

Abstract
The hepatic endoplasmic reticulum (ER)-anchored cytochromes P450 (P450s) are mixed-function oxidases engaged in the biotransformation of physiologically relevant endobiotics as well as of myriad xenobiotics of therapeutic and environmental relevance. P450 ER-content and hence function is regulated by their coordinated hemoprotein syntheses and proteolytic turnover. Such P450 proteolytic turnover occurs through a process known as ER-associated degradation (ERAD) that involves ubiquitin-dependent proteasomal degradation (UPD) and/or autophagic-lysosomal degradation (ALD). Herein, on the basis of available literature reports and our own recent findings of in vitro as well as in vivo experimental studies, we discuss the therapeutic and pathophysiological implications of altered P450 ERAD and its plausible clinical relevance. We specifically (i) describe the P450 ERAD-machinery and how it may be repurposed for the generation of antigenic P450 peptides involved in P450 autoantibody pathogenesis in drug-induced acute hypersensitivity reactions and liver injury, or viral hepatitis; (ii) discuss the relevance of accelerated or disrupted P450-ERAD to the pharmacological and/or toxicological effects of clinically relevant P450 drug substrates; and (iii) detail the pathophysiological consequences of disrupted P450 ERAD, contributing to non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH) under certain synergistic cellular conditions.
Key words: Cytochromes P450, Endoplasmic reticulum-associated degradation, CHIP E3 ubiquitin ligase, gp78/AMFR E3 ubiquitin ligase, JNK1, AMPK1, Non-alcoholic fatty liver disease, Non-alcoholic steatohepatitis
Abbreviations: ACC1, acetyl-CoA carboxylase 1; ACC2, acetyl-CoA carboxylase 2; ACHE, acetylcholinesterase; ACOX1, acyl-CoA oxidase 1; ATF2, activating transcription factor 2; AP-1, activator protein 1; AdipoR1, gene of adiponectin receptor 1; ASK1, apoptosis signal-regulating kinase; AAA, ATPases associated with various cellular activities; gp78/AMFR, autocrine motility factor receptor; Atg14, autophagy-related 14; ALD, autophagic-lysosomal degradation; CBZ, carbamazepine; CHIP, carboxy-terminus of Hsc70-interacting protein; Mcp1, chemokine (C–C motif) ligand 1; Fas, fatty acid synthase; FOXO, forkhead box O; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Insig1, insulin-induced gene 1; IRS1, insulin receptor substrate 1; Il-1β, interleukin 1 β; Il-6, interleukin 6; INH, isoniazid; Lpl, lipoprotein lipase; 3MA, 3-methyladenine; Pgc1, peroxisome proliferator-activated receptor coactivator 1; shRNAi, shRNA interference; SREBP1c, sterol regulatory element binding transcription factor 1c; Scd1, stearoyl-coenzyme A desaturase; Tnf, tumor necrosis factor; Ub, ubiquitin; UPD, ubiquitin (Ub)-dependent proteasomal degradation
Graphical abstract
In this article, we discuss the physiological, therapeutic and pathophysiological relevance of cytochrome P450 protein degradation. Employing mice with genetic ablation of CHIP and gp78/AMFR E3 ubiquitin-ligases that stabilize hepatic CYP2E1, we document why this stabilization not always leads to non-alcoholic fatty liver disease: It requires concomitantly enhanced hepatic lipogenesis.

1. Introduction
The ubiquitous hemoproteins cytochromes P450 (P450s or CYPs; MW ∼50 kDa) are enzymes engaged both in synthetic and degradative functions critically important to the cellular integrity, physiology and defense1, 2. In the mammalian liver, the endoplasmic reticulum (ER)-anchored P450s function together with their redox partners cytochrome P450 reductase (CPR) and cytochrome b5 in the oxidative metabolism and elimination of numerous endobiotics (arachidonic acid, retinoic acid, steroids, vitamin D) as well as xenobiotics (pharmacological and recreational drugs, carcinogens, toxins and other foreign substances of dietary or environmental origin)1, 2. Hepatic P450 content and function can be modulated not only through substrate-mediated transcriptional and translational regulation as widely recognized3, 4, 5, but also, as now increasingly appreciated, through their enhanced or reduced proteolytic turnover6, 7, 8, 9, 10. The latter by altering hepatic P450 content also affects P450 function, thereby significantly influencing P450-dependent therapeutics and pathophysiology, the theme of this review.
