

Abstract
Marine natural products have proven, over the last half‐century, to be effective biological modulators. These molecules have revealed new targets for cancer therapy as well as dissimilar modes of action within typical classes of drugs. In this scenario, innovation from marine‐based pharmaceuticals has helped advance cancer chemotherapy in many aspects, as most of these are designated as first‐in‐class drugs. Here, by examining the path from discovery to development of clinically approved drugs of marine origin for cancer treatment—cytarabine (Cytosar‐U®), trabectedin (Yondelis®), eribulin (Halaven®), brentuximab vedotin (Adcetris®), and plitidepsin (Aplidin®)— together with those in late clinical trial phases—lurbinectedin, plinabulin, marizomib, and plocabulin—the present review offers a critical analysis of the contributions given by these new compounds to cancer pharmacotherapy.
Abbreviations
- ADC
antibody‐drug conjugate
- ALCL
anaplastic large cell lymphoma
- ALL
acute lymphocytic leukaemia
- AML
acute myeloid leukaemia
- CLL
chronic lymphocytic leukaemia
- CML
chronic myeloid leukaemia
- CT‐L
chymotrypsin‐like
- DLBCL
diffuse large B‐cell lymphoma
- DLT
dose‐limiting toxicity
- LPS
liposarcoma
- LMS
leiomyosarcoma
- MDR
multidrug resistance
- MF
mycosis fungoides
- MM
multiple myeloma
- MNPs
marine natural products
- NHL
non‐Hodgkin's lymphoma
- NSCLC
non‐small cell lung cancer
- ORR
objective response rate
- OS
overall survival
- PFS
progression‐free survival
- Pol II
RNA polymerase II
- PSN
peripheral sensory neuropathy
- SCLC
small cell lung cancer
- STS
soft tissue sarcoma
- TAM
tumour‐associated macrophages
- TC‐NER
transcription coupled‐nucleotide excision repair
- TME
tumour micro‐environment
- TNBC
triple negative breast cancer.
1. INTRODUCTION
Natural products have long been used in the treatment of human maladies and set a strong foundation upon which modern pharmacology has been erected. Particularly for cancer treatment, chemotherapeutic agents of natural origin have remarkably impacted the field, not only for their clinical importance but also for allowing the expansion of knowledge in cancer pharmacology. Natural agents like doxorubicin, paclitaxel, vincristine, and vinblastine are commonly used as first‐line treatments for several cancers. The compounds of marine origin have a more recent history. Nevertheless, studies have shown their unique chemical structures and new cellular targets and modes of actions, which are bringing significant innovation to the field.
This review starts with a description of the chemical features that make the marine natural products versatile as biological modulators, provided in Section 2. Then, it will examine the contribution of marine molecules to cancer pharmacology using the discovery and development of clinically approved drugs as foundation for the discussion. Most of these are actually described as “first‐in‐class” pharmaceuticals, and the novelty in their pharmacology will be emphasized.
To date, eight marine‐derived drugs have been approved for clinical use: https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4827 (Cytosar‐U®), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4806 (Vira‐A®; US discontinued), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2774 (Yondelis®), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2536 (Prialt®), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6813 (Halaven®), plitidepsin (Aplidin®), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6772 (Adcetris®), and omega‐3‐acid ethyl esters (Lovaza®; Jiménez, 2018, http://marinepharmacology.midwestern.edu/clinical_pipeline.html). Five of them are used in different anticancer treatments and those will be discussed in depth, under Sections 3 and 4. Moreover, numerous antibody‐drug conjugates that use a marine compound as their cytotoxic principle are also undergoing clinical trials, and those will be sorted through in Section 4, when presenting the first approved drug in this class, brentuximab vedotin (Adcetris). Furthermore, Section 5 will afford grounds for understanding the potential of marine molecules in cancer therapeutics in the near future. In this section, compounds that are currently in advanced stages of clinical trials for cancer, such as lurbinectedin (Zepsyre®), plinabulin, marizomib, and plocabulin, will be surveyed.
2. CHEMICAL FEATURES OF MARINE COMPOUNDS: HOW THEIR UNIQUE STRUCTURES AFFECT MOLECULAR INTERACTIONS
The structural complexity of natural products differs from synthetic drug‐like compounds in numerous ways. Natural products have a higher number of chiral centres, stereocentres, sp3‐hybridized bridgehead atoms, and hydrogen‐bond donors than synthetic drugs. On average, they also contain higher carbon, hydrogen, and oxygen content and less nitrogen content than synthetic drug molecules. Additionally, natural products have much fewer aromatic rings than synthetic medicinal agents, as aliphatic systems appear to be favoured in nature, a fact that may improve selectivity when binding to stereo‐defined sites (Grabowski & Schneider, 2007). They may also present original ring systems—for example, macrocyclizations, fused‐rings, ether crosslinks, and extensive conjugation—with appropriate geometries for spatial side‐chain substitution and conformations that bind to specific biological targets (Clardy & Walsh, 2004; Rodrigues, Reker, Schneider, & Schneider, 2016).
Natural products often disobey the drug‐likeness Lipinski's “rule of five” (Ro5), once they present molecular masses above 500 Da and high polarities—for example, rapamycin is 914 Da with a polar surface area of 195 Å2, and paclitaxel is 854 Da and possesses a polar surface area of 221 Å2 (Chai & Mátyus, 2016). In fact, 18% of the natural products from the Dictionary of Natural Products database, 31% of the natural products from the Traditional Chinese Medicine database, 15% of the DrugBank, and 9% of the ChEMBL database do not comply with the Ro5 (Chai & Mátyus, 2016).
Several approaches may potentiate natural products discovery programs and improve the number of natural product‐derived drugs making it to the market. The use of moderate, instead of hard cut‐offs for drug‐likeness property rules during the in silico selection of drug‐lead candidates, is reasonable (Chai & Mátyus, 2016). Furthermore, advances in synthetic biology and synthetic drug design may also pave the way towards that direction. Structurally complex frameworks of natural products can be dissociated into smaller and complex fragment‐like scaffolds bearing high ligand efficiency to simplify drug design. Moreover, approaches founded on fragment‐based de novo design using natural product‐derived fragments to infer the bioactivities and biomolecular targets of such molecules can support the development of natural product‐derived drugs (Rodrigues et al., 2016).
Marine natural products (MNPs) are a well‐established subfield of NPs, which have been gaining increased attention in the last decades. Marine environments are complex systems with variations in luminosity, temperature, oxygen levels, pH, salinity, and pressure. Therefore, marine organisms had to adapt and evolve differently from their terrestrial counterparts. They developed distinct biosynthetic pathways, which produced novel bioactive secondary metabolites with unmatched and complex structures (Hu, Ying, Zhang, Qiu, & Lu, 2018). Many marine organisms—such as sponges, tunicates, and shell‐less molluscs, as well as microorganisms, such as fungi, bacteria, and cyanobacteria—have provided secondary metabolites with interesting pharmacological properties (Blunt et al., 2018).
MNPs present much novelty in chemical scaffolds, compared to terrestrial natural products (Kong, Jiang, & Zhang, 2010). In this context, a preclinical cytotoxicity screening from the USA National Cancer Institute revealed 10 times higher incidence of antitumour properties for marine samples than for terrestrial samples (Munro et al., 1999). These compounds usually present unique structures due to the specific carbon arrangements with several stereochemical features for the presence of non‐essential amino acids; they also have interesting ring systems, besides the incorporation of halogen atoms, such as chloride, bromide, iodine, and fluorine, which are covalently attached to organic compounds.
MNPs also present appropriate geometries for spatial side‐chain substitution and conformations that bind to specific biological targets (Clardy & Walsh, 2004; Rodrigues et al., 2016). Figure 1 shows the chemical structures discussed in this review; trabectedin and lurbinectedin have been boxed and highlighted for some of the chemical features. Lurbinectedin is a derivative of trabectedin used as an example to illustrate those unique chemical features found in compounds from marine sources, such as several heteroatoms (oxygen, red; nitrogen, green; and sulphur, orange) and many stereo sites (blue circles).
Figure 1.

Marine natural products and their derivatives of pharmacological relevance. In the box, the tetrahydroisoquinoline rings (pink)—in which both A and B rings have been proven essential for bioactivity due to their involvement in the interaction with DNA minor groove—are highlighted in the chemical structure of trabectedin. For lurbinectedin, further noteworthy chemical features particularly found in marine natural products are emphasized, such as stereochemical carbons (blue spheres), complex intra‐cyclization, and a high number of heteroatoms generating different chemical functions comprising oxygen (red), nitrogen (green), and sulphur (orange) atoms
This structural complexity of MNPs has attracted the attention of organic chemists, as if it was an interesting puzzle to be solved. The elucidation of chemical structures from marine sources is often difficult, considering the structural intricacy of several of these compounds. The structure of trabectedin (ecteinascidin 743, ET‐743)—the bioactive compound responsible for anticancer activity from extracts of the tunicate Ecteinascidia turbinata—took over 20 years to be fully elucidated (Rinehart et al., 1990; Wright et al., 1990).
The chemical scaffolds of MNPs support prolific bioactivities and preferred structural ligand–protein binding motifs to interact with biological space. This is not fully understood, although attempts to explain this singularity are based on hypotheses that consider co‐evolution, synergistic effects, xenohormesis, natural selection, and enzymatic mode of generation of MNPs (Howitz & Sinclair, 2008; Ji, Li, & Zhang, 2009). Undoubtedly, there is still much to discover in the field of MNPs. Adopting drug discovery strategies such as “multi‐component therapeutics” rather than “one‐disease–one‐target–one‐drug” may benefit the discovery of cancer therapies and prevent the development of resistance against anti‐infective, antimalarial, and anticancer drugs.
3. THE EARLIEST DAYS OF MARINE PHARMACOLOGY: CYTARABINE INTRODUCED MARINE DRUGS AS A GROUND‐BREAKING CANCER THERAPY
Throughout the 1940s and 1950s, Werner Bergmann, a researcher from Yale University, and his collaborators published several articles in a series called “Contributions to the Study of Marine Products” (Bergmann & Feeney, 1951; Lester & Bergmann, 1941). The best known among these works, in 1951, described the isolation of two arabinonucleosides, which were named spongothymidine and spongouridine, from the Caribbean marine sponge Tectitethya crypta (Bergmann & Feeney, 1951).
The discovery of such odd nucleosides unfolded into, at least, two important scientific elaborations. The first challenged the current paradigm, which argued that a nucleoside would only have a biological function if it contained ribose or deoxyribose in its structure. And the second introduced the pharmacological concept of antimetabolites in the context of anticancer chemotherapy, thus guiding the synthesis of analogues, such as cytosine arabinose (Ara‐C or cytarabine; Figure 1).
An antimetabolite drug has a similar structure to a naturally produced metabolite, but it lacks the ability to perform its function, thereby misleading normal cell metabolism. Within the cell, cytarabine is converted into its respective triphosphate arabino‐nucleoside through sequential phosphorylation. The cytarabine triphosphate then becomes a substrate of DNA polymerase and, in place of a cytidine, is subsequently incorporated into the DNA strand. Because arabinose has replaced deoxyribose, a phosphodiester bond between the two pentose residues cannot be established and, therefore, hinders the extension of the strand, thus interrupting the process of DNA synthesis and repair (Schwartsmann, da Rocha, Berlinck, & Jimeno, 2001).
Efforts in producing modified nucleosides were already being carried out before cytarabine. However, the unconventional nucleosides from the marine sponge transformed how the rational design of unnatural nucleosides was being conceived and handled by proving that both the sugar and the nitrogenous base, could be substituted to achieve the projected antimetabolite effect. Moreover, besides anticancer, the principle of taking a non‐functional nucleoside to inhibit the elongation of the DNA strand was also used strategically in other chemotherapeutic treatments, such as antiviral, for example the use of AZT as an anti‐HIV drug.
Cytarabine, the first drug in this series, was approved for clinical use as Cytosar‐U in 1969, and it is still widely used in the treatment of various types of leukaemia, such as acute lymphocytic leukaemia (ALL), acute myeloid leukaemia (AML), chronic myeloid leukaemia (CML), and non‐Hodgkin's lymphoma (NHL). Although this molecule is a synthetic analogue and not the natural product itself, cytarabine is historically reported as the first example of a commercially available marine drug. Moreover, the extensive list of nucleoside analogues currently undergoing trials and the ones that have been recently approved for clinical use demonstrate the value of this therapy (Jordheim, Durantel, Zoulim, & Dumontet, 2013). One such example, nelarabine (Ara‐GTP), a synthetic arabinonucleoside, was approved in October 2005 for the treatment of the rare T‐cell lymphoblastic leukaemia (Buie, Epstein, & Lindley, 2007), whereas thiarabine (T‐Ara‐C) is an arabinonucleoside in Phase I clinical trials for haematological malignancies and solid tumours (Parker, Waud, & Secrist, 2015).
4. FIFTY YEARS LATER: CLINICALLY APPROVED ANTICANCER DRUGS OF MARINE ORIGIN
4.1. Trabectedin (Yondelis): Beyond DNA alkylation
Trabectedin (Figure 1), also referred to as ecteinascidin or ET‐743, is an alkaloid isolated from extracts of the tunicate E. turbinata. It took over 20 years from the detection of biological activity in these extracts to the complete structural elucidation of the compound (Rinehart et al., 1990; Wright et al., 1990), as the description of the three fused tetrahydroisoquinoline rings, designated A, B, and C (highlighted in pink in Figure 1), posed a major challenge.
The peculiar and complex mode of action (Figure 2) for trabectedin has been established. This compound binds to the DNA minor grooves (D'Incalci & Galmarini, 2010) while conventional alkylating drugs generally bind to guanine at N‐7 or O‐6, trabectedin binds to guanine at N2 in GC‐rich sequences (Pommier et al., 1996). Both the A and B rings interact with DNA minor grooves, and the carbinolamine moiety present in ring A is mandatory for its activity. Ring C, in turn, is not responsible for the cytotoxicity of trabectedin (Erba et al., 2004); however, it bends DNA towards the major groove, providing a unique feature for trabectedin, when compared to other minor groove binders (Zewail‐Foote & Hurley, 1999).
Figure 2.
