

Abstract
Background and Purpose
Nox4 is the major isoform of NADPH oxidase found in the kidney and contributes to the pathogenesis of diabetic nephropathy. However, the molecular mechanisms of increased Nox4 expression induced by hyperglycaemia remain to be elucidated. Here, the role of the connexin32–Nox4 signalling axis in diabetic nephropathy and its related mechanisms were investigated.
Experimental Approach
Diabetes was induced in mice by low‐dose streptozotocin (STZ) combined with a high‐fat diet. Effects of connexin32 on Nox4 expression and on renal function and fibrosis in STZ‐induced diabetic mice were investigated using adenovirus‐overexpressing connexin32 and connexin32‐deficient mice. Interactions between connexin32 and Nox4 were analysed by co‐immunoprecipitation and immunofluorescence assays.
Key Results
Connexin32 was down‐regulated in the kidneys of STZ‐induced diabetic mice. Overexpression of connexin32 reduced expression of Nox4 and improved renal function and fibrosis in diabetic mice, whereas connexin32 deficiency had opposite effects. Down‐regulation of fibronectin expression by connexin32 was not dependent on gap junctional intercellular communication involving connexin32. Connexin32 interacted with Nox4 and reduced the generation of hydrogen peroxide, leading to the down‐regulation of fibronectin expression. Mechanistically, connexin32 decreased Nox4 expression by promoting its K48‐linked polyubiquitination. Interestingly, Smurf1 overexpression inhibited K48‐linked polyubiquitination of Nox4. Furthermore, connexin32 interacted with Smurf1 and inhibited its expression.
Conclusion and Implications
Connexin32 ameliorated renal fibrosis in diabetic mice by promoting K48‐linked Nox4 polyubiquitination and degradation via inhibition of Smurf1 expression. Targeting the connexin32–Nox4 signalling axis may contribute to the development of novel treatments for diabetic nephropathy.
Abbreviations
- Ad‐Cx32
Cx32 adenovirus
- Ad‐V
vector adenovirus
- Cx32
connexin32
- ECM
extracellular matrix
- FBG
fasting blood glucose
- FN
fibronectin
- GJIC
gap junctional intercellular communication
- GMCs
glomerular mesangial cells
- Nox4
NADPH oxidase 4
- Smurf1
smad ubiquitylation regulatory factor‐1
- STZ
streptozotocin
- Ub
ubiquitins
- WT
wild type
- α‐SMA
α smooth muscle actin
1. What is already known
The NADPH oxidase isoform, Nox4, contributes to the pathogenesis of diabetic nephropathy .
Connexin32 inhibits oxidative stress in experimental non‐alcoholic steatohepatitis.
2. What this study adds
Connexin32 reduced Nox4 expression and improved renal function and fibrosis in STZ‐induced diabetic mice.
Connexin32 increased K48‐linked polyubiquitination of Nox4 via the inhibition of Smurf1 expression.
3. What is the clinical significance
Targeting the connexin32–Nox4 signalling axis may provide new approaches to treatment of diabetic nephropathy.
1. INTRODUCTION
Diabetic nephropathy is one of the major microvascular complications of diabetes and an important cause of end‐stage renal diseases (Kanwar, Sun, Xie, Liu, & Chen, 2011; Schena & Gesualdo, 2005). Although the clinical management of diabetic nephropathy, such as intensive blood glucose and blood pressure control of patients, can delay the occurrence and deterioration of disease to a certain extent, a large proportion of diabetic patients eventually develop renal failure (ADVANCE Collaborative Group et al., 2008; Zoungas et al., 2014). Therefore, it is extremely important to find effective new pharmacological targets to prevent and treat diabetic nephropathy.
Connexin32 (Cx32, also known as β1 connexin), a gap junction protein, is extensively expressed in liver, kidney, and myocardial tissue (Oyamada, Takebe, & Oyamada, 2013). Recently, attention has been focused on the regulation of Cx32 in oxidative stress during the course of diseases. More pronounced oxidative stress and liver damage were observed in Cx32‐deficient mice than those in wild‐type (WT) animals in experimental non‐alcoholic steatohepatitis (Tiburcio et al., 2017). The expression of Cx32 was significantly down‐regulated in the perineurium of streptozotocin (STZ)‐induced diabetic rats with severe peripheral neuropathy (Pitre, Seifert, & Bauer, 2001). However, the role of Cx32 in diabetic renal fibrosis remains unclear. Because Cx32 can effectively regulate oxidative stress, which is the key factor in the development of diabetic renal fibrosis, we assessed the participation of Cx32 in the pathological process of diabetic renal fibrosis.
NADPH oxidase (Nox), a multicomponent enzyme, has been identified as one of the key sources of ROS in the diabetic kidney (Jha et al., 2014; Jha et al., 2016; Jha et al., 2017; Jha, Banal, Chow, Cooper, & Jandeleit‐Dahm, 2016). https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=3004, the major NADPH isoform in the kidney, produces mainly hydrogen peroxide. ROS produced by Nox4 play an important role in mediating inflammation and fibrosis as well as subsequent kidney injury in diabetic mice. Knockout (KO) of Nox4 attenuates glomerulosclerosis, mesangial expansion, and the accumulation of extracellular matrix (ECM) proteins (Jha et al., 2014; Jha, Thallas‐Bonke, et al., 2016; Thallas‐Bonke et al., 2014). Under high glucose (HG) conditions, the expression of Nox4 in mesangial cells was up‐regulated, promoting the pathological process of renal fibrosis (Rhee, 2016). Additionally, Nox4, which is localized on the mitochondrial inner membrane, inhibited the activity of the mitochondrial respiratory chain complex I, leading to mitochondrial dysfunction and ROS overproduction (Ago et al., 2010; Block, Gorin, & Abboud, 2009; Koziel et al., 2013). Moreover, Nox4 protein stability is closely regulated by K48‐linked polyubiquitination and subsequent proteasomal degradation (Martyn, Frederick, von Loehneysen, Dinauer, & Knaus, 2006; Palumbo et al., 2017; Tsubouchi et al., 2017). However, the molecular mechanism of the up‐regulation of Nox4 expression induced by HG in glomerular mesangial cells (GMCs) remains to be elucidated.
Smad ubiquitylation regulatory factor‐1 (Smurf1), a HECT type E3 ubiquitin‐protein ligase, has been linked to multiple biological processes, including cell growth, migration, bone formation, embryonic development, and tumourigenesis (Cao & Zhang, 2013). Additionally, it was demonstrated that Smurf1 inhibited K48‐linked polyubiquitination of Keap1 and oestrogen receptor alpha (ERα) and increased the substrate stability (Gong et al., 2018; Yang et al., 2018). However, whether Smurf1 can inhibit the K48‐linked polyubiquitination of Nox4 remains unclear.
The study investigated whether Cx32 reduced oxidative stress in the kidney of diabetic mice by inhibiting Nox4 expression, thereby ameliorating diabetic renal fibrosis and dysfunction, and further explore whether the underlying mechanism involved the inhibition of Smurf1 expression and promotion of Nox4 K48‐linked polyubiquitination.
2. METHODS
2.1. Animals
All animal care and experimental procedures complied with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85‐23, revised 1996) and the China Animal Welfare Legislation, and were approved by the Ethics Committee on the Care and Use of Laboratory Animals of Sun Yat‐sen University, Guangzhou, China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology.
C57BL/6 mice (male, 6–8 weeks old, weighing 20 ± 2 g, specific pathogen‐free [SPF] grade, and certification No. 44008500016251, MGI Cat# 5656552, RRID:MGI:5656552) were obtained from the Laboratory Animal Centre, Sun Yat‐sen University, Guangzhou, China. Cx32+/− mice (European Mouse Mutant Archive, ID: 00243, Italy, IMSR Cat# EM:00243, RRID:IMSR_EM:00243) in the C57BL/6 background originally constructed by K Willecke Laboratories (Nelles et al., 1996) were kindly provided by Professor Ziqing Hei from the Third Affiliated Hospital of Sun Yat‐sen University, Guangzhou, China. Cx32+/− mice were backcrossed with C57BL/6 mice to generate Cx32+/+ (WT) and Cx32−/− (KO) mice. Genotyping was performed by PCR analysis of DNA from mouse tail tips as previously described (Moennikes, Buchmann, Ott, Willecke, & Schwarz, 1999). Cx32+/+ (WT) and Cx32−/− (KO) mice (male, 6–8 weeks old, weighing 20 ± 2 g, SPF grade) were used for subsequent experiments. All animals were housed under SPF conditions and in a temperature‐controlled (20–25°C) and humidity‐controlled (40–70%) barrier system with a 12‐hr:12‐hr light and dark cycle.
2.2. Mouse model of diabetic nephropathy
Diabetic mice were induced as previously described (Nath, Ghosh, & Choudhury, 2017). Briefly, male mice were given a commercial high‐fat diet (58% fat, 25% protein, and 17% carbohydrate, as a percentage of total kcal; Guangdong Medical Laboratory Animal Center, Foshan City, Guangdong Province, China) for 2 weeks, followed by the i.p. injection of freshly prepared STZ (40 mg·kg−1, dissolved in freshly prepared ice‐cold 0.1‐M citrate buffer, pH 4.5) once a day for five consecutive days in continuation with a high‐fat diet. The control mice were provided with a normal standard balanced diet, followed by only an equal volume of vehicle—that is, 0.1‐M citrate buffer, pH 4.5 administered i.p. The fasting blood glucose (FBG) of STZ‐induced mice was measured 2 weeks after STZ injection, and mice with FBG more than 11.1 mM were regarded as diabetic mice.
2.3. Tail vein injection of recombinant Cx32 adenovirus
Recombinant Cx32 adenovirus (Ad‐Cx32; Genbank ID NM_001302496, Genechem, Shanghai, China) and diabetic C57BL/6 mice were randomly divided into two groups: STZ + vector adenovirus (Ad‐V; n = 9) and STZ + Ad‐Cx32 (n = 9); similarly, control C57BL/6 mice were randomly divided into two groups: Ctrl + Ad‐V (n = 9) and Ctrl + Ad‐Cx32 (n = 9). Our laboratory previously found that treatment with 2 × 109 viral particles (approximately 2 × 107 plaque forming unit [PFU]) of CKIP‐1 adenovirus could effectively infect the mice (Gong et al., 2017). However, in order to determine the optimal amount of Ad‐Cx32 injection in mice, we conducted a preliminary experiment with three doses (2 × 106, 2 × 107, and 2 × 108 PFU, respectively) and found that 2 × 108 PFU was the most effective. Therefore, the dosage of 2 × 108 PFU was used in these experiments. The mice were administered Ad‐Cx32 (2 × 108 PFU, 200 μl per mouse) or negative control adenovirus (Ad‐V; 2 × 108 PFU, 200 μl per mouse) once a week via tail vein injection. Mouse FBG was measured weekly using a OneTouch blood glucose meter. After treatment with adenovirus and a high‐fat diet for 8 weeks, all the mice were placed into metabolic cages overnight to collect urine. The next day, body weights were recorded, and then the mice were anaesthetized by isoflurane and then sodium pentobarbital (50 mg·kg−1, i.p.), and a blood sample was obtained by cardiac puncture. Finally, the animals were killed by cervical dislocation, and both kidneys were quickly removed and weighed.