2. P450 proteolytic turnover via ERAD
The hepatic ER-anchored P450s, in common with luminal and other membrane-integrated ER-proteins, incur proteolytic turnover via a vital physiological process termed “ER-associated degradation (ERAD)”11, 12, 13. This ERAD process is critical not just for protein quality control required to mitigate the unfolded protein response (UPR) following ER-stress and/or other cellular/oxidative stresses, but also for normal physiological ER-protein turnover11, 12, 13. Physiological P450 ERAD involves either ubiquitin (Ub)-dependent proteasomal degradation (UPD) or autophagic-lysosomal degradation (ALD) or both14, 15, 16, 17 (and references therein). Thus, while some P450s predominantly incur UPD, others ALD and yet others incur both14, 15, 16, 17 (and references therein). This basal physiological P450 ERAD is however greatly accelerated upon P450 inactivation9, 10, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24.
2.1. P450 ERAD via UPD
Hepatic P450s are typical Type I monotopic ER-membrane proteins with their N-terminal signal anchor integrated in the ER-membrane and their globular catalytic domain embedded in the ER-membrane while largely exposed to the cytosol25, 26. In spite of this common monotopic ER-topology, the individual lifespans of hepatic P450s vary with t1/2s ranging from 7 to 38 h14, 15, 16, 17. By some criteria27, P450s would qualify as long-lived proteins. The shorter t1/2s are attributed to the propensity of these P450s to engender injurious ROS during their futile catalytic cycling occurring in the absence of substrates. ROS oxidatively damage active sites and/or surface residues of the P450 catalytic domains. Mechanism-based inactivators can also structurally damage P450 catalytic domains. This dual feature of their specific ER-topology coupled with structural lesions in their cytosolic domains renders these P450s excellent substrates of ERAD/UPD11, 12, 13, 17, specifically the “ERAD-Cytosol (C)” pathway28, 29, 30. Accordingly, the dominant human liver/intestinal CYP3A4 and human liver CYP2E1 are found to be prototypic ERAD-C substrates. CYP3A4, accounting for ∼30% of the hepatic P450 content, is engaged in the metabolism of >50% of clinically relevant drugs and other xenobiotics1, 2, whereas CYP2E1 biotransforms clinically relevant drugs (acetaminophen, halothane), EtOH, and other xenobiotics, carcinogens (nitrosamines), endogenous acetone and fatty acids (arachidonic acid) to toxic/reactive intermediates1, 31, 32, 33. Substrates such as troleandomycin (TAO), ketoconazole, and clotrimazole that bind tightly to CYP3A prosthetic heme and/or active site stabilize the P450 protein, reduce its oxidative turnover and extend its t1/26, 34, 35. Furthermore, reduced futile oxidative cycling upon conditional deletion of the liver CPR gene elevates the constitutive hepatic CYP3A content in mice36, 37, plausibly via protein stabilization. Hepatic CYP2E1 similarly exhibits a high propensity for ROS generation and is labile in the absence of relevant substrates and/or inducers that stabilize the protein38, 39, 40. Additional P450s established as ERAD/UPD target substrates include CYPs 2B6 and 2C917.