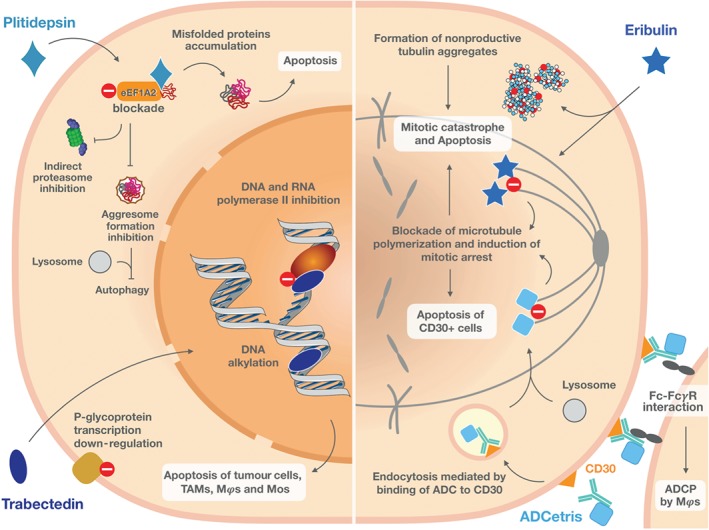
Overview of the multiple mechanisms of action of anticancer marine drugs in clinical use. Inhibition of the respective molecular targets by plitidepsin (upper left), trabectedin (lower left), eribulin (upper right), and ADCetris® (lower right) triggers tumour cell stress, which culminates in cell death by apoptosis. Plitidepsin hinders the pro‐oncogenic function of eukaryotic elongation factor 1A2 (eEF1A2), which impairs transportation of misfolded proteins to the proteasome and causes accumulation of non‐functional proteins. Additionally, by inhibiting eEF1A2, it further blocks activation of the aggresome and, therefore, prevents protein digestion by autophagy. The marked accumulation of non‐functional proteins leads to cell death by apoptosis. Trabectedin is a DNA‐alkylating agent that specifically inhibits the activity of RNA polymerase II, thus not affecting basal transcription. The selective inhibition of transcription induced by trabectedin decreases the levels of P‐glycoprotein, assuring a potent cytotoxicity against resistant tumours. Modulation of tumour micro‐environment is a valuable and interesting feature of this compound, which is attained by the specific depletion of macrophages (Mϕs), tumour‐associated macrophages (TAMs), and circulating monocytes (Mos) by apoptosis. Eribulin is an antitubulin agent that blocks tubulin polymerization, further leading to cell cycle arrest and mitotic catastrophe. Additionally, eribulin induces the formation of non‐functional tubulin aggregates and triggers apoptosis. ADCetris, an antibody‐drug conjugate (ADC), is selectively recognized and internalized by CD30+ cells through endocytosis. The warhead, monomethyl auristatin E, is released into the cytosol after digestion in endolysosomes and blocks tubulin polymerization, causing persistent mitotic arrest and induction of apoptosis. Furthermore, ADCetris also induces antibody‐dependent cellular phagocytosis (ADCP) by Mϕs via Fc‐FcγR binding
Additionally, trabectedin inhibits transcription by binding to transcribing RNA polymerase II (Pol II) and blocking its activity (Feuerhahn et al., 2011). This causes rapid degradation of Pol II, a process dependent on the transcription factor called transcription coupled‐nucleotide excision repair (TC‐NER; Aune et al., 2008). TC‐NER deficient cells were found resistant to trabectedin (Zewail‐Foote & Hurley, 1999), revealing the dependency on this system for activity of the compound. Furthermore, TC‐NER activity is increased in cell lines resistant to platinum compounds, which justifies the later approval of trabectedin as second‐line treatment for patients with platinum resistant tumours (Soares et al., 2011).
Trabectedin was the first compound described with the ability to specifically displace an oncogenic transcription factor from its target promoters (D'Incalci, 2013). Bonfanti et al. (1999) showed trabectedin displaced NF‐Y from its DNA consensus sequence at μM concentrations, suggesting its potential to inhibit multidrug resistance (MDR) activation, which was later demonstrated by Jin, Gorfajn, Faircloth, and Scotto (2000) using human colon carcinoma cell line SW620. In fact, inhibition of activated https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=768, but not constitutive MDR1 induced by trabectedin, involves a complex mechanism that eventually blocks https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=768 transcription, as demonstrated in human epidermoid KB carcinoma cells (Kanzaki et al., 2002).
The biological activity of trabectedin was demonstrated in a variety of cell lines and xenograft models (Table 1). In the NCI‐60 cell line panel, trabectedin (NSC 648766) presented a mean GI50 of 1.94 nM (https://dtp.cancer.gov/, accessed 04 August 2019). However, the NCI screening does not include sarcoma cells, and it was against these tumours that trabectedin proved to be more effective than other treatments (Li et al., 2001). The efficacy of trabectedin in sarcoma subtypes, such as myxoid liposarcoma and Ewing's sarcomas, is also related to its ability to inhibit transcription by decreasing the transactivating ability of chimeric proteins, such as FUS‐CHOP, EWS‐CHOP, and EWSFli1, and eventually leading to adipocytic differentiation and apoptosis (D'Incalci & Galmarini, 2010; Forni et al., 2009).
Table 1.
Preclinical and clinical data on approved marine drugs for cancer treatment or under clinical trials
| Drug | Class | Preclinical evidences | Clinical evidences |
|---|---|---|---|
| Trabectedin |
DNA‐alkylating agent |
In vitro studies and cells lines
In vivo studies
|
|
| Eribulin |
Microtubule‐target agent |
In vitro studies and cell lines
In vivo studies
|
|
| Brentuximab vedotin | Antibody‐drug conjugate (ADC)/microtubule stabilizer |
In vitro studies and cell lines
In vivo studies
|
|
| Plitidepsin | Inhibitor of protein synthesis by targeting the eukaryotic elongation factor 1A2 (eEF1A2) |
In vitro studies and cell lines
In vivo studies
|
|
| Lurbidectedin |
DNA‐alkylating agent |
In vitro studies and cell lines
In vivo studies
|
|
| Plinabulin |
Microtubule‐target agent |
In vitro studies and cell lines
In vivo studies
|
|
| Marizomib | Proteasome inhibitor |
In vitro studies and cell lines
In vivo studies
|
|
| Plocabulin |
Microtubule‐target agent |
In vitro studies and cell lines
In vivo studies Antitumour activity and low toxicity observed in NCI‐H460 lung tumour cells xenografted mice treated during three consecutive weeks at 0.08, 8, and 16 mg·kg−1·day−1. Plocabulin‐treated mice showed up to 65% decrease in tumour vasculature (Galmarini et al., 2018) |
|
Abbreviations: ALCL, anaplastic large cell lymphoma; ALL, acute lymphocytic leukaemia; AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia; CLL, chronic lymphocytic leukaemia; HL, Hodgkin's lymphoma; MM, multiple myeloma; NHL, non‐Hodgkin's lymphoma; PTCL, peripheral T‐cell lymphoma; TNBC, triple negative breast cancer.
Preclinical in vivo data reinforced the potential of trabectedin and other ecteinascidins (ET‐729 and ET‐722) as anticancer compounds since the first description of the activity on murine melanoma B16 and leukaemia P388 tumours (Rinehart et al., 1990; Sakai, Rinehart, Guan, & Wang, 1992). Many studies using human xenograft models in nude mice confirmed its in vivo activity using different schedules of drug administration, from twice a day during three consecutive days to once a day every 4 days on Days 0, 4, and 8; and doses ranging from 0.025 to the maximum tolerated dose 0.2 mg·kg−1. On B‐16 and osteosarcoma models, trabectedin showed significant in vivo activity especially when associated with dexamethasone (revised by Van Kesteren et al., 2003).
Clinical evidence of trabectedin first suggested its use for soft tissue sarcoma (STS) with multiple Phase I studies covering its isolated use or in combination with other chemotherapies, such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7069 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5343, and suggested the recommended dose of 1.5 mg·m−2 as a 24‐hr continuous intravenous infusion every 3 weeks (Taamma et al., 2001). Phase II studies demonstrated positive results, especially for liposarcoma (LMS) and leiomyosarcoma (LPS), and determined a median overall survival (OS) of 9.2 months (Monk et al., 2012). Phase III studies concluded that trabectedin was superior in comparison to dacarbazine in patients with advanced LPS and LMS after failure of prior chemotherapy (Demetri et al., 2017). A summarized description of the clinical evidence for ovarian carcinoma is found in Table 1.
The European Medicines Agency (EMEA) approved trabectedin for therapeutic use and commercial trading to PharmaMar (S.A., Madrid, Spain) under the name Yondelis in 2007, when it became the first marine drug identical to the original natural product to obtain marketing authorization. At that time, the European Commission allowed the use of trabectedin for the treatment of patients with advanced STS after failure of anthracyclines and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7201. Two years later, the drug was also approved for the treatment of patients with relapsed platinum‐sensitive ovarian cancer in combination with pegylated liposomal doxorubicin (Puyo, Montaudon, & Pourquier, 2014). Still, it was nearly a decade later, in 2015, that the United States Agency Food and Drug Administration (FDA) authorized Yondelis for clinical use (available from https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm430302.htm, accessed 19 February 2019). A timeline between the discovery of trabectedin and the full development of Yondelis is shown in Figure 3.
Figure 3.
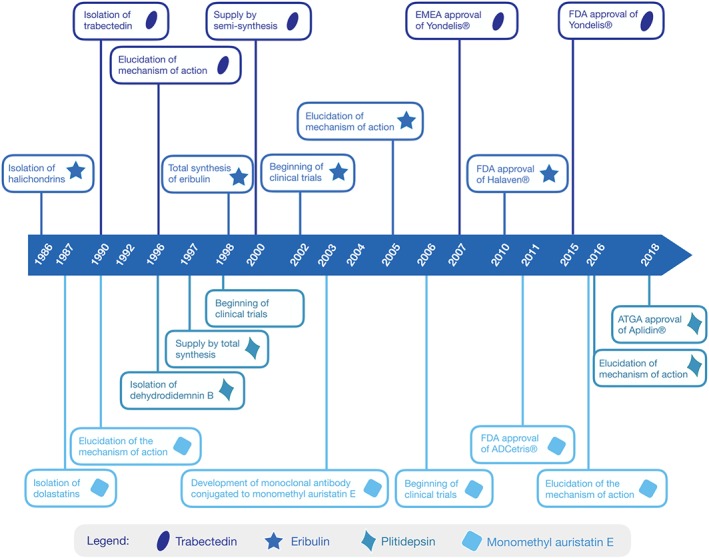
Milestones of anticancer drugs from marine origin on the market. The length of time between the isolation of the natural molecule and the approval of the commercial product was 17 years for trabectedin (Yondelis®), 22 years for dihydrodidemin B (Aplidin®), 24 years for halichondrin (Halaven®), and 24 years for dolastatin (ADCetris®)
A recent study evaluated treatment options for STS and demonstrated that patients receiving trabectedin presented a median OS of 21.3 months (Le Cesne, 2018). Additionally, 53% of patients showed long‐term tumour control when trabectedin was administered early in the treatment scheme, and, even more encouraging, 65.5% of patients achieved disease control (Le Cesne, 2018). In order to evaluate the 10‐year period of trabectedin use after approval for STS treatment, a cohort of 86 patients was accessed, and the main results indicated that trabectedin was chosen by the majority as a second‐ or third‐line treatment. Only 22% of cases presented side effects, mainly related to myelosuppression and no treatment‐related deaths were observed (Shamai & Merimsky, 2017).
Despite the approved clinical status for Yondelis, further studies have been conducted to better understand this molecule and its activity on different cancer models. Until now, other four effects of ET‐743 were discovered, explaining its effective anticancer activity. First, trabectedin modulates the tumour micro‐environment (TME). It specifically depletes monocytes and tumour‐associated macrophages (TAM), decreases indirectly the production of inflammatory mediators, such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=821, and https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=963, and compromises the expression of genes involved in extracellular matrix (Galmarini, D'Incalci, & Allavena, 2014). Second, trabectedin also appears to be involved in alternative lengthening of telomeres (ALT), a preventive strategy for telomere shortening that assures immortality of cancer cells. Such changes are found in 10% to 15% of tumours, and ALT positive cells seem to be more sensitive to trabectedin (Pompili, Leonetti, Biroccio, & Salvati, 2017). Third, a three‐stranded nucleic acid structure referred as R‐loops induces genome instability caused by DNA damage dependent of RNA–DNA hybrid assemblies. Researchers have demonstrated that high levels of R‐loops may enhance the activity of trabectedin, opening prospects for treatment of cancers with high levels of these events (Tumini et al., 2018). Finally, the involvement of trabectedin in the repression of genes regarding cancer stem cell phenotype, and their related pathways was also documented (Martinez‐Cruzado et al., 2017).
4.2. Eribulin mesylate (Halaven): Do we still need one more antitubulin agent?
Eribulin mesylate (Figure 1) is a synthetic analogue of the natural product halichondrin B (Figure 1), initially identified by Hirata and Uemura (1986) in extracts from the marine sponge Halichondria okadai, found in the Miura Peninsula, in Japan. Subsequently, halichondrin B was also isolated from other species of marine sponges, such as Axinella sp., (Pettit et al., 1991), Phakellia carteri (Pettit et al., 1993), and Lissodendoryx sp. (Litaudon, Hart, Blunt, Lake, & Munro, 1994). The natural compound belongs to a class of polyether macrolides and showed a potent in vitro and in vivo antitumour activity (Hirata & Uemura, 1986). Eribulin, in turn, is a halichondrin B derivative based on pharmacophoric‐driven structural simplification, but it still represents the most complex drug supplied through total synthesis (Yu, Kishi, & Littlefield, 2011). The eribulin synthesis route mostly repeated the macrocyclic ring cytotoxic core of halichondrin B, but the side chain was omitted, thus losing almost half of the original molecular weight.
Eribulin is a potent microtubule‐destabilizing anticancer agent. This molecule interferes in the mitosis phase of the cell cycle through a peculiar mechanism of action that differs from other antimitotic agents, such as the vinca alkaloids. A non‐taxane microtubule dynamics inhibitor, eribulin predominantly binds with high affinity to the plus ends of the microtubules, preventing polymerization of https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=858 without affecting depolymerization (Figure 2). However, this molecule can also bind to the soluble α and β tubulin, thus decreasing the availability of the subunits for polymerization. Thus, suppression of the microtubule dynamics induces a cell cycle arrest at the G2/M phase, which then signals for mitotic catastrophe, further leading to cell death by apoptosis (Eslamian, Wilson, & Young, 2017; Okouneva, Azarenko, Wilson, Littlefield, & Jordan, 2008).
In preclinical cancer models, eribulin has shown antitumour activity against colon cancer, glioblastoma, head and neck cancer, melanoma, non‐small cell lung cancer (NSCLC), ovarian cancer, pancreatic cancer, and small cell lung cancer (Towle et al., 2012). It has been shown to go beyond the inhibition of microtubule dynamics, as it participates in other important regulatory processes of tumour cell biology, such as epithelial‐to‐mesenchymal transition (EMT), vascularization, and hypoxia, which shows the promising potential of this drug to contribute to clinical benefits. Table 1 summarizes the preclinical and clinical data obtained for eribulin.