2.4. Induction of diabetes in Cx32‐deficient mice
Cx32 WT mice were randomly divided into two groups: WT + Vehicle (n = 8) and WT + STZ (n = 8). Cx32 KO mice were also randomly divided into two groups: KO + Vehicle (n = 8) and KO + STZ (n = 8). After diabetes was successfully induced, the FBG of mice was measured weekly using a OneTouch blood glucose meter (Johnson, USA) in continuation with a high‐fat diet for 8 weeks. Next, all the mice were placed into metabolic cages overnight to collect urine. The next day, body weights were recorded, and then the mice were anaesthetized by isoflurane inhalation and then sodium pentobarbital (50 mg·kg−1, i.p.), and a blood sample was obtained by cardiac puncture. Finally, the animals were killed by cervical dislocation, and both kidneys were quickly removed and weighed.
2.5. Culture of primary rat mesangial cells
Primary GMCs were isolated from the glomeruli cortex fragments of young Sprague–Dawley rats (male, weighing 150–180 g) using the previously described protocol (Chen et al., 2017). Briefly, cortex fragments being chopped into 1‐ to 2‐mm pieces were mechanically sieved and harvested by iterative selection on specific mesh sizes (175, 147, and finally 74 μm). The tissue (containing the glomeruli) was collected, washed with PBS, and digested with 0.1% collagenase (Type IV; U·ml−1) in DMEM for 15–25 min at 37°C. The glomeruli were resuspended in growth media (20% FBS, 0.66 U·ml−1 of insulin, 2‐mM L‐glutamine, 100 U·ml−1 of penicillin, and 100 U·ml−1 of streptomycin in DMEM), seeded in culture flasks, and incubated at 37°C in 5% CO2 in air. The medium was not replaced until the fourth day and was renewed every 2 or 3 days. The cultures were examined morphologically for contaminating cell types. Additionally, the cultures were identified by immunofluorescence for specific cell marker proteins. Finally, >98% of the cells were stained for vimentin and desmin but were non‐reactive to antibodies against cytokeratin (glomerular epithelial cells; Figure S1).
GMCs (Passages 5–12) were used for subsequent experiments. The cells were maintained in DMEM supplemented with 10% FBS at 37°C in 5% CO2 in air. At 70–80% confluence, the cells were growth arrested with serum‐free medium for 12 hr and incubated with either normal glucose (NG, 5.6 mM) or HG (30 mM) for the times indicated.
2.6. Transfections of plasmids and small‐interfering RNA
Plasmids: mEmerald‐Cx32‐7 (RRID:Addgene_54054), mEmerald‐N1 (RRID:Addgene_53976), pcDNA3.1‐hNox4 (RRID:Addgene_69352), pcDNA3.1 (RRID:Addgene_52535), pRK5‐HA‐Ub (RRID:Addgene_17608), pRK5‐HA‐Ub‐KO (RRID:Addgene_17603), pRK5‐HA‐Ub‐K48 (RRID:Addgene_17605), pRK5‐HA‐Ub‐K63 (RRID:Addgene_17606), pRK‐Myc‐Smurf1 (RRID:Addgene_13676), and pRK‐Myc‐Smurf1 delHECT (RRID:Addgene_13677) were obtained from Addgene (Watertown, MA, USA). GMCs were grown in 35‐mm plates and transfected with 2 μg of relevant plasmids according to the instructions using the transfection reagent LTX & PLUS at 70–80% confluence. Three short hairpin RNAs (shRNAs) targeting Nox4 were synthesized by Genechem. The sequences were as follows: shRNA‐4: sense: GCTTCTACCTATGCAATAA, antisense: TTATTGCATAGGTAGAAGC; shRNA‐5: sense: GGAGTCACTGAACTATGAA, antisense: TTCATAGTTCAGTGACTCC; and shRNA‐6: sense: TCCCTCAGATGTCATGGAA, antisense: TTCCATGACATCTGAGGGA.
Small‐interfering RNA (siRNA) targeting Cx32 was synthesized by GenePharma (Shanghai, China). The sequences were as follows: sense: CACCAACAACACAUAGAAATT and antisense: UUUCUAUGUGUUGUUGGUGTT. GMCs, grown in 35‐mm plates, were transfected with 5 μl of siRNA (50 nM), using RNAiMAX according to the manufacturer's protocol at 70–80% confluence. After further treatment, the cells were harvested and western blot assay was performed.
2.7. Western blot analysis
The antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. HRP‐conjugated secondary antibodies were obtained from Promega Corporation (Madison, WI, USA). Western blot analysis was performed as previously described (Chen et al., 2017) and complied with BJP guidelines and the editorial on reporting of such studies (Alexander et al., 2018). Briefly, the total proteins were extracted using RIPA (Tris [pH 7.4], 150‐mM Nacl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, etc., with protease inhibitor cocktail, phosphatase inhibitor A and B). After centrifuging at 12,000 g for 15 min at 4°C, the protein concentration was determined using a BCATM Protein Assay Kit (Pierce, USA). Equal amounts of protein lysates were separated by SDS‐PAGE and were transferred onto PVDF membranes (Bio‐Rad Laboratories, USA). The blots were visualized with a GE ImageQuant LAS4000mini (GE healthcare, Waukesha, WI, USA, ImageQuant, RRID:SCR_014246) and analysed using Quantity One Protein Analysis Software (Bio‐Rad, Hercules, CA, USA; Version 4.6.2, RRID:SCR_016622).
2.8. Immunofluorescence staining
Immunofluorescence staining was performed as previously described (Chen, Chen, et al., 2017). Briefly, GMCs were grown on glass coverslips in 24‐well plates and were treated with corresponding stimuli when the confluence was up to 60%. After treatment, the cells were washed with PBS and were fixed with 4% paraformaldehyde for 15 min at room temperature and were permeabilized using 0.1% Triton X‐100 in PBS for another 10 min at room temperature. The cells were then blocked with 10% goat serum for 45 min at room temperature and were incubated with primary antibodies, including rabbit antibody against Nox4 and mouse antibody against Cx32 in 10% goat serum overnight at 4°C. Coverslips were washed with PBS and incubated with a secondary antibody (Alexa Fluor 488 and Alexa Fluor 594, Invitrogen, Carlsbad, CA) in the dark at room temperature for 1 hr. The nuclei were co‐labelled with DAPI dihydrochloride (Cell Signaling Technology, Cat# 4083) for 10 min at room temperature. At the end, the coverslips were mounted on slides with antifade mounting medium (Beyotime, Cat# P0126). The images were collected using a Zeiss LSM 510 laser confocal fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
2.9. Immunoprecipitation assay
Immunoprecipitation assays were performed as previously described (Chen, Xie, et al., 2017). GMCs were lysed with immunoprecipitation lysate buffer (supplemented with protease inhibitor cocktail, phosphatase inhibitor A and B) on ice for 30 min and then centrifuged at 12,000 g for 15 min at 4°C. The whole‐cell lysates (300 μg) were incubated with 15 μl of protein agarose A/G beads (Pierce, Rockford, IL, USA) before immunoprecipitation to reduce nonspecific combination, followed by transient centrifugation to collect the supernatants. The supernatants were then incubated with 2 μg of IgG or Nox4 or Smurf1 antibodies overnight at 4°C with shaking. Protein agarose A/G beads (20 μl) were added to the supernatants for 2 hr at 4°C with shaking. SDS loading buffer (20 μl) was added to the beads after washing three times with immunoprecipitation buffer. The immunoprecipitates were analysed by western blot assay with respective antibodies.
2.10. Scrape‐loading/dye transfer assays
Gap junction intercellular communication (GJIC) was assessed using the scrape‐loading/dye transfer technique as previously described (el‐Fouly, Trosko, & Chang, 1987; Xie et al., 2013). It employs scrape loading to introduce a low MW fluorescent dye, lucifer yellow CH (MW 457.2) into cells in culture and allows the monitoring of its transfer into contiguous cells. Scrape loading was performed by scraping the cell layer with a broken razor blade in DMEM medium containing lucifer yellow (1 mg·ml−1). After incubation at 37°C in the dark for 5 min, the images were collected using a Zeiss LSM 510 laser confocal fluorescence microscope (Carl Zeiss).
2.11. Amplex Red/HRP assay
Amplex Red (Invitrogen), a molecular probe, is widely applied to measure hydrogen peroxide released from cultured cells and tissue homogenates (Miwa et al., 2016). Therefore, the production of hydrogen peroxide by Nox4 in GMCs was detected using Amplex Red as previously reported (Jaquet et al., 2011). Briefly, GMCs were plated at a density of 2 × 105 cells per well into six‐well plates. After treatment, the cells were collected by trypsinization, centrifuged for 5 min at 200 g at 4°C, washed with HBSS, counted, and resuspended in HBSS at 500,000 cells·ml−1. The cells (100 μl) were added into 96‐well microplates placed on ice at a density of 50,000 cells. Then, the Amplex Red reaction mixture was added to give final concentrations of 0.005 U·ml−1 HRP and 25‐μM Amplex Red. The plate was placed in the fluorescence reader (Flex Station 3, Moleculardevices, USA) prewarmed to 37°C and the fluorescence intensity recorded every 1 min during 1 hr with excitation and emission wavelengths of 550 and 600 nm respectively. The amount of hydrogen peroxide generated was calculated from a hydrogen peroxide standard curve ranging from 0 to 5 μM.
2.12. Measurement of mitochondrial superoxide production
MitoSOX Red, a mitochondria‐targeted superoxide fluorescent probe, was used for the detection of mitochondrial ROS by live cell imaging; 5‐mM MitoSOX Red was diluted into 5 μM with PBS, added to the cells, and incubated at 37°C for 10 min in the dark. Subsequently, the cells were gently washed three times with PBS. Then, the fluorescence was detected and quantified using a high‐content system (ArrayScan VTI 600 plus).