Systematic dissection of the hepatic CYP3A and CYP2E1 ERAD-C process employing various in vivo and in vitro reconstituted eukaryotic systems has revealed that it involves initial post-translational phosphorylation by cytosolic protein kinases A and C of P450 Ser/Thr residues40, 41, 42, 43, 44, 45. These phosphorylated pSer/pThr residues are either contiguous or proximal to Asp/Glu residues on surface loops or disordered regions, engendering discrete acidic/negatively charged pSer/pThr/Asp/Glu surface clusters46. These P450 clusters serve as “linear or conformational phosphodegrons” for its molecular recognition by positively charged residues of the E2/E3 complexes46. Upon molecular recognition of P450 pSer/pThr/Asp/Glu clusters by the E3 Ub-ligases and their cognate E2 Ub-conjugating enzymes, P450-Lys residues vicinal to these clusters are ubiquitinated17, 44, 45, 46, 47. The polyubiquitinated P450s, in common with polytopic transmembrane and/or luminal ER-proteins48, 49, 50, 51, 52, are then extracted out of the ER-membrane into the cytosol by the p97 AAA ATPase-Npl4-Ufd1 chaperone complex19, 53, 54, and delivered to the 26S proteasome for subsequent degradation (Fig. 1)9, 18, 21.
Figure 1.

CYP3A4 ERAD-UPD. For details see the text.
2.1.1. P450-ubiquitination machinery
In ER-protein degradation, Ub-conjugation is essential for targeting proteins to the 26S proteasome55, 56, 57, 58, 59, 60 or to autophagic receptors61, 62. Because Ub is a ubiquitous, highly conserved, albeit inert ∼8.63 kDa molecule, its conjugation requires its ATP-dependent activation by one of the two Ub-activation E1-enzymes to form a reactive, high energy thioester, which is then relayed onto an active-site Cys-residue of one of the 27 or so Ub-conjugating E2-enzymes55, 56, 57, 58, 59, 60. The E2 may then relay this Ub-molecule one at a time onto the N-terminal α-NH2 or most commonly an ε-NH2 of a Lys-residue (or less commonly to Cys, Thr or Ser-residues63, 64, 65) of a target substrate corralled by the E3 Ub-ligase, or even to the Cys of a cognate E355, 56, 57, 58, 59, 60. A processive ordered chain consisting of 4–20 Ub-molecules is then elaborated, wherein the C-terminal G76COOH of the second Ub, is coupled via an isopeptide bond to the K48 of the first Ub in a signature herring bone pattern, involved in targeting the ubiquitinated protein to the 26S proteasome55, 56, 57, 58, 59, 60. Alternatively, the E2 can first elaborate the K48-linked polyUb-chain and then transfer it “en bloc” onto the E3-targeted substrate66, 67. Additionally, K63 (or any of the 7 internal Ub-Lys residues) can also be similarly targeted, resulting in a differently configured Ub-chain, with other cellular functional roles68. Apparently, such chain-processing is not mutually exclusive, as some P450s exhibit both K48- and K63-Ub-linkages, albeit possibly elaborated on different P450 Lys-residues17. E3 Ub-ligases play a critical role in protein ubiquitination, as they not only foster interactions of E2s with target proteins, but also confer exquisite substrate specificity through recognition of specific structural determinant(s) or degron(s), thereby preempting promiscuous 26S proteasomal attacks on cellular proteins.
2.1.2. E2/E3 complexes involved in P450 ERAD
In vitro functional reconstitution studies17, 44, 45, 46, 47, 69, 70 of E1/E2/E3-mediated CYP3A4 and CYP2E1 ubiquitination have identified UbcH5a/Hsc70/Hsp40/CHIP and UBC7/AMFR/gp78 complexes as two relevant E2/E3 systems in CYP3A4 and CYP2E1 ubiquitination:
-
(i)
CHIP (carboxy-terminus of Hsc70-interacting protein), a cytoplasmic Hsc70-cochaperone, functions with its cognate UbcH5a E2 and Hsc70/Hsp40 co-chaperones in substrate ubiquitination71, 72, 73, 74, 75. CHIP contains a catalytic U-Box with a cross-brace structure, resembling the cross-brace structure of the RING (really interesting new gene) finger, albeit lacking the canonical Zn-binding His and Cys residues75. Instead, it folds through salt-bridges and hydrophobic interactions. CHIP also contains a classical N-terminal tetratricopeptide (TPR) domain for Hsc70/Hsp40 or Hsp90 recruitment. CHIP was thought to largely ubiquitinate inactive, unfolded proteins corralled by Hsp70 upon exposure of their hydrophobic residues76. However, CHIP ubiquitinates native P450 proteins in vitro, albeit less extensively than inactivated P450s, and Chip-knockdown or knockout (KO) results in the stabilization of functionally active P450s, suggesting that it also targets native P450s.
