

Abstract
Background and Purpose
Considerable effort has recently been directed at developing multifunctional opioid drugs to minimize the unwanted side effects of opioid analgesics. We have developed a novel multifunctional opioid agonist, DN‐9. Here, we studied the analgesic profiles and related side effects of peripheral DN‐9 in various pain models.
Experimental Approach
Antinociceptive effects of DN‐9 were assessed in nociceptive, inflammatory, and neuropathic pain. Whole‐cell patch‐clamp and calcium imaging assays were used to evaluate the inhibitory effects of DN‐9 to calcium current and high‐K+‐induced intracellular calcium ([Ca2+]i) on dorsal root ganglion (DRG) neurons respectively. Side effects of DN‐9 were evaluated in antinociceptive tolerance, abuse, gastrointestinal transit, and rotarod tests.
Key Results
DN‐9, given subcutaneously, dose‐dependently produced antinociception via peripheral opioid receptors in different pain models without sex difference. In addition, DN‐9 exhibited more potent ability than morphine to inhibit calcium current and high‐K+‐induced [Ca2+]i in DRG neurons. Repeated treatment with DN‐9 produced equivalent antinociception for 8 days in multiple pain models, and DN‐9 also maintained potent analgesia in morphine‐tolerant mice. Furthermore, chronic DN‐9 administration had no apparent effect on the microglial activation of spinal cord. After subcutaneous injection, DN‐9 exhibited less abuse potential than morphine, as was gastroparesis and effects on motor coordination.
Conclusions and Implications
DN‐9 produces potent analgesia with minimal side effects, which strengthen the candidacy of peripherally acting opioids with multifunctional agonistic properties to enter human studies to alleviate the current highly problematic misuse of classic opioids on a large scale.
What is already known
DN‐9 is a novel multifunctional agonist for opioid and neuropeptide FF receptors.
What this study adds
Subcutaneous DN‐9 produces potent antinociceptive effects via peripheral opioid receptors in several preclinical pain models.
DN‐9 shows less antinociceptive tolerance, constipation, motor impairment, and reward/abuse liability compared with morphine.
What is the clinical significance
DN‐9 may serve as a potential candidate for treating inflammatory and neuropathic pain diseases.
Abbreviations
- DRG
dorsal root ganglion
- NPFF
neuropeptide FF
- RF9
1‐adamantanecarbonyl‐RF‐NH2
1. INTRODUCTION
Pain is an evolution‐rooted sensory mechanism that protects the integrity of the organism. However, chronic pain that is refractory to therapy remains a clinical challenge that can derail patients' lives in all of its aspects including social and professional life (Birnbaum et al., 2011). Opioid analgesics have become widely used to treat acute and chronic pain of multiple origins, but the negative side effects, including constipation, abuse, and tolerance, adversely affect patient outcome (Stein, 2013a). Progress has been made in the development of opioid peptide analgesics to separate analgesia from unwanted effects. The dermorphin‐derived analogue [Dmt1]DALDA exhibits more potent and longer lasting antinociception than https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 without respiratory depression in rodents and shows no cross‐tolerance to morphine in morphine‐tolerant mice (Schiller, 2005). The https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1623 analogue ZH853 produces greater antinociceptive effects than morphine with reduced side effects on tolerance, respiratory depression, and abuse liability (Zadina et al., 2016). In addition, multifunctional opioid compounds have been proposed as a strategy for drug development. The mixed https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319&familyId=50&familyType=GPCR and κ‐ opioid receptor agonist Dmt‐c[D‐Lys‐Phe‐D‐1‐Nal‐Asp]‐NH2 attenuates experimental nociceptive pain in a dose‐dependent manner (Perlikowska et al., 2016). The mixed opioid/nociceptin receptor agonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9294 and AT‐121 exhibited powerful analgesia without abuse liability in primates (Ding et al., 2016; Ding et al., 2018). Therefore, the discovery of bifunctional opioid peptides provides an attractive strategy to address and mitigate the ongoing large scale opioid misuse.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1479 (NPFF) is an endogenous opioid‐modulating peptide that binds and activates the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=300 1 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=301 2 receptors (Mouledous, Mollereau, & Zajac, 2010). NPFF and related peptides exert complex modulatory effects on opioid analgesia depending on the routes of administration (Fang, Jiang, Li, Han, & Wang, 2011; Kontinen & Kalso, 1995). Recently, our group developed multifunctional peptides that can activate both the opioid and NPFF receptors. DN‐9 behaved as a multifunctional agonist at μ, δ and κ opioid receptors and at NPFF1 and NPFF2 receptors in a cAMP assay (Wang et al., 2016). Supraspinal DN‐9 administration exhibited potent non‐tolerance‐forming antinociception in both the tail–flick test and complete Freund's adjuvant (CFA)‐induced inflammatory pain model (Wang et al., 2016).
It is well known that the systemic antinociceptive effects of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 is mainly mediated by the μ receptors in the brain. However, numerous studies have demonstrated that peripheral opioid receptors are also involved in opioid‐induced analgesia, especially under pathological pain conditions (Hervera, Negrete, Leanez, Martin‐Campos, & Pol, 2011; Stein, Schafer, & Hassan, 1995). Pharmacological evidence indicates that activation of the peripheral opioid receptors exhibits significant antinociception in inflammatory, visceral and formalin‐induced pain models (Al‐Khrasani et al., 2012; Balogh et al., 2018; Furst et al., 2005; Lacko et al., 2016; Stein, 2013b). In theory, peripherally acting opioids may produce potent analgesia with reduced central side effects because they do not readily cross the blood–brain barrier (BBB) and activate opioid receptors in the brain. Earlier results have indicated that chronic morphine treatment does not lead to antinociceptive tolerance at peripheral μ receptors in an inflammatory pain model (Zollner et al., 2008). In addition, both the peripherally acting μ receptor agonist DALDA and the κ receptor agonist JT09 exhibit efficient antinociception with reduced side effects (Beck, Reichel, Helke, Bhadsavle, & Dix, 2019; Tiwari et al., 2016).
Here, we have recorded the antinociceptive effects of peripheral DN‐9 and systematically studied its side effects in mice to expand our knowledge of peripherally acting opioids, especially opioid ligands targeting multiple receptors. Subcutaneous administration of DN‐9 reduced nociceptive, inflammatory, and neuropathic pain via peripheral opioid receptors. DN‐9 exhibited greater inhibitory effects on Ca2+ current and high‐K+‐induced intracellular calcium ([Ca2+]i) in cultured dorsal root ganglion (DRG) neurons than morphine. Our data demonstrated that peripheral administration of DN‐9 produced antinociceptive effects that were greater than those of morphine, but with less antinociceptive tolerance, constipation, motor impairment, and reward/abuse liability.
2. METHODS
2.1. Animals
All animal care and experimental protocols complied with the guidelines established by the European Community (2010/63/EU) and were approved by the Ethics Committee and Institutional Animal Care and Use Committee of Lanzhou University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. Male and female Kunming strain mice (18–22 g) were obtained from the Experimental Animal Centre of Lanzhou University. The μ receptor‐deficient (MOR−/−) mice on C57BL/6 background (Schuller et al., 1999) were kindly provided by Dr Pintar (Rutgers Robert Wood Johnson Medical School). Mice were housed in a constant temperature (22 ± 1°C) and 12/12‐hr dark/light cycle environment with food and water available ad libitum. The wild‐type (WT) mice on a C57BL/6 background were used as control for MOR−/− mice on a C57BL/6 background. Kunming strain mice were used in all of the other experiments.
In all experiments, animals were randomly assigned into treatment groups. The numbers of mice in each group are designated in the individual figures. The sample size in each group was determined based on our previous studies with similar experimental protocols. The studies were blinded to treatment assignment and outcome assessment.
2.2. Tail–flick test
The mouse tail–flick test was used as an acute pain model to evaluate the antinociceptive potency of drugs. Briefly, Kunming mice were gently stabilized by hand, and a radiant heat source was focused on the dorsal surface of the tail approximately 3 cm from the tip. Tail–flick latency was defined as the time from the start of heat source exposure to a quick movement of the tail away from the heat source. The radiant heat intensity was calibrated to produce a baseline latency of approximately 3–5 s in naïve mice. A cut‐off time was set at 10 s to minimize tissue damage. The latency was determined before and after drug administration. Antinociceptive effects were quantified using the tail–flick latency and the AUC of the percent maximal possible effect (MPE%). MPE (%) = 100 × [(T1 − T2)/(10 − T2)], where T1 and T2 represented the post‐drug and baseline responsive latencies respectively. The AUCs were calculated using trapezoidal rules based on total % MPE during a 60‐min (morphine) or 90‐min (DN‐9) period.
2.3. Metabolic stability
Stability analysis of DN‐9 was performed using 100% mouse plasma as described in previous studie (Gentry et al., 1999). Briefly, blood was collected from an eye socket, and plasma was obtained after centrifugation for 20 min at 2,000 g. Approximately 0.1 μmol of drug (10 μl) was incubated with mouse plasma at 37°C. At 0, 5, 10, 15, 30, and 60 min after incubation, 20 μl of the mixture was removed, and enzyme activity was terminated by placing the sample on ice. Acetonitrile (20 μl) was added to the sample to precipitate proteins. Samples were centrifuged for 15 min at 13,000 g. The supernatant of the sample (20 μl) was collected for RP‐HPLC analysis. The degradation t 1/2 in mouse plasma was calculated using linear regression.
2.4. Inflammatory pain model
To evaluate the anti‐allodynic effects and tolerance of DN‐9 on inflammatory pain (Du, Wang, Chi, & Li, 2017), carrageenan‐ and CFA‐induced inflammatory pain were used in this study. Baseline responses to mechanical stimulation were determined prior to injection. Mice were s.c. injected with 20 μl of carrageenan (2% in sterile water, Sigma‐Aldrich) or CFA (each ml contained 1‐mg heat‐killed and dried Mycobacterium tuberculosis, 0.85‐ml paraffin oil, and 0.15‐ml mannide monooleate, Sigma‐Aldrich) into the plantar surface of the right hindpaw. All behaviour experiments were performed on Day 1 after carrageenan and Day 4 after CFA injections.
2.5. Neuropathic pain model
To evaluate the anti‐allodynic and anti‐hyperalgesic effects of DN‐9 on neuropathic pain (Du et al., 2017), a mouse sciatic nerve chronic constriction injury (CCI) model was used in this study. Baseline thresholds for mechanical, cold, or thermal stimulation were determined before surgery. Mice were anaesthetized with sodium pentobarbital (i.p., 80 mg·kg−1). An incision was made in the lateral skin of the right thigh, parallel to the sciatic nerve. The sciatic nerve was exposed by separating the biceps femoris and the gluteus superficialis. Three surgical chorda chirurgicalis (8/0 silk) were tied loosely around the sciatic nerve at a 1‐mm spacing until a brief twitch was observed in the right hindlimb. The wound was closed with surgical suture, and erythromycin ointment was applied externally. All behavioural experiments were initiated 7 days after surgery.