2.13. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All the studies were designed to generate groups of equal size, using randomization and blinded analysis. Statistical analysis was undertaken only for studies where each group size was at least n = 5 using GraphPad Prism 5.0 (GraphPad Prism, RRID:SCR_002798). The values were expressed as mean ± SD. Sample sizes in each group subjected to statistical analysis were determined based on our previous studies, preliminary results, and the power analysis. Additionally, group size is the number of independent values and that statistical analyses used these independent values (i.e., not treating technical replicates as independent values). The values of the control group were normalized to 100% in western blotting analysis to reduce unwanted sources of variation, which was analysed by non‐parametric tests (Kruskal–Wallis test followed by Dunn's multiple comparison). The Y‐axis shows the percentage of the treated group to that of the corresponding matched control values. Outliers were excluded according to a predefined and defensible set of exclusion criteria (such as the maximum and minimum) in the animal experiments. Unpaired Student's t‐test was used to compare two groups. For multiple comparisons, the data were analysed by one‐way ANOVA with post hoc multiple comparisons. Post hoc tests were further conducted only if the F was significant and there was no significant variance inhomogeneity. P < .05 was considered to be statistically significant, and this P value was not varied later.
2.14. Materials
D‐Glucose (Cat# 0188) was obtained from AMRESCO (Solon, OH, USA). Other reagents were from Sigma–Aldrich Corporation (St. Louis, MO, USA; STZ [Cat# S0130], penicillin–streptomycin [Cat# V900929], HRP [Cat# P8375], and 18‐α‐glycyrrhetinic acid (18‐α‐GA; Cat# G8503) and from Thermo Fisher Scientific (Rockford, IL, USA; FBS [Cat# 10099141]), RNAiMAX transfection reagent (Cat# 13778075), LTX reagent with PLUS™ reagent (Cat# 15338030), lucifer yellow (Cat# L453), MitoSOX™ Red (Cat# M36008), Amplex Red (Cat# A22188), and DMEM (Cat# 41965062). MG132 (Cat# S2619) was purchased from Selleck Chemicals (Houston, Texas, USA). Total SOD (Cat# S0101) Assay Kit with WST‐8 and Lipid Peroxidation MDA (Cat# S0131) Assay Kit were purchased from Beyotime (Haimen, China). An enhanced chemiluminescence substrate (Cat# WP20005) for the detection of HRP was obtained from Thermo Fisher Scientific, Inc.
Antibodies against fibronectin (FN; Proteintech Cat# 15613‐1‐AP, RRID:AB_2105691), Nox4 (Proteintech Cat# 14347‐1‐AP, RRID:AB_10638146), α smooth muscle actin (α‐SMA; Proteintech Cat# 14395‐1‐AP, RRID:AB_2223009), TGF‐β1 (Proteintech Cat# 21898‐1‐AP, RRID:AB_2811115), and Smurf1 (Proteintech Cat# 55175‐1‐AP, RRID:AB_10859079) were purchased from Proteintech Group (Chicago, IL, USA); α‐Tubulin (Sigma‐Aldrich Cat# T8203, RRID:AB_1841230) was purchased from Sigma–Aldrich Corporation; Cx32 (Abcam Cat# ab66613, RRID:AB_1141052) was from Abcam (Cambridge, MA, USA); Cx32 (Novus Cat# NBP2‐53381, RRID:AB_2811114) was from Novus Biologicals (Centennial, USA); and ubiquitin (Ub; Cell Signaling Technology Cat# 3936, RRID:AB_331292) was from Cell Signaling Technology (Danvers, USA).
2.15. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Cidlowski et al., 2017; Alexander, Fabbro, et al., 2017; Alexander, Kelly, et al., 2017a; Alexander, Kelly, et al., 2017b).
3. RESULTS
3.1. Overexpression of Cx32 ameliorates renal function and fibrosis in STZ‐induced diabetic mice
Firstly, we found that Cx32 expression was down‐regulated in the STZ‐induced diabetic kidney (Figure S2A) and HG‐induced GMCs (Figure S2B), suggesting that Cx32 may be involved in the pathogenesis of diabetic nephropathy.
To evaluate the role of Cx32 in diabetic renal fibrosis, we investigated the effects of adenovirus‐mediated Cx32 overexpression (Ad‐Cx32) on renal function and fibrosis in STZ‐induced diabetic mice, giving the adenovirus by tail vein injection. Compared with diabetic mice (STZ + Ad‐V), overexpression of Cx32 (STZ + Ad‐Cx32) reduced the FBG (Figure 1b), kidney weight/body weight ratio (Figure 1e), serum creatinine (Figure 1f), blood urea nitrogen (Figure 1h), and 24‐hr urine protein (Figure 1i) of diabetic mice, and increased the body weight (Figure 1c) of diabetic mice, suggesting that Cx32 overexpression could improve renal function in diabetic mice.
Figure 1.
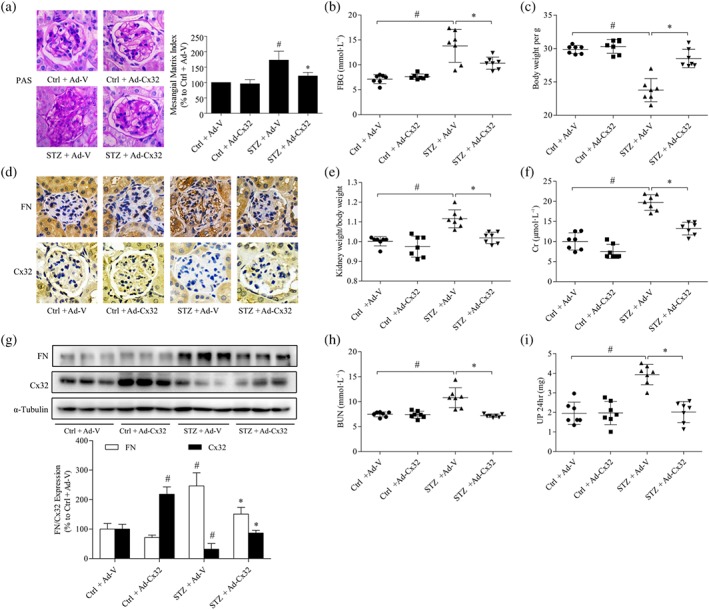
Overexpression of Cx32 ameliorates renal function and fibrosis in STZ‐induced diabetic mice. (a) Glomerular histopathology analysis was performed by periodic acid‐Schiff (PAS) staining (400× magnification). (b,c) The FBG and body weight of diabetic mice were analysed. (d) The expression levels of fibronectin (FN) and Cx32 in the glomeruli were shown by immunohistochemical staining (400× magnification). (e) The kidney weight/body weight ratio was calculated to evaluate the kidney hypertrophy index. (f) Serum creatinine (Cr) was detected to assess renal injury. (g) The expression of Cx32 and fibronectin in the kidney of mice were measured using the western blot assay. (h,i) Serum BUN and 24‐hr UP were detected to assess renal injury. The data are presented as the means ± SD; n = 7 (two outliers were excluded). # P < .05, significantly different from Ctrl + Ad‐V; * P < .05, significantly different from STZ + Ad‐V. Ad‐Cx32, Cx32 adenovirus; Ad‐V, vector adenovirus; BUN, blood urea nitrogen; Cr, serum creatinine; Ctrl, control; FBG, fasting blood glucose; STZ, streptozocin; UP 24 hr, urine protein for 24 hr
Along with these changes, overexpression of Cx32 also improved the mesangial expansion and glomerular basement membrane thickening as well as deposition of ECM in the kidney of diabetic mice (STZ + Ad‐Cx32), as shown by PAS staining analysis (Figure 1a) and immunohistochemical staining (Figure 1d). Masson staining also revealed that overexpression of Cx32 decreased the generation of collagen fibres in the kidney of diabetic mice (Figure S4A). Western blotting results confirmed that Ad‐Cx32 treatment increased the expression of Cx32 and inhibited the up‐regulated expression of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6754 (Figure 1g), TGF‐β1, and α‐SMA (Figure S4C) in the kidney of diabetic mice (STZ + Ad‐Cx32), compared with that in diabetic mice (STZ + Ad‐V). The above results revealed that overexpression of Cx32 could improve renal function and fibrosis in the kidneys of diabetic mice.
3.2. Cx32 deficiency potentiates renal dysfunction and fibrosis in the kidney of STZ‐induced diabetic mice
To further investigate the effects of Cx32 on renal function and fibrosis in the kidney of diabetic mice, Cx32‐deficient mice were used. Genotyping was performed by PCR analysis of DNA from mouse tail tips (Figure S3), and homozygous mice (such as the fifth mouse) were used for the subsequent experiments. The results showed that FBG (Figure 2b), kidney weight/body weight (Figure 2e), serum creatinin (Figure 2f), blood urea nitrogen (Figure 2h), and 24‐hr urine protein (Figure 2i) of Cx32‐deficient diabetic mice (KO + STZ) were increased compared with those in diabetic mice (WT + STZ), and the body weight (Figure 2c) was reduced, suggesting that Cx32 deficiency could worsen renal function in diabetic mice.
Figure 2.