-
(ii)
gp78/AMFR (autocrine motility factor receptor) is a polytopic, transmembrane cell surface77, as well as an ER-integral protein with a C-terminal cytoplasmic domain (309–643 residues) containing a RING-finger and other subdomains critical to its recruitment of its cognate UBC7/Ube2g2 E2 and catalytic E3-ligase-mediated ubiquitination function78, 79, 80, 81. Its cytosolic domain is composed of the following subdomains: a RING-finger (RF), an oligomerization site (OS) to enable the recruitment of multiple E2s in sufficiently close proximity to elaborate the Ub-chain, a coupling of Ub to ER-degradation (Cue)-domain for recognition of the E2-elaborated polyUb chain, a G2BR-domain whose binding of its cognate E2 (UBC7 or Ube2g2) significantly enhances the E2's affinity for the RF and hence unloading of activated Ub79, 81, as well as a C-terminal VCP-interaction motif (VIM) for recruitment of p97/VCP-AAA ATPase50 to the ER-integrated polyubiquitinated target.
The individual roles of CHIP and gp78 in CYP3A and CYP2E1 ubiquitination and ERAD were verified upon lentiviral shRNA interference (shRNAi) analyses targeted individually against each of these E3s in cultured rat hepatocytes17, 70. Thus, upon CHIP-knockdown, CYP3A was stabilized largely as the parent (55 kDa) species along with a minor CYP3A fraction consisting of its high molecular mass (HMM) ubiquitinated species. Upon gp78 knockdown, CYP3A was largely found as its parent species along with a significant amount of its ubiquitinated species70. However, upon either E3-knockdown, the functionally active CYP3A fraction not only was proportional to the relative amount of the stabilized unmodified parent species, but also considerably greater than that in the corresponding non-targeting shRNA-treated control hepatocytes70, indicating that CYP3A-ERAD disruption could be therapeutically relevant. Furthermore, each system by itself was capable of CYP3A4-ubiquitination as determined upon in vitro reconstitution studies of each individual CYP3A4 E2/E3-ubiquitination system46. However, CYP3A4 ubiquitination was both greatly accelerated and magnified when both E2/E3 systems were present simultaneously46. The CYP3A4 ubiquitination profile observed upon sequential introduction of each E2/E3 in the reconstituted system suggested that UbcH5a/CHIP most likely serves as the E3, with UBC7/gp78 serving as the E4 involved in the elongation of CYP3A4 polyUb chains46.
2.1.3. P450 ER-extraction into the cytosol
ER luminal proteins and domains of polytopic ER-proteins shielded from the cytosolic ubiquitination machinery require retrotranslocation into the cytosol for their ubiquitination before their subsequent delivery to the 26S proteasome48, 49, 50, 51, 52, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96. This extraction from the ER-membrane is accomplished primarily by the p97/Npl4/Ufd1 chaperone complex, with subsequent dislocation into the cytosol potentially by the 19S proteasomal lid Rpt AAA ATPase subunits, as documented for HMG-CoA reductase97. p97, albeit incorrectly also known as VCP (valosin-containing protein), or Cdc48p (yeast), is an abundant cytosolic AAA ATPase (ATPases associated with various cellular activities) involved in multiple cellular functional processes98, 99, 100, 101, 102. p97 is a barrel-shaped homohexameric protein with each subunit structurally composed of two ATPase domains stacked on top of each other103, 104, 105. It functions as a heterotrimeric complex in concert with two additional heterodimeric adapters, Ufd1p and Npl4p, which bind to its N-terminal domain82, 83, 84, 85, 86, 89. These adapters, by engaging the polyUb chains of the target substrate, assist in the recruitment of the polyubiquitinated substrates by the p97-complex82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96. This heterotrimeric p97-complex was documented to be required for CYP3A UPD in Saccharomyces cerevisiae53. Through chemical (formaldehyde)-crosslinking, immune affinity-purification and LC–MS/MS analyses, its close cellular association with ubiquitinated CYP3A23 in cultured rat hepatocytes was also documented19, a finding subsequently verified by p97-targeted lentiviral shRNAi analyses in cultured rat hepatocytes54. Lentiviral shRNAi-elicited p97-knockdown led to the accumulation of parent and ubiquitinated CYP3A23 species that remained largely anchored to the ER-membrane, unlike those from control shRNA-treated hepatocytes that were largely extracted into the cytosol54. Besides UPD, p97 also functions in diverse cellular processes including homotypic membrane fusion, vesicular transport, and nonUPD-mediated degradation of cytosolic proteins98, 99, 100. Additionally, in yeast, Cdc48 (the p97 homolog) together with its other adapter p47 also reportedly functions in autophagosome biogenesis101, 102. Thus, p97 may also play a role in mammalian P450 ALD.