2.6. Primary cultures of DRG neurons
DRG neurons were cultured as previously reported (Bang et al., 2018). DRGs were aseptically removed from mice or rats (5–8 weeks old) and then incubated with collagenase (1.25 mg·ml−1, Roche, Penzberg, Germany)/dispase‐II (2.4 units·ml−1, Roche, Penzberg, Germany) at 37°C for 90 min. After incubation, cells were dissociated with a pipette and passed through a 70‐μm filter. After centrifugation, DRG neurons were plated on 0.5 mg·ml−1 of poly‐d‐lysine–coated glass coverslips and grown in neurobasal medium supplemented with 10% FBS, 2% B‐27 supplement, and 1% penicillin/streptomycin for 24–48 hr before use.
2.7. Whole‐cell patch‐clamp recording in DRG neurons
Whole‐cell patch‐clamp recordings were performed at room temperature using an Axopatch‐200B amplifier (Axon Instruments, Inc., Foster, CA, USA) with a Digidata 1440A (Axon Instruments). The patch pipettes were pulled from borosilicate capillaries (World Precision Instruments, Inc., New Haven, CT, USA) using a P‐97 Flaming/Brown micropipette puller (Sutter Instrument Co., Novato, CA, USA). Pipette resistance was 4–6 MΩ. The calcium current was evoked by a 40‐ms step depolarization to −10 from −80 mV. The pipette solution contained 126‐mM CsCl, 5‐mM Mg‐ATP, 10‐mM EGTA, and 10‐mM HEPES, adjusted to a pH of 7.3 with CsOH. The external solution contained 135‐mM TEA‐Cl, 1‐mM CaCl2, 10‐mM HEPES, 4‐mM MgCl2, and 0.1‐μM TTX, adjusted to a pH of 7.4 with TEA‐OH.
2.8. Calcium imaging
Calcium imaging studies were performed in rat DRG neuron cultures using a Zeiss confocal microscope (LSM710, Carl Zeiss, Thornwood, NY, USA) with a 20× objective. Cells were loaded with FluoForte® Reagent (2 μM, Enzo, Farmingdale, NY, USA) for 30 min at 37°C and subsequently incubated in fresh buffer for a further 30 min. The buffer contained (in mM) 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES. The pH was adjusted to 7.30 and BSA (0.1%, Solarbio Life Science, Beijing, China) was added. FluoForte® Reagent was excited at 490 nm and emitted light measured at 520 nm. DRG neurons were depolarized with a high‐K+ solution, in which KCl was raised to 50 mM and NaCl reduced to maintain osmotic pressure. Data are expressed as the relative change in fluorescence (F/F0), where F is the control increase of fluorescence during the high‐K+ depolarization and F0 is the basal fluorescence.
2.9. Mechanical allodynia (electronic von Frey measurement)
Mice were individually placed in Plexiglas cages positioned on a wire‐mesh floor. Mice were allowed to acclimate to the environment for approximately 30 min prior to testing. A von Frey filament connected to a force transducer (IITC, Woodland Hills, CA, USA) was applied to the midplantar surface of the right hindpaw. Mechanical threshold was considered the maximum force that elicited paw withdrawal or licking, and paw withdrawal thresholds were tested before and 15, 30, 45, 60, and 90 min after injection. The von Frey filament was applied to the plantar surface three times at every time point, and the time interval between the three consecutive stimulations was approximately 2 min.
2.10. Thermal hyperalgesia (Hargreaves's test)
Sensitivity to thermal stimulation was evaluated using a thermal stimulator (PL‐200, Chengdu Technology & Market Co., Ltd., Chengdu, China). Briefly, mice were individually placed in Plexiglas cages with a glass floor to acclimate for approximately 30 min. A radiant beam of light (20% intensity) was focused on the surface of the right hindpaw, and the paw withdrawal response was determined as a quick movement of the hindpaw away from the radiant source. A cut‐off time was set at 25 s to avoid tissue damage. The latency was recorded as the average of three measurements with approximately 3‐min intervals between measurements.
2.11. Acetone evaporation assay
The acetone evaporation assay was performed as in a previous report (Bankar et al., 2018). Mice were individually placed on a wire‐mesh floor to acclimate to the environment for approximately 30 min. Then, 40‐μl acetone was applied on the plantar surface of the hindpaw. The time that the animal spent on flicking and licking the paw was recorded for 120 s. The cold sensitivity of mice was determined 1 day prior to CCI surgery and 7 days post‐surgery.
2.12. Development of tolerance to antinociception
Acute (tail–flick), inflammatory (CFA), and neuropathic (CCI) pain models were used to evaluate the development of antinociceptive tolerance. Mice were s.c. injected with saline, DN‐9 (9.48 μmol·kg−1) or morphine (31.08 μmol·kg−1) once daily for 7 or 8 days in acute or chronic pain models. DN‐9 (9.48 μmol·kg−1) was injected s.c. in the right flank of mice in inflammatory and neuropathic pain models on Day 8 to evaluate its effects on the nociceptive latency in mouse tolerated to morphine. The tail–flick latency was tested 30 min after morphine injection and 15 min after DN‐9 injection as these time points correlate to the observation of the maximal effects of the drugs. Similarly, mechanical allodynia or thermal hyperalgesia of mice was recorded 30 min post‐injection.
2.13. Immunofluorescence
The immunofluorescence procedures and analysis were performed according to the BJP guidelines (Alexander et al., 2018). Mice were anaesthetized with pentobarbital sodium (100 mg·kg−1, i.p.) and perfused with PBS (0.01 M) followed by 4% paraformaldehyde after 7 days of treatment with saline or drugs. The L4‐L5 lumbar segment of the spinal cord was excised, post‐fixed overnight in 4% paraformaldehyde, and placed in 20% sucrose at 4°C for cryoprotection. Tissue was embedded in OCT medium (Tissue‐Tek, Sakura, CA, USA), frozen at −80°C, and cut into 30‐μm transverse sections using a cryostat (CM3050S, Leica Microsystems, Nussloch, German). Non‐specific adsorption was minimized by incubating the sections in 5% normal donkey serum (Solarbio Life Science) in 0.01% Triton‐X 100 for 45 min. Sections were incubated overnight with the primary antibody against Iba‐1 (1:200, Wako, Tokyo, Japan, Cat #: 019‐19741, RRID:AB_839504). The appropriate fluorescently conjugated secondary antibody (1:1000, Thermo Fisher Scientific, Franklin, MA, USA, Cat #: A11012, RRID:AB_141359) was incubated with the sections for 2 hr. Images were randomly captured using a fluorescence microscope (BX53, Olympus Co., Tokyo, Japan), and the mean fluorescent pixels of spinal cord dorsal horn were measured by Image J (RRID:SCR_003070).
2.14. Open‐field test
Mouse locomotor activity was assessed using the open‐field test. A Plexiglas arena (black walls and floor, 50 cm × 50 cm × 40 cm) was equipped with a video tracking system (PMT‐100, Chengdu Technology & Market Co., Ltd.) to monitor mouse activity. The arena was wiped with 75% ethanol before and after each test to eliminate scents left by other mice. Mice were individually placed in the centre of the arena at the beginning of each measurement and allowed to explore freely for 30 min to confirm baseline activity. Animals were s.c. injected with saline, DN‐9 (9.48 μmol·kg−1) or morphine (31.08 μmol·kg−1), and the locomotor activity was continuously monitored for 150 min.
2.15. Place conditioning experiment
Conditioned place preference (CPP) testing was performed in a three‐chambered CPP apparatus in a sound‐attenuating environment (Lu, Wu, Zhang, Ai, & Li, 2011). The chambers consisted of two choice compartments (20 cm × 20 cm × 20 cm) on either side and a centre narrow compartment (5 cm × 20 cm × 20 cm) with a rectangular corridor (5 cm × 5 cm × 5 cm). The two choice compartments were visually and tactilely different (white walls with a rough floor vs. black walls with a smooth floor, about 20 lux for blank and 50 lux for white).
Mice had free access to the choice compartments for 15 min on the preconditioning test (Day 1), and the time spent in each compartment was recorded. Mice that spent over 60% of their time in one compartment were excluded from the experiments. Mice received two sessions daily in the conditioning test (Days 2–4) for 3 days. Mice were s.c. injected with drugs (saline, DN‐9, or morphine) and placed in one of the choice compartments for 15 min in the morning. Animals received a s.c. injection of saline approximately 6 hr later and were placed in the other compartment for 15 min. Mice had free access to the compartments during the 15‐min testing trial on the post‐conditioning test (Day 5), and the time spent in each compartment was measured manually. CPP score = T1 − T2, where the T1 and T2 were the time spent in the drug‐associated compartment on Day 5 and Day 1 respectively.
2.16. Naloxone‐precipitated withdrawal test
The main behavioural symptom of naloxone‐precipitated withdrawal response, jumping, was evaluated according to previous studies (Lin, Kao, Day, Chang, & Chen, 2016). Briefly, mice were injected with increasing doses of drug at an injection interval of approximately 8 hr. Then, mice were injected with naloxone (s.c., 10 mg·kg−1) 2 hr after the last challenge (Seth, Upadhyaya, Moghe, & Ahmad, 2011) and placed in a separate cylinder at a height of 32 cm and diameter of 12 cm. The number of jumps was recorded for 30 min.
2.17. Gastrointestinal transit test
The gastrointestinal transit (GIT) test was performed as we previously reported (Li et al., 2016). Briefly, mice fasted for 16 hr with free access to water prior to experiments. Saline, DN‐9, or morphine was s.c. injected 15 min before mice received an p.o. suspension of charcoal meal (5% charcoal in 10% gum arabic) at a volume of 0.1 ml per 10‐g body weight. Animals were killed 30 min later. The small intestine was carefully removed from the pyloric junction to the caecum, and the length of total small intestine and the distance the charcoal meal traveled were recorded. The results were expressed as the percent of GIT (GIT = (L1/L2) × 100; L1 and L2 were the distance travelled by the charcoal and the total distance of small intestine, respectively).
2.18. Rotarod test
Motor coordination, ataxia, and equilibrium were measured using a rotarod apparatus (ZB‐200, Chengdu Technology & Market Co., Ltd.) as previously reported (Li et al., 2016). Briefly, mice were trained in three consecutive trials for 2 days, and only the animals that were able to remain on the rod (16 rpm) for at least 180 s (cut‐off time 300 s) were used in the study. Tests were performed at 10, 20, 30, and 40 min after s.c. administration of saline, DN‐9 (9.48 μmol·kg−1), and morphine (31.08 μmol·kg−1). The time each mouse remained on the rod was recorded at each time point.
2.19. Data and statistical analysis
The data and statistical analysis comply with the recommendations of British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All data are presented as the mean ± SEM. Samples subjected to statistical analysis were from at least 5 animals per group (n = 5), where n = number of independent values. The exclusion criteria for mice in the tail–flick experiment and statistical analysis was the basal response did not range between 3 and 5 s. Data obtained from our study, excluding the tolerance development test and CPP test, were analysed using one‐way ANOVA followed by Dunnett's or Bonferroni's post hoc test. The post hoc tests were conducted only if F in ANOVA achieved P < .05 and there was no significant variance inhomogeneity. The ED50 values for the antinociceptive effects of drugs were determined using GraphPad Prism (RRID:SCR_002798). Data from the tolerance development test were analysed using one‐way ANOVA followed by Tukey's HSD test. Data from the CPP test were analysed using unpaired t tests. Two‐way ANOVA was used to analyse the time courses of the effects of DN‐9 and morphine. For immunofluorescence quantification, the results were normalized to the mean of the control value, and the control value was set as 1. The criterion of statistical significance was a probability less than 5%.