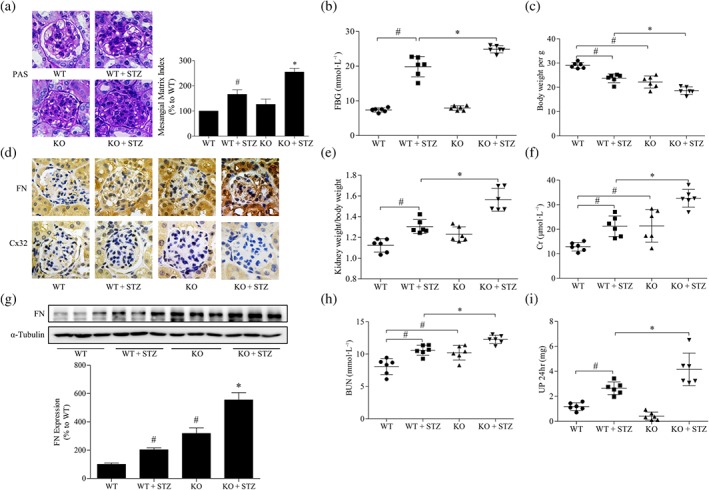
Cx32 deficiency exacerbates renal fibrosis in STZ‐induced diabetic mice. (a) Glomerular histopathology analysis by periodic acid‐Schiff (PAS) staining (400× magnification). (b,c) The FBG and body weight of diabetic mice were analysed. (d) The expression of fibronectin (FN) and Cx32 in the glomeruli were shown by immunohistochemical staining (400× magnification). (e) The kidney weight/body weight ratio was calculated to evaluate the kidney hypertrophy index. (f) Serum Cr was detected to assess renal injury. (g) The expression of fibronectin in the kidney of mice were measured by western blot assay. (h,i) Serum BUN and 24‐hr UP were assayed to assess renal injury. The data are presented as the means ± SD; n = 6 (two outliers were excluded). # P < .05, significantly different from WT; * P < .05, significantly different from WT + STZ. BUN, blood urea nitrogen; Cr, serum creatinine; FBG, fasting blood glucose; KO, knockout; STZ, streptozocin; UP 24 hr, urine protein for 24 hr; WT, wild type
PAS staining analysis showed that mesangial expansion and glomerular basement membrane thickening occurred in the kidney of Cx32‐deficient mice compared with that in WT mice. Masson staining also revealed that Cx32 deficiency promoted the generation of collagen fibres in the kidney of diabetic mice (Figure S4B). Moreover, the glomerular pathological changes of Cx32‐deficient diabetic mice (KO + STZ) were more severe than those of diabetic mice (WT + STZ; Figure 2a). Further deposition of the ECM protein fibronectin was observed in the glomeruli of Cx32‐deficient diabetic mice (KO + STZ) compared with that in diabetic mice (WT + STZ), as revealed by immunohistochemistry staining (Figure 2d). Western blot analysis showed that compared with those in WT mice, the expressions of fibronectin (Figure 2g), TGF‐β1, and α‐SMA (Figure S4D) in the kidney of Cx32‐deficient mice were increased. Additionally, fibronectin and TGF‐β1 expression were further increased in the kidney of Cx32‐deficient diabetic mice (KO + STZ) compared with that in diabetic mice (WT + STZ). However, Cx32 deficiency only slightly increased the protein expression of α‐SMA in the kidney of diabetic mice, with no statistical difference. This may be due to the large individual differences in Cx32 deficiency diabetic mice. Taken together, these data suggest that Cx32 deficiency aggravated diabetic nephropathy in mice, induced by STZ.
3.3. The regulation of fibronectin expression by Cx32 in GMCs is not dependent on GJIC
To further investigate the roles of Cx32 in diabetic renal fibrosis, we examined the expression of markers of fibrosis, such as fibronectin, in GMCs. As shown in Figure 3a, the protein expression of Cx32 was significantly increased by the transfection of the Cx32 plasmid in GMCs, whereas the vector had no effects, indicating that the Cx32 plasmid was successfully transfected. Meanwhile, HG stimulation for 24 hr significantly increased the levels of fibronectin in GMCs compared with NG, while the overexpression of Cx32 significantly reduced the protein level of fibronectin (Figure 3b), and the vector had no effects. Additionally, we found that the transfection of Cx32 siRNA in HG‐cultured GMCs further reduced Cx32 protein expression (Figure 3c), resulting in a significant increase in HG‐induced fibronectin (Figure 3d) expression, compared with HG, supporting that Cx32 ameliorates diabetic renal fibrosis.
Figure 3.
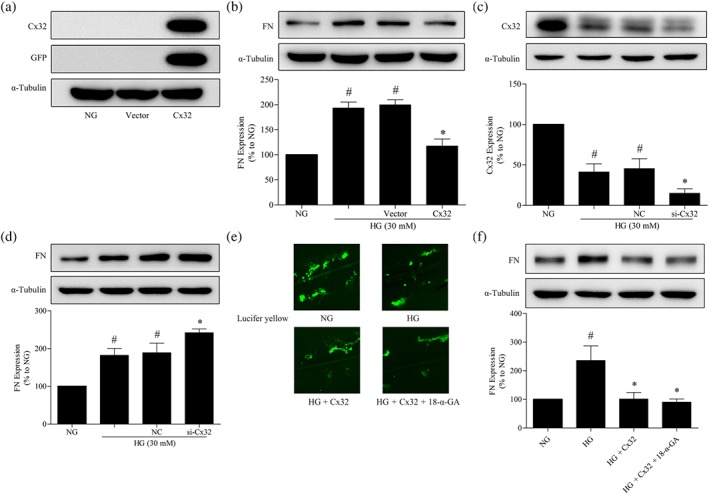
Regulation of fibronectin expression by Cx32 in GMCs is not dependent on GJIC. (a) The expression of Cx32 and GFP‐tag were measured by western blotting to confirm that the plasmids were successfully transfected into GMCs. (b) Cx32 overexpression inhibited fibronectin expression in HG‐induced GMCs. The transfection of Cx32 siRNA decreased Cx32 (c) expression and up‐regulated fibronectin (d) expression. (e) Photomicrographs obtained after Lucifer yellow scrape loading in GMCs transfected with the Cx32 plasmid with or without 18‐α‐GA (10 μM). (f) Treatment with 18‐α‐GA did not block the down‐regulation of fibronectin expression by Cx32. The data are presented as the means ± SD; n = 5. # P < .05, significantly different from NG; * P < .05, significantly different from HG,
To investigate whether the regulation of Cx32 in fibronectin expression in GMCs depends on GJIC, we used 18‐α‐GA (10‐μM), a GJIC inhibitor; Taylor, Chaytor, Evans, & Griffith, 1998; Yang et al., 2014) to block the gap junction formed by Cx32. As expected, treatment with 18‐α‐GA (10 μM) significantly inhibited GJIC involving Cx32 (Figure 3e). However, 18‐α‐GA did not block the down‐regulation of fibronectin expression by Cx32 (Figure 3f), suggesting that the regulation of fibronectin expression by Cx32 in GMCs was not dependent on GJIC.
3.4. Cx32 resists oxidative stress in the kidney of diabetic mice and HG‐induced GMCs
We observed the effects of Cx32 on oxidative stress in the kidney of diabetic mice. Additionally, SOD and MDA are commonly used biomarkers for oxidative stress (Amin et al., 2018; Hajiluian, Abbasalizad Farhangi, Nameni, Shahabi, & Megari‐Abbasi, 2018). The results showed that the total SOD activity in the kidney of diabetic mice treated with Ad‐Cx32 (STZ + Ad‐Cx32) was increased (Figure 4a) compared with that in diabetic mice treated with negative control adenovirus (STZ + Ad‐V), and the MDA levels were reduced (Figure 4b). Compared with diabetic mice (WT + STZ), the total SOD activity in the kidney of Cx32 knockdown diabetic mice (KO + STZ) was further reduced (Figure 4c), while the MDA levels were further increased (Figure 4d), suggesting that Cx32 inhibits oxidative stress in the kidneys of diabetic mice.
Figure 4.
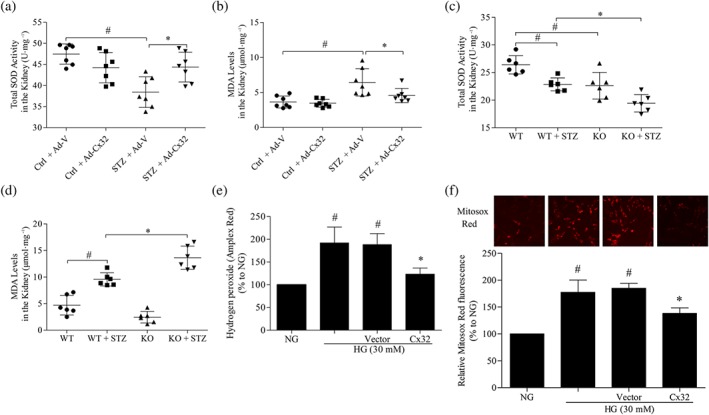
Cx32 decreases oxidative stress in the kidney of diabetic mice and HG‐induced GMCs. Treatment with Cx32 adenovirus increased total SOD activity (a) and decreased the MDA levels (b) in the kidneys of diabetic mice. The data are presented as the means ± SD; n = 7. # P < .05, significantly different from Ctrl + Ad‐V; * P < .05, significantly different from STZ + Ad‐V. Cx32 deficiency decreased total SOD activity (c) and increased the MDA levels (d) in the kidney of diabetic mice. The data are presented as the means ± SD; n = 6. # P < .05, significantly different from WT; * P < .05, significantly different from WT + STZ. KO, knockout; WT, wild type. (e) The hydrogen peroxide released from HG‐cultured GMCs was measured using Amplex Red. Overexpression of Cx32 inhibited the generation of hydrogen peroxide in GMCs induced by HG for 12 hr. (f) The mitochondrial superoxide levels were detected using the MitoSOX Red probe. The data are presented as the means ± SD; n = 5. # P < .05, significantly different from NG; * P < .05, significantly different from HG. Ad‐Cx32, Cx32 adenovirus; Ad‐V, vector adenovirus; Ctrl, control; MDA, malonaldehyde; STZ, streptozocin.
To further confirm the involvement of Cx32 in HG‐induced ROS generation, Amplex Red and MitoSOX Red were used to detect the levels of hydrogen peroxide released from HG‐cultured GMCs and mitochondrial superoxide respectively. Compared with NG, HG (30 mM) treatment for 12 hr elevated the levels of hydrogen peroxide and mitochondrial superoxide in GMCs. Meanwhile, overexpression of Cx32 restrained the overproduction of intracellular hydrogen peroxide (Figure 4e) as well as mitochondrial superoxide (Figure 4f) in HG‐induced GMCs, confirming the antioxidative stress effects of Cx32 at the cellular levels.
3.5. Nox4 promotes the expression of fibronectin in GMCs
Consistent with the results reported previously (Sedeek et al., 2010), we found that the protein expression of Nox4 was increased in GMCs under hyperglycaemic conditions (Figure 5a). Transfection of the Nox4 plasmid under NG conditions significantly increased the expressions of Nox4 and fibronectin in GMCs (Figure 5b). Additionally, transfection of the Nox4 plasmid under hyperglycaemic conditions further promoted HG‐induced increases in Nox4 (Figure 5c) and fibronectin expression (Figure 5d). Furthermore, GMCs were transfected with the negative control and three shRNAs of Nox4 for 72 hr under normal conditions, followed by harvesting and western blot analysis. The results showed that all three shRNAs could reduce the expression of Nox4, and shRNA‐4 had the best interference efficiency (Figure 5e). Therefore, the shRNA‐4 of Nox4 was used for subsequent experiments. We found that Nox4 depletion clearly decreased HG‐induced fibronectin expression in GMCs (Figure 5f).
Figure 5.