2.1.4. P450 proteasomal degradation
The “constitutive” 26S proteasome, a 2000 kDa multifunctional protease, is composed of a 20S proteolytic core capped at either or both ends by the 19S/PA700 regulatory subunit complex consisting of thirteen Rpn (non-ATPase) lid subunits including Ub-chain recognition/deubiquitinating Rpn subunits and six Rpt AAA ATPase base subunits for unfolding and threading the targeted protein into the 20S cavity106, 107, 108, 109, 110, 111, 112, 113, 114, 115. The 20S proteasomal core is composed of a stack of four concentric heptameric rings: The two innermost heptameric β-subunit rings are juxtaposed to each of the outer heptameric α-subunit rings, creating the 20S proteolytic core, lined by chymotrypsin-like (β5), peptidyl-glutamyl-peptide hydrolase (caspase-like; β1), and trypsin-like (β2)-subunit proteases within each β-subunit ring106, 107, 108, 109, 110, 111, 112, 113. These proteolytic β-subunits autocatalyze the cleavage of a short precursor peptide to expose a catalytically active N-terminal Thr-residue that participates in the isopeptide bond cleavage, and thus the target of irreversible proteasomal inhibition111. Each α-subunit ring consists of 7 different subunits, each with a uniquely defined N-terminal sequence110, 111, 112, 113, 116. The 20S inner cylindrical core has an 13–14 Å diameter orifice, incapable of accepting most folded proteins without prior unfolding. Furthermore, in its resting state, the 20S proteasome assumes a “closed gate” structure, wherein the N-terminal residues of α2, α3 and α4 subunits through H-bonds and van der Waals interactions seal the central aperture, thereby creating a closed α-antechamber at each end of the 20S proteasomal core that effectively sequesters the β-proteolytic subunits from the cytosolic solvent106, 107, 109, 110, 111, 112, 113, 114, 115, 116.
Upon delivery of an ubiquitinated protein, the 19S lid Rpn10 and Rpn13 subunits, strategically situated just above the 19S base, serve as receptors to tether its Ub-chain, enabling the Rpn11 deubiquitinase to cleave off the substrate-attached proximal Ub-molecule of the tethered chain, thereby detaching the Ub-chain109, 110, 111, 112, 113. The chain is subsequently dismantled by the 19S deubiquitylating cysteine proteases (Ubp6 and UCH37) and the Ub-molecules returned to the cellular Ub-pool for recycling110, 111, 112, 113. The 19S base Rpt AAA ATPases then unfold the deubiquitinated protein for insertion into the 20S core for degradation110, 111, 112, 113, 114, 115. The substrate proteins are generally degraded into peptide fragments of 2–3, 8–10 (most commonly) and 20–30 residues long, depending on the rate of proteasomal degradation, the target protein length and its dwelling time within the 20S proteasomal core, controlled by the opening and closure of its central aperture110, 111, 112, 113.