2.20. Materials
DN‐9 was obtained from Hybio Pharmaceutical Co., Ltd. (Shenzhen, China). NPFF and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1486 were synthesized on Rink MBHA resin using the N‐fluorenylmethoxycarbonyl (Fmoc)‐based solid‐phase synthesis (Wang et al., 2014) and purified using preparative reversed‐phase HPLC with a gradient of 30–75% solvent A (solvent A: 0.1% CF3COOH in CH3CN/water 9:1) and solvent B (solvent B: 0.1% CF3COOH in water). The purities of these compounds were confirmed using analytical HPLC and an electrospray ionization mass spectrometer (ESI‐Q‐TOF maXis‐4G, Bruker Daltonics, Bremen, Germany).
The opioid receptors antagonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638 and naloxone methiodide were purchased from Sigma‐Aldrich (St. Louis, MO, USA), and the selective antagonists https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1631 hydrochloride (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1631), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1641 (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1641), and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1642 dihydrochloride (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1642) were purchased from Tocris (Bristo, UK). Morphine hydrochloride was produced by Shenyang First Pharmaceutical Factory (Shenyang, China). All drugs were dissolved in physiological saline (0.9% NaCl) to the required concentrations and stored at −20°C.
2.21. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
3. RESULTS
3.1. Antinociceptive effects of DN‐9
3.1.1. DN‐9 produced antinociceptive effects in the mouse tail–flick test
As shown in Figure 1a,b, s.c. administration of graded doses of DN‐9 and morphine in male mice significantly increased the tail–flick latency. Similarly, peripheral administration of DN‐9 also produced equipotent antinociception in female mice (Figure 1c), suggesting that the effects of DN‐9 on pain management are not sex dependent. As shown in Table 1, DN‐9 was approximately 24‐fold more potent than morphine. The duration of analgesia for DN‐9 and morphine was approximately 90 and 60 min respectively (Figure 1a–c).
Figure 1.
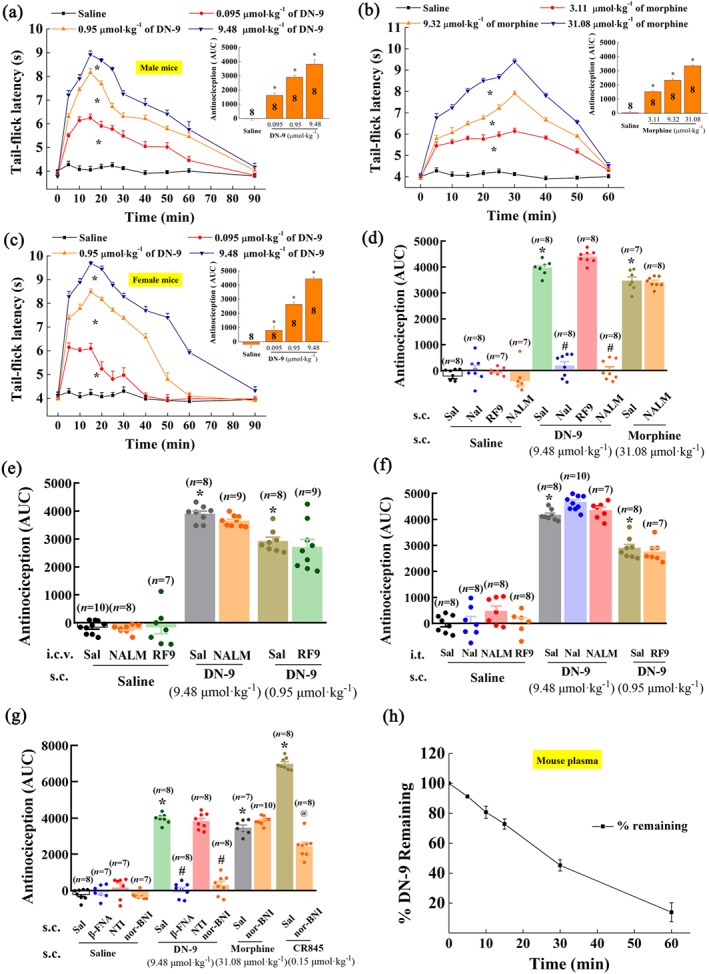
Antinociceptive effects of DN‐9 and morphine in the mouse tail–flick test. Time‐response curves for the antinociception induced by 0.095, 0.95, and 9.48 μmol·kg−1 of DN‐9 (a), 3.11, 9.32, and 31.08 μmol·kg−1 of morphine (b) in male mice, and 0.095, 0.95, and 9.48 μmol·kg−1 of DN‐9 in female mice (c) after s.c. administration. The AUC values of MPE % during the observed period from these data were statistically analysed and are presented in the insert. * P < .05, significantly different from saline group; one‐way ANOVA followed by Dunnett's post hoc test. (d) Effects of peripheral administration of the opioid receptor antagonists naloxone (Nal, 10 mg·kg−1) and naloxone methiodide (NALM, 10 mg·kg−1) and NPFF receptor antagonist RF9 (5 mg·kg−1) on the antinociception induced by DN‐9 and morphine. Effect of i.c.v. (e) and i.t. (f) administration of the opioid receptor antagonists naloxone (5 nmol) and NALM (5 nmol) and NPFF receptor antagonist RF9 (10 nmol) on the antinociception induced by DN‐9. (g) Effects of peripheral administration of the selective opioid receptor antagonists β‐FNA (1 mg·kg−1), NTI (1 mg·kg−1), and nor‐BNI (1 mg·kg−1) on the antinociception induced by DN‐9, morphine, and CR845.Each data point represents the mean ± SEM, n = 7–10 mice per group. * P < .05, significantly different from saline + saline group, # P < .05, significantly different from saline + DN‐9 group, @ P < .05, significantly different from saline + CR845 group; one‐way ANOVA followed by Bonferroni's post hoc test. (h) in vitro plasma stability assessment of DN‐9 in mouse plasma. Each data point represents the mean ± SEM, n = 4 independent experiments. RF9, 1‐adamantanecarbonyl‐RF‐NH2
Table 1.
Antinociceptive effects of DN‐9 and morphine in various pain models
| Pain models | Acute pain | Inflammatory pain | Neuropathic pain | |||
|---|---|---|---|---|---|---|
| Mechanical | Thermal | Cold | ||||
| ED50 for DN‐9 (μmol·kg−1) | Male | 0.22 (0.14–0.31) | 3.08 (2.55–3.67) | 3.32 (2.96–3.69) | 2.96 (2.43–3.56) | 0.25 (0.08–0.66) |
| Female | 0.22 (0.17–0.28) | 4.04 (3.12–5.05) | 3.42 (2.15–4.69) | 3.15 (1.67–4.40) | 0.29 (0.14–0.59) | |
| ED50 for Morphine (μmol·kg−1) | Male | 5.24 (4.57–5.95) | 8.05 (5.93–10.53) | 10.96 (3.31–46.35) | 10.13 (4.28–28.41) | 4.20 (3.08–5.26) |
Note. DN‐9 was injected s.c. in mice. The ED50 values were determined at the time of peak effects using non‐linear regression analysis.
Our previous results demonstrated that DN‐9 functions as a multifunctional agonist of opioid and NPFF receptors (Wang et al., 2016). To further determine whether opioid and NPFF receptors are involved in DN‐9‐induced peripheral antinociception, mice were pretreated with the opioid receptor antagonist naloxone or the peripherally acting opioid antagonist naloxone methiodide (Mika, Wawrzczak‐Bargiela, Osikowicz, Makuch, & Przewlocka, 2009). DN‐9‐induced peripheral antinociception was completely blocked by s.c. administration of naloxone (10 mg·kg−1) or naloxone methiodide (10 mg·kg−1) but not spinal or supraspinal administration of naloxone methiodide (5 nmol; Figure 1d–f). The doses of naloxone and naloxone methiodide were as used in our previous study (Zhang et al., 2017). In contrast, morphine‐induced antinociception (31.08 μmol·kg−1, s.c.) was not affected by s.c. administration of naloxone methiodide (10 mg·kg−1). The results showed in Figure 1g indicated that the μ receptor antagonist β‐FNA (1 mg·kg−1, s.c.) and the κ receptor antagonist nor‐BNI (1 mg·kg−1, s.c.), but not the δ receptor antagonist NTI (1 mg·kg−1, s.c.), significantly reduced the antinociceptive effects of DN‐9. In addition, the antinociception induced by s.c. CR845 (0.15 μmol·kg−1), a peripherally acting κ receptor agonist (Janecka, Perlikowska, Gach, Wyrebska, & Fichna, 2010), but not morphine (31.08 μmol·kg−1) was significantly blocked by the peripheral administration of nor‐BNI (1 mg·kg−1, s.c.). Both peripheral (5 mg·kg−1, s.c.) and central (10 nmol, i.t. and i.c.v.) injection of RF9, an antagonist of NPFF receptors, had no significant influence on DN‐9‐induced peripheral antinociception (Figure 1d‐f).
As shown in Figure 1h, our experiments demonstrate that the degradation t 1/2 of DN‐9 in mouse plasma was 21.8 ± 3.6 min, which basically correlates with the peak analgesic effects of DN‐9.
3.1.2. DN‐9 produced antinociceptive effects in an inflammatory pain model
The mean baseline value for mechanical stimulation was 4.99 ± 0.04 g prior to carrageenan injection and decreased significantly to 1.20 ± 0.04 g at 24 hr post‐injection (Figure 2a–c). Both DN‐9 and morphine dose‐dependently increased the withdrawal thresholds in male mice (Figure 2a,b). Furthermore, there was no sex dimorphism of the anti‐allodynic effects of DN‐9 (Figure 2c,). The anti‐allodynic ED50 values for DN‐9 and morphine are presented in Table 1.
Figure 2.