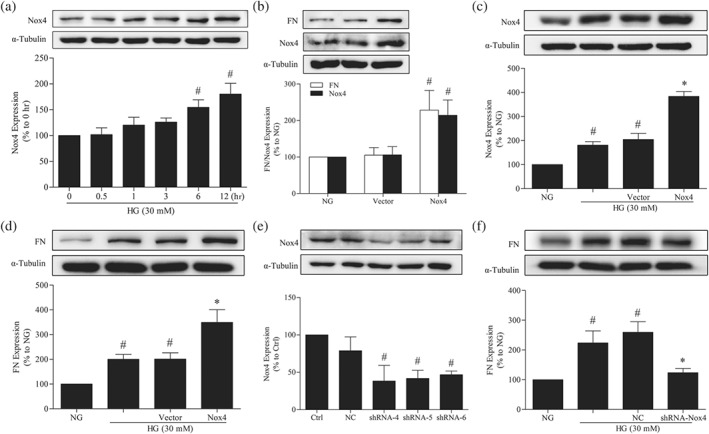
Nox4 promotes the expression of fibronectin (FN) in GMCs. (a) The expression of Nox4 was up‐regulated in GMCs under HG conditions. # P < .05, significantly different from 0 hr. (b) Nox4 overexpression enhanced the expression of fibronectin in GMCs under normal conditions. # P < .05, significantly different from NG. (c) Transfection of the Nox4 plasmid further increased Nox4 protein levels in GMCs under HG conditions. # P < .05, significantly different from NG; * P < .05, significantly different from HG. (d) Nox4 overexpression further enhanced the expression of fibronectin in GMCs under HG conditions. # P < .05, significantly different from NG; * P < .05, significantly different from HG. (e) GMCs were transfected with the negative control and three shRNAs of Nox4 for 72 hr under normal conditions and then were harvested and subjected to western blot analysis. # P < .05, significantly different from Ctrl. (f) Effects of Nox4 depletion on fibronectin expression. # P < .05, significantly different from NG; * P < .05, significantly different from HG. The data are presented as the means ± SD; n = 5.
3.6. Nox4, at least partly, mediates the regulation by Cx32 of fibronectin expression in HG‐induced GMCs
The immunohistochemical results showed that the protein expression of Nox4 was reduced in the glomeruli of diabetic mice treated with Ad‐Cx32 (STZ + Ad‐Cx32) compared with that in diabetic mice treated with negative control adenovirus (STZ + Ad‐V; Figure 6a). Additionally, the protein expression of Nox4 was further increased in the glomeruli of Cx32‐deficient diabetic mice (KO + STZ) compared with that in diabetic mice (WT + STZ; Figure 6b). Next, we examined the effects of Cx32 on the expression of Nox4 protein in HG‐induced GMCs. Overexpression of Cx32 reduced the HG‐induced expression of Nox4 in GMCs under HG conditions (Figure 6c), while the knockdown of Cx32 further increased Nox4 expression (Figure 6d), as shown by western blotting. To investigate the molecular mechanism by which Cx32 regulates Nox4 expression, immunofluorescence and immunoprecipitation assays were performed. We observed that Cx32 was co‐localized with Nox4 in GMCs under normal conditions, as revealed by immunofluorescence assay (Figure 6e). Moreover, immunoprecipitation assays showed that Cx32 interacted with Nox4 in GMCs under normal conditions (Figure 6f). To confirm the involvement of Nox4‐mediated hydrogen peroxide production in the regulation of Cx32 in renal fibrosis, we simultaneously overexpressed Cx32 and Nox4 under HG conditions. The results demonstrated that Nox4 overexpression efficiently blocked the regulation by Cx32 of fibronectin expression (Figure 6g), suggesting that Nox4, at least partly, mediates the regulation of fibronectin expression by Cx32 in GMCs.
Figure 6.
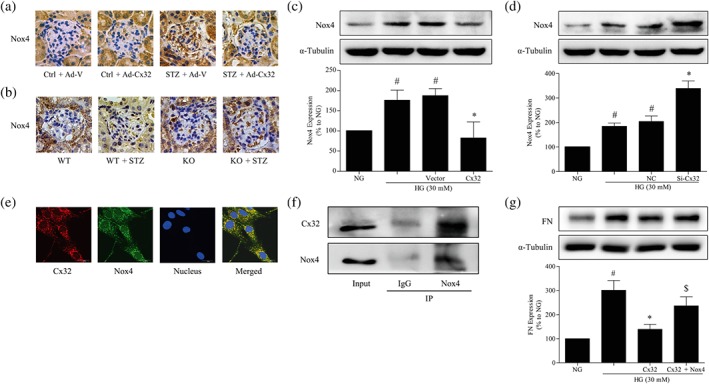
Nox4, at least partly, mediates the down‐regulation of fibronectin expression by Cx32 in HG‐induced GMCs. (a and b) The expression of Nox4 in the glomeruli was shown by immumohistochemical staining (400× magnification). (c) Cx32 overexpression decreased the expression of Nox4. # P < .05, significantly different from NG; * P < .05, significantly different from HG. (d) Cx32 knockdown increased Nox4 expression. # P < .05, significantly different from NG; * P < .05, significantly different from HG. (e) Cx32 co‐localized with Nox4 in GMCs under normal conditions, as revealed by immunofluorescence assay. Red fluorescence indicates the localization of Cx32. Green fluorescence indicates Nox4. Blue fluorescence indicates nuclei. (f) Immunoprecipitation analysis was used to detect the interaction between Cx32 and Nox4 in GMCs. (g) Overexpression of Nox4 under HG conditions inhibited the down‐regulation effects of Cx32 in the expression of fibronectin . # P < .05, significantly different from NG; * P < .05, significantly different from HG; $ P < .05, significantly different from HG + Cx32. The data are presented as the means ± SD; n = 5.
3.7. Cx32 decreases the protein levels of Nox4 by promoting Nox4 K48‐linked polyubiquitination
HA‐Ub K0 is a mutant plasmid of ubiquitin (HA‐Ub), seven lysine residues of which are converted into arginine, leading to the failure to form ubiquitin chains. The proteasome inhibitor MG132 was used to block the degradation of ubiquitinated proteins. We found that co‐transfection of the WT ubiquitin plasmid (HA‐Ub) significantly increased the ubiquitination of Nox4 in HEK‐293A (RRID:CVCL_6910) cells exposed to MG132 (5 μM), while the mutant ubiquitin plasmid (HA‐Ub‐K0) had no effects (Figure 7a), suggesting that Nox4 could indeed be polyubiquitinated.
Figure 7.
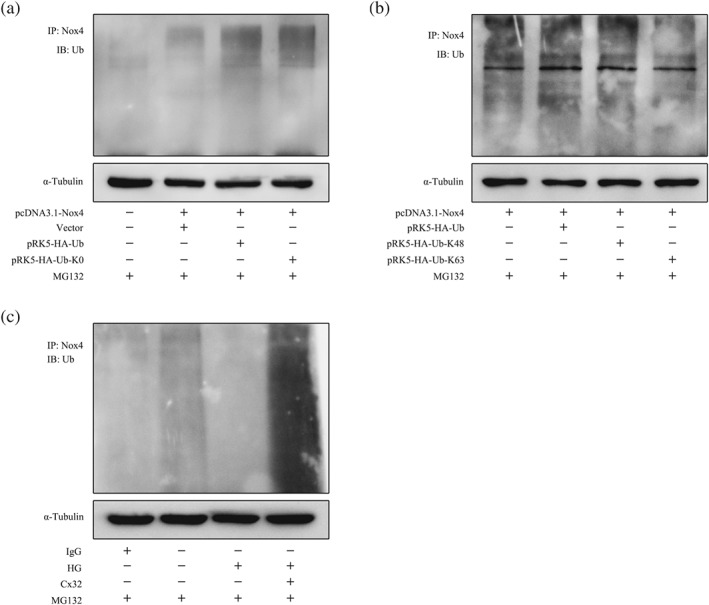
Cx32 decreases the protein levels of Nox4 by promoting K48‐linked polyubiquitination. (a) Co‐transfection of the plasmids pcDNA3.1‐Nox4 and HA‐Ub (wild type) or HA‐Ub‐K0 (mutant type) in HEK‐293A cells and western blotting results showed that Nox4 could be polyubiquitinated. (b) The plasmid pcDNA3.1‐Nox4 was co‐transfected with plasmids HA‐Ub or K48‐only ubiquitin (HA‐Ub‐K48) or K63‐only ubiquitin (HA‐Ub‐K63) in HEK‐293A cells exposed to MG132 (5 μM). (c) Overexpression of Cx32 increased the polyubiquitination of Nox4 in GMCs induced by HG.
To explore which ubiquitin chains modified Nox4, plasmid pcDNA3.1‐Nox4 was co‐transfected with plasmids HA‐Ub or K48‐only ubiquitin (HA‐Ub‐K48) or K63 only ubiquitin (HA‐Ub‐K63) in HEK‐293A cells exposed to MG132 (5 μM). The results showed that the ubiquitination of Nox4 after the co‐transfection of HA‐Ub‐K48 was comparable to that of HA‐Ub. However, the co‐transfection of HA‐Ub‐K63 failed to increase the ubiquitination of Nox4 (Figure 7b), suggesting that Nox4 is mainly modified by K48‐linked polyubiquitination.
Next, we observed the effects of Cx32 on the polyubiquitination of Nox4 in GMCs. As shown in Figure 7c, the polyubiquitination of Nox4 was reduced under HG conditions and was promoted by Cx32 overexpression in GMCs exposed to MG132 (5 μM). The results indicated that Cx32 increased the K48‐linked, but not K63‐linked, polyubiquitination of Nox4 (Figure 9).
Figure 9.