Several reversible and/or irreversible 20S proteasomal inhibitors [MG132, MG262, epoxomicin, lactacystin, vinylsulfone, PS-341 (bortezomib/velcade), carfilzomib, ixazomib, marizomib and oprazomib], are available and used as diagnostic markers of ERAD/UPD, although they cannot distinguish 20S from 26S proteasomal degradation117, 118, 119, 120, 121. Some of these are FDA-approved and in clinical use in multiple myelomas and other hematologic cancers121. The irreversible inhibitors such as lactacystin and epoxomicin target the proteolytic β-subunit N-terminal Thr-residue118, 119, 120. These inhibitors are not all equally potent and differentially inhibit each pair of the three 20S proteasomal proteases. Because these inhibitors act at a step post protein-ubiquitination, assessment of their inhibitory potential requires monitoring the stabilization of both the parent protein substrate and its HMM ubiquitinated species.
The naked 20S proteasomal core is believed to be the most abundant cellular species122. It is reportedly capable of degrading some oxidized, nonubiquitinated proteins in the absence of ATP, by itself or with the assistance of an alternate cytosolic 11S/PA28α,β proteasomal activator122, 123, 124. Antigen-processing cells also generate a specialized variant of the constitutive 26S proteasome termed the immunoproteasome or i-proteasome, whose assembly is triggered by proinflammatory cytokines, interferon-γ, TNFα, and/or oxidative stress122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132. The constitutive 20S proteasomal β1-, β2-and β5-subunits are substituted in the i-proteasome with the inducible iβ1 (LMP2), iβ2 (MECL1) and iβ5 (LMP7)-subunits that are relatively much more efficient in generating peptides competent in binding the MHC class I complex122, 127, 128, 129, 130, 131. It too can be activated by 11S/PA28α,β and is efficient in degrading oxidized/nonubiquitinated proteins that may be relevant to the generation of pathogenic “P450-autoantibodies”, see below. While all these cellular proteasomal species co-exist, asymmetric “hybrid” proteasomes also exist with a 19S activator at one end of the 20S and an 11S/PA28α,β at the other, i.e., 19S–20S–11S complex that is more efficient in tri- and tetrapeptide hydrolyses than the 26S proteasome133, 134.
Although the central pore of the naked 20S proteasome or the 11S–20S proteasomal complex is incapable of accepting substrates of the globular size of P450s, it can more readily accept oxidized proteins than the 26S species123. This is so, because upon oxidation, the proteins are chemically modified and conformationally altered so as to expose hydrophobic domains that are recognized by the 20S proteasome, promoting the opening of the α-subunit antechamber aperture for insertion of the oxidized protein into the inner proteolytic β-subunit core. Several lines of evidence support a role for the 11S–20S proteasome complex rather than the 26S complex in the degradation of oxidized proteins: (i) oxidized proteins do not require ATP or Ub for their selective 20S proteasomal degradation, and upon size-subfractionation generally do not segregate with the pool of ubiquitinated proteins123; (ii) oxidative stress triggers the association of the Ecm29-protein scaffold with the 19S-activator with subsequent Hsp70-chaperoned 19S-dissociation from the 26S proteasome to free up the 20S core for selective oxidized protein degradation135; (iii) cellular Hsp70 is induced upon oxidative stress136; and (iv) oxidative stress inactivates the 19S cap subunits along with the SH-sensitive Ub-machinery, thus further disabling the cellular capacity for protein ubiquitination and minimizing 26S proteasome-catalyzed degradation of ubiquitinated proteins136, 137, 138. Similar oxidized protein handling by the 11S-immunoproteasome complex may generate peptides with hydrophobic C-termini required for MHC class I antigen presentation122. Although P450s are quite globular proteins, it is unclear to what extent their oxidation by incipiently generated ROS and/or insults inflicted upon generation of reactive drug metabolites would unfold them exposing hydrophobic domains that render them susceptible to 20S proteasomal/immunoproteasomal degradation. Normal physiological degradation of some native P450 isoforms however, apparently involves UPD as evidenced by (i) the marked accumulation of HMM ubiquitinated P450 species upon proteasomal inhibition9, 17, 18 and (ii) their significant functional stabilization upon impairment of their ubiquitination through shRNAi or genetic deletion of the participating E3-ligases70, 139, 140.