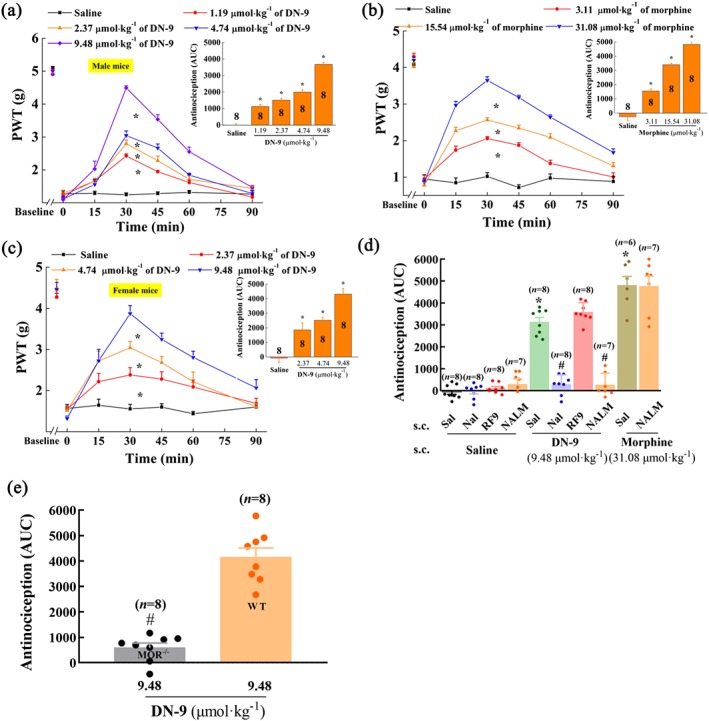
Effects of s.c. injection of DN‐9 and morphine on carrageenan‐induced inflammatory pain in mice. Time‐response curves for the anti‐allodynic effects induced by 1.19, 2.37, 4.74, and 9.48 μmol·kg−1 of DN‐9 (a), 3.11, 15.54, and 31.08 μmol·kg−1 of morphine (b) in male mice, and 2.37, 4.74, and 9.48 μmol·kg−1 DN‐9 in female mice (c). The AUC values of MPE % during the observed period from these data were statistically analysed and presented in the insert. * P < .05, significantly different from saline group; one‐way ANOVA followed by Dunnett's post hoc test. (d) Effects of peripheral administration of the opioid receptor antagonists naloxone (Nal, 10 mg·kg−1) and naloxone methiodide (NALM, 10 mg·kg−1) and NPFF receptor antagonist RF9 (5 mg·kg−1) on the anti‐allodynic effects of DN‐9 and morphine. * P < .05, significantly different from saline + saline group, # P < .05, significantly different from saline + DN‐9 group; one‐way ANOVA followed by Bonferroni's post hoc test. (e) The different anti‐allodynic effects between WT and MOR−/− mice. # P < .05, significantly different from WT mice; unpaired t test. Each data point represents the mean ± SEM, n = 6–8 mice per group
In addition, pretreatment with s.c. naloxone (10 mg·kg−1) and naloxone methiodide (10 mg·kg−1) abolished the anti‐allodynic effects of s.c. DN‐9 (Figure 2d). This is in line with the results obtained from MOR−/− mice on a C57BL/6 background in which DN‐9‐induced anti‐allodynic effects were significantly attenuated compared with the WT mice on a C57BL/6 background (Figure 2e). However, pretreatment with s.c. naloxone methiodide (10 mg·kg−1) did not affect the anti‐allodynic effects of morphine (Figure 2d). In contrast, pretreatment with the NPFF receptor antagonist RF9 (5 mg·kg−1, s.c.) did not modify the anti‐allodynic effects of DN‐9 in this pain model (Figure 2d).
3.1.3. DN‐9 produced antinociceptive effects in a neuropathic pain model
Sciatic nerve ligation induced significantly nociceptive responses to mechanical, thermal, and cold stimulations. In male mice, the nociceptive responses to all three modes of stimulation were markedly reduced by s.c. DN‐9 (Figure 3a,d,g) or morphine (Figure 3b,e,g) in a dose‐dependent manner. Our results showed that the analgesic potency of morphine was decreased in the CCI neuropathic pain model as described earlier (Dworkin et al., 2010; Kalso, Edwards, Moore, & McQuay, 2004; Popiolek‐Barczyk, Makuch, Rojewska, Pilat, & Mika, 2014). Similarly, in female mice, s.c. DN‐9 also produced equipotent antinociception to male mice (Figure 3c,f,h). The antinociceptive ED50 values for DN‐9 are presented in Table 1.
Figure 3.
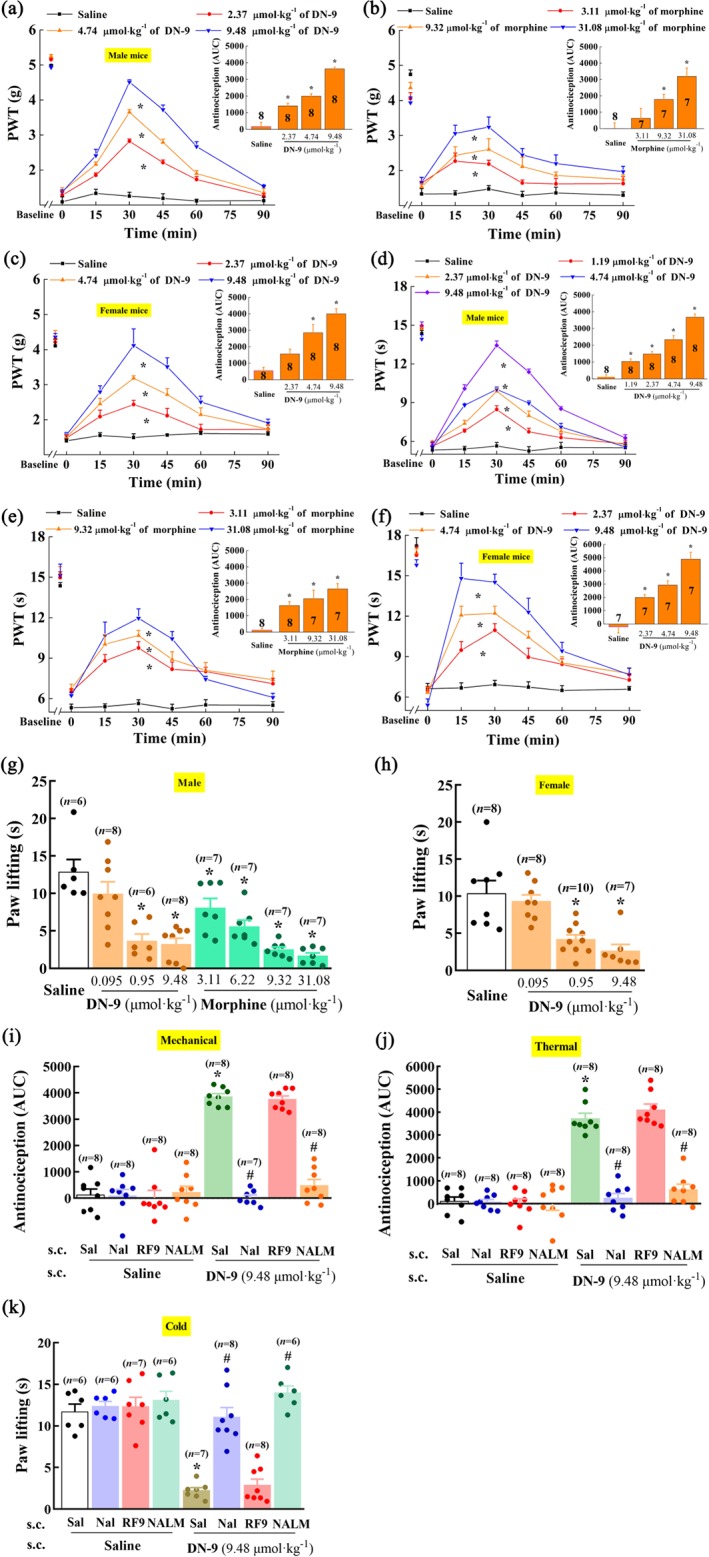
Effects of s.c. injection of DN‐9 and morphine on CCI‐induced neuropathic pain in mice. Time‐response curves for the anti‐allodynic effects induced by 2.37, 4.74, and 9.48 μmol·kg−1 of DN‐9 in male (a) and female (c) mice and 3.11, 9.32, and 31.08 μmol·kg−1 of morphine in male mice (b); anti‐hyperalgesic effects induced by 1.19, 2.37, 4.74, and 9.48 μmol·kg−1 of DN‐9 (d) and 3.11, 9.32, and 31.08 μmol·kg−1 of morphine (e) in male mice and 2.37, 4.74, and 9.48 μmol·kg−1 of DN‐9 in female mice (f). The AUC values of MPE % during the observed period from these data were statistically analysed and presented in the insert. The anti‐allodynic effects induced by 0.095, 0.95, and 9.48 μmol·kg−1 of DN‐9 in male (g) and female mice (h) and 3.11, 6.22, 9.32, and 31.08 μmol·kg−1 of morphine in male mice (g) in an acetone evaporation assay. * P < .05, significantly different from saline group; one‐way ANOVA followed by Dunnett's post hoc test. Effects of peripheral administrations of the opioid receptor antagonists naloxone (Nal, 10 mg·kg−1) and naloxone methiodide (NALM, 10 mg·kg−1) and NPFF receptor antagonist RF9 (5 mg·kg−1) on the anti‐allodynic [(i) for mechanical stimuli and (k) for cold stimuli] and anti‐hyperalgesic effects (j) induced by DN‐9. * P < .05, significantly different from saline + saline group, # P < .05, significantly different from saline + DN‐9 group; one‐way ANOVA followed by Bonferroni's post hoc test, n = 6–10 mice per group
Opioid and NPFF receptor antagonists were used to identify the roles of opioid and NPFF receptors in DN‐9‐induced analgesia in the CCI neuropathic pain model. Figure 3i–k demonstrates that pretreatment with naloxone (10 mg·kg−1, s.c.) and naloxone methiodide (10 mg·kg−1, s.c.) completely blocked the peripheral antinociceptive effects of DN‐9. However, the NPFF receptor antagonist RF9 (5 mg·kg−1, s.c.) did not affect the antinociceptive effects of DN‐9.
3.2. DN‐9 decreases the current of voltage‐gated Ca2+ channels and high‐K+‐induced [Ca2+]i in the cultured DRG neurons
Small‐sized DRG neurons are predominant nociceptors involved in pain processing (Snider & McMahon, 1998). We examined the inhibitory effects of DN‐9 and morphine to calcium current on small‐sized neurons using whole‐cell patch clamp. A step depolarization (−40 ms) from −80 to −10 mV triggered the voltage‐dependent calcium currents (Figure 4a). We found that both DN‐9 and morphine significantly decreased the Ca2+ current during the recording period (Figure 4a). However, 0.1 μM of DN‐9 was as effective as 10 μM of morphine (Figure 4b), implying that DN‐9 was more potent than morphine in this assay.
Figure 4.