In fact, recent studies using breast and sarcoma tumours have shown the non‐antimitotic effects of eribulin on tumour biology and in the TME, such as reversal of EMT, decreased capacity for migration and invasion, and vascular remodelling and perfusion (Funahashi et al., 2014; Yoshida et al., 2014). Moreover, preclinical studies in triple negative breast cancer (TNBC) cell cultures showed that treatment with eribulin increased gene expression of epithelial markers (i.e., CDH1 and KRT18) and downgraded the levels of mesenchymal markers (i.e., CDH2, VIM, TWIST1, SNAI2, ZEB1, and ZEB2), leading to the reversal of EMT after 7 days of exposure to eribulin (Yoshida et al., 2014). Combination of two antitubulin agents—eribulin and paclitaxel—resulted in a synergistic antiproliferative effect on TNBC cells in vitro. Exposure to eribulin increased E‐cadherin expression and decreased the expression of mesenchymal markers in MDA‐MB‐231 and Hs578T cells, showing a clear phenotypic switch from mesenchymal to epithelial state (Oba & Ito, 2018). Loss of phenotypic characteristics of mesenchymal cells and gain of epithelial phenotypic characteristics indicate a possible decrease in malignancy and, consequently, in tumour aggressiveness. Findings reported by Funahashi et al. (2014) showed that a single dose of eribulin increased tumour perfusion and generated changes in vascularization. The authors further observed that microvessel density and proportion of small vessels both increased. These changes were associated with reduced hypoxia and, subsequently, with the prevention of drug resistance and metastasis (Funahashi et al., 2014).
Clinical studies supported the use of eribulin treatment for advanced or metastatic breast cancer and also suggested that it could help in the treatment of solid tumours resistant to other classes of microtubule dynamics inhibitors. The recommended doses for an injection of eribulin are slightly different in the United States (1.4 mg·m−2 of the salt, eribulin mesylate) from those recommended in Europe (1.23 mg·m−2 of the active substance,eribulin). The administration of eribulin is scheduled on the first and eighth day of a 21‐day cycle (Goel et al., 2009; Tan et al., 2009). In this setting, eribulin has demonstrated efficacy in Phase II studies involving patients who previously received both, an anthracycline and a taxane (Aogi et al., 2012; McIntyre et al., 2014; Vahdat et al., 2009), or this combination with the addition of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6799 (Cortes et al., 2010). Advanced or recurrent HER2‐positive breast cancer has been treated with eribulin combined with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5082 (Sakaguchi et al., 2018; Wilks et al., 2014) or https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5046 (Araki et al., 2017), which presented an acceptable treatment option supported by a high objective response rate (ORR), a prolonged median progression‐free survival (PFS), and an acceptable safety profile. The most common drug‐related grades 3–4 toxicities were neutropenia, leukopenia, peripheral neuropathy, and febrile neutropenia (Aogi et al., 2012; McIntyre et al., 2014; Vahdat et al., 2009).
In 2011, the first non‐randomized multicentre Phase II study involving eribulin and previously treated progressive or high‐grade STS was published (Schöffski et al., 2011), which yielded median PFS of 2.1 to 2.9 months in patients with different types of STS (Schöffski et al., 2011). Another open‐label, multicentre, and non‐randomized Phase II trial evaluated patients with advanced or metastatic STS. This study found a median PFS and ORR for LPS/LMS patients of 5.5 and 17.0 months, respectively, against 2.0 months for both parameters in other subtypes (Kawai et al., 2017). Furthermore, a Phase III study comparing eribulin (1.4 mg·m−2 intravenously on Days 1 and 8) with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9075 (850 mg·m−2, 1,000 mg·m−2 intravenously on Day 1) every 21 days in patients with advanced LPS or LMS showed an improvement in OS and PFS for eribulin‐treated patients versus those who received dacarbazine alone (Demetri et al., 2017).
In November 2010, Halaven was approved by the FDA as a third‐line therapy for advanced or metastatic breast cancer patients who were previously treated with an anthracycline and a taxane, and, in January 2016, it became the second line of treatment for unresectable or metastatic liposarcoma (available from https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm430302.htm, accessed 10 August 2019). In fact, eribulin was the first drug approved for patients with advanced, metastatic, or inoperable STS that demonstrated an improvement in overall survival. A timeline from the discovery of halichondrin B to the full development of Halaven is shown in Figure 3.
4.3. Warhead drugs (brentuximab vedotin, Adcetris): Special delivery to overcome toxicity
In 1970, extracts obtained from the mollusc Dolabella auricularia, found in the Indian Ocean, stood out for their pronounced anticancer activity (Pettit, Day, Hartwell, & Wood, 1970). The active principles, a series of peptides called dolastatins, were highlighted for their potent antiproliferative activities against several tumour cell lines, especially dolastatins 10 and 15, with IC50 values in the pM‐range (Pettit et al., 1987). It was then found that tubulin is the main target of action of these peptides, inducing blockade of microtubule polymerization and inhibiting cell proliferation (Pettit, 1997). Despite the potent in vitro activity of dolastatins, none of these peptides—or their synthetic derivatives, such as auristatin (soblidotine), cematodine, and synthatodine—have progressed into clinical trials beyond Phase II (Perez et al., 2005; Vaishampayan et al., 2000) due to toxicity and lack of efficacy (Newman & Cragg, 2017). However, monomethylauristatin E (MMAE), an auristatin derivative, successfully entered the composition of the antibody‐drug conjugate (ADC) brentuximab vedotin, which was further developed by Seattle Genetics (Doronina et al., 2003; Senter & Sievers, 2012).
ADCs are recent formulations that seek to improve selectivity and overcome the toxicity of anticancer therapy by delivering the pharmacological agent to specific tumour cells. Such systems comprise three main components: a monoclonal antibody that will guide the ADC to the target cells; a potent cytotoxic compound (dubbed a payload or a warhead), which should kill the tumour cells; and a linker that will covalently connect both parts and is further accountable for releasing the payload inside the target cell (Ducry & Stump, 2010; Storz, 2015). In the specific case of brentuximab vedotin, the monoclonal antibody cAC10 is directed to the human https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1877 membrane protein, a TNF receptor, and is also recognized as a tumour marker for some lymphomas (Younes, 2011). In fact, CD30 had previously shown to be a promising antigen for anticancer immunotherapy based on in vitro and in vivo preclinical evidence generated by exposure to cAC10. However, in clinical trials, this kind of therapy displayed only limited efficacy (Wahl et al., 2002).
Vedotin (vcMMAE) is the second part of the conjugate and comprises the linker and the cytotoxic principle, MMAE. Linker technology is important for the success of ADCs, as it must guarantee stability of the system under physiological conditions and allow enough time for the conjugate to reach the target cell, but it should also be specifically cleaved to free the payload inside the cell. For brentuximab vedotin, a cathepsin cleavable valine‐citrulline linker was chosen, as it met these criteria and, moreover, allowed for better water solubility and satisfactory pharmacokinetics of the drug (Doronina et al., 2003). Recognition of the brentuximab antibody by CD30 protein leads to the internalization of the ADC which, within the cell, releases vedotin—nearly four units of the agent per complex—by a selective proteolytic cleavage carried out by lysosome cysteine proteases. In turn, free MMAE reaches its target, blocks cell division in G2/M by disrupting microtubule dynamics, and, ultimately, induces cell death (Doronina et al., 2003; Senter & Sievers, 2012).
Preclinical studies demonstrated that conjugation with MMAE did not alter the affinity of the CD30 antibody with the surface antigen. In fact, brentuximab vedotin showed enhanced cytotoxicity when assessed against CD30+ cell lines originating from Hodgkin's lymphoma and systemic anaplastic large cell lymphoma (ALCL), with IC50 values in the pM range. In turn, in CD30− cells lines, the conjugate showed a reduced toxicity by nearly 1,000 times, thus displaying an antigen‐dependent effect (Francisco et al., 2003; Yi, Kim, & Kim, 2017). Further in vivo findings showed favourable outcomes in mice bearing xenograft tumours of human Hodgkin's lymphoma and ALCL cells, with tumour regression, and prolonged survival of treated animals. Moreover, in a single‐dose treatment scheme with the ADC, xenograft mice models injected with 1 to 3 mg·kg−1 exhibited regressed and even cured tumours. Such efficacy allowed for a wide therapeutic window, once the maximum tolerated dose was established at 100 mg·kg−1 (Senter & Sievers, 2012). Table 1 summarizes preclinical and clinical data for brentuximab vedotin.
In a clinical trial conducted on CD30‐positive haematological cancers, a large percentage of patients had a complete response to treatment with brentuximab vedotin. A Phase I trial that used escalating doses of intravenously administered ADC in 45 patients every 3 weeks showed an ORR in ~40% of patients, including complete remission for ~25% (Younes, 2011). Furthermore, Phase II trials in patients with Hodgkin's lymphoma and ALCL showed ORR of 73% (with 32% complete responses) and 86% (with 57% complete responses) respectively (Younes, Yasothan, & Kirkpatrick, 2012).
ADCetris (brentuximab vedotin) was approved by the FDA in 2011 for the treatment of Hodgkin's lymphoma and systemic ALCL, types of cancer distinguished by the high expression of CD30. Recently, between 2017 and 2018, the FDA expanded the approval of ADCetris for treatment of CD30‐expressing mycosis fungoides (MF), a cutaneous T‐cell lymphoma, and for CD30‐expressing peripheral T‐cell lymphomas (PTCL; available at https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm601935.htm, accessed 22 February 2019).
Furthermore, as shown in Table 2, MMAE and other auristatin derivatives, mainly their phenylalanine variant monomethyl auristatin F (MMAF), are being used in the composition of several ADCs that are currently undergoing clinical trials in which the antibody of the complex is directed to a profusion of relevant membrane discriminators of assorted tumour types. For https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8404, an ADC undergoing Phase III clinical trials, the warhead MMAE is linked to the monoclonal antibody anti‐https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2852, a specific B lymphocyte antigen receptor, which is highly expressed in the majority of B‐cell lymphomas and NHL (Newman & Cragg, 2017). Also, in Phase III clinical trials, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7970, which carries MMAF as a payload, and a monoclonal antibody directed at the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1797 (EGFR). This ADC is being tried for the treatment of brain tumours, mainly against glioblastoma carrying amplification of the EGFR gene, which may be present in ~50% of glioblastoma patients (Lassman et al., 2019).
Table 2.
Marine‐derived warheads in antibody‐drug conjugates (ADCs) under clinical trials
| ADC name | Warhead | Antibody target | Condition | Clinical trial status | Developer |
|---|---|---|---|---|---|
|
Depatuxizumab mafodotin (ABT‐414) |
MMAF | EGFR | Glioblastoma, gliosarcoma, malignant glioma | Phase III | AbbVie |
|
Polatuzumab vedotin (DCDS‐4501A) |
MMAE | CD79b | NHL, CLL, B‐cell lymphoma, follicular lymphoma | Phase III | Genentech (Roche) |
|
Belantamab mafodotin (GSK2857916) |
MMAF | BCMA | MM | Phase II | GlaxoSmithKline |
|
Cirmtuzumab vedotin (UC‐961ADC3) |
MMAE | ROR1 | Solid tumours, CLL, B‐cell lymphomas, acute leukaemia, breast | Phase II | UC San Diego |
|
Enapotamab vedotin (HuMax®‐AXL‐ADC) |
MMAE | AXL | Solid tumours, ovarian, NSCLC, endometrial, cervical, thyroid | Phase II | GenMab |
|
Enfortumab vedotin (ASG‐22ME) |
MMAE | Nectin‐4 | Urothelial (M) | Phase II | Seattle Genetics |
|
https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8926 (CDX‐011) |
MMAE | GPNMB | Melanoma (M), breast (M), TNBC | Phase II | Celldex Therapeutics |
| Hertuzumab vedotin | MMAE | HER2 | Breast, gastric (HER2+) | Phase II | Rongchang Pharmaceuticals and MabPlex |
|
https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8718 (MLN‐0264) |
MMAE | GCC | Gastrointestinal, pancreatic | Phase II | Takeda/Millennium Pharmaceuticals |
|
Ladiratuzumab vedotin (SGN‐LIV1A) |
MMAE | LIV‐1 | Breast | Phase II | Seattle Genetics |
|
https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8405 (DNIB0600A) |
MMAE | NaPi2b | Ovarian, NSCLC | Phase II | Genentech (Roche) |
|
Pinatuzumab vedotin (DCDT‐2980S) |
MMAE | CD22 | NHL, DLBCL, CLL, B‐cell lymphomas | Phase II | Genentech (Roche) |
|
Sofituzumab vedotin (DMUC5754A) |
MMAE | MUC16 | Ovary, fallopian tube, pancreas, peritoneal | Phase II | Genentech (Roche) |
|
Telisotuzumab vedotin (ABBV‐399) |
MMAE | c‐Met | Solid tumours | Phase II | AbbVie |
|
Tisotumab vedotin (HuMax®‐TF‐ADC) |
MMAE | TM | Ovary, cervix, endometrium, bladder, CRPC, SCCHN, oesophagus, NSCLC | Phase II | GenMab |
| AGS‐16C3F | MMAF | ENPP3 | RCC | Phase II | Agensys and Astellas Pharma |
|
CAB‐ROR2 (BA‐3021) |
MMAE | ROR2 | Solid tumours, NSCLC, STS, TNBC | Phase II | BioAtla |
| CX‐2029 | MMAE | CD71 | Solid tumours, head and neck, NSCLC, pancreatic, DLBCL | Phase II | AbbVie and CitomX |
| RC‐48 | MMAE | HER2 | Solid tumours, breast, urothelial, gastric (and HER2 overexpressing) | Phase II | RemeGen |
| W0101 | MMAE | IGF1R | Solid tumours (M) | Phase II | Pierre‐Fabre |
|
Azintuxizumab vedotin (ABBV‐838) |
MMAE | SLAMF7 (CD319) | MM, plasma cell myeloma | Phase I | AbbVie |
|
Denintuzumab mafodotin (SGN‐CD19A) |
MMAF | CD19 | ALL, NHL | Phase I | Seattle Genetics |
|
Iladatuzumab vedotin (DCDS0780A) |
MMAE | CD79b | NHL | Phase I | Genentech (Roche) |
|
Losatuxizumab vedotin (ABBV‐221) |
MMAE | EGFR | Solid tumours (overexpressing EGFR) | Phase I | AbbVie |
|
Lupartumab amadotin (BAY 1129980) |
Auristatin W derivative | LYPD3 | Solid tumours | Phase I | Bayer |
|
Samrotamab vedotin (ABBV‐085) |
MMAE | LRRC15 | Solid tumours | Phase I | AbbVie |
|
Sirtratumab vedotin (ASG‐15ME) |
MMAE | SLITRK6 | Urothelial (M) | Phase I | Astellas and Seattle Genetics |
| ALT‐P7 | MMAE | HER2 | Breast, gastric | Phase I | 3SBio and Alteogen |
| ARX‐788 | MMAF | HER2 | Breast, gastric | Phase I | Ambrex and Zhejiang Medicine |
| ASG‐5ME | MMAE | SLC44A4 | Gastric, pancreas, prostate | Phase I | Astellas and Seattle Genetics |
| ASG‐67E | MMAE | CD37 | Lymphoid malignancy (R) | Phase I | Astellas and Seattle Genetics |
| CDX‐014 | MMAE | TIM‐1 | RCC | Phase I | Celldex Therapeutics |
|
DMOT4039A (RG 7600) |
MMAE | MSLN | Ovarian, pancreas, digestive system | Phase I | Genentech (Roche) |
| PF‐06804103 | Auristatin 0101 | HER2 | Breast, stomach, oesophagogastric junction, NSCLC | Phase I | Pfeizer and AbbVie |
| SGN‐CD48A | MMAF | CD48 | MM | Phase I | Seattle Genetics |
| XMT‐1522 | AF‐HPA | HER2 | Breast, gastric, NSCLC | Phase I | Mersana Therapeutics and Takeda |
| XMT‐1536 | AF‐HPA | NaPi2b | Solid tumours | Phase I | Mersana Therapeutics |
| ZW‐49 | MMAE variant | HER2 | HER2 overexpressing cancers | Phase I | Zymeworks and BeiGene |
Abbreviations: ADC, antibody‐drug conjugate; AF‐HPA, auristatin F‐hydroxypropylamide; AXL, AXL receptor TK; BCMA, B‐cell maturation antigen; c‐Met, tyrosine‐protein kinase met; CD19, cluster of differentiation 19, also known as B lymphocyte surface antigen B4; CD22, cluster of differentiation 22, also known as sialic acid binding Ig‐like lectin 2; CD37, cluster of differentiation 37, also known as Tetraspanin 26 (TSPAN26) or Glycoprotein 52‐40 (GP52‐40); CD48, cluster of differentiation 48, also known as B‐lymphocyte activation marker 1 (BLAST‐1) or signalling lymphocytic activation molecule 2 (SLAMF2); CD71, cluster of differentiation 71, also known as transferrin receptor 1 (TfR‐1); CD79b, cluster of differentiation 79b; CLL, chronic lymphocytic leukaemia; CRPC, castration‐resistant prostate cancers; DLBCL, diffuse large B‐cell lymphoma; EGFR, epidermal growth factor receptor; ENPP3, ectonucleotide pyrophosphatase/PDE family member 3; GPNMB, glycoprotein nonmetastatic B; HER2, human EGF receptor‐type 2; IGF1R, insulin‐like growth factor receptor 1; LIV‐1, solute carrier family 39, member 6 (SLC39A6); LRRC15, leucine‐rich repeat containing 15; LY6E, lymphocyte antigen 6 complex, locus E; LYPD3, LY6/PLAUR domain containing 3; (M), metastatic; MM, multiple myeloma; MMAE, monomethylauristatin E; MMAF, monomethylauristatin F; MSLN, mesothelin; MUC16, mucin 16, cell surface associated; NaPi2b, sodium‐dependent phosphate transport protein 2b; NHL, non‐Hodgkin's lymphoma; NSCLC, non‐small cell lung cancer; (R), refractory; RCC, renal cell carcinoma; ROR1, receptor tyrosine‐kinase like orphan receptor 1; SCCHN, squamous cell carcinoma of the head and neck; SLAMF7 (CD319), SLAM family member 7; SLC44A4, solute carrier family 44 member 4; SLITRK6, SLIT and NTRK‐like protein 6; STS, soft tissue sarcoma; TIM‐1, T‐cell immunoglobulin mucin domain 1; TNBC, triple negative breast cancer; TM, tissue factor.