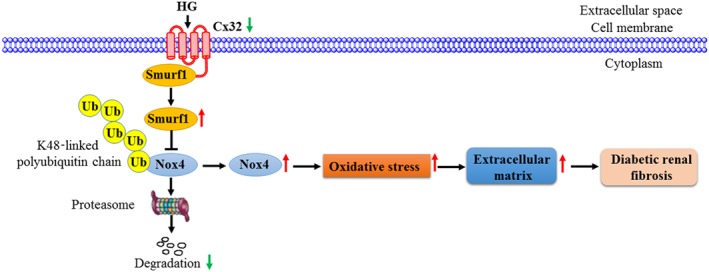
Diagram of the pathways involved in the effects of Cx32 on kidneys of diabetic mice. Cx32 ameliorates diabetic renal fibrosis by promoting Nox4 polyubiquitination and degradation via the inhibition of Smurf1 expression. Under normal conditions, Smurf1 is bound to the carboxyl terminal of Cx32 on the membrane of GMCs. Also, Nox4 is mainly modified by K48‐linked polyubiquitination and subsequently degraded by the proteasome. Under diabetic or HG conditions, the protein expression of Cx32 was decreased, leading to the up‐regulation of Smurf1 expression. Overexpression of Smurf1 reduces K48‐linked polyubiquitination of Nox4 and increases its protein levels, resulting in intracellular H2O2 overproduction. Increased oxidative stress further promoted extracellular matrix protein (fibronectin) expression, leading to exacerbation of diabetic renal fibrosis
3.8. Cx32 increases K48‐linked polyubiquitination of Nox4 via the inhibition of Smurf1 expression
To clarify the mechanisms underlying enhanced Nox4‐specific ubiquitination, we focused on the role of Smurf1, a HECT‐type E3 ubiquitin‐protein ligase, which is associated with multiple biological processes (Cao & Zhang, 2013). Therefore, we next explored whether Smurf1 is involved in the regulation of Cx32 on K48‐linked polyubiquitination of Nox4 in GMCs. The expression of Smurf1 was increased at 24 hr after HG stimulation, compared with NG, while the overexpression of Cx32 reduced Smurf1 expression (Figure 8a). Conversely, Cx32 knockdown further increased Smurf1 expression under HG conditions (Figure 8b). Additionally, Cx32 interacted with Smurf1 in GMCs, as shown by the co‐immunoprecipitation assay (Figure 8c). PRK‐Myc‐Smurf1 or pRK‐Myc‐Smurf1 del HECT plasmid was transfected into GMCs to analyse the effects of Smurf1 on Nox4 expression. We found that transfection of Smurf1 plasmid (WT) increased Nox4 expression compared with NG, whereas transfection of Smurf1 plasmid (HECT domain deleted) had no effects (Figure 8d), indicating that Smurf1 regulates the expression of Nox4 via its HECT domain. Moreover, co‐transfection of plasmids HA‐Ub‐K48 and pRK‐Myc‐Smurf1 was performed in GMCs. We observed that the overexpression of Smurf1 reduced K48‐linked polyubiquitination of Nox4, to a similar extent as that of Nox4 under HG conditions (Figure 8e). These data suggested that Cx32 increases K48‐linked polyubiquitination of Nox4 probably via inhibition of Smurf1 expression (Figure 9).
Figure 8.
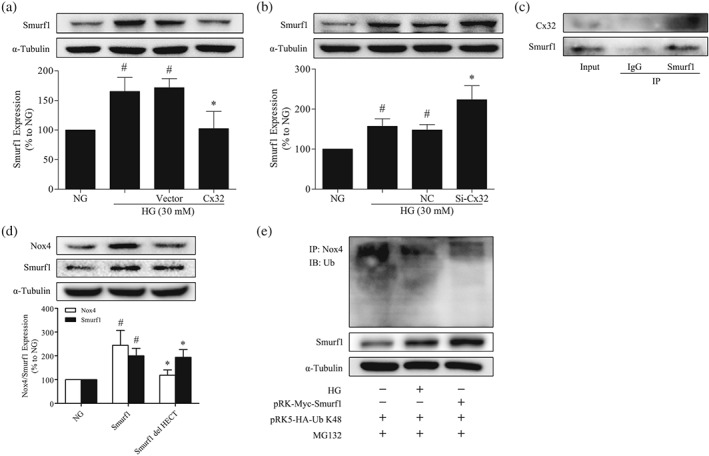
Cx32 increases K48‐linked polyubiquitination of Nox4 via the inhibition of Smurf1 expression. (a) Cx32 overexpression decreased the expression of Smurf1, # P < .05, significantly different from NG; * P < .05, significantly different from HG. (b) Cx32 knockdown increased Smurf1 expression, # P < .05, significantly different from NG; * P < .05, significantly different from HG. (c) Cx32 interacted with Smurf1 in GMCs. (d) Smurf1 increased Nox4 expression through its HECT domain, # P < .05, significantly different from NG; * P < .05, significantly different from Smurf1. The data are presented as the means ± SD; n = 5. (e) Co‐transfection of plasmids HA‐Ub‐K48 and pRK‐Myc‐Smurf1 was performed in GMCs
4. DISCUSSION
In diabetic nephropathy, reduction of cell–cell communication mediated by connexins within the nephron may serve as an early marker of disease development. However, our current understanding of the roles of connexins in diabetic kidney remains minimal (Hills, Price, & Squires, 2015).
In the present study, we showed that the expression of Cx32 was significantly down‐regulated in the kidney of STZ‐induced diabetic mice and HG‐induced GMCs, a finding that was consistent with the previous study in which a significant reduction in Cx32 expression was observed in the perineurium of STZ‐induced diabetic rats (Pitre et al., 2001), suggesting that Cx32 may be involved in the pathological process of diabetic nephropathy.
To evaluate the role of Cx32 in diabetic renal fibrosis, we investigated the effects of Cx32 overexpression via adenovirus infection on renal function and fibrosis in STZ‐induced diabetic mice by tail vein injection. The results showed that overexpression of Cx32 improved the abnormal FBG and renal function in diabetic mice. Additionally, Cx32 overexpression ameliorated the diabetes‐induced increase in renal hypertrophy, mesangial expansion, ECM deposition, and fibrosis. A multifactorial increase in renal oxidative stress is the key link in the pathogenesis of diabetic renal fibrosis, which has gradually become a consensus (Forbes, Coughlan, & Cooper, 2008; Lee, Yu, Yang, Jiang, & Ha, 2003; Ruiz, Pergola, Zager, & Vaziri, 2013). Here, we found that Cx32 overexpression decreased oxidative stress in the kidney of diabetic mice. However, Cx32 deficiency had opposite effects, suggesting that Cx32 ameliorates diabetic renal fibrosis by suppressing oxidative stress in the kidneys of diabetic mice.
GMCs, intrinsic cells of the glomerulus, play an important role in the structure and physiological functions of the kidney. The accumulation of ECM proteins, such as fibronectin, promotes renal fibrosis, eventually leading to diabetic nephropathy (Abboud, 2012; Cove‐Smith & Hendry, 2008; Schlondorff & Banas, 2009). Thus, inhibiting the up‐regulation of fibronectin expression induced by oxidative stress in GMCs is of great significance to prevent and treat diabetic nephropathy. We found that Cx32 could reduce the generation of hydrogen peroxide, as well as suppress the up‐regulation of fibronectin , a marker of fibrosis, in HG‐induced GMCs, suggesting that Cx32 indeed ameliorates diabetic renal fibrosis via decreasing oxidative stress.
To determine whether the regulation of Cx32 in renal fibrosis depends on GJIC comprising Cx32, we used 18‐α‐GA, a GJIC blocker. 18‐α‐GA did not block the down‐regulation of fibronectin expression by Cx32 in GMCs, indicating that this effect of CX32 was not dependent on GJIC function. Indeed, a significant body of data demonstrated that connexin proteins also fulfil their biological functions by the formation of hemichannels in non‐gap junction regions or by the regulation of specific gene expression, or by the interaction with other cytoplasmic and membrane proteins, or as adhesion molecules without the need for functional gap junctions (Cotrina, Lin, & Nedergaard, 2008; Herve, Bourmeyster, & Sarrouilhe, 2004; Wei, Xu, & Lo, 2004).
To clarify how Cx32 regulates oxidative stress in the kidney of diabetic mice, we focused on the source of ROS in the diabetic kidney. Nox4 is the predominant Nox isoform expressed in the kidney. The major sources of ROS in the diabetic kidney are the mitochondrial electron transport chain and Nox, especially Nox4 (Gorin & Block, 2013; Rhee, 2016). Nox4, which localizes to membranes and mitochondria, inhibits the activity of the mitochondrial respiratory chain complex I, leading to mitochondrial dysfunction and excessive ROS production (Ago et al., 2010; Block et al., 2009; Koziel et al., 2013). Additionally, Nox4 deletion reduces oxidative stress in diabetic nephropathy (Thallas‐Bonke et al., 2014). Therefore, consistent with the previous report (Sedeek et al., 2010), we found that under HG conditions, the expression of Nox4 in GMCs was increased. Additionally, Nox4 depletion inhibited the expression of fibronectin in GMCs. Furthermore, Cx32 reduced the expression of Nox4. Meanwhile, Cx32 interacted with Nox4 in GMCs, indicating that Cx32 may reduce oxidative stress in the kidney of diabetic mice by inhibiting Nox4 expression. Importantly, Nox4 overexpression blocked the down‐regulation of fibronectin expression by Cx32 in HG‐induced GMCs. Taken together, our data suggest that Nox4 at least partly mediates the down‐regulation of fibronectin expression by Cx32 induced by oxidative stress in GMCs.
Regarding Nox4 activity, its protein level is a major point of regulation, and the proteasome is responsible for its degradation (Desai et al., 2014; Gil Lorenzo et al., 2015). Therefore, we attempted to investigate the possible mechanism by which Cx32 down‐regulates Nox4 protein expression, focusing on the ubiquitination/proteasome pathways. Interestingly, our data showed that Cx32 promoted K48‐linked polyubiquitination of Nox4. Our findings support that Cx32 reduces Nox4 expression and hydrogen peroxide overproduction, eventually inhibiting fibronectin expression, probably by promoting K48‐linked polyubiquitination of Nox4 as well as its proteasomal degradation.
Smurf1, a HECT‐type E3 ligase (Cao & Zhang, 2013), was reported to inhibit K48‐linked polyubiquitination of Keap1 and ERα and increase substrate stability (Gong et al., 2018; Yang et al., 2018). In our study, we observed that overexpression of Smurf1 inhibited the K48‐linked polyubiquitination of Nox4 and increased the expression of Nox4, whereas transfection of Smurf1 (HECT domain deleted) plasmid had no effects, being slightly contradictory with the traditional role of Smurf1 in targeting substrates for ubiquitin‐proteasome degradation. It remains to be determined why Smurf1 exerts different protein stability effects on its substrates. Furthermore, Cx32 interacted with Smurf1 as well as reducing its expression in HG‐induced GMCs, indicating that Cx32 might enhance the K48‐linked polyubiquitination of Nox4 through inhibiting Smurf1 expression.