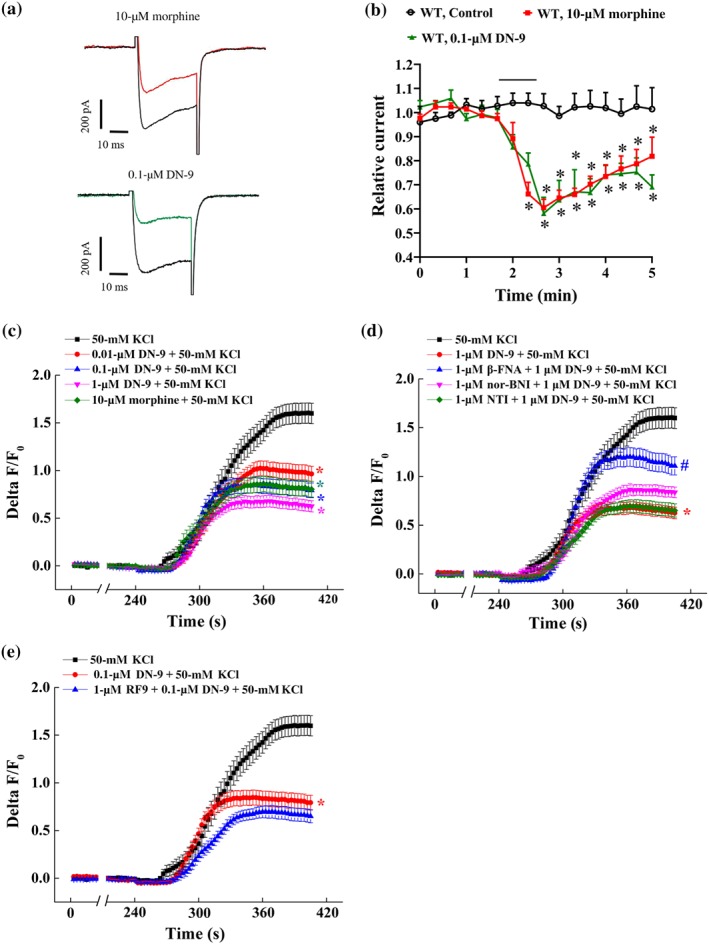
Effects of DN‐9 and morphine on calcium currents and [Ca2+]i in DRG neurons. (a) Trace of calcium currents before and after morphine (10 μM) or DN‐9 (0.1 μM) treatment in small‐sized DRG neurons from WT mice. (b) Time‐course of relative calcium currents before and after morphine (10 μM) or DN‐9 (0.1 μM) perfusion. Each data point represents the mean ± SEM, n = 5–6 neurons per group. * P < .05, significantly different from control group; one‐way ANOVA, followed by Bonferroni's post hoc test. (c) Calcium responses of DRG neurons (as Δ F/Fo) to 50 mM of high K+ after 1 min of DN‐9 (0.01, 0.1, and 1 μM) or morphine (10 μM) perfusion. Calcium responses of DRG neurons to 50‐mM high K+ after 2 min of β‐FNA (1 μM), NTI (1 μM), and nor‐BNI (1 μM) plus 1 min of DN‐9 (1 μM) perfusion (d) and after 2 min of RF9 (1 μM) plus 1 min of DN‐9 (0.1 μM) perfusion. Each data point represents the mean ± SEM, n > 50 neurons per group. * P < .05, significantly different from control group, # P < .05, significantly different from DN‐9 + KCl group; one‐way ANOVA, followed by Bonferroni's post hoc test
In addition, we examined the effects of DN‐9 and morphine on high‐K+‐induced Ca2+ increases in rat DRG neurons using a calcium‐imaging assay (Figure 4c). As reported, the rates of Ca2+ fluorescence increased following high KCl (50 mM) perfusion (Chen & Huang, 2017). DN‐9 was found to block the high‐K+‐induced [Ca2+]I in a dose‐dependent manner. Similarly, 0.1 μM of DN‐9 (48.8%) was equi‐potency compared to 10 μM of morphine (47.1%), which is consistent with the results obtained from whole‐cell patch clamp.
To investigate the role of opioid and NPFF receptors‐related events in DN‐9‐induced Ca2+ changes, neurons were pretreated with the selective opioid receptors antagonists β‐FNA (1 μM), NTI (1 μM), and nor‐BNI (1 μM) and the NPFF receptors antagonist RF9 (1 μM; Fitting et al., 2014). DN‐9‐induced inhibition was significantly antagonized by β‐FNA but not NTI (Figure 4d). In addition, nor‐BNI perfusion exhibited a slight but not significant reverse on DN‐9‐induced inhibition. In contrast, pretreatment with RF9 prior to DN‐9 perfusion resulted in a slight but not significant potentiation of inhibition by DN‐9, compatible with a previous report that NPFF counteracts the effects of activation of opioid receptors in the DRG neurons (Roumy & Zajac, 1999).
3.3. DN‐9 produced non‐tolerance‐forming antinociception in multiple pain models
In the tail–flick test, mice were daily treated with a single dose of DN‐9 or morphine for eight consecutive days. DN‐9‐induced analgesia (9.48 μmol·kg−1, s.c.) did not decrease during the 8‐day time course, which indicates that s.c. injection of DN‐9 produces a non‐tolerance‐forming analgesic effect in this acute pain model (Figure 5a). In contrast, the antinociceptive potency of morphine (31.08 μmol·kg−1, s.c.) gradually declined during chronic exposure from Day 4 to Day 8. Then, we investigated the effects of pharmacological blockade of NPFF receptors on the tolerance development of DN‐9. As shown in Figure 5a, daily treatment with RF9 (5 mg·kg−1, s.c.) prior to DN‐9 had no significant influence on DN‐9‐induced antinociception during the 8‐day time course.
Figure 5.
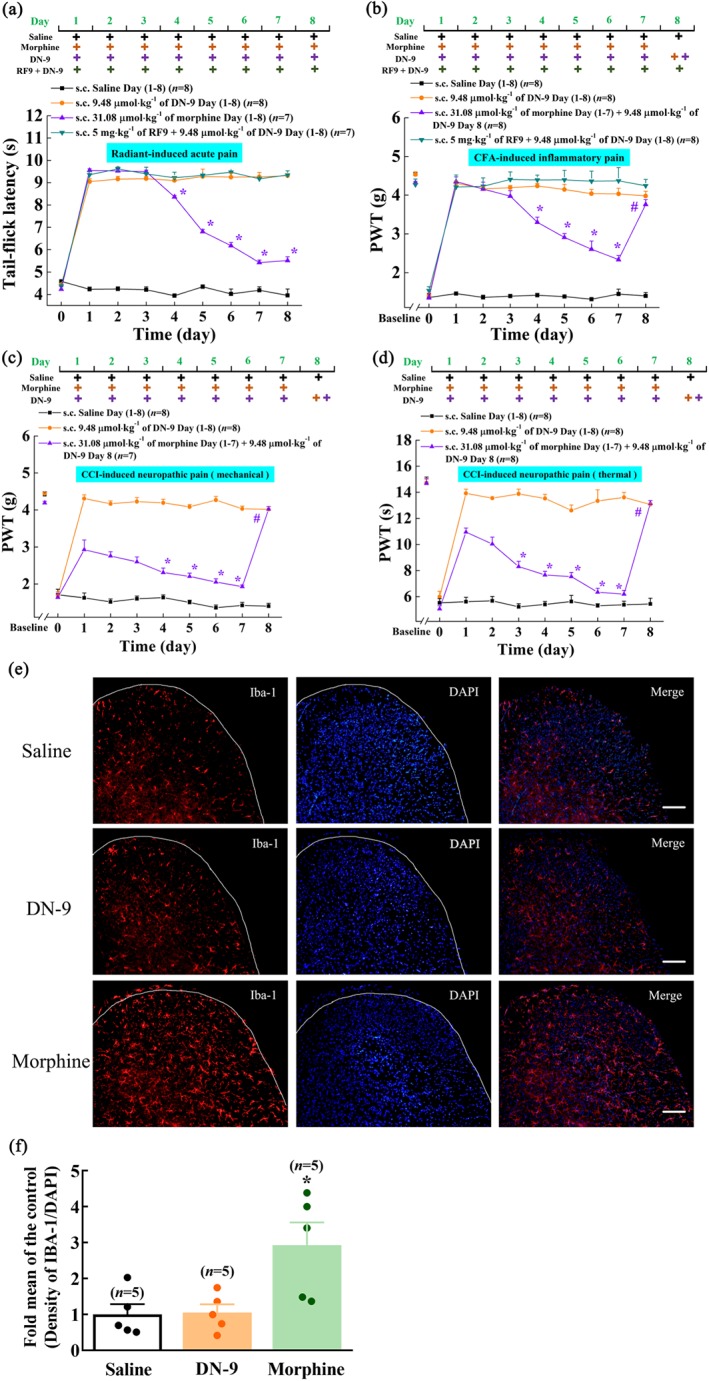
Tolerance evaluation of DN‐9 and morphine in different pain models. Antinociceptive effects of repeated administration of DN‐9 (9.48 μmol·kg−1, s.c.) and morphine (31.08 μmol·kg−1, s.c.) on acute (a), inflammatory (b), and neuropathic pain models [(c) anti‐allodynia; (d) anti‐hyperalgesia]. To evaluate the role of NPFF receptor on the tolerance development of DN‐9, RF9 was administrated prior to DN‐9 daily treatment in acute (a) and inflammatory (b) pain models. * P < .05, significantly different from the nociceptive latency on Day 1; one‐way ANOVA followed by Tukey's HSD post hoc test. To evaluate the effects of DN‐9 on the nociceptive latency in mice tolerance to morphine (31.08 μmol·kg−1, once daily for 7 days), DN‐9 (9.48 μmol·kg−1) was s.c. administered on Day 8 in inflammatory (b) and neuropathic (c,d) pain models. Each data point represents the mean ± SEM, n = 7–8 mice per group. # P < .05, indicates significant difference between latencies on Day 7 and Day 8; paired t test. (e,f) Effects of chronic DN‐9 and morphine administration on the expression of microglial cells in spinal cord. Each data point represents the mean ± SEM, n = 5, 5–6 images per animal. * P < .05, significantly different from saline group; one‐way ANOVA followed by Bonferroni's post hoc test. Scale bar equals 50 μm
Repeated administration of DN‐9 (9.48 μmol·kg−1, s.c.) produced similar, consistent anti‐allodynic effect over an 8‐day treatment course in CFA‐induced inflammatory pain model (Figure 5b). In contrast, while morphine (31.08 μmol·kg−1, s.c.) produced a potent anti‐allodynic effect from Day 1 to Day 3, its efficacy gradually declined from Day 4 to Day 7 (Figure 5b). Subcutaneous administration of DN‐9 (9.48 μmol·kg−1, s.c.) on Day 8 produced effective anti‐allodynia in morphine‐tolerant mice (Figure 5b). In addition, we also found that daily s.c. RF9 (5 mg·kg−1) prior to DN‐9 treatment had no significant influence on DN‐9‐induced anti‐allodynic effect in this inflammatory pain model.
Morphine (31.08 μmol·kg−1, s.c.) produced a significant decrease of antinociceptive potency from Day 4 to Day 7 in CCI‐induced neuropathic pain (Figure 5c,d). In contrast to morphine, the potencies of antinociceptive effects induced by repeated administration of DN‐9 (9.48 μmol·kg−1, s.c.) were sustained for 8 days (Figure 5c,d). As in the CFA pain model, the administration on Day 8 of DN‐9 (9.48 μmol·kg−1, s.c.) to morphine‐tolerant mice resulted in potent antinociception in the CCI pain model (Figure 5c,d).
Microglial activation in the spinal cord contributes to the development and maintenance of opioid tolerance (Qu et al., 2017). As shown in Figure 5e,f, morphine (31.08 μmol·kg−1, s.c.) but not DN‐9 (9.48 μmol·kg−1, s.c.) significantly up‐regulated the expression of microglial cells after daily treatment for 7 days.
3.4. DN‐9 exhibited low abuse liability
The abuse liability of peripheral DN‐9 was evaluated in three experimental paradigms: the classical responses of opioids to acute hyperlocomotion, CPP, and naloxone‐precipitated withdrawal.
The addictive properties of opioid analgesics are partially associated with activation of the dopaminergic reward circuits, which induces an acute hyperlocomotive response (Funada, Suzuki, Narita, Misawa, & Nagase, 1993; Siegfried, Filibeck, Gozzo, & Castellano, 1982). Figure 6 demonstrates that treatment with morphine (31.08 μmol·kg−1, s.c.) enhanced the locomotor effect in the open‐field assay and significantly increased the total distance moved, compared to treatment with saline. In contrast, DN‐9 (9.48 μmol·kg−1, s.c.) had no apparent effect on locomotion (71.4 ± 20.6 m). Subcutaneous morphine, but not DN‐9, induced acute hyperlocomotion with a stereotypic cycling movement (Figure 6c).
