholar]
- 74.Liti G. The fascinating and secret wild life of the budding yeast S. cerevisiae. Elife. 2015;4:1–9. [DOI] [PMC free article] [PubMed]
- 75.Blanc-Mathieu R, Verhelst B, Derelle E, Rombauts S, Bouget FY, Carre I, et al. An improved genome of the model marine alga Ostreococcus tauri unfolds by assessing Illumina de novo assemblies. BMC Genom. 2014;15:1103. doi: 10.1186/1471-2164-15-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hirakawa MP, Martinez DA, Sakthikumar S, Anderson MZ, Berlin A, Gujja S, et al. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res. 2015;25:413–25. doi: 10.1101/gr.174623.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Filatov DA. Extreme lewontin’s paradox in ubiquitous marine phytoplankton species. Mol Biol Evol. 2019;36:4–14. doi: 10.1093/molbev/msy195. [DOI] [PubMed] [Google Scholar]
- 78.Mock T, Otillar RP, Strauss J, McMullan M, Paajanen P, Schmutz J, et al. Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature. 2017;541:536–40. doi: 10.1038/nature20803. [DOI] [PubMed] [Google Scholar]
- 79.Read BA, Kegel J, Klute MJ, Kuo A, Lefebvre SC, Maumus F, et al. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature. 2013;499:209–13. doi: 10.1038/nature12221. [DOI] [PubMed] [Google Scholar]
- 80.Koester JA, Berthiaume CT, Hiranuma N, Parker MS, Iverson V, Morales R, et al. Sexual ancestors generated an obligate asexual and globally dispersed clone within the model diatom species Thalassiosira pseudonana. Sci Rep. 2018;8:10492. doi: 10.1038/s41598-018-28630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patil S, Moeys S, von Dassow P, Huysman MJ, Mapleson D, De Veylder L, et al. Identification of the meiotic toolkit in diatoms and exploration of meiosis-specific SPO11 and RAD51 homologs in the sexual species Pseudo-nitzschia multistriata and Seminavis robusta. BMC Genomics. 2015;16:930. doi: 10.1186/s12864-015-1983-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Allen DE, Lynch M. The effect of variable frequency of sexual reproduction on the genetic structure of natural populations of a cyclical parthenogen. Evolution. 2012;66:919–26. doi: 10.1111/j.1558-5646.2011.01488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Becker C, Hagmann J, Muller J, Koenig D, Stegle O, Borgwardt K, et al. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature. 2011;480:245–9. doi: 10.1038/nature10555. [DOI] [PubMed] [Google Scholar]
- 84.Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. doi: 10.1038/ng.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010;20:883–9. doi: 10.1101/gr.104695.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huff JT, Zilberman D. Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell. 2014;156:1286–97. doi: 10.1016/j.cell.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Hirohashi S. Aberrant DNA methylation precedes loss of heterozygosity on chromosome 16 in chronic hepatitis and liver cirrhosis. Cancer Lett. 2000;148:73–80. doi: 10.1016/s0304-3835(99)00316-x. [DOI] [PubMed] [Google Scholar]
- 88.Clark Darren R., Flynn Kevin J., Owens Nicholas J.P. The large capacity for dark nitrate-assimilation in diatoms may overcome nitrate limitation of growth. New Phytologist. 2002;155(1):101–108. doi: 10.1046/j.1469-8137.2002.00435.x. [DOI] [PubMed] [Google Scholar]
- 89.Ivanikova NV, McKay R, Bullerjahn GS. Construction and characterization of a cyanobacterial bioreporter capable of assessing nitrate assimilatory capacity in freshwaters. Limnol Oceanogr. 2005;3:86–93. [Google Scholar]
- 90.Li Weiguo, Wang Jianbo. Influence of light and nitrate assimilation on the growth strategy in clonal weed Eichhornia crassipes. Aquatic Ecology. 2010;45(1):1–9. [Google Scholar]
- 91.Pickett-Heaps JD, Forer A. Pac-Man does not resolve the enduring problem of anaphase chromosome movement. Protoplasma. 2001;215:16–20. doi: 10.1007/BF01280300. [DOI] [PubMed] [Google Scholar]
- 92.Doyle JJaJLD. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytoch Bull. 1987;19:11–15. [Google Scholar]
- 93.Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA. 1979;76:5269–73. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vartanian M, Descles J, Quinet M, Douady S, Lopez PJ. Plasticity and robustness of pattern formation in the model diatom Phaeodactylum tricornutum. New Phytol. 2009;182:429–42. doi: 10.1111/j.1469-8137.2009.02769.x. [DOI] [PubMed] [Google Scholar]
- 95.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, et al. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seibel PN, Muller T, Dandekar T, Schultz J, Wolf M. 4SALE–a tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinf. 2006;7:498. doi: 10.1186/1471-2105-7-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Auton A, McVean G. Recombination rate estimation in the presence of hotspots. Genome Res. 2007;17:1219–27. doi: 10.1101/gr.6386707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Etherington GJ, Dicks J, Roberts IN. Recombination Analysis Tool (RAT): a program for the high-throughput detection of recombination. Bioinformatics. 2005;21:278–81. doi: 10.1093/bioinformatics/bth500. [DOI] [PubMed] [Google Scholar]
- 100.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 101.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinf. 2011;12:246. doi: 10.1186/1471-2105-12-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strainw1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Z, Li J, Zhao XQ, Wang J, Wong GK, Yu J. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteom Bioinform. 2006;4:259–63. doi: 10.1016/S1672-0229(07)60007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cheong WH, Tan YC, Yap SJ, Ng KP. ClicO FS: an interactive web-based service of Circos. Bioinformatics. 2015;31:3685–7. doi: 10.1093/bioinformatics/btv433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.