Nevertheless, based on the encouraging data generated by these trailblazing studies, there are more entries of ADCs in clinical testing, as seen by the broad list of such drugs in earlier trial phases, which is also shown in Table 2. Moreover, this technology, a combination of immunotherapy and chemotherapy, is attracting various pharmaceutical companies to undertake the development of auristatin microtubule‐disruptor ADCs, which is further evidence of their ample potential and success in cancer treatment. It is worth mentioning that some biological targets have been well explored within the anticancer immunotherapy arsenal, such as https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2019, and are recurrent in this ADC setting yet are functionalized with a cytotoxic compound connected with a perfected linker. This conjugation has shown to improve the efficacy of immunotherapy and add specificity to chemotherapy.
4.4. Plitidepsin (Aplidin): Targeting protein synthesis
The cyclic depsipeptides plitidepsin (dehydrodidemnin B) and didemnin B are chemically related compounds; the only structural difference is the oxidation (carbonyl group) of the hydroxyl group at the lactoyl‐proline in didemnin B (Figure 1). Whereas didemnin B was isolated from the Caribbean tunicate Tridedmnum solidum back in 1981 (Rinehart et al., 1981), plitidepsin was isolated from the Mediterranean tunicate Aplidium albicans over 10 years later (Sakai et al., 1996).
One of the greatest challenges related to this class of molecules was the elucidation of their mechanism of action. The first studies focused on describing the ability of didemnin B in inhibiting protein synthesis and on the strong correlation of this effect with antiproliferative properties (Li et al., 1984). However, inhibition of cell proliferation was observed at much lower concentrations (nM range) than those required to inhibit protein synthesis (μM range; Ahuja et al., 2000). The elongation factor eEF1A1 was then suggested as a target of didemnin B, but the affinity of such molecules for this component was still assessed in the μM range (Crews, Collins, Lane, Snapper, & Schreiber, 1994). Elongation factors are ubiquitous in most cells, as they deliver aminoacyl‐tRNA to the ribosome during the elongation cycle of protein biosynthesis (Mateyak & Kinzy, 2010). Despite similarities between the two isoforms of eEF1A, their expression is mutually exclusive and they are attributed a different functionality. Thus while the first isoform is widespread in mammalian tissues and is responsible for the elongation function itself, the second presents non‐canonical pro‐oncogenic properties and has a more limited distribution (Sun et al., 2014). In this context, new evidence concerning the molecular mechanism underlying protein synthesis inhibition triggered by didemnins pointed to a crucial role for the non‐canonical elongation factor—eEF1A2—in such processes (SirDeshpande & Toogood, 1995).
The observed affinity constant (K D) for molecular interaction for plitidepsin and eEF1A2 is 80 nM, with a target residence of 9 min. Moreover, sensitivity to plitidepsin is correlated with eEF1A2 expression. While resistant cells exhibited reduced eEF1A2 expression, ectopic expression of eEF1A2 restored sensitivity in these cells. The biological effects resulting from this interaction include inhibition of the transportation of misfolded proteins to the proteasome, which leads to an accumulation of toxic proteins in the tumour cells (Losada et al., 2016). It is further suggested that plitidepsin also inhibits the activation and subsequent destruction of the aggresome in the lysosome, which also accumulates an excess of misfolded proteins in the cells, which then triggers apoptosis (available from https://www.pharmamar.com, accessed 22 February 2019). Figure 2 illustrates the mechanism of action for plitidepsin in tumour cells.
Preclinical data demonstrated in vitro activity of plitidepsin in several cancer cell lines in the sub‐nM range. Haematological cell lines were the most sensitive ones, with IC50 means of 28 nM for ALL, 24 nM for AML, 74 nM for CML, 77 nM for multiple myeloma (MM), and 78 nM for NHL (Depenbrock et al., 1998). In vivo assays were carried out in xenograft models mainly with haematological tumours including MM, T‐cell lymphoma, diffuse large B‐cell lymphoma (DLBCL), and Burkitt lymphoma, with promising results (revised by Alonso‐Álvarez et al., 2017). Table 1 summarizes preclinical and clinical data with plitidepsin.
In 1986, didemnin B was the first marine natural product to enter clinical trials in patients with advanced cancer, but due to neuromuscular toxicity and low efficacy, its further development was interrupted (Vera & Joullié, 2002). Despite the similarities of didemin B and plitidepsin and the failure of didemnin B in clinical trials, the increased potency of ptilidepsin and a better pharmacokinetic profile (Alonso‐Álvarez et al., 2017) stimulated the beginning of Phase I trials with ptilidepsin by PharmaMar in 1998. It is important to point out that although didemnin B showed a very short half‐life, as it was converted to an unidentified metabolite (Dorr, Kuhn, Phillips, & von Hoff, 1988), plitidepsin was found to have a longer half‐life, low clearance, and a high volume of distribution (Nalda‐Molina et al., 2009).
Clinical studies with plitidepsin were conducted in groups of patients with haematological cancer and, although the results for use as a single drug were not very encouraging, the combination of plitidepsin with other drugs was promising, especially for patients with MM (Alonso‐Álvarez et al., 2017; Mateos et al., 2010; Ocio et al., 2016). The addition of dexamethasone in the treatment of patients with relapsed or refractory MM receiving plitidepsin improves the overall response rate (ORR) from 13% to 22% (Mateos et al., 2010). The concomitant treatment of MM patients with infusion of plitidepsin during 4 weeks at 4–5.0 mg·m−2, dexamethasone (oral) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6391 (s.c.) provided an ORR of 56% (Ocio et al., 2016). The observed toxicity was manageable, including anaemia and thrombocytopenia among the most severe haematological effects, and muscle toxicity was a dose‐limiting side effect (Faivre et al., 2005; Mateos et al., 2010; Ocio et al., 2016). The ADMYRE Phase III study was conducted with 255 patients with relapsed/refractory MM, comparing the efficacy of plitidepsin in combination with dexamethasone to that of dexamethasone alone (available from https://www.clinicaltrials.gov, accessed 25 January 2019). The results available so far indicate a 35% lower risk of disease progression or death in patients treated with the drug combination (Alonso‐Álvarez et al., 2017).
Considering other haematological malignancies, such as leukaemia and lymphoma, plitipesin has been tested in at least four different clinical studies, but ORR is variable, from non‐objective response to a maximum of 20.7% for non‐cutaneous peripheral T‐cell lymphoma (Aspeslagh et al., 2017; Ribrag et al., 2013). Studies conducted in patients bearing solid tumours—including melanoma, renal, advanced colorectal, prostate, thyroid, and NSCLC, among others—were not very promising, and the ORRs were generally not significant (revised by Alonso‐Álvarez et al., 2017).
The treatment of refractory MM with the combination of plitidepsin and dexamethasone in patients that relapse after three lines of treatment, was approved by the Australian regulatory agency in December 2018 (available from https://www.ebs.tga.gov.au, accessed 20 February 2019). The EMEA, on the other hand, refused marketing authorization for Aplidin (plitidepsin) in Europe. According to the re‐examination carried out by EMEA's Committee for Medicinal Products for Human Use, the benefits of plitidepsin do not outweigh the risks, since toxicity is observed at higher frequencies and there is merely a modest increase in patient response compared to that of dexamethasone alone (available from https://www.ema.europa.eu, accessed 21 February 2019). Still, Aplidin has received orphan drug designation in the European Union and in the United States. The timeline between the discovery of plitidepsin and the full development of Aplidin is showed in Figure 3.
5. WHAT IS NEXT? MARINE NATURAL PRODUCTS IN CLINICAL TRIALS
5.1. Lurbinectedin
Based on the unique features of trabectedin, PharmaMar designed a synthetic analogue, lurbinectedin (Figure 1; Zepsyre, PM01183). The main difference is attributed to the substitution of the tetrahydroisoquinoline with a tetrahydro β‐carboline, resulting in increased antitumour activity of lurbinectedin (Leal et al., 2010). The mechanism of action of lurbidectadin is similar to that of trabectedin, as it also binds to CG‐rich sequences in the DNA minor groove, inhibits active transcription, and degrades Pol II through the ubiquitin/proteasome system (Santamaria Nuñez et al., 2016). Furthermore, lurbinectedin can cause a detachment of transcription factors, such as ASCL1, NeuroD1 and NFIB in small cell lung cancer (SCLC), from their target promoters, inducing blockade of their transactivating activity (Farago et al., 2019) and exerting important effects on TME (Belgiovine et al., 2017) as well as upon cancer stem cells (Yokoi et al., 2018).
Lurbinectedin has also shown positive results against other tumour types, such as ovary carcinoma, Ewing sarcoma, and pancreatic ductal adenocarcinoma (Harlow et al., 2016; Takahashi et al., 2016; Zhang, Feng, & Kennedy, 2017). As with trabectedin, TC‐NER deficient cells showed resistance to lurbinectedin, whereas those deficient in homologous recombination were markedly more sensitive. Lurbinectedin also showed good antitumour activity in several in vivo tumours and xenografts models, including MNMCA1 mouse fibrosarcoma and HOC18 human ovarian carcinoma, however with a different spectrum of activity and distinct pharmacokinetics properties when compared to the parent compound (Romano et al., 2013).
A Phase I study that evaluated the efficacy of lurbinectedin and doxorubicin in patients with relapsed SCLC observed durable response rates in 91.7% of patients with sensitive disease and in 33.3% and 20.0% of resistant disease as second‐ and third‐line treatments respectively. Additionally, tumour shrinkage was maintained in 88% of patients who switched to lurbinectedin alone (Calvo et al., 2017). A Phase II trial conducted with patients that received different chemotherapy‐free intervals showed a 52% partial response when intervals were above 90 days. Additionally, 40% of cases presented disease stabilization, with a progression‐free survival of 4.2 months (Perez et al., 2018). A Phase III trial involving 600 patients who had failed one prior platinum‐containing treatment is currently under investigation (ATLANTIS NCT02566993); they are being treated with a combination of doxorubicin and lurbinectedin, versus topotecan or the VCR (cyclophosphamide, doxorubicin, and vincristine) combination (Farago et al., 2019; Table 1). With this evidence, the FDA granted lurbinectedin, in August 2018, orphan drug status for SCLC.
5.2. Plinabulin
Plinabulin (NPI‐2358, Figure 1) is a synthetic tert‐butyl analogue related to the naturally occurring fungal diketopiperazine halimide, also known as phenylahistin (Gomes, Lefranc, Kijjoa, & Kiss, 2015). In fact, halimide was isolated by Fenical, Jensen, and Cheng (1999) from the fermentation broth of a marine fungus, Aspergillus sp., found in association with the algae Halimeda sp., collected in the Philippines. At the same time, phenylahistin was described as a fungal metabolite obtained from the terrestrial Aspergilus ustus (Kanoh et al., 1997, 1999).
The natural product is a potent cytotoxic compound, as it arrests cell cycle in mitosis by inhibiting tubulin polymerization through interaction with the colchicine‐binding site (Kanoh et al., 1997). An in vivo study using P388 leukaemia and Lewis lung carcinoma showed very promising results, increasing the mean survival of mice bearing the haematological cancer in 51% of cases at 30 mg·kg−1 and reduced tumour growth by 81% in mice with the solid tumour at 100 mg·kg−1. These results stimulated the synthesis of more potent analogues (Kanoh et al., 1999). The establishment of essential structural requirements for eliciting effective cytotoxicity by (−)‐phenylahistin contributed to the design of the clinical candidate, plinabulin, in 2006 (Nicholson et al., 2006). The new analogue demonstrated a potent activity against human colon adenocarcinoma (HT‐29), prostate adenocarcinomas (PC‐3 and DU 145), breast adenocarcinoma (MDA‐MB‐231), NSCLC (NCI‐H292), and T‐cell leukaemia (Jurkat) cell lines, including those with MDR profiles. Additionally, plinabulin promoted an increased permeability in HUVECs, which is consistent with a tumour vascular collapse. Such evidence showed plinabulin to be a vascular disrupting agent (Nicholson et al., 2006).