Figure 6.
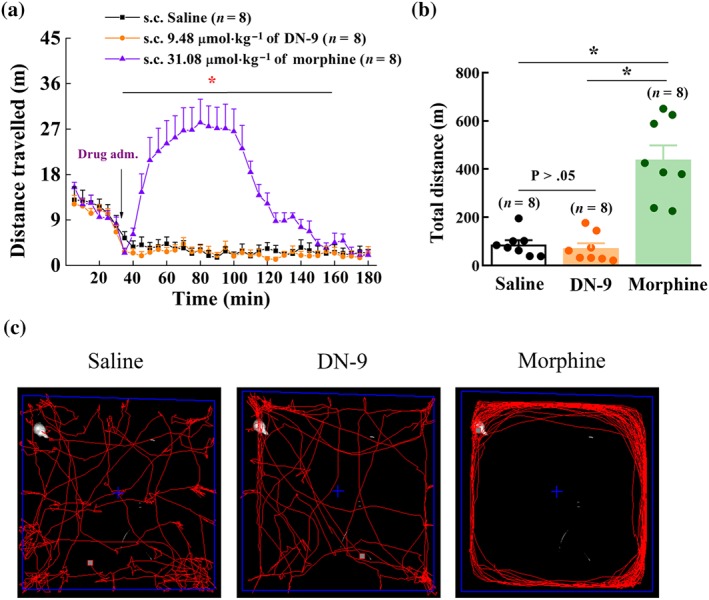
Effects of DN‐9 and morphine on locomotor activity in mice. (a) Time‐response curve of DN‐9 (9.48 μmol·kg−1) and morphine (31.08 μmol·kg−1) after s.c. administration. (b) The total distance after drug injection. Each data point represents the mean ± SEM, n = 8 mice per group. * P < .05, significantly different from saline group; one‐way ANOVA followed by Bonferroni's post hoc test. (c) Morphine, but not DN‐9, caused stereotypical circling after s.c. injection
Figure 7a presents the results of CPP experiments. Mice treated with saline (control group) did not exhibit a place preference change (4.7 ± 16.2 s), which indicates that s.c. injection of saline exerts no effect in an unbiased CPP paradigm. Subcutaneous morphine (31.08 μmol·kg−1) produced a significantly increased CPP score compared with the saline group. In contrast, DN‐9 (9.48 μmol·kg−1, s.c.) evoked a significantly weaker CPP response than morphine in mice, which indicates that s.c. DN‐9 caused less CPP change than morphine at equipotent analgesic dose.
Figure 7.
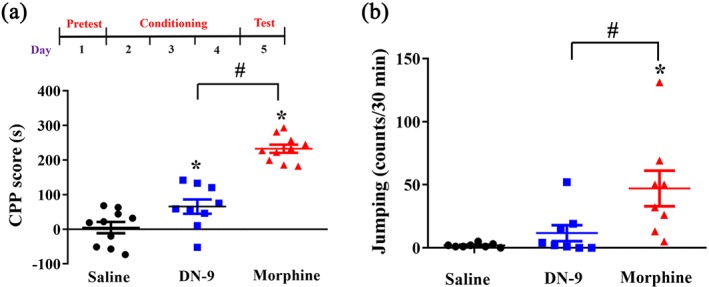
Dependence evaluation of DN‐9 and morphine in mice. (a) Effects of s.c. administration of DN‐9 (9.48 μmol·kg−1) and morphine (31.08 μmol·kg−1) on place conditioning in mice. The results are expressed as CPP score (time spent in drug‐paired side on post‐conditioning day minus the time on preconditioning day). * P < .05, significantly different from saline group, # P < .05, significant difference between DN‐9 group and morphine group; unpaired t test. (b) The jumping counts of naloxone‐precipitated withdrawal after DN‐9 and morphine treatment in mice. Each data point represents the mean ± SEM, n = 8–10 mice per group. * P < .05, significantly different from saline group, # P < .05, significant difference between DN‐9 group and morphine group; one‐way ANOVA followed by Dunnett's post hoc test
Jumping is the main behavioural symptom of naloxone‐precipitated withdrawal response (Zarrindast & Torkaman‐Boutorabi, 2003). Figure 7b demonstrates that morphine challenge significantly increased the jumping counts after naloxone injection compared to the saline group. In contrast, the jumping counts of the DN‐9 group were not significantly different from the saline group, indicating that DN‐9 mice exhibit no surrogates of withdrawal. Student's t test revealed a significant difference between the DN‐9 and morphine groups.
3.5. DN‐9 had a less potent effect on side effects like GIT and motor coordination
DN‐9 produced a significant, dose‐dependent delay in upper GIT when compared to vehicle treatment (Figure 8a), with an ED50 value (95% confidence limits) of 9.13 (3.62–19.69) μmol·kg−1. Treatment with morphine (31.08 μmol·kg−1) significantly reduced GIT, exhibiting a greater inhibition of GIT than DN‐9, at equi‐analgesic doses.
Figure 8.

Effects of s.c. administration of DN‐9 and morphine on gastrointestinal transit and motor coordination. (a) Effects of s.c. administration of DN‐9 (0.95, 9.48, 28.45, and 94.83 μmol·kg−1) and morphine (31.08 μmol·kg−1) on gastrointestinal transit in mice. * P < .05, significantly different from saline group, # P < .05, significantly different from morphine group; one‐way ANOVA followed by Dunnett's post hoc test. (b) Effects of s.c. injection of 9.48 μmol·kg−1 of DN‐9 and 31.08 μmol·kg−1 of morphine on motor coordination in mice. Each data point represents the mean ± SEM, n = 8–9 mice per group. No significant difference between drug group and saline group; one‐way ANOVA followed by Dunnett's post hoc test
Lastly, s.c. injection of DN‐9 (9.48 μmol·kg−1) or morphine (31.08 μmol·kg−1) did not affect the endurance time on the rotating rod, even 40 min after injection, and there were no significant differences when compared with saline‐treated mice (Figure 8b).
4. DISCUSSION
Morphine plays an important role in treating moderate to severe pain. There is an urgent need for compounds that can provide opioid‐like analgesia without the unwanted side effects. The recent development of multifunctional peptide‐based approaches that target opioid and other neuropeptide receptors have emerged as an attractive approach for innovative pain management in the age of the “opioid epidemic.” In addition, peripherally acting opioids may elicit limited central side effects due to their poor BBB penetration. However, several studies indicate that the therapeutic utility of peripherally restricted opioids can still be limited by adverse events, including inhibition of gastrointestinal motility, less potent antinociception to acute pain, and increased urine output (Albert‐Vartanian et al., 2016; DeHaven‐Hudkins & Dolle, 2004).
In this study, our results demonstrated that the antinociception induced by s.c. DN‐9 was blocked by peripheral but not central administration of the opioid receptor antagonists naloxone and naloxone methiodide, whereas the s.c. antinociception of morphine was not modified by peripheral administration of naloxone methiodide, as described in the previous study (Zhang et al., 2017). Thus, these findings suggest that DN‐9 did not penetrate the BBB and produced antinociception via peripheral opioid receptors. In addition, i.c.v. or i.t. injection of the NPFF receptor antagonist RF9 did not modify the antinociception induced by s.c. DN‐9, implying it is independent of the central NPFF receptors. In contrast, earlier studies indicated that the central antinociception of DN‐9 was significantly augmented by i.c.v. administration of RF9 (Wang et al., 2016); these findings also support the inability of DN‐9 to cross the BBB. Furthermore, DN‐9‐induced peripheral antinociception was selectively blocked by the antagonists of μ and κ receptors, suggesting that this antinociception is mainly via peripheral μ and κ receptors. Consistent with our findings, the previous reports demonstrated that the mixed μ and κ receptor agonists produced potent antinociceptive effects after peripheral administration (Perlikowska et al., 2016). A recent study has shown that the peripheral κ receptors play important roles in pain management (Snyder et al., 2018). In fact, s.c. administration of the selective κ receptor agonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9044 produced peripheral antinociception, as previously described (Janecka et al., 2010). Strikingly, DN‐9 elicits potent analgesia in various preclinical mouse models for acute, inflammatory, and neuropathic pain with minimal side effects after s.c. administration.
Addiction to opioid analgesics is at least partly associated with the activation of the dopaminergic reward circuits in the CNS, which contributes to CPP and induces an acute hyperlocomotive response upon withdrawal (Funada et al., 1993). DN‐9 was less potent than morphine in assays designed to measure CPP and withdrawal response. Previous studies reported that κ receptor agonists exerted potent antinociceptive effects with reduced abuse potential and antagonized μ receptor‐mediated reward (Hunter et al., 1990; Pan, 1998). Therefore, our data suggest that s.c. injection of DN‐9 exhibits reduced abuse liability, which may be associated with its κ receptor agonism and poor CNS penetration. Another unwanted effect of classic opioid analgesics is gastro‐paresis, which leads to not only inconvenient constipation but also significant co‐morbidity. DN‐9 produced less severe inhibition of GIT compared to morphine, consistent with our previous finding for BN‐9 (Li et al., 2016). Moreover, in the rotarod assay, peripherally applied DN‐9 did not elicit motor effects, underscoring its benign safety profile.
Antinociceptive tolerance complicates clinical management strategies and facilitates opioid side effects. Our results indicate that there was no development of tolerance to the antinociceptive effects of DN‐9 during 8‐day treatment in various pain models. However, in acute and inflammatory pain, daily treatment with the NPFF receptor antagonist RF9 had no significant effects on DN‐9‐induced antinociception. These results suggest that NPFF system is not involved in DN‐9‐induced non‐tolerance‐forming antinociception. In contrast, tolerance developed to morphine‐induced analgesia in various mouse models no later than Day 3/4 as previously described (Harada, Nakamoto, & Tokuyama, 2013; Li et al., 2016). The activation of microglial cells has been reported to play a crucial role in the development and maintenance of opioid tolerance (Qu et al., 2017). Unlike morphine, chronic treatment with DN‐9 did not significantly activate microglial cells in the spinal cord, which might account for the non‐tolerance‐forming antinociception of DN‐9. In addition, we are highly encouraged by the observation that the antinociceptive effects of DN‐9 in inflammatory or neuropathic pain models were also observed in morphine‐tolerant mice, which suggests that mice do not experience cross‐tolerance to DN‐9 following exposure to morphine. These findings are in line with the previous pharmacological results showing that the peripherally acting κ receptor agonist lacks cross‐tolerance to morphine (Walker, Catheline, Guilbaud, & Kayser, 1999). Thus, DN‐9 is an ideal candidate in the search for a therapeutic molecule that induces potent analgesia but does not cause tolerance in the animal.
Several studies indicate that morphine exhibits more potent antinociceptive effects in male mice compared with female mice (Mitrovic et al., 2003; Wang, Traub, & Murphy, 2006). Of note, DN‐9‐induced peripheral antinociception showed no sex difference in mouse models for acute, inflammatory, and neuropathic pain. Consistent with our findings, activation of peripheral μ receptors produced antinociception in a neuropathic pain model without sex dimorphism (Tiwari et al., 2016). Therefore, the non‐gender‐related antinociception of DN‐9 might be associated with its peripheral μ receptor agonism.