2.2. P450 ERAD via ALD
As noted above, in spite of their common type I ER-topology, not all P450s are typical ERAD-C substrates, some incur ALD17, 141, 142, 143, 144. Thus, CYP2B1 and CYP2C11, in cultured rat hepatocytes treated with ALD inhibitors [3-methyladenine (3MA)/NH4Cl] or heterologously expressed in a vacuolar (lysosomal) degradation-deficient S. cerevisiae strain, are stabilized relative to those expressed in wild-type (WT) or proteasomal-subunit defective strains17, 143, 144. CYP2E1 degradation, on the other hand, is biphasic, exhibiting a rapid phase (t1/2, 7 h) and a slow phase (t1/2, 37 h) presumed to reflect its ERAD via UPD and ALD, respectively7, 8, 14. Upon substrate [isoniazid (INH) or acetone] induction, it largely incurs ALD7, 17, 139. Functional inactivation of its catalytic partner CPR abolishes all redox-flux, thus minimizing oxidative damage and stabilizing CYP2E1 by prolonging its t1/238, 39. Notably, this dual CYP2E1 proteolytic mode is conserved upon its heterologous expression in yeast, even though P450 redox flux and consequent ROS-generating potential are greatly minimized by the poor yeast CPR content45. Thus, to what extent redox flux contributes to its UPD is unknown and remains to be determined. Specific UPD/ALD probes reveal that CYP2D6 and CYP2A5 also similarly turn over via both pathways17, 140.
2.2.1. Multiple lysosomal degradation pathways
Most relevant to P450 protein clearance is classical macroautophagy (ALD) involving target ubiquitination, multiple autophagy-related (ATG) genes including the autophagosome marker microtubule-associated protein light chain 3 (LC3), and autophagic cargo receptors, i.e., p62/SQSTM1 (Sequestosome) and NBR-1 (neighbor of Braca 1 gene)61, 62, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158. Briefly, upon induction of an isolation membrane (phagophore, derived from various sources including the ER), its surface phosphatidylethanolamine (PE) is conjugated to LC3-I by the ATG5/ATG12/ATG16 complex to form LC3-II, which recruits p62/NBR1 receptors via their LC3-interacting regions (LIR), along with their ubiquitinated protein cargo corralled through their Ub-associated (UBA) domains61, 62, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158. It then expands into a double membrane vesicle, the autophagosome that fuses with the lysosome for proteolytic digestion of its cargos (Fig. 2). Additional protein degradation routes to the lysosome also exist159, 160, 161, 162, 163, although it is unclear to what extent they transport P450 cargo. For instance, upon cessation of phenobarbital induction, CYP2B10 is known to be degraded along with its ER-membrane roost in an autophagic process known as ER-phagy or reticulophagy, involving p62 and/or possibly other reticulon adapters164, 165, 166, 167.
Figure 2.

P450 ERAD-ALD. For details see the text.
3. Therapeutic and pathophysiological implications of P450-turnover
3.1. Therapeutic relevance of P450 ERAD
Human hepatic P450s play a major role in the metabolism and elimination of ∼74% of therapeutically relevant drugs and are thus involved in many drug–drug interactions (DDIs) and drug-related toxicities1, 2. Thus, altered P450 ERAD/UPD can have a major influence on clinical therapeutics. At a very fundamental level, P450 ERAD/UPD regulates hepatic ER P450 content and function by determining the levels of nascent P450 molecules that are incorporated into the ER-membrane and thus functional, as most (∼70%) of freshly synthesized P450 molecules are cleared via ERAD/UPD18, 19. Furthermore, many chemical agents including dietary agents, drugs, and other xenobiotics that qualify as P450 substrates modulate hepatic P450 isoform content, diversity and/or function not only through increased protein synthesis/expression1, 2, 3, 4, 5, but also through protein stabilization, i.e., half-life (t1/2) prolongation6, 7, 8. By contrast, “suicide” substrates/inactivators dramatically shorten the t1/2s of certain P450s by accelerating their proteolytic turnover9, 10, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24. Such regulation of hepatic P450 content and hence function influences both the pharmacological effectiveness and the therapeutic duration of the drug response, thus contributing to the severity and the time course of many pharmacokinetic/pharmacodynamic DDIs35, 168, 169, 170, 171. The serious, life-threatening DDIs observed with the CYP3A4 mechanism-based inactivators grapefruit juice furanocoumarins and terfenadine, astemizole or amiodarone are some illustrative examples172, 173.