Based on these data mentioned above, it is plausible that Cx32 reduces oxidative stress in the kidney of diabetic mice by inhibiting Nox4 expression, thereby ameliorating diabetic renal fibrosis and dysfunction, and its mechanism may be related to the inhibition of Smurf1 expression and promotion of Nox4 K48‐linked polyubiquitination (Figure 9). Targeting the Cx32‐Nox4 signalling axis may contribute to the development of novel therapeutic agents for diabetic nephropathy.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
C.Z.Q. conceived and designed the study, performed the experiments, collected the data, analysed and interpreted the data, and drafted the manuscript. S.X.H. and C.Q.H. contributed to data collection and some of the experiments. H.K.P. contributed to improving the language. X.H.M., L.Z.Y., and Y.Y. contributed to the analysis and interpretation of data. L.P.Q. contributed to the revision of the manuscript. L.T. and H.H.Q. contributed to the research design and supervision of the project.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Fig. S1. Immunofluorescence identification of primary GMCs. Antibodies of Desmin (A) and Vimentin (C), markers of GMCs, as well as Cytokeratin (B), a marker of epithelial cells were used for the immunofluorescence assays.
Figure S2. The expression of Cx32 is downregulated in the kidney of diabetic mice and HG‐induced GMCs. The expression of Cx32 was detected using western blotting analysis. (A) Cx32 expression in the kidney of STZ‐induced diabetic mice. # P < 0.01 vs. C57BL/6, n = 5. (B) Cx32 expression in HG‐induced GMCs. # P < 0.05 vs. 0 h, n = 5. The data are presented as the means ± SD.
Figure S3. Genotyping is performed by PCR analysis of DNA from mice tail‐tips. Primers used for detection of the Cx32 WT alleles generate a PCR product of 550 base pairs. Primers used for detection of the Cx32 KO alleles generate a PCR product of 414 base pairs.
Figure S4. The effects of Cx32 on renal fibrosis in STZ‐induced diabetic mice. (A and B) Glomerular histopathology in the kidney of diabetic mice was analysed by masson stain (400 × magnification), blue indicates collagen fibers. (C) Overexpression of Cx32 reduced the expressions of TGF‐β1 and α‐SMA in the kidney of STZ‐induced diabetic mice as shown by western blotting. # P < 0.05 vs. Ctrl + Ad‐V; *P < 0.05 vs. STZ + Ad‐V, n = 5. (D) Cx32 deficiency promoted the expression of TGF‐β1 in the kidney of STZ‐induced diabetic mice. #P < 0.05 vs. WT; *P < 0.05 vs. WT + STZ, n = 5. The data are presented as the means ± SD.
ACKNOWLEDGEMENTS
The authors thank Professor Ziqing Hei for kindly providing the Cx32 deficient mice. This work was supported by research grants from the National Natural Science Foundation of China (Grants 81573477, 81770816, and 81973375), the Key Project of Natural Science Foundation of Guangdong Province, China (Grant 2017A030311036), Guangdong Provincial Key Laboratory of Construction Foundation (Grant 2017B030314030), the Natural Science Foundation of Guangdong Province, China (Grant 2016A030310152), and the Science and Technology Planning Project of Guangdong Province, China (Grant 2016A020215219).
Chen Z, Sun X, Chen Q, et al. Connexin32 ameliorates renal fibrosis in diabetic mice by promoting K48‐linked NADPH oxidase 4 polyubiquitination and degradation. Br J Pharmacol. 2020;177:145–160. 10.1111/bph.14853
Contributor Information
Tian Lan, Email: lantian@gdpu.edu.cn.
Heqing Huang, Email: huangheq@mail.sysu.edu.cn.
REFERENCES
- Abboud, H. E. (2012). Mesangial cell biology. Experimental Cell Research, 318, 979–985. 10.1016/j.yexcr.2012.02.025 [DOI] [PubMed] [Google Scholar]
- ADVANCE Collaborative Group , Patel, A. , MacMahon, S. , Chalmers, J. , Neal, B. , Billot, L. , … Travert, F. (2008). Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. The New England Journal of Medicine, 358, 2560–2572. [DOI] [PubMed] [Google Scholar]
- Ago, T. , Kuroda, J. , Pain, J. , Fu, C. , Li, H. , & Sadoshima, J. (2010). Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circulation Research, 106, 1253–1264. 10.1161/CIRCRESAHA.109.213116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise guide to pharmacology 2017/18: Enzymes. British Journal of Pharmacology, 174(Suppl 1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017b). The Concise guide to pharmacology 2017/18: Other ion channels. British Journal of Pharmacology, 174(Suppl 1), S195–S207. 10.1111/bph.13881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise guide to pharmacology 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174, S208–S224. 10.1111/bph.13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017a). The Concise guide to pharmacology 2017/18: Other proteins. British Journal of Pharmacology, 174, S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, M. M. , Rafiei, N. , Poursafa, P. , Ebrahimpour, K. , Mozafarian, N. , Shoshtari‐Yeganeh, B. , … Kelishadi, R. (2018). Association of benzene exposure with insulin resistance, SOD, and MDA as markers of oxidative stress in children and adolescents. Environmental Science and Pollution Research International, 25, 34046–34052. 10.1007/s11356-018-3354-7 [DOI] [PubMed] [Google Scholar]
- Block, K. , Gorin, Y. , & Abboud, H. E. (2009). Subcellular localization of Nox4 and regulation in diabetes. Proceedings of the National Academy of Sciences of the United States of America, 106, 14385–14390. 10.1073/pnas.0906805106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, Y. , & Zhang, L. (2013). A Smurf1 tale: Function and regulation of an ubiquitin ligase in multiple cellular networks. Cellular and Molecular Life Sciences, 70, 2305–2317. 10.1007/s00018-012-1170-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Chen, Q. , Huang, J. , Gong, W. , Zou, Y. , Zhang, L. , … Huang, H. (2017). CK2α promotes advanced glycation end products‐induced expressions of fibronectin and intercellular adhesion molecule‐1 via activating MRTF‐A in glomerular mesangial cells. Biochemical Pharmacology, 148, 41–51. [DOI] [PubMed] [Google Scholar]
- Chen, Z. , Xie, X. , Huang, J. , Gong, W. , Zhu, X. , Chen, Q. , … Huang, H. (2017). Connexin43 regulates high glucose‐induced expression of fibronectin, ICAM‐1 and TGF‐β1 via Nrf2/ARE pathway in glomerular mesangial cells. Free Radical Biology & Medicine, 102, 77–86. 10.1016/j.freeradbiomed.2016.11.015 [DOI] [PubMed] [Google Scholar]
- Cotrina, M. L. , Lin, J. H. , & Nedergaard, M. (2008). Adhesive properties of connexin hemichannels. Glia, 56, 1791–1798. 10.1002/glia.20728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cove‐Smith, A. , & Hendry, B. M. (2008). The regulation of mesangial cell proliferation. Nephron. Experimental Nephrology, 108, e74–e79. 10.1159/000127359 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai, L. P. , Zhou, Y. , Estrada, A. V. , Ding, Q. , Cheng, G. , Collawn, J. F. , & Thannickal, V. J. (2014). Negative regulation of NADPH oxidase 4 by hydrogen peroxide‐inducible clone 5 (Hic‐5) protein. The Journal of Biological Chemistry, 289, 18270–18278. 10.1074/jbc.M114.562249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el‐Fouly, M. H. , Trosko, J. E. , & Chang, C. C. (1987). Scrape‐loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Experimental Cell Research, 168, 422–430. 10.1016/0014-4827(87)90014-0 [DOI] [PubMed] [Google Scholar]
- Forbes, J. M. , Coughlan, M. T. , & Cooper, M. E. (2008). Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes, 57, 1446–1454. 10.2337/db08-0057 [DOI] [PubMed] [Google Scholar]
- Gil Lorenzo, A. F. , Costantino, V. V. , Appiolaza, M. L. , Cacciamani, V. , Benardon, M. E. , Bocanegra, V. , & Vallés, P. G. (2015). Heat shock protein 70 and CHIP promote Nox4 ubiquitination and degradation within the losartan antioxidative effect in proximal tubule cells. Cellular Physiology and Biochemistry, 36, 2183–2197. 10.1159/000430184 [DOI] [PubMed] [Google Scholar]
- Gong, W. , Chen, Z. , Zou, Y. , Zhang, L. , Huang, J. , Liu, P. , & Huang, H. (2017). CKIP‐1 affects the polyubiquitination of Nrf2 and Keap1 via mediating Smurf1 to resist HG‐induced renal fibrosis in GMCs and diabetic mice kidneys. Free Radical Biology & Medicine, 115, 338–350. [DOI] [PubMed] [Google Scholar]
- Gong, W. , Chen, Z. , Zou, Y. , Zhang, L. , Huang, J. , Liu, P. , & Huang, H. (2018). CKIP‐1 affects the polyubiquitination of Nrf2 and Keap1 via mediating Smurf1 to resist HG‐induced renal fibrosis in GMCs and diabetic mice kidneys. Free Radical Biology & Medicine, 115, 338–350. 10.1016/j.freeradbiomed.2017.12.013 [DOI] [PubMed] [Google Scholar]
- Gorin, Y. , & Block, K. (2013). Nox4 and diabetic nephropathy: With a friend like this, who needs enemies? Free Radical Biology & Medicine, 61, 130–142. 10.1016/j.freeradbiomed.2013.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajiluian, G. , Abbasalizad Farhangi, M. , Nameni, G. , Shahabi, P. , & Megari‐Abbasi, M. (2018). Oxidative stress‐induced cognitive impairment in obesity can be reversed by vitamin D administration in rats. Nutritional Neuroscience, 21, 744–752. 10.1080/1028415X.2017.1348436 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herve, J. C. , Bourmeyster, N. , & Sarrouilhe, D. (2004). Diversity in protein‐protein interactions of connexins: Emerging roles. Biochimica et Biophysica Acta, 1662, 22–41. 10.1016/j.bbamem.2003.10.022 [DOI] [PubMed] [Google Scholar]
- Hills, C. E. , Price, G. W. , & Squires, P. E. (2015). Mind the gap: Connexins and cell‐cell communication in the diabetic kidney. Diabetologia, 58, 233–241. 10.1007/s00125-014-3427-1 [DOI] [PubMed] [Google Scholar]