To characterize the differences between the analgesic effects of DN‐9 and morphine, we compared them in various pain models. The ED50 value for DN‐9 was 0.22 μmol·kg−1 in the tail–flick assay, suggesting it is 24‐fold more potent than morphine. Furthermore, we showed that DN‐9 was more potent than morphine with regard to depolarization‐induced Ca2+ transients and high‐K+‐induced [Ca2+]i in the DRG neurons, which implies that DN‐9 has powerful opioid agonistic activities in the peripheral nervous system. In fact, our previous study also demonstrated that, in the tail–flick assay, DN‐9 was 62‐fold more potent than morphine (0.0163 vs. 1.02 nmol) via μ and κ receptors after supraspinal injection (Li et al., 2016; Wang et al., 2016). Taken together, DN‐9 exerted potent antinociception at both central and peripheral levels.
Subcutaneous DN‐9 produced dose‐dependent antinociception in carrageenan‐induced inflammatory pain and CCI‐induced neuropathic pain, which was approximately 2.6‐ to 17‐fold more potent than morphine. We demonstrated that these antinociceptive effects were mediated by peripheral opioid receptors. In addition, our recent results demonstrating that BN‐9, a mixed opioid/NPFF receptor agonist, similarly produced peripheral anti‐allodynia after intraperitoneal administration (Zhang et al., 2017). DN‐9‐induced anti‐allodynic effects in inflammatory pain were significantly decreased in MOR−/− mice on a C57BL/6 background compared to WT mice on a C57BL/6 background, suggesting the involvement of μ receptors in the peripheral anti‐allodynia of DN‐9. These results correlate with those from our earlier study of pharmacological blockade with the selective opioid antagonists in tail flick assay. In addition, in the calcium imaging assay, the inhibitory effects of DN‐9 to high‐K+‐induced intracellular calcium were mainly mediated by μ receptors. Subcutaneous DN‐9 produced antinociceptive responses that were 2.6‐ to 24‐fold greater than those produced by morphine. This diverse antinociceptive potency between DN‐9 and morphine might be explained by the different pain models used in this study. Similarly, the previous results showed that the peripheral opioid agonists exhibited different analgesic profiles in mouse pain models (Lacko et al., 2016; Tiwari et al., 2016).
Our previous data demonstrated that DN‐9 acted as a mixed agonist at opioid/NPFF receptors in vitro and in vivo (Wang et al., 2016). Interestingly, we here found that DN‐9‐induced peripheral analgesia was not modified by the NPFF receptor antagonist RF9, which suggests that the peripheral antinociceptive effects of DN‐9 do not critically rely on the NPFF receptor system. Consistent with these data, in the calcium imaging assay, the inhibitory effects of DN‐9 were not significantly modified by RF9.
Taken together, our present work indicates that s.c. administration of DN‐9 produced potent antinociceptive effects via peripheral opioid receptors in several preclinical pain models without sex difference. In addition, at the peripheral level, DN‐9 showed less antinociceptive tolerance and abuse liability compared with morphine, which might be associated with its κ receptor agonism and peripherally acting properties. The electrophysiology and calcium imaging data also support the ability of DN‐9 to act as a potent agonist in the peripheral nervous system. Moreover, DN‐9 exhibited less severe constipation and motor impairment after s.c. administration.
Therefore, our data support DN‐9 as a potential candidate for translational‐medical development in the near future. This compound represents a technical innovation that positions us to respond to the “opioid‐crisis” without contributing to the associated addiction and mortality epidemics or losing the required potent analgesia.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
B.X., M.Z., X.S., R.Z., D.C., Y.C., Z.W., Y.Q., T.Z., K.X., and X.Z. performed the research. Q.F. and R.W. designed the research study. B.X., M.Z., and X.S. analysed the data. Q.F., B.X., W.L., and R.Z. wrote the paper.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
ACKNOWLEDGEMENTS
This study was supported by grants from the National Natural Science Foundation of China (81673282 and 81273355), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R27), and the Fundamental Research Funds for the Central Universities (lzujbky‐2018‐ot02).
Xu B, Zhang M, Shi X, et al. The multifunctional peptide DN‐9 produced peripherally acting antinociception in inflammatory and neuropathic pain via μ‐ and κ‐opioid receptors. Br J Pharmacol. 2020;177:93–109. 10.1111/bph.14848
Contributor Information
Wolfgang Liedtke, Email: wolfgang@neuro.duke.edu.
Rui Wang, Email: wangrui@lzu.edu.cn.
Quan Fang, Email: fangq@lzu.edu.cn.
REFERENCES
- Albert‐Vartanian, A. , Boyd, M. R. , Hall, A. L. , Morgado, S. J. , Nguyen, E. , Nguyen, V. P. , … Raffa, R. B. (2016). Will peripherally restricted κ‐opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential? Journal of Clinical Pharmacy and Therapeutics, 41, 371–382. 10.1111/jcpt.12404 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The concise guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(Suppl 1), S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Khrasani, M. , Lackó, E. , Riba, P. , Király, K. , Sobor, M. , Timár, J. , … Fürst, S. (2012). The central versus peripheral antinociceptive effects of μ‐opioid receptor agonists in the new model of rat visceral pain. Brain Research Bulletin, 87, 238–243. 10.1016/j.brainresbull.2011.10.018 [DOI] [PubMed] [Google Scholar]
- Balogh, M. , Zádori, Z. S. , Lázár, B. , Karádi, D. , László, S. , Mousa, S. A. , … Al‐Khrasani, M. (2018). The peripheral versus central antinociception of a novel opioid agonist: Acute inflammatory pain in rats. Neurochemical Research, 43, 1250–1257. 10.1007/s11064-018-2542-7 [DOI] [PubMed] [Google Scholar]
- Bang, S. , Yoo, J. , Gong, X. , Liu, D. , Han, Q. , Luo, X. , … Ji, R. R. (2018). Differential inhibition of Nav1.7 and neuropathic pain by hybridoma‐produced and recombinant monoclonal antibodies that target Nav1.7: Differential activities of Nav1.7‐targeting monoclonal antibodies. Neuroscience Bulletin, 34(1), 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankar, G. , Goodchild, S. J. , Howard, S. , Nelkenbrecher, K. , Waldbrook, M. , Dourado, M. , … Cohen, C. J. (2018). Selective NaV1.7 antagonists with long residence time show improved efficacy against inflammatory and neuropathic pain. Cell Reports, 24, 3133–3145. 10.1016/j.celrep.2018.08.063 [DOI] [PubMed] [Google Scholar]
- Beck, T. C. , Reichel, C. M. , Helke, K. L. , Bhadsavle, S. S. , & Dix, T. A. (2019). Non‐addictive orally‐active κ opioid agonists for the treatment of peripheral pain in rats. European Journal of Pharmacology, 856, 172396 10.1016/j.ejphar.2019.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum, H. G. , White, A. G. , Schiller, M. , Waldman, T. , Cleveland, J. M. , & Roland, C. L. (2011). Societal costs of prescription opioid abuse, dependence, and misuse in the United States. Pain Medicine, 12, 657–667. 10.1111/j.1526-4637.2011.01075.x [DOI] [PubMed] [Google Scholar]
- Chen, Y. , & Huang, L. M. (2017). A simple and fast method to image calcium activity of neurons from intact dorsal root ganglia using fluorescent chemical Ca2+ indicators. Molecular Pain, 13, 1744806917748051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven‐Hudkins, D. L. , & Dolle, R. E. (2004). Peripherally restricted opioid agonists as novel analgesic agents. Current Pharmaceutical Design, 10, 743–757. 10.2174/1381612043453036 [DOI] [PubMed] [Google Scholar]
- Ding, H. , Czoty, P. W. , Kiguchi, N. , Cami‐Kobeci, G. , Sukhtankar, D. D. , Nader, M. A. , … Ko, M. C. (2016). A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proceedings of the National Academy of Sciences of the United States of America, 113, E5511–E5518. 10.1073/pnas.1605295113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, H. , Kiguchi, N. , Yasuda, D. , Daga, P. R. , Polgar, W. E. , Lu, J. J. , … Ko, M. C. (2018). A bifunctional nociceptin and μ opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Science Translational Medicine, 10, eaar3483 10.1126/scitranslmed.aar3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, K. , Wang, X. , Chi, L. , & Li, W. (2017). Role of Sigma‐1 receptor/p38 MAPK inhibition in acupoint catgut embedding‐mediated analgesic effects in complete Freund's adjuvant‐induced inflammatory pain. Anesthesia and Analgesia, 125, 662–669. 10.1213/ANE.0000000000001857 [DOI] [PubMed] [Google Scholar]
- Dworkin, R. H. , O'Connor, A. B. , Audette, J. , Baron, R. , Gourlay, G. K. , Haanpaa, M. L. , … Wells, C. D. (2010). Recommendations for the pharmacological management of neuropathic pain: An overview and literature update. Mayo Clinic Proceedings, 85, S3–S14. 10.4065/mcp.2009.0649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Q. , Jiang, T. N. , Li, N. , Han, Z. L. , & Wang, R. (2011). Central administration of neuropeptide FF and related peptides attenuate systemic morphine analgesia in mice. Protein and Peptide Letters, 18, 403–409. 10.2174/092986611794654012 [DOI] [PubMed] [Google Scholar]
- Fitting, S. , Knapp, P. E. , Zou, S. , Marks, W. D. , Bowers, M. S. , Akbarali, H. I. , & Hauser, K. F. (2014). Interactive HIV‐1 Tat and morphine‐induced synaptodendritic injury is triggered through focal disruptions in Na+ influx, mitochondrial instability, and Ca2+ overload. The Journal of Neuroscience, 34, 12850–12864. 10.1523/JNEUROSCI.5351-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funada, M. , Suzuki, T. , Narita, M. , Misawa, M. , & Nagase, H. (1993). Modification of morphine‐induced locomotor activity by pertussis toxin: biochemical and behavioral studies in mice. Brain Research, 619, 163–172. 10.1016/0006-8993(93)91608-U [DOI] [PubMed] [Google Scholar]
- Furst, S. , Riba, P. , Friedmann, T. , Timar, J. , Al‐Khrasani, M. , Obara, I. , … Schmidhammer, H. (2005). Peripheral versus central antinociceptive actions of 6‐amino acid‐substituted derivatives of 14‐O‐methyloxymorphone in acute and inflammatory pain in the rat. The Journal of Pharmacology and Experimental Therapeutics, 312, 609–618. 10.1124/jpet.104.075176 [DOI] [PubMed] [Google Scholar]