Evaluation of the effects of plinabulin on MM demonstrated that besides the anti‐angiogenic potential on vascular endothelial cells, it induced cell death through caspase activation and showed an obligatory role of https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=518 during plinabulin‐induced MM cell death (Singh et al., 2011). An immune‐mediated mechanism of action was recently proposed, supported by an increase in dendritic cell maturation and subsequent cytokine release, which occurs through activation of the guanine nucleotide exchange factor (GEF‐H1; available from https://www.beyondspringpharma.com/en/pipeline/, accessed 24 February 2019).
Efficacy of plinabulin in Phase I trials was shown for different human tumours. However, a meaningful response was obtained in murine NSCLC, either alone or synergistically with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6809 (Millward et al., 2012). Moreover, plinabulin presented a favourable safety profile evaluated by its half‐life, clearance, and distributive volume in NSCLC patients (Mita et al., 2010). A Phase II study demonstrated that combined treatment of plinabulin and docetaxel promoted an increased OS rate and PFS in patients with advanced NSCLC, and more recently, the association of plinabulin with the anti‐hPD1 https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7335 was tested for the treatment of five patients with recurrent or metastatic NSCLC, but the study was prematurely terminated because the immunotherapy was approved for NSCLC in the first‐line setting (http://www.clinicaltrials.gov, accessed 10 August 2019).
5.3. Marizomib
Salinosporamide A (Figure 1), also named NPI‐0052 or marizomib, is a cytotoxic β‐lactone‐γ‐lactam produced by the strictly marine bacterium Salinispora tropica. It was isolated in 2003 and its chemical structure, cytotoxic activity, and pharmacological targets described by Feling et al. (2003). The compound exhibited low‐nM GI50 values on the NCI 60 cell line panel (Feling et al., 2003), and preclinical development was strongly favoured by the supply ensured by saline fermentation of the S. tropica wild strain itself, an unprecedented process for sourcing an active pharmaceutical ingredient (Tsueng, Teisan, & Lam, 2008). Genome sequencing of S. tropica allowed disclosure of the salinosporamide A peculiar biosynthetic pathway (Udwary et al., 2007), uncovering a new chlorination mechanism and a unique starter unit in polyketide biosynthesis, resulting in the production of this densely functionalized molecule (Eustáquio, Pojer, Noel, & Moore, 2008).
Salinosporamide A was then shown to be a novel https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2410 inhibitor, differentiated from other drugs in this class. It also has interesting advantages that fulfil unmet clinical needs, such as enhanced potency, wider inhibition spectrum, and effectiveness against tumour cells resistant to clinically available proteasome inhibitors (Potts et al., 2011). The ubiquitin‐26S proteasome complex contains a proteolytic 20S core comprising three pairs of catalytic subunits, which have caspase‐, trypsin‐, and chymotrypsin‐like (CT‐L) activities, where protein degradation occurs (Lowe et al., 1995; Sala et al., 2018). Salinosporamide A irreversibly inhibited all three catalytic functions of the 20S proteasome, with IC50 values ranging from low to mid nM (Feling et al., 2003; Potts et al., 2011). Peptide boronic acids, for example, bortezomib (Velcade) and delanzomib, block only the CT‐L activity and in a slow and reversible fashion (Groll, Berkers, Ploegh, & Ovaa, 2006).
Translational studies have demonstrated the activity of marizomib against solid tumours and haematological malignancies as a single agent or in combination with clinically used drugs. As a single agent in in vivo models for haematological malignancies, such as MM, NHL, Waldenstrom's macroglobulinemia, ALL, AML, and chronic lymphocytic leukaemia (CLL), as well as in solid tumours, such as colon, glioma, and pancreatic cancer, this compound has been achieving good results (Potts et al., 2011; Sala et al., 2018). Studies in animal models using marizomib in combination with standard of care drugs confirmed the broad activity spectrum of this compound. Moreover, marizomib re‐sensitizes resistant MM cells to apoptosis induced by chemotherapeutic and immunotherapeutic drugs. Particularly interesting results indicate an enhanced anti‐MM activity of low‐dose treatment of marizomib combined with bortezomib (Chauhan, Hideshima, & Anderson, 2006), revealing that this vertical combination targeting one oncogenic pathway at several levels provided greater inhibition. Additionally, marizomib displayed better results than bortezomib when used in combination with other treatments, for example, with immunotherapy and https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=848 inhibitors for ALL (Chauhan et al., 2010; Miller et al., 2009).
Marizomib entered clinical trials just 3 years after its discovery. To date, nine clinical trials, either alone or in combination with other cancer therapies, are listed with marizomib, among which four are still ongoing and wish to establish optimal practices for this agent. These studies include MM, refractory MM, relapsed MM, NSCLC, pancreatic cancer, melanoma, lymphomas, ependymoma, glioblastoma, and newly diagnosed glioblastoma (available from https://www.clinicaltrials.gov, accessed 25 January 2019), which seem to demonstrate this drug is well tolerated as monotherapy and in combination with other chemotherapy or radiotherapy. Marizomib exhibits wider, faster, and more durable proteasome inhibition than bortezomib without the limiting toxicities associated with the latter and other clinically approved proteasome inhibitors, such as neutropenia, thrombocytopenia, and peripheral neuropathy. Most side effects occurred in the early cycles of therapy and neither new nor cumulative toxicities were produced with longer periods of treatments (Potts et al., 2011; Richardson et al., 2016). The most common side effects related to marizomib administration were fatigue, nausea, vomiting, dizziness, diarrhoea, and pain at the infusion site (Harrison et al., 2016; Richardson et al., 2016). Recently, a consortium comprising Celgene Corporation, Triphase Accelerator Corporation, and the European Organization for Research and Treatment of Cancer has sponsored Phase I and II clinical trials with marizomib in glioblastoma patients. Ongoing Phase II and III trials focuses on glioblastoma and MM (available from https://www.clinicaltrials.gov, accessed 25 January 2019).
5.4. Plocabulin
Plocabulin, or PM060184 (Figure 1), is a polyketide currently under three clinical studies for cancer treatment. This tubulin inhibitor and its chlorinated analogue, PM050489, were isolated from the Madagascan sponge Lithoplocamia lithistoides (Martín et al., 2013; Pera et al., 2013). Based on the high success of anticancer drugs targeting tubulin, added to its powerful tubulin inhibition rates, this marine molecule has become a promising alternative to cancer treatment, and with some advantages over current antitubulin drugs. Plocabulin showed potent antitumour activity in a panel of tumour xenograft models, including MDA‐MB‐231 (breast), HCT‐116 (colon), H‐460 (lung), 22RV1 (prostate), HGC‐27 (gastric) and Caki‐1 (renal), and displayed particularly high cytotoxic against P‐glycoprotein overexpressing cell lines. In these latter cells, paclitaxel and vinblastine lacked or had lower potency compared to PM060184. This feature supports the use of plocabulin in tumours resistant to antitubulin agents (Martínez‐Díez et al., 2014).
Plocabulin binds, in a sub‐ or low‐nM range, to a site different from than that of vinblastine, although it still affects binding of this drug. During mitosis, cells display multipolar figures and lagging chromosomes in the metaphase plate (Martínez‐Díez et al., 2014; Pera et al., 2013). Still, upon interphase cells, these undergo disorganization and fragmentation of the microtubule network, along with the inhibition of cell migration. Additionally, PM060184 inhibits angiogenesis both in vitro and in vivo (Galmarini et al., 2018; Martínez‐Díez et al., 2014).
Currently, there is one Phase I clinical trial completed and two ongoing studies in Phases I and II with PM060184 alone or in combination with other antitumour drugs in patients with advanced solid tumours (available from https://www.clinicaltrials.gov, accessed 25 January 2019). The finalized clinical study highlighted mild to moderate toxicities, such as abdominal pain, myalgia, fatigue, nausea, vomiting, and disease stabilization. Furthermore, myelosuppression was transient and manageable and the main dose‐limiting toxicity (DLT) reported was an expected grade 3 peripheral sensory neuropathy (PSN). Other DLTs were tumour lysis syndrome and cardiac failure and myalgia (Elez et al., 2019). PM060184 has a half‐life of ~4 hr and extensive diffusion to peripheral body tissues. Furthermore, antitumour responses were observed in cervix carcinoma and in pretreated metastatic NSCLC patients. Disease stabilization for longer than 3 months was observed in patients with colorectal, breast, thymic, and gastrointestinal stromal tumours, among others (Elez et al., 2019).
It is worth mentioning that the recent success of ADCs in the treatment of cancer has revitalized the interest in antitubulin drugs. As this strategy combines specificity and a dramatic decrease of toxicity, it enables the use of highly potent and otherwise unacceptably toxic compounds. In this sense, although PM060184 studies are only just beginning, they already have shown many properties and possibilities for a successful use in cancer treatment, in the future.
6. CONCLUDING REMARKS
There is no doubt the oceans will play a fundamental role in society for the next decades and will provide new tools to face the health challenges affecting the world's population in the years to come. We have argued that MNPs may represent a renewed hope in drug discovery, as these have been applied for pharmacologically orphaned cancers, for the treatment of cancers with particular characteristics and have also overcome cases of tumour drug resistance. It is rather noticeable that four of the five marine‐derived drugs that make up the anticancer arsenal have been approved over the past 12 years. This is, most certainly, a reflection of the advances of science and technology in multidisciplinary fields of knowledge, particularly in analytical chemistry and biology. Even though this review only discussed the successes, these marine molecules definitely illustrate the pharmacological potential that lies beneath the salty waters of our globe, which actually accounts for the largest section of our planet.
A global estimate of the value of anticancer marine pharmaceuticals yet to be discovered ranged from U$ 563 billion to U$ 5.69 trillion, considering the projected marine biodiversity in the oceans (Erwin, López‐Legentil, & Schuhmann, 2010). It is still soon to presume whether this estimate is accurate, because, as discussed here, most of the marketed drugs were launched in recent years. For example, the sales of Yondelis amounted to €74.2 million in 2018 compared to €84.6 million in 2017, showing indeed a retraction of 12% (Annual Report 2018 available at https://www.pharmamar.com, accessed 10 August 2019), while Adcetris net product sales increased from U$ 307.6 million in 2017 to U$ 476.9 million in 2018, an increase of 55% (Annual Report 2018 available at http://www.seattlegenetics.com, accessed 10 August 2019). Halaven sales are also growing as the number of sarcoma patients using this therapy increased, especially in Japan (http://www.elsai.com, accessed 10 August 2019). As Aplidin was only approved on December 2018, data on yearly sales are not yet available.
As promising as these molecules may be, research and development of marine drugs have been afflicted by their share of difficulties. Collecting natural samples is not a simple task, as marine environments pose many abiotic and physiological restrictions to human assessment. Also, from a legal point of view, the collection of samples in international oceanic waters has been often discussed. In this context, the adoption of the Nagoya Protocol in 2010, established under the Convention of Biological Diversity from 1992, was an attempt to legitimize access to marine genetic resources, exploration, and benefit‐sharing between the signatory countries (Lallier et al., 2014).
Natural samples and their derived extracts are chemically complex samples, as these consist of many different compounds from distinct chemical classes. Therefore, the yields of pure compounds that can be isolated from natural sources may be quite low. Besides, marine natural products come, generally, in structural arrangements of high complexity, making the elucidation and synthesis or even semi‐synthesis of such molecules a complicated process. Therefore, the supply of sufficient amounts of such compounds for bioassays and structure–activity relationships studies or for clinical trials is certainly one of most challenging issues to overcome. Additionally, the intrinsic toxicity of marine compounds is also an important issue to cope with. Understanding the pharmacophoric structural requirements of bioactive molecules is key for the development of improved anticancer drugs, furnished with enhanced efficacy, better selectivity, and minimum adverse effects. In this context, technology is probably the best ally in the effort to reduce toxicity, as witnessed in the successful stories of the ADCs (Senter & Sievers, 2012).
Still, to make a study ecologically viable, conscious researchers must be careful to collect a sufficient amount of each sample without endangering the existence of the species. Yet information on the conservation status for most marine species assessed in a drug discovery perspective is scarce, as these weigh largely towards fish and larger metazoans (Arrieta, Arnaud‐Haond, & Duarte, 2010). To illustrate this point, an online search was done on The Red List of Endangered Species of the International Union for Conservation of Nature (IUCN) for species mentioned in the scope of this review—E. turbinata, H. okadai, D. auricularia, and A. albicans—and returned no results for any of these entries (Available from https://www.iucnredlist.org/, accessed 5 March 2019).
Nevertheless, the predatory nature of field sampling of marine invertebrates used in the development of new medicines has significantly reduced their natural population due to oversampling. One important example is the halichondrin‐producing sponges, where 600 kg of H. okadai was collected off the coast of Aburatsubo, in the Miura Peninsula of Japan (Hirata & Uemura, 1986); then, another 200 kg of Lissodendoryx sp. was taken from the Kaikoura Peninsula in New Zealand (Litaudon et al., 1994). In this context, there is increasing evidence of the important roles of the microbiota in the production of secondary metabolites by holobiont systems, including corals, ascidians, and sponges.
All these issues make marine‐based drug development a time‐ and money‐consuming endeavour. From compound discovery to marketing of the finished product has taken between 17 and 29 years, with an average of 23 years (examples in Figure 3). Molecules derived from microbial sources seem to speed up this process, as large‐scale fermentation allows for the necessary supply to warrant preclinical and clinical studies, as seen with marizomib (Feling et al., 2003; Tsueng et al., 2008). Still, this shows the importance of investing in science as means to find creative and sustainable strategies to overcome these difficulties, as well as the disproportionate requirements for identifying a marketable product as a condition for granting support for MNP bioprospecting projects in the field of health sciences, once these projects have the duration of 3 to 5 years.
MNP research is improving and advancing, but it is still a risky, time‐consuming, and costly endeavour. However, once even more life‐saving drugs are added to this equation, the potential gains will surely outweigh the risks.
6.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017a, 2017b; Alexander, Kelly et al., 2017a, 2017b).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
The authors are grateful to FAPESP (Grants 2015/17177‐6; 2017/09022‐8, and 2017/17648‐4), CNPQ, ArboControl Brasil Project (Grants FNS/UnB TED74/2016 and TED42/2017), FCT (Grant IF/00700/2014 to S.P.G.), and National Institute of Science and Technology – INCTBioNat (Grant 465637/2014‐0). This work was supported by the Applied Molecular Biosciences Unit (UCIBIO), which is financed by national funds from FCT/MCTES (Grant UID/Multi/04378/2019).