- Jaquet, V. , Marcoux, J. , Forest, E. , Leidal, K. G. , McCormick, S. , Westermaier, Y. , … Bedard, K. (2011). NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. British Journal of Pharmacology, 164, 507–520. 10.1111/j.1476-5381.2011.01439.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha, J. C. , Banal, C. , Chow, B. S. , Cooper, M. E. , & Jandeleit‐Dahm, K. (2016). Diabetes and kidney disease: Role of oxidative stress. Antioxidants & Redox Signaling, 25, 657–684. 10.1089/ars.2016.6664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha, J. C. , Banal, C. , Okabe, J. , Gray, S. P. , Hettige, T. , Chow, B. S. M. , … Jandeleit‐Dahm, K. (2017). NADPH oxidase Nox5 accelerates renal injury in diabetic nephropathy. Diabetes, 66, 2691–2703. 10.2337/db16-1585 [DOI] [PubMed] [Google Scholar]
- Jha, J. C. , Gray, S. P. , Barit, D. , Okabe, J. , El‐Osta, A. , Namikoshi, T. , … Jandeleit‐Dahm, K. A. (2014). Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long‐term diabetic nephropathy. Journal of the American Society of Nephrology, 25, 1237–1254. 10.1681/ASN.2013070810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha, J. C. , Thallas‐Bonke, V. , Banal, C. , Gray, S. P. , Chow, B. S. , Ramm, G. , … Jandeleit‐Dahm, K. A. (2016). Podocyte‐specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia, 59, 379–389. 10.1007/s00125-015-3796-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar, Y. S. , Sun, L. , Xie, P. , Liu, F. Y. , & Chen, S. (2011). A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annual Review of Pathology, 6, 395–423. 10.1146/annurev.pathol.4.110807.092150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & NC3Rs Reporting Guidelines Working Group (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziel, R. , Pircher, H. , Kratochwil, M. , Lener, B. , Hermann, M. , Dencher, N. A. , & Jansen‐Dürr, P. (2013). Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. The Biochemical Journal, 452, 231–239. 10.1042/BJ20121778 [DOI] [PubMed] [Google Scholar]
- Lee, H. B. , Yu, M. R. , Yang, Y. , Jiang, Z. , & Ha, H. (2003). Reactive oxygen species‐regulated signaling pathways in diabetic nephropathy. Journal of the American Society of Nephrology, 14, S241–S245. 10.1097/01.ASN.0000077410.66390.0F [DOI] [PubMed] [Google Scholar]
- Martyn, K. D. , Frederick, L. M. , von Loehneysen, K. , Dinauer, M. C. , & Knaus, U. G. (2006). Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cellular Signalling, 18, 69–82. 10.1016/j.cellsig.2005.03.023 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa, S. , Treumann, A. , Bell, A. , Vistoli, G. , Nelson, G. , Hay, S. , & von Zglinicki, T. (2016). Carboxylesterase converts Amplex red to resorufin: Implications for mitochondrial H2O2 release assays. Free Radical Biology & Medicine, 90, 173–183. 10.1016/j.freeradbiomed.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moennikes, O. , Buchmann, A. , Ott, T. , Willecke, K. , & Schwarz, M. (1999). The effect of connexin32 null mutation on hepatocarcinogenesis in different mouse strains. Carcinogenesis, 20, 1379–1382. 10.1093/carcin/20.7.1379 [DOI] [PubMed] [Google Scholar]
- Nath, S. , Ghosh, S. K. , & Choudhury, Y. (2017). A murine model of type 2 diabetes mellitus developed using a combination of high fat diet and multiple low doses of streptozotocin treatment mimics the metabolic characteristics of type 2 diabetes mellitus in humans. Journal of Pharmacological and Toxicological Methods, 84, 20–30. 10.1016/j.vascn.2016.10.007 [DOI] [PubMed] [Google Scholar]
- Nelles, E. , Butzler, C. , Jung, D. , Temme, A. , Gabriel, H. D. , Dahl, U. , … Willecke, K. (1996). Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32‐deficient mice. Proceedings of the National Academy of Sciences of the United States of America, 93, 9565–9570. 10.1073/pnas.93.18.9565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyamada, M. , Takebe, K. , & Oyamada, Y. (2013). Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochimica et Biophysica Acta, 1828, 118–133. 10.1016/j.bbamem.2011.12.031 [DOI] [PubMed] [Google Scholar]
- Palumbo, S. , Shin, Y. J. , Ahmad, K. , Desai, A. A. , Quijada, H. , Mohamed, M. , … Hecker, L. (2017). Dysregulated Nox4 ubiquitination contributes to redox imbalance and age‐related severity of acute lung injury. American Journal of Physiology. Lung Cellular and Molecular Physiology, 312, L297–L308. 10.1152/ajplung.00305.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitre, D. A. , Seifert, J. L. , & Bauer, J. A. (2001). Perineurium inflammation and altered connexin isoform expression in a rat model of diabetes related peripheral neuropathy. Neuroscience Letters, 303, 67–71. 10.1016/S0304-3940(01)01696-2 [DOI] [PubMed] [Google Scholar]
- Rhee, E. P. (2016). NADPH oxidase 4 at the nexus of diabetes, reactive oxygen species, and renal metabolism. Journal of the American Society of Nephrology, 27, 337–339. 10.1681/ASN.2015060698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz, S. , Pergola, P. E. , Zager, R. A. , & Vaziri, N. D. (2013). Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney International, 83, 1029–1041. 10.1038/ki.2012.439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena, F. P. , & Gesualdo, L. (2005). Pathogenetic mechanisms of diabetic nephropathy. Journal of the American Society of Nephrology, 16(Suppl 1), S30–S33. 10.1681/ASN.2004110970 [DOI] [PubMed] [Google Scholar]
- Schlondorff, D. , & Banas, B. (2009). The mesangial cell revisited: No cell is an island. Journal of the American Society of Nephrology, 20, 1179–1187. 10.1681/ASN.2008050549 [DOI] [PubMed] [Google Scholar]
- Sedeek, M. , Callera, G. , Montezano, A. , Gutsol, A. , Heitz, F. , Szyndralewiez, C. , … Hébert, R. L. (2010). Critical role of Nox4‐based NADPH oxidase in glucose‐induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. American Journal of Physiology. Renal Physiology, 299, F1348–F1358. 10.1152/ajprenal.00028.2010 [DOI] [PubMed] [Google Scholar]
- Taylor, H. J. , Chaytor, A. T. , Evans, W. H. , & Griffith, T. M. (1998). Inhibition of the gap junctional component of endothelium‐dependent relaxations in rabbit iliac artery by 18‐α glycyrrhetinic acid. British Journal of Pharmacology, 125, 1–3. 10.1038/sj.bjp.0702078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thallas‐Bonke, V. , Jha, J. C. , Gray, S. P. , Barit, D. , Haller, H. , Schmidt, H. H. , … Jandeleit‐Dahm, K. A. (2014). Nox‐4 deletion reduces oxidative stress and injury by PKC‐α‐associated mechanisms in diabetic nephropathy. Physiological Reports, 2(11), e12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiburcio, T. C. , Willebrords, J. , da Silva, T. C. , Pereira, I. V. , Nogueira, M. S. , Crespo Yanguas, S. , … Cogliati, B. (2017). Connexin32 deficiency is associated with liver injury, inflammation and oxidative stress in experimental non‐alcoholic steatohepatitis. Clinical and Experimental Pharmacology & Physiology, 44, 197–206. 10.1111/1440-1681.12701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi, K. , Araya, J. , Minagawa, S. , Hara, H. , Ichikawa, A. , Saito, N. , … Kuwano, K. (2017). Azithromycin attenuates myofibroblast differentiation and lung fibrosis development through proteasomal degradation of NOX4. Autophagy, 13, 1420–1434. 10.1080/15548627.2017.1328348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, C. J. , Xu, X. , & Lo, C. W. (2004). Connexins and cell signaling in development and disease. Annual Review of Cell and Developmental Biology, 20, 811–838. 10.1146/annurev.cellbio.19.111301.144309 [DOI] [PubMed] [Google Scholar]
- Xie, X. , Lan, T. , Chang, X. , Huang, K. , Huang, J. , Wang, S. , … Huang, H. (2013). Connexin43 mediates NF‐κB signalling activation induced by high glucose in GMCs: Involvement of c‐Src. Cell Communication and Signaling: CCS, 11, 38 10.1186/1478-811X-11-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Yu, N. , Xu, J. , Ding, X. , Deng, W. , Wu, G. , … Zhuang, T. (2018). SMURF1 facilitates estrogen receptor a signaling in breast cancer cells. Journal of Experimental & Clinical Cancer Research, 37, 24 10.1186/s13046-018-0672-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Qin, S. K. , Wu, Q. , Wang, Z. S. , Zheng, R. S. , Tong, X. H. , … He, X. D. (2014). Connexin‐dependent gap junction enhancement is involved in the synergistic effect of sorafenib and all‐trans retinoic acid on HCC growth inhibition. Oncology Reports, 31, 540–550. 10.3892/or.2013.2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoungas, S. , Chalmers, J. , Neal, B. , Billot, L. , Li, Q. , Hirakawa, Y. , … Woodward, M. (2014). Follow‐up of blood‐pressure lowering and glucose control in type 2 diabetes. The New England Journal of Medicine, 371, 1392–1406. 10.1056/NEJMoa1407963 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunofluorescence identification of primary GMCs. Antibodies of Desmin (A) and Vimentin (C), markers of GMCs, as well as Cytokeratin (B), a marker of epithelial cells were used for the immunofluorescence assays.
Figure S2. The expression of Cx32 is downregulated in the kidney of diabetic mice and HG‐induced GMCs. The expression of Cx32 was detected using western blotting analysis. (A) Cx32 expression in the kidney of STZ‐induced diabetic mice. # P < 0.01 vs. C57BL/6, n = 5. (B) Cx32 expression in HG‐induced GMCs. # P < 0.05 vs. 0 h, n = 5. The data are presented as the means ± SD.
Figure S3. Genotyping is performed by PCR analysis of DNA from mice tail‐tips. Primers used for detection of the Cx32 WT alleles generate a PCR product of 550 base pairs. Primers used for detection of the Cx32 KO alleles generate a PCR product of 414 base pairs.
Figure S4. The effects of Cx32 on renal fibrosis in STZ‐induced diabetic mice. (A and B) Glomerular histopathology in the kidney of diabetic mice was analysed by masson stain (400 × magnification), blue indicates collagen fibers. (C) Overexpression of Cx32 reduced the expressions of TGF‐β1 and α‐SMA in the kidney of STZ‐induced diabetic mice as shown by western blotting. # P < 0.05 vs. Ctrl + Ad‐V; *P < 0.05 vs. STZ + Ad‐V, n = 5. (D) Cx32 deficiency promoted the expression of TGF‐β1 in the kidney of STZ‐induced diabetic mice. #P < 0.05 vs. WT; *P < 0.05 vs. WT + STZ, n = 5. The data are presented as the means ± SD.