- Gentry, C. L. , Egleton, R. D. , Gillespie, T. , Abbruscato, T. J. , Bechowski, H. B. , Hruby, V. J. , & Davis, T. P. (1999). The effect of halogenation on blood‐brain barrier permeability of a novel peptide drug. Peptides, 20, 1229–1238. 10.1016/S0196-9781(99)00127-8 [DOI] [PubMed] [Google Scholar]
- Harada, S. , Nakamoto, K. , & Tokuyama, S. (2013). The involvement of midbrain astrocyte in the development of morphine tolerance. Life Sciences, 93, 573–578. 10.1016/j.lfs.2013.08.009 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervera, A. , Negrete, R. , Leanez, S. , Martin‐Campos, J. M. , & Pol, O. (2011). Peripheral effects of morphine and expression of μ‐opioid receptors in the dorsal root ganglia during neuropathic pain: nitric oxide signaling. Molecular Pain, 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, J. C. , Leighton, G. E. , Meecham, K. G. , Boyle, S. J. , Horwell, D. C. , Rees, D. C. , & Hughes, J. (1990). CI‐977, a novel and selective agonist for the κ‐opioid receptor. British Journal of Pharmacology, 101, 183–189. 10.1111/j.1476-5381.1990.tb12110.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecka, A. , Perlikowska, R. , Gach, K. , Wyrebska, A. , & Fichna, J. (2010). Development of opioid peptide analogs for pain relief. Current Pharmaceutical Design, 16, 1126–1135. 10.2174/138161210790963869 [DOI] [PubMed] [Google Scholar]
- Kalso, E. , Edwards, J. E. , Moore, R. A. , & McQuay, H. J. (2004). Opioids in chronic non‐cancer pain: Systematic review of efficacy and safety. Pain, 112, 372–380. 10.1016/j.pain.2004.09.019 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & Group NCRRGW (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontinen, V. K. , & Kalso, E. A. (1995). Differential modulation of α2‐adrenergic and μ‐opioid spinal antinociception by neuropeptide FF. Peptides, 16, 973–977. 10.1016/0196-9781(95)00068-U [DOI] [PubMed] [Google Scholar]
- Lacko, E. , Riba, P. , Giricz, Z. , Varadi, A. , Cornic, L. , Balogh, M. , … al‐Khrasani, M. (2016). New morphine analogs produce peripheral antinociception within a certain dose range of their systemic administration. The Journal of Pharmacology and Experimental Therapeutics, 359, 171–181. 10.1124/jpet.116.233551 [DOI] [PubMed] [Google Scholar]
- Li, N. , Han, Z. L. , Wang, Z. L. , Xing, Y. H. , Sun, Y. L. , Li, X. H. , … Wang, R. (2016). BN‐9, a chimeric peptide with mixed opioid and neuropeptide FF receptor agonistic properties, produces nontolerance‐forming antinociception in mice. British Journal of Pharmacology, 173, 1864–1880. 10.1111/bph.13489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. T. , Kao, S. C. , Day, Y. J. , Chang, C. C. , & Chen, J. C. (2016). Altered nociception and morphine tolerance in neuropeptide FF receptor type 2 over‐expressing mice. European Journal of Pain, 20, 895–906. 10.1002/ejp.814 [DOI] [PubMed] [Google Scholar]
- Lu, G. Y. , Wu, N. , Zhang, Z. L. , Ai, J. , & Li, J. (2011). Effects of D‐cycloserine on extinction and reinstatement of morphine‐induced conditioned place preference. Neuroscience Letters, 503, 196–199. 10.1016/j.neulet.2011.08.034 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika, J. , Wawrzczak‐Bargiela, A. , Osikowicz, M. , Makuch, W. , & Przewlocka, B. (2009). Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain, Behavior, and Immunity, 23, 75–84. 10.1016/j.bbi.2008.07.005 [DOI] [PubMed] [Google Scholar]
- Mitrovic, I. , Margeta‐Mitrovic, M. , Bader, S. , Stoffel, M. , Jan, L. Y. , & Basbaum, A. I. (2003). Contribution of GIRK2‐mediated postsynaptic signaling to opiate and α2‐adrenergic analgesia and analgesic sex differences. Proceedings of the National Academy of Sciences of the United States of America, 100, 271–276. 10.1073/pnas.0136822100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouledous, L. , Mollereau, C. , & Zajac, J. M. (2010). Opioid‐modulating properties of the neuropeptide FF system. BioFactors, 36, 423–429. 10.1002/biof.116 [DOI] [PubMed] [Google Scholar]
- Pan, Z. Z. (1998). μ‐Opposing actions of the κ‐opioid receptor. Trends in Pharmacological Sciences, 19, 94–98. 10.1016/S0165-6147(98)01169-9 [DOI] [PubMed] [Google Scholar]
- Perlikowska, R. , Piekielna, J. , Gentilucci, L. , De Marco, R. , Cerlesi, M. C. , Calo, G. , … Janecka, A. (2016). Synthesis of mixed MOR/KOR efficacy cyclic opioid peptide analogs with antinociceptive activity after systemic administration. European Journal of Medicinal Chemistry, 109, 276–286. 10.1016/j.ejmech.2015.12.012 [DOI] [PubMed] [Google Scholar]
- Popiolek‐Barczyk, K. , Makuch, W. , Rojewska, E. , Pilat, D. , & Mika, J. (2014). Inhibition of intracellular signaling pathways NF‐κB and MEK1/2 attenuates neuropathic pain development and enhances morphine analgesia. Pharmacological Reports, 66, 845–851. 10.1016/j.pharep.2014.05.001 [DOI] [PubMed] [Google Scholar]
- Qu, J. , Tao, X. Y. , Teng, P. , Zhang, Y. , Guo, C. L. , Hu, L. , … Liu, W. T. (2017). Blocking ATP‐sensitive potassium channel alleviates morphine tolerance by inhibiting HSP70‐TLR4‐NLRP3‐mediated neuroinflammation. Journal of Neuroinflammation, 14, 228 10.1186/s12974-017-0997-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumy, M. , & Zajac, J. (1999). Neuropeptide FF selectively attenuates the effects of nociceptin on acutely dissociated neurons of the rat dorsal raphe nucleus. Brain Research, 845, 208–214. 10.1016/S0006-8993(99)01965-4 [DOI] [PubMed] [Google Scholar]
- Schiller, P. W. (2005). Opioid peptide‐derived analgesics. The AAPS Journal, 7, E560–E565. 10.1208/aapsj070356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller, A. G. , King, M. A. , Zhang, J. , Bolan, E. , Pan, Y. X. , Morgan, D. J. , … Pintar, J. E. (1999). Retention of heroin and morphine‐6 β‐glucuronide analgesia in a new line of mice lacking exon 1 of MOR‐1. Nature Neuroscience, 2, 151–156. 10.1038/5706 [DOI] [PubMed] [Google Scholar]
- Seth, V. , Upadhyaya, P. , Moghe, V. , & Ahmad, M. (2011). Role of calcium in morphine dependence and naloxone‐precipitated withdrawal in mice. Journal of Experimental Pharmacology, 3, 7–12. 10.2147/JEP.S15240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried, B. , Filibeck, U. , Gozzo, S. , & Castellano, C. (1982). Lack of morphine‐induced hyperactivity in C57BL/6 mice following striatal kainic acid lesions. Behavioural Brain Research, 4, 389–399. 10.1016/0166-4328(82)90063-8 [DOI] [PubMed] [Google Scholar]
- Snider, W. D. , & McMahon, S. B. (1998). Tackling pain at the source: New ideas about nociceptors. Neuron, 20, 629–632. 10.1016/S0896-6273(00)81003-X [DOI] [PubMed] [Google Scholar]
- Snyder, L. M. , Chiang, M. C. , Loeza‐Alcocer, E. , Omori, Y. , Hachisuka, J. , Sheahan, T. D. , … Ross, S. E. (2018). κ opioid receptor distribution and function in primary afferents. Neuron, 99, 1274–1288 e1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, C. (2013a). Opioids, sensory systems and chronic pain. European Journal of Pharmacology, 716, 179–187. 10.1016/j.ejphar.2013.01.076 [DOI] [PubMed] [Google Scholar]
- Stein, C. (2013b). Targeting pain and inflammation by peripherally acting opioids. Frontiers in Pharmacology, 4, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, C. , Schafer, M. , & Hassan, A. H. (1995). Peripheral opioid receptors. Annals of Medicine, 27, 219–221. 10.3109/07853899509031962 [DOI] [PubMed] [Google Scholar]
- Tiwari, V. , Yang, F. , He, S. Q. , Shechter, R. , Zhang, C. , Shu, B. , … Raja, S. N. (2016). Activation of peripheral μ‐opioid receptors by dermorphin [D‐Arg2, Lys4] (1‐4) amide leads to modality‐preferred inhibition of neuropathic pain. Anesthesiology, 124, 706–720. 10.1097/ALN.0000000000000993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, J. , Catheline, G. , Guilbaud, G. , & Kayser, V. (1999). Lack of cross‐tolerance between the antinociceptive effects of systemic morphine and asimadoline, a peripherally‐selective κ‐opioid agonist, in CCI‐neuropathic rats. Pain, 83, 509–516. 10.1016/S0304-3959(99)00158-X [DOI] [PubMed] [Google Scholar]
- Wang, X. , Traub, R. J. , & Murphy, A. Z. (2006). Persistent pain model reveals sex difference in morphine potency. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 291, R300–R306. 10.1152/ajpregu.00022.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. L. , Fang, Q. , Han, Z. L. , Pan, J. X. , Li, X. H. , Li, N. , … Wang, R. (2014). Opposite effects of neuropeptide FF on central antinociception induced by endomorphin‐1 and endomorphin‐2 in mice. PLoS ONE, 9, e103773 10.1371/journal.pone.0103773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. L. , Pan, J. X. , Song, J. J. , Tang, H. H. , Yu, H. P. , Li, X. H. , … Wang, R. (2016). Structure‐based optimization of multifunctional agonists for opioid and neuropeptide FF receptors with potent nontolerance forming analgesic activities. Journal of Medicinal Chemistry, 59, 10198–10208. 10.1021/acs.jmedchem.6b01181 [DOI] [PubMed] [Google Scholar]
- Zadina, J. E. , Nilges, M. R. , Morgenweck, J. , Zhang, X. , Hackler, L. , & Fasold, M. B. (2016). Endomorphin analog analgesics with reduced abuse liability, respiratory depression, motor impairment, tolerance, and glial activation relative to morphine. Neuropharmacology, 105, 215–227. 10.1016/j.neuropharm.2015.12.024 [DOI] [PubMed] [Google Scholar]
- Zarrindast, M. R. , & Torkaman‐Boutorabi, A. (2003). Effects of imipramine on the expression and development of morphine dependence in mice. European Journal of Pharmacology, 473, 19–25. 10.1016/S0014-2999(03)01913-7 [DOI] [PubMed] [Google Scholar]
- Zhang, R. , Xu, B. , Zhang, M. N. , Zhang, T. , Wang, Z. L. , Zhao, G. , … Wang, R. (2017). Peripheral and central sites of action for anti‐allodynic activity induced by the bifunctional opioid/NPFF receptors agonist BN‐9 in inflammatory pain model. European Journal of Pharmacology, 813, 122–129. 10.1016/j.ejphar.2017.07.044 [DOI] [PubMed] [Google Scholar]
- Zollner, C. , Mousa, S. A. , Fischer, O. , Rittner, H. L. , Shaqura, M. , Brack, A. , … Schäfer, M. (2008). Chronic morphine use does not induce peripheral tolerance in a rat model of inflammatory pain. The Journal of Clinical Investigation, 118, 1065–1073. 10.1172/JCI25911 [DOI] [PMC free article] [PubMed] [Google Scholar]