Jimenez PC, Wilke DV, Branco PC, et al. Enriching cancer pharmacology with drugs of marine origin. Br J Pharmacol. 2020;177:3–27. 10.1111/bph.14876
REFERENCES
- Ahuja, D. , Vera, M. D. , SirDeshpande, B. V. , Morimoto, H. , Williams, P. G. , Joullié, M. M. , et al. (2000). Inhibition of protein synthesis by didemnin B: How EF‐1α mediates inhibition of translocation. Biochemistry, 39, 4339–4346. [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. British Journal of Pharmacology, 174, S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. British Journal of Pharmacology, 174, S360–S446. 10.1111/bph.13883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso‐Álvarez, S. , Pardal, E. , Sánchez‐Nieto, D. , Navarro, M. , Caballero, M. D. , Mateos, M. V. , et al. (2017). Plitidepsin: Design, development, and potential place in therapy. Drug Design, Development and Therapy, 11, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aogi, K. , Iwata, H. , Masuda, N. , Mukai, H. , Yoshida, M. , Rai, Y. , et al. (2012). A phase II study of eribulin in Japanese patients with heavily pretreated metastatic breast cancer. Annals of Oncology, 23, 1441–1448. [DOI] [PubMed] [Google Scholar]
- Araki, K. , Fukada, I. , Yanagi, H. , Kobayashi, K. , Shibayama, T. , Horii, R. , et al. (2017). First report of eribulin in combination with pertuzumab and trastuzumab for advanced HER2‐positive breast cancer. The Breast, 35, 78–84. [DOI] [PubMed] [Google Scholar]
- Arrieta, J. M. , Arnaud‐Haond, S. , & Duarte, C. M. (2010). What lies underneath: Conserving the oceans' genetic resources. Proceedings of the National Academy of Sciences, 107, 18318–18324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspeslagh, S. , Stein, M. , Bahleda, R. , Hollebecque, A. , Salles, G. , Gyan, E. , et al. (2017). Phase I dose‐escalation study of plitidepsin in combination with sorafenib or gemcitabine in patients with refractory solid tumors or lymphomas. Anti‐Cancer Drugs, 28, 341–349. [DOI] [PubMed] [Google Scholar]
- Aune, G. J. , Takagi, K. , Sordet, O. , Guirouilh‐Barbat, J. , Antony, S. , Bohr, V. A. , et al. (2008). Von Hippel‐Lindau‐coupled and transcription‐coupled nucleotide excision repair‐dependent degradation of RNA polymerase II in response to trabectedin. Clinical Cancer Research, 14, 6449–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgiovine, C. , Bello, E. , Liguori, M. , Craparotta, I. , Mannarino, L. , Paracchini, L. , et al. (2017). Lurbinectedin reduces tumour‐associated macrophages and the inflammatory tumour microenvironment in preclinical models. British Journal of Cancer, 117, 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann, W. , & Feeney, R. J. (1951). Contributions to the study of marine products. XXXII. The nucleosides of sponges. I. 1. The Journal of Organic Chemistry, 16, 981–987. [Google Scholar]
- Blunt, J. W. , Carroll, A. R. , Copp, B. R. , Davis, R. A. , Keyzers, R. A. , & Prinsep, M. R. (2018). Marine natural products. Natural Product Reports, 35, 8–53. [DOI] [PubMed] [Google Scholar]
- Bonfanti, M. , La Valle, E. , Fernandez Sousa Faro, J. M. , Faircloth, G. , Caretti, G. , Mantovani, R. , & D'Incalci, M. (1999). Effect of ecteinascidin‐743 on the interaction between DNA binding proteins and DNA. Anti‐Cancer Drug Design, 14, 179–186. [PubMed] [Google Scholar]
- Buie, L. W. , Epstein, S. S. , & Lindley, C. M. (2007). Nelarabine: A novel purine antimetabolite antineoplastic agent. Clinical Therapeutics, 29, 1887–1899. [DOI] [PubMed] [Google Scholar]
- Calvo, E. , Moreno, V. , Flynn, M. , Holgado, E. , Olmedo, M. E. , Lopez Criado, M. P. , et al. (2017). Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small‐cell lung cancer: Results from a phase I study. Annals of Oncology, 28, 2559–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai, C. L. , & Mátyus, P. (2016). One size does not fit all: Challenging some dogmas and taboos in drug discovery. Future Medicinal Chemistry, 8, 29–38. [DOI] [PubMed] [Google Scholar]
- Chauhan, D. , Hideshima, T. , & Anderson, K. C. (2006). A novel proteasome inhibitor NPI‐0052 as an anticancer therapy. British Journal of Cancer, 95, 961–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan, D. , Singh, A. V. , Ciccarelli, B. , Richardson, P. G. , Palladino, M. A. , & Anderson, K. C. (2010). Combination of novel proteasome inhibitor NPI‐0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood, 115, 834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clardy, J. , & Walsh, C. (2004). Lessons from natural molecules. Nature, 432, 829–837. [DOI] [PubMed] [Google Scholar]
- Cortes, J. , Vahdat, L. , Blum, J. L. , Twelves, C. , Campone, M. , Roché, H. , et al. (2010). Phase II study of the halichondrin B analog eribulin mesylate in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline, a taxane, and capecitabine. Journal of Clinical Oncology, 28, 3922–3928. [DOI] [PubMed] [Google Scholar]
- Crews, C. M. , Collins, J. L. , Lane, W. S. , Snapper, M. L. , & Schreiber, S. L. (1994). GTP‐dependent binding of the antiproliferative agent didemnin to elongation factor 1α. The Journal of Biological Chemistry, 269, 15411–15414. [PubMed] [Google Scholar]
- Demetri, G. D. , Schöffski, P. , Grignani, G. , Blay, J.‐Y. , Maki, R. G. , Van Tine, B. A. , et al. (2017). Activity of eribulin in patients with advanced liposarcoma demonstrated in a subgroup analysis from a randomized phase III study of eribulin versus dacarbazine. Journal of Clinical Oncology, 35, 3433–3439. [DOI] [PubMed] [Google Scholar]
- Depenbrock, H. , Peter, R. , Faircloth, G. T. , Manzanares, I. , Jimeno, J. , & Hanauske, A. R. (1998). In vitro activity of aplidine, a new marine‐derived anti‐cancer compound, on freshly explanted clonogenic human tumour cells and haematopoietic precursor cells. British Journal of Cancer, 78, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Incalci, M. (2013). Trabectedin mechanism of action: What's new? Future Oncology, 9, 5–10. [DOI] [PubMed] [Google Scholar]
- D'Incalci, M. , & Galmarini, C. M. (2010). A review of trabectedin (ET‐743): A unique mechanism of action. Molecular Cancer Therapeutics, 9, 2157–2163. [DOI] [PubMed] [Google Scholar]
- Doronina, S. O. , Toki, B. E. , Torgov, M. Y. , Mendelsohn, B. A. , Cerveny, C. G. , Chace, D. F. , et al. (2003). Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nature Biotechnology, 21, 778–784. [DOI] [PubMed] [Google Scholar]
- Dorr, F. A. , Kuhn, J. G. , Phillips, J. , & von Hoff, D. D. (1988). Phase I clinical and pharmacokinetic investigation of didemnin B, a cyclic depsipeptide. European Journal of Cancer and Clinical Oncology, 24, 1699–1706. [DOI] [PubMed] [Google Scholar]
- Ducry, L. , & Stump, B. (2010). Antibody−drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chemistry, 21, 5–13. [DOI] [PubMed] [Google Scholar]
- Elez, E. , Gomez‐Roca, C. , Soto Matos‐Pita, A. , Argiles, G. , Valentin, T. , Coronado, C. , et al. (2019). First‐in‐human phase I study of the microtubule inhibitor plocabulin in patients with advanced solid tumors. Investigational New Drugs, 37, 674–683. [DOI] [PubMed] [Google Scholar]
- Erba, E. , Cavallaro, E. , Damia, G. , Mantovani, R. , Di Silvio, A. , Di Francesco, A. M. , et al. (2004). The unique biological features of the marine product YondelisTM (ET‐743, trabectedin) are shared by its analog ET‐637, which lacks the C ring. Oncology Research, 14, 579–587. [DOI] [PubMed] [Google Scholar]
- Erwin, P. M. , López‐Legentil, S. , & Schuhmann, P. W. (2010). The pharmaceutical value of marine biodiversity for anti‐cancer drug discovery. Ecological Economics, 70, 445–451. [Google Scholar]
- Eslamian, G. , Wilson, C. , & Young, R. J. (2017). Efficacy of eribulin in breast cancer: A short report on the emerging new data. OncoTargets and Therapy, 10, 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustáquio, A. S. , Pojer, F. , Noel, J. P. , & Moore, B. S. (2008). Discovery and characterization of a marine bacterial SAM‐dependent chlorinase. Nature Chemical Biology, 4, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre, S. , Chièze, S. , Delbaldo, C. , Ady‐Vago, N. , Guzman, C. , Lopez‐Lazaro, L. , et al. (2005). Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. Journal of Clinical Oncology, 23, 7871–7880. [DOI] [PubMed] [Google Scholar]
- Farago, A. F. , Drapkin, B. J. , Lopez‐Vilarino de Ramos, J. A. , Galmarini, C. M. , Núñez, R. , Kahatt, C. , et al. (2019). ATLANTIS: A Phase III study of lurbinectedin/doxorubicin versus topotecan or cyclophosphamide/doxorubicin/vincristine in patients with small‐cell lung cancer who have failed one prior platinum‐containing line. Future Oncology, 15, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feling, R. H. , Buchanan, G. O. , Mincer, T. J. , Kauffman, C. A. , Jensen, P. R. , & Fenical, W. (2003). Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angewandte Chemie, International Edition, 42, 355–357. [DOI] [PubMed] [Google Scholar]
- Fenical, W. , Jensen, P.R. , Cheng, X.C. (1999). Halimide, a cytotoxic marine natural product, and derivatives thereof. International Publication Number NO99/48889.
- Feuerhahn, S. , Giraudon, C. , Martínez‐Díez, M. , Bueren‐Calabuig, J. A. , Galmarini, C. M. , Gago, F. , et al. (2011). XPF‐Dependent DNA breaks and RNA polymerase II arrest induced by antitumor DNA interstrand crosslinking‐mimetic alkaloids. Chemistry & Biology, 18, 988–999. [DOI] [PubMed] [Google Scholar]
- Forni, C. , Minuzzo, M. , Virdis, E. , Tamborini, E. , Simone, M. , Tavecchio, M. , et al. (2009). Trabectedin (ET‐743) promotes differentiation in myxoid liposarcoma tumors. Molecular Cancer Therapeutics, 8, 449–457. [DOI] [PubMed] [Google Scholar]
- Francisco, J. A. , Cerveny, C. G. , Meyer, D. L. , Mixan, B. J. , Klussman, K. , Chace, D. F. , et al. (2003). cAC10‐vcMMAE, an anti‐CD30‐monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood, 102, 1458–1465. [DOI] [PubMed] [Google Scholar]
- Funahashi, Y. , Okamoto, K. , Adachi, Y. , Semba, T. , Uesugi, M. , Ozawa, Y. , et al. (2014). Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Science, 105, 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmarini, C. , D'Incalci, M. , & Allavena, P. (2014). Trabectedin and plitidepsin: Drugs from the sea that strike the tumor microenvironment. Marine Drugs, 12, 719–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmarini, C. M. , Martin, M. , Bouchet, B. P. , Guillen‐Navarro, M. J. , Martínez‐Diez, M. , Martinez‐Leal, J. F. , et al. (2018). Plocabulin, a novel tubulin‐binding agent, inhibits angiogenesis by modulation of microtubule dynamics in endothelial cells. BMC Cancer, 18, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Carbonero, R. , Supko, J. G. , Maki, R. G. , Manola, J. , Ryan, D. P. , Harmon, D. , et al. (2005). Ecteinascidin‐743 (ET‐743) for chemotherapy‐naive patients with advanced soft tissue sarcomas: Multicenter phase II and pharmacokinetic study. Journal of Clinical Oncology, 23, 5484–5492. [DOI] [PubMed] [Google Scholar]
- Goel, S. , Mita, A. C. , Mita, M. , Rowinsky, E. K. , Chu, Q. S. , Wong, N. , et al. (2009). A Phase I study of eribulin mesylate (E7389), a mechanistically novel inhibitor of microtubule dynamics, in patients with advanced solid malignancies. Clinical Cancer Research, 15, 4207–4212. [DOI] [PubMed] [Google Scholar]
- Gomes, N. , Lefranc, F. , Kijjoa, A. , & Kiss, R. (2015). Can some marine‐derived fungal metabolites become actual anticancer agents? Marine Drugs, 13, 3950–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, E. M. , Sankhala, K. K. , Chawla, N. , & Chawla, S. P. (2016). Trabectedin for Soft Tissue Sarcoma: Current Status and Future Perspectives. Advances in Therapy, 33, 1055–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski, K. , & Schneider, G. (2007). Properties and architecture of drugs and natural products revisited. Current Chemical Biology, 1, 115–127. [Google Scholar]
- Grignani, G. , D'Ambrosio, L. , Pignochino, Y. , Palmerini, E. , Zucchetti, M. , Boccone, P. , et al. (2018). Trabectedin and olaparib in patients with advanced and non‐resectable bone and soft‐tissue sarcomas (TOMAS): An open‐label, phase 1b study from the Italian Sarcoma Group. The Lancet Oncology, 19, 1360–1371. [DOI] [PubMed] [Google Scholar]
- Groll, M. , Berkers, C. R. , Ploegh, H. L. , & Ovaa, H. (2006). Crystal structure of the boronic acid‐based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. St Alexander, S. P. H., Kelly, E., Marrion, N. V., Peters, J. A., Faccenda, E., Harding, S. D., Pawson, A. J., Sharman, J. L., Southan, C., Buneman, O. P., Cidlowski, J. A., Christopoulos, A., Davenport, A. P., Fabbro, D., Spedding, M., Striessnig, J., Davies, J. A., and CGTP Collaborators (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Other proteins. British Journal of Pharmacology, 174, S1–S16. doi: 10.1111/bph.13882. Alexander, S. P. H., Kelly, E., Marrion, N. V., Peters, J. A., Faccenda, E., Harding, S. D., Pawson, A. J., Sharman, J. L., Southan, C., Buneman, O. P., Cidlowski, J. A., Christopoulos, A., Davenport, A. P., Fabbro, D., Spedding, M., Striessnig, J., Davies, J. A., and CGTP Collaborators (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Other proteins. British Journal of Pharmacology, 174, S1–S16. 10.1111/bph.13882 ructure 14: 451–456 [DOI] [Google Scholar]
- Hamblett, K. J. , Senter, P. D. , Chace, D. F. , Sun, M. M. C. , Lenox, J. , Cerveny, C. G. , … Francisco, J. A. (2004). Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clinical Cancer Research, 10, 7063–7070. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , et al. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow, M. L. , Maloney, N. , Roland, J. , Guillen Navarro, M. J. , Easton, M. K. , Kitchen‐Goosen, S. M. , et al. (2016). Lurbinectedin inactivates the Ewing sarcoma oncoprotein EWS‐FLI1 by redistributing it within the nucleus. Cancer Research, 76, 6657–6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, S. J. , Mainwaring, P. , Price, T. , Millward, M. J. , Padrik, P. , Underhill, C. R. , et al. (2016). Phase I clinical trial of marizomib (NPI‐0052) in patients with advanced malignancies including multiple myeloma: Study NPI‐0052‐102 final results. Clinical Cancer Research, 22, 4559–4566. [DOI] [PubMed] [Google Scholar]
- Hirata, Y. , & Uemura, D. (1986). Halichondrins—Antitumor polyether macrolides from a marine sponge. Pure and Applied Chemistry, 58, 701–710. [Google Scholar]
- Howitz, K. T. , & Sinclair, D. A. (2008). Xenohormesis: Sensing the chemical cues of other species. Cell, 133, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, L. , Ying, J. , Zhang, M. , Qiu, X. , & Lu, Y. (2018). Antitumor potential of marine natural products: A mechanistic investigation. Anti‐Cancer Agents in Medicinal Chemistry, 18, 702–718. [DOI] [PubMed] [Google Scholar]
- Ji, H.‐F. , Li, X.‐J. , & Zhang, H.‐Y. (2009). Natural products and drug discovery. Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Reports, 10, 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez, C. (2018). Marine natural products in medicinal chemistry. ACS Medicinal Chemistry, 9, 959–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, S. , Gorfajn, B. , Faircloth, G. , & Scotto, K. W. (2000). Ecteinascidin 743, a transcription‐targeted chemotherapeutic that inhibits MDR1 activation. Proceedings of the National Academy of Sciences, 97, 6775–6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordheim, L. P. , Durantel, D. , Zoulim, F. , & Dumontet, C. (2013). Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nature Reviews. Drug Discovery, 12, 447–464. [DOI] [PubMed] [Google Scholar]
- Kanoh, K. , Kohno, S. , Asari, T. , Harada, T. , Katada, J. , Muramatsu, M. , et al. (1997). (−)‐Phenylahistin: A new mammalian cell cycle inhibitor produced by Aspergillus ustus . Bioorganic & Medicinal Chemistry Letters, 7, 2847–2852. [Google Scholar]
- Kanoh, K. , Kohno, S. , Katada, J. , Hayashi, Y. , Muramatsu, M. , & Uno, I. (1999). Antitumor activity of phenylahistin in vitro and in vivo . Bioscience, Biotechnology, and Biochemistry, 63, 1130–1133. [DOI] [PubMed] [Google Scholar]
- Kanzaki, A. , Takebayashi, Y. , Ren, X.‐Q. , Miyashita, H. , Mori, S. , Akiyama, S. , & Pommier, Y. (2002). Overcoming multidrug drug resistance in P‐glycoprotein/MDR1‐overexpressing cell lines by ecteinascidin 743. Molecular Cancer Therapeutics, 1, 1327–1334. [PubMed] [Google Scholar]
- Kawai, A. , Araki, N. , Naito, Y. , Ozaki, T. , Sugiura, H. , Yazawa, Y. , et al. (2017). Phase 2 study of eribulin in patients with previously treated advanced or metastatic soft tissue sarcoma. Japanese Journal of Clinical Oncology, 47, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, D.‐X. , Jiang, Y.‐Y. , & Zhang, H.‐Y. (2010). Marine natural products as sources of novel scaffolds: Achievement and concern. Drug Discovery Today, 15, 884–886. [DOI] [PubMed] [Google Scholar]
- Lallier, L. E. , McMeel, O. , Greiber, T. , Vanagt, T. , Dobson, A. D. W. , & Jaspars, M. (2014). Access to and use of marine genetic resources: Understanding the legal framework. Natural Product Reports, 31, 612‐616. [DOI] [PubMed] [Google Scholar]
- Lassman, A. B. , van den Bent, M. J. , Gan, H. K. , Reardon, D. A. , Kumthekar, P. , Butowski, N. , et al. (2019). Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR‐amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro‐Oncology, 21, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Cesne, A. (2018). Making the best of available options for optimal sarcoma treatment. Oncology, 95, 11–20. [DOI] [PubMed] [Google Scholar]
- Leal, J. , Martínez‐Díez, M. , García‐Hernández, V. , Moneo, V. , Domingo, A. , Bueren‐Calabuig, J. , et al. (2010). PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti‐tumour activity. British Journal of Pharmacology, 161, 1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester, D. , & Bergmann, W. (1941). Contributions to the study of marine products. VI. The occurrence of cetyl palmitate in corals. The Journal of Organic Chemistry, 6, 120–122. [Google Scholar]
- Li, L. H. , Timmins, L. G. , Wallace, T. L. , Krueger, W. C. , Prairie, M. D. , & Im, W. B. (1984). Mechanism of action of didemnin B, a depsipeptide from the sea. Cancer Letters, 23, 279–288. [DOI] [PubMed] [Google Scholar]
- Li, W. W. , Takahashi, N. , Jhanwar, S. , Cordon‐Cardo, C. , Elisseyeff, Y. , Jimeno, J. , … Bertino, J. R. (2001). Sensitivity of soft tissue sarcoma cell lines to chemotherapeutic agents: Identification of ecteinascidin‐743 as a potent cytotoxic agent. Clinical Cancer Research, 7, 2908–2911. [PubMed] [Google Scholar]
- Litaudon, M. , Hart, J. B. , Blunt, J. W. , Lake, R. J. , & Munro, M. H. G. (1994). Isohomohalichondrin B, a new antitumor polyether macrolide from the New Zealand deep‐water sponge Lissodendoryx sp. Tetrahedron Letters, 35, 9435–9438. [Google Scholar]
- Losada, A. , Muñoz‐Alonso, M. J. , García, C. , Sánchez‐Murcia, P. A. , Martínez‐Leal, J. F. , Domínguez, J. M. , et al. (2016). Translation elongation factor eEF1A2 is a novel anticancer target for the marine natural product plitidepsin. Scientific Reports, 6, e35100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, J. , Stock, D. , Jap, B. , Zwicki, P. , Baumeister, W. , & Huber, R. (1995). Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 angstrom resolution. Science, 268, 533–539. [DOI] [PubMed] [Google Scholar]
- Martín, M. J. , Coello, L. , Fernández, R. , Reyes, F. , Rodríguez, A. , Murcia, C. , et al. (2013). Isolation and first total synthesis of PM050489 and PM060184, two new marine anticancer compounds. Journal of the American Chemical Society, 135, 10164–10171. [DOI] [PubMed] [Google Scholar]
- Martinez‐Cruzado, L. , Tornin, J. , Rodriguez, A. , Santos, L. , Allonca, E. , Fernandez‐Garcia, M. T. , et al. (2017). Trabectedin and campthotecin synergistically eliminate cancer stem cells in cell‐of‐origin sarcoma models. Neoplasia, 19, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Díez, M. , Guillén‐Navarro, M. J. , Pera, B. , Bouchet, B. P. , Martínez‐Leal, J. F. , Barasoain, I. , et al. (2014). PM060184, a new tubulin binding agent with potent antitumor activity including P‐glycoprotein over‐expressing tumors. Biochemical Pharmacology, 88, 291–302. [DOI] [PubMed] [Google Scholar]
- Mateos, M. V. , Cibeira, M. T. , Richardson, P. G. , Prosper, F. , Oriol, A. , & la Rubia, J. de, et al. (2010). Phase II clinical and pharmacokinetic study of plitidepsin 3‐hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clinical Cancer Research, 16, 3260–3269. [DOI] [PubMed] [Google Scholar]
- Mateyak, M. K. , & Kinzy, T. G. (2010). eEF1A: Thinking outside the ribosome. The Journal of Biological Chemistry, 285, 21209–21213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre, K. , O'Shaughnessy, J. , Schwartzberg, L. , Glück, S. , Berrak, E. , Song, J. X. , et al. (2014). Phase 2 study of eribulin mesylate as first‐line therapy for locally recurrent or metastatic human epidermal growth factor receptor 2‐negative breast cancer. Breast Cancer Research and Treatment, 146, 321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metaxas, Y. , Cathomas, R. , Mark, M. , & von Moos, R. (2016). Combination of cisplatin and lurbinectedin as palliative chemotherapy in progressive malignant pleural mesothelioma: Report of two cases. Lung Cancer, 102, 136–138. [DOI] [PubMed] [Google Scholar]
- Miller, C. P. , Rudra, S. , Keating, M. J. , Wierda, W. G. , Palladino, M. , & Chandra, J. (2009). Caspase‐8 dependent histone acetylation by a novel proteasome inhibitor, NPI‐0052: A mechanism for synergy in leukemia cells. Blood, 113, 4289–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward, M. , Mainwaring, P. , Mita, A. , Federico, K. , Lloyd, G. K. , Reddinger, N. , et al. (2012). Phase 1 study of the novel vascular disrupting agent plinabulin (NPI‐2358) and docetaxel. Invest. New Drugs, 30, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Mita, M. M. , Spear, M. A. , Yee, L. K. , Mita, A. C. , Heath, E. I. , Papadopoulos, K. P. , et al. (2010). Phase 1 first‐in‐human trial of the vascular disrupting agent plinabulin (NPI‐2358) in patients with solid tumors or lymphomas. Clinical Cancer Research, 16, 5892–5899. [DOI] [PubMed] [Google Scholar]
- Monk, B. J. , Blessing, J. A. , Street, D. G. , Muller, C. Y. , Burke, J. J. , & Hensley, M. L. (2012). A phase II evaluation of trabectedin in the treatment of advanced, persistent, or recurrent uterine leiomyosarcoma: A gynecologic oncology group study. Gynecologic Oncology, 124, 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro, M. H. G. , Blunt, J. W. , Dumdei, E. J. , Hickford, S. J. H. , Lill, R. E. , Li, S. , et al. (1999). The discovery and development of marine compounds with pharmaceutical potential. Journal of Biotechnology, 70, 15–25. [DOI] [PubMed] [Google Scholar]
- Nalda‐Molina, R. , Valenzuela, B. , Ramon‐Lopez, A. , Miguel‐Lillo, B. , Soto‐Matos, A. , & Perez‐Ruixo, J. J. (2009). Population pharmacokinetics meta‐analysis of plitidepsin (Aplidin) in cancer subjects. Cancer Chemotherapy and Pharmacology, 64, 97–108. [DOI] [PubMed] [Google Scholar]
- Newman, D. J. , & Cragg, G. M. (2017). Current status of marine‐derived compounds as warheads in anti‐tumor drug candidates. Marine Drugs, 15, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, B. , Lloyd, G. K. , Miller, B. R. , Palladino, M. A. , Kiso, Y. , Hayashi, Y. , et al. (2006). NPI‐2358 is a tubulin‐depolymerizing agent: In‐vitro evidence for activity as a tumor vascular‐disrupting agent. Anti‐Cancer Drugs, 17, 25–31. [DOI] [PubMed] [Google Scholar]
- Oba, T. , & Ito, K. I. (2018). Combination of two anti‐tubulin agents, eribulin and paclitaxel, enhances anti‐tumor effects on triple‐negative breast cancer through mesenchymal‐epithelial transition. Oncotarget, 9, 22986–23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Cearbhaill, R. E. , Wolfer, A. , Disilvestro, P. , O'Malley, D. M. , Sabbatini, P. , Shohara, L. , et al. (2018). 945PA phase I/II study of chemo‐immunotherapy with durvalumab (durva) and pegylated liposomal doxorubicin (PLD) in platinum‐resistant recurrent ovarian cancer (PROC). Annals of Oncology, 29, mdy285.153 [Google Scholar]
- Ocio, E. M. , Mateos, M.‐V. , Prosper, F. , Martin, J. , Rocafiguera, A. O. , Jarque, I. , et al. (2016). Phase I study of plitidepsin in combination with bortezomib and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Journal of Clinical Oncology, 34, 8006–8006. [Google Scholar]
- Okouneva, T. , Azarenko, O. , Wilson, L. , Littlefield, B. A. , & Jordan, M. A. (2008). Inhibition of centromere dynamics by eribulin (E7389) during mitotic metaphase. Molecular Cancer Therapeutics, 7, 2003–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, W. B. , Waud, W. R. , & Secrist, J. A. (2015). Thiarabine, 1‐(4‐Thio‐β‐D‐arabinofuranosyl) cytosine. A deoxycytidine analog with excellent anticancer activity. Current Medicinal Chemistry, 22, 3881–3896. [DOI] [PubMed] [Google Scholar]
- Pera, B. , Barasoain, I. , Pantazopoulou, A. , Canales, A. , Matesanz, R. , Rodriguez‐Salarichs, J. , et al. (2013). New interfacial microtubule inhibitors of marine origin, PM050489/PM060184, with potent antitumor activity and a distinct mechanism. ACS Chemical Biology, 8, 2084–2094. [DOI] [PubMed] [Google Scholar]
- Perez, E. A. , Hillman, D. W. , Fishkin, P. A. , Krook, J. E. , Tan, W. W. , Kuriakose, P. A. , et al. (2005). Phase II trial of dolastatin‐10 in patients with advanced breast cancer. Investigational New Drugs, 23, 257–261. [DOI] [PubMed] [Google Scholar]
- Perez, J. M. T. , Leary, A. , Besse, B. , Castellano, D. E. , Ponce Aix, S. , Arrondeau, J. , et al. (2018). Efficacy and safety of lurbinectedin (PM1183) in small cell lung cancer (SCLC): Results from a phase 2 study. Journal of Clinical Oncology, 36, 8570–8570. [Google Scholar]
- Pérez‐López, M. E. , Curiel, T. , Gómez, J. G. , & Jorge, M. (2007). Role of pegylated liposomal doxorubicin (Caelyx) in the treatment of relapsing ovarian cancer. Anti‐Cancer Drugs, 18, 611–617. [DOI] [PubMed] [Google Scholar]
- Pettit, G. R. (1997). The dolastatins. Fortschritte der Chemie Organischer Naturstoffe, 70, 1–79. [DOI] [PubMed] [Google Scholar]
- Pettit, G. R. , Day, J. F. , Hartwell, J. L. , & Wood, H. B. (1970). Antineoplastic components of marine animals. Nature, 227, 962–963. [DOI] [PubMed] [Google Scholar]
- Pettit, G. R. , Herald, C. L. , Boyd, M. R. , Leet, J. E. , Dufresne, C. , Doubek, D. L. , et al. (1991). Antineoplastic agents. 219. Isolation and structure of the cell growth inhibitory constituents from the western Pacific marine sponge Axinella sp. Journal of Medicinal Chemistry, 34, 3339–3340. [DOI] [PubMed] [Google Scholar]
- Pettit, G. R. , Kamano, Y. , Herald, C. L. , Tuinman, A. A. , Boettner, F. E. , Kizu, H. , et al. (1987). The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. Journal of the American Chemical Society, 109, 6883–6885. [Google Scholar]
- Pettit, G. R. , Tan, R. , Williams, M. D. , Tackett, L. , Schmidt, J. M. , Cerny, R. L. , et al. (1993). Isolation and structure of phakellistatin 2 from the eastern Indian Ocean marine sponge Phakellia carteri . Bioorganic & Medicinal Chemistry Letters, 3, 2869–2874. [Google Scholar]
- Pfisterer, J. , Plante, M. , Vergote, I. , du Bois, A. , Hirte, H. , Lacave, A. J. , et al. (2006). Gemcitabine plus carboplatin compared with carboplatin in patients with platinum‐sensitive recurrent ovarian cancer: An intergroup trial of the AGO‐OVAR, the NCIC CTG, and the EORTC GCG. Journal of Clinical Oncology, 24, 4699–4707. [DOI] [PubMed] [Google Scholar]
- Pommier, Y. , Kohlhagen, G. , Bailly, C. , Waring, M. , Mazumder, A. , & Kohn, K. W. (1996). DNA sequence‐ and structure‐selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the caribbean tunicate Ecteinascidia turbinata . Biochemistry, 35, 13303–13309. [DOI] [PubMed] [Google Scholar]
- Pompili, L. , Leonetti, C. , Biroccio, A. , & Salvati, E. (2017). Diagnosis and treatment of ALT tumors: Is trabectedin a new therapeutic option? Journal of Experimental & Clinical Cancer Research, 36, e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts, B. C. , Albitar, M. X. , Anderson, K. C. , Baritaki, S. , Berkers, C. , Bonavida, B. , et al. (2011). Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Current Cancer Drug Targets, 11, 254–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puyo, S. , Montaudon, D. , & Pourquier, P. (2014). From old alkylating agents to new minor groove binders. Critical Reviews in Oncology/Hematology, 89, 43–61. [DOI] [PubMed] [Google Scholar]
- Ribrag, V. , Caballero, D. , Ferme, C. , Zucca, E. , Arranz, R. , Briones, J. , et al. (2013). Multicenter phase II study of plitidepsin in patients with relapsed/refractory non‐Hodgkin's lymphoma. Haematologica, 98, 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, P. G. , Zimmerman, T. M. , Hofmeister, C. C. , Talpaz, M. , Chanan‐Khan, A. A. , Kaufman, J. L. , et al. (2016). Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI‐0052‐101 Part 1. Blood, 127, 2693–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart, K. L. , Gloer, J. B. , Hughes, R. G. , Renis, H. E. , McGovren, J. P. , Swynenberg, E. B. , et al. (1981). Didemnins: Antiviral and antitumor depsipeptides from a caribbean tunicate. Science, 212, 933–935. [DOI] [PubMed] [Google Scholar]
- Rinehart, K. L. , Holt, T. G. , Fregeau, N. L. , Stroh, J. G. , Keifer, P. A. , Sun, F. , et al. (1990). Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata . The Journal of Organic Chemistry, 55, 4512–4515. [Google Scholar]
- Rodrigues, T. , Reker, D. , Schneider, P. , & Schneider, G. (2016). Counting on natural products for drug design. Nature Chemistry, 8, 531–541. [DOI] [PubMed] [Google Scholar]
- Romano, M. , Frapolli, R. , Zangarini, M. , Bello, E. , Porcu, L. , Galmarini, C. M. , et al. (2013). Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). International Journal of Cancer, 133, 2024–2033. [DOI] [PubMed] [Google Scholar]
- Sakaguchi, K. , Nakatsukasa, K. , Koyama, H. , Kato, M. , Sakuyama, A. , Matsuda, T. , et al. (2018). Phase II clinical trial of first‐line eribulin plus trastuzumab for advanced or recurrent HER2‐positive breast cancer. Anticancer Research, 38, 4073–4081. [DOI] [PubMed] [Google Scholar]
- Sakai, R. , Rinehart, K. L. , Guan, Y. , & Wang, A. H.‐J. (1992). Additional antitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo. Proceedings of the National Academy of Sciences, 89, 11456–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, R. , Rinehart, K. L. , Kishore, V. , Kundu, B. , Faircloth, G. , Gloer, J. B. , et al. (1996). Structure−activity relationships of the didemnins 1,2. Journal of Medicinal Chemistry, 39, 2819–2834. [DOI] [PubMed] [Google Scholar]
- Sala, G. D. , Agriesti, F. , Mazzoccoli, C. , Tataranni, T. , Costantino, V. , & Piccoli, C. (2018). Clogging the ubiquitin‐proteasome machinery with marine natural products: Last decade update. Marine Drugs, 16, e467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria Nuñez, G. , Robles, C. M. G. , Giraudon, C. , Martinez‐Leal, J. F. , Compe, E. , Coin, F. , et al. (2016). Lurbinectedin specifically triggers the degradation of phosphorylated RNA polymerase II and the formation of DNA breaks in cancer cells. Molecular Cancer Therapeutics, 15, 2399–2412. [DOI] [PubMed] [Google Scholar]
- Schafer, E. S. , Rau, R. E. , Berg, S. , Liu, X. , Minard, C. G. , D'Adamo, D. , et al. (2018). A phase 1 study of eribulin mesylate (E7389), a novel microtubule‐targeting chemotherapeutic agent, in children with refractory or recurrent solid tumors: A children's oncology group phase 1 consortium study (ADVL1314). Pediatric Blood & Cancer, 65, e27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöffski, P. , Ray‐Coquard, I. L. , Cioffi, A. , Bui, N. B. , Bauer, S. , Hartmann, J. T. , et al. (2011). Activity of eribulin mesylate in patients with soft‐tissue sarcoma: A phase 2 study in four independent histological subtypes. The Lancet Oncology, 12, 1045–1052. [DOI] [PubMed] [Google Scholar]
- Schwartsmann, G. , da Rocha, A. B. , Berlinck, R. G. , & Jimeno, J. (2001). Marine organisms as a source of new anticancer agents. The Lancet Oncology, 2, 221–225. [DOI] [PubMed] [Google Scholar]
- Senter, P. D. , & Sievers, E. L. (2012). The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nature Biotechnology, 30, 631–637. [DOI] [PubMed] [Google Scholar]
- Shamai, S. , & Merimsky, O. (2017). Trabectedin for advanced soft tissue sarcoma: Ten‐year real‐life perspective. Journal of Clinical Oncology, 35, 11060–11060. [PubMed] [Google Scholar]
- Singh, A. V. , Bandi, M. , Raje, N. , Richardson, P. , Palladino, M. A. , Chauhan, D. , et al. (2011). A novel vascular disrupting agent plinabulin triggers JNK‐mediated apoptosis and inhibits angiogenesis in multiple myeloma cells. Blood, 117, 5692–5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SirDeshpande, B. V. , & Toogood, P. L. (1995). Mechanism of protein synthesis inhibition by didemnin B in vitro . Biochemistry, 34, 9177–9184. [DOI] [PubMed] [Google Scholar]
- Soares, D. G. , Machado, M. S. , Rocca, C. J. , Poindessous, V. , Ouaret, D. , Sarasin, A. , et al. (2011). Trabectedin and its C subunit modified analogue PM01183 attenuate nucleotide excision repair and show activity toward platinum‐resistant cells. Molecular Cancer Therapeutics, 10, 1481–1489. [DOI] [PubMed] [Google Scholar]
- Storz, U. (2015). Antibody‐drug conjugates: Intellectual property considerations. MAbs, 7, 989–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y. , Du, C. , Wang, B. , Zhang, Y. , Liu, X. , & Ren, G. (2014). Up‐regulation of eEF1A2 promotes proliferation and inhibits apoptosis in prostate cancer. Biochemical and Biophysical Research Communications, 450, 1–6. [DOI] [PubMed] [Google Scholar]
- Taamma, A. , Misset, J. L. , Riofrio, M. , Guzman, C. , Brain, E. , Lopez‐Lazaro, L. , … Cvitkovic, E. (2001). Phase I and pharmacokinetic study of ecteinascidin‐743, a new marine compound, administered as a 24‐h continuous infusion in patients with solid tumors. Journal of Clinical Oncology, 19, 1256–1265. [DOI] [PubMed] [Google Scholar]
- Takahashi, R. , Mabuchi, S. , Kawano, M. , Sasano, T. , Matsumoto, Y. , Kuroda, H. , et al. (2016). Preclinical investigations of PM01183 (lurbinectedin) as a single agent or in combination with other anticancer agents for clear cell carcinoma of the ovary. PLoS ONE, 11, e0151050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, A. R. , Rubin, E. H. , Walton, D. C. , Shuster, D. E. , Wong, Y. N. , Fang, F. , et al. (2009). Phase I study of eribulin mesylate administered once every 21 days in patients with advanced solid tumors. Clinical Cancer Research, 15, 4213–4219. [DOI] [PubMed] [Google Scholar]
- Towle, M. J. , Nomoto, K. , Asano, M. , Kishi, Y. , Yu, M. J. , & Littlefield, B. A. (2012). Broad spectrum preclinical antitumor activity of eribulin (Halaven®): Optimal effectiveness under intermittent dosing conditions. Anticancer Research, 32, 1611–1619. [PubMed] [Google Scholar]
- Tsueng, G. , Teisan, S. , & Lam, K. S. (2008). Defined salt formulations for the growth of Salinispora tropica strain NPS21184 and the production of salinosporamide A (NPI‐0052) and related analogs. Applied Microbiology and Biotechnology, 78, 827–832. [DOI] [PubMed] [Google Scholar]
- Tumini, E. , Herrera‐Moyano, E. , San Martín‐Alonso, M. , Barroso, S. , Galmarini, C. M. , & Aguilera, A. (2018). The antitumor drugs trabectedin and lurbinectedin induce transcription‐dependent replication stress and genome instability. Molecular Cancer Research, 17, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udwary, D. W. , Zeigler, L. , Asolkar, R. N. , Singan, V. , Lapidus, A. , Fenical, W. , et al. (2007). Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica . Proceedings of the National Academy of Sciences, 104, 10376–10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahdat, L. T. , Pruitt, B. , Fabian, C. J. , Rivera, R. R. , Smith, D. A. , Tan‐Chiu, E. , et al. (2009). Phase II study of eribulin mesylate, a halichondrin B analog, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. Journal of Clinical Oncology, 27, 2954–2961. [DOI] [PubMed] [Google Scholar]
- Vaishampayan, U. , Glode, M. , Du, W. , Kraft, A. , Hudes, G. , Wright, J. , et al. (2000). Phase II study of dolastatin‐10 in patients with hormone‐refractory metastatic prostate adenocarcinoma. Clinical Cancer Research, 6, 4205–4208. [PubMed] [Google Scholar]
- Van Kesteren, C. , de Vooght, M. M. M. , López‐Lázaro, L. , Mathôt, R. A. A. , Schellens, J. H. M. , Jimeno, J. M. , & Beijnen, J. H. (2003). Yondelis® (trabectedin, ET‐743): The development of an anticancer agent of marine origin. Anti‐Cancer Drugs, 14(7), 487–502. [DOI] [PubMed] [Google Scholar]
- Vera, M. D. , & Joullié, M. M. (2002). Natural products as probes of cell biology: 20 years of didemnin research. Medicinal Research Reviews, 22, 102–145. [DOI] [PubMed] [Google Scholar]
- Wahl, A. F. , Klussman, K. , Thompson, J. D. , Chen, J. H. , Francisco, L. V. , Risdon, G. , et al. (2002). The anti‐CD30 monoclonal antibody SGN‐30 promotes growth arrest and DNA fragmentation in Vitro and affects antitumor activity in models of Hodgkin's disease. Cancer Research, 62, 3736–3742. [PubMed] [Google Scholar]
- Wilks, S. , Puhalla, S. , O'Shaughnessy, J. , Schwartzberg, L. , Berrak, E. , Song, J. , et al. (2014). Phase 2, multicenter, single‐arm study of eribulin mesylate with trastuzumab as first‐line therapy for locally recurrent or metastatic HER2‐positive breast cancer. Clin. Breast Cancer, 14, 405–412. [DOI] [PubMed] [Google Scholar]
- Wright, A. E. , Forleo, D. A. , Gunawardana, G. P. , Gunasekera, S. P. , Koehn, F. E. , & McConnell, O. J. (1990). Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata . The Journal of Organic Chemistry, 55, 4508–4512. [Google Scholar]
- Yi, J. H. , Kim, S. J. , & Kim, W. S. (2017). Brentuximab vedotin: Clinical updates and practical guidance. Blood Research, 52(4), 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi, E. , Mabuchi, S. , Shimura, K. , Komura, N. , Kozasa, K. , Kuroda, H. , et al. (2018). Lurbinectedin (PM01183), a selective inhibitor of active transcription, effectively eliminates both cancer cells and cancer stem cells in preclinical models of uterine cervical cancer. Investigational New Drugs, 37, 818‐827. 10.1007/s10637-018-0686-6 [DOI] [PubMed] [Google Scholar]
- Yoshida, T. , Ozawa, Y. , Kimura, T. , Sato, Y. , Kuznetsov, G. , Xu, S. , et al. (2014). Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial–mesenchymal transition (EMT) to mesenchymal–epithelial transition (MET) states. British Journal of Cancer, 110, 1497–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes, A. (2011). CD30‐targeted antibody therapy. Current Opinion in Oncology, 23, 587–593. [DOI] [PubMed] [Google Scholar]
- Younes, A. , Yasothan, U. , & Kirkpatrick, P. (2012). Brentuximab vedotin. Nature Reviews. Drug Discovery, 11, 19–20. [DOI] [PubMed] [Google Scholar]
- Yu, M. , Kishi, Y. , & Littlefield, B. (2011). Discovery of E7389, a fully synthetic macrocyclic ketone analog of halichondrin B In Anticancer agents from natural products (Second ed.) (pp. 317–346). Boca Raton: CRC Press. [Google Scholar]
- Zewail‐Foote, M. , & Hurley, L. H. (1999). Ecteinascidin 743: A minor groove alkylator that bends DNA toward the Major Groove. Journal of Medicinal Chemistry, 42, 2493–2497. [DOI] [PubMed] [Google Scholar]
- Zhang, Q. , Feng, Y. , & Kennedy, D. (2017). Multidrug‐resistant cancer cells and cancer stem cells hijack cellular systems to circumvent systemic therapies, can natural products reverse this? Cellular and Molecular Life Sciences, 74, 777–780. [DOI] [PMC free article] [PubMed] [Google Scholar]