Altered ERAD susceptibility of P450 genetic variants also contributes to DDIs35 (and references therein). Indeed, ERAD via UPD plays a major role in the clinically relevant polymorphic expression of human liver P450s by eliminating various defective, misfolded and/or proteolytically susceptible P450 variants35 (and references therein). This quality control is critical to forestalling proteotoxic stress triggered by the intracellular accumulation of abnormal/aberrant and/or structurally damaged proteins. However, accelerated ERAD of some P450 allelic variants can also be beneficial. For instance, the prompt clearance of the CYP1B1.8∗ variant via ERAD is believed to confer a lower incidence of endometrial cancer in female carriers, putatively by obviating its capacity for carcinogenic bioactivation174.
On the other hand, stabilization of functionally active hepatic P450s upon disruption of their ERAD through genetic ablation in vivo of key participating E3 Ub-ligases can also influence therapeutic or toxic outcomes. Accordingly, hepatic stabilization of CYP2A5 upon liver-specific genetic ablation of gp78 E3-ligase in mice is shown to enhance their hepatic metabolism of drugs such as coumarin and nicotine140. Similar hepatic stabilization of CYP3A-enzymes in these mice can significantly enhance the bioactivation of the breast cancer chemotherapeutic prodrug tamoxifen to its pharmacologically active end-product endoxifen140. This finding suggests that short-term inhibition of E3-ligases participating in P450 ERAD may be therapeutically desirable for potentiation of P450-mediated bioactivation of cancer chemotherapeutic prodrugs140.
Hepatic CYP2E1 stabilization upon liver-specific genetic ablation of gp78 E3-ligase in mice enhances acetaminophen-metabolism to its reactive N-acetyl-p-quinoneimine, thereby predisposing the hepatocytes to drug-induced hepatotoxicity at relatively innocuous concentrations140. On the other hand, hepatic CYP2E1 stabilization through disruption of its ERAD via genetic CHIP-ablation contributes to pathogenic NASH (non-alcoholic steatohepatitis) syndrome139 (see below). Hepatic P450 ERAD is thus both therapeutically and pathophysiologically relevant.
3.2. Pathophysiologic relevance of P450-degradation
3.2.1. Pathogenesis of P450 autoantibodies
Drug-induced liver injury (DILI), hepatitis, acute hypersensitivity reactions and/or idiosyncrasies as well as viral infections are often clinically associated with detectable drug-induced serum immunoreactive P450 peptide autoantibodies175, 176, 177, 178, 179. Basal physiological protein ERAD via the constitutive proteasome is apparently too slow and inefficient a process for the generation of antigenic peptides128, 130. Thus, this route may be ineffectual for the generation of antigenic peptides from native P450s. Antigenic P450 peptides are most likely derived from structurally altered P450 proteins either upon direct drug-adduction (hapten generation) or drug-induced conformational alterations that predispose such modified P450 proteins to accelerated ERADe. Such ERAD would have to involve the immunoproteasomal proteolytic subunits in order to generate either P450 peptides containing hydrophobic C-termini, or P450 precursor peptides that upon further trimming by ancillary cellular peptidases expose their hydrophobic C-termini128, 130. Hydrophobic C-termini of peptides are important because they can readily fit into the groove of MHC class I molecules for antigenic presentation to cytotoxic T lymphocytes127, 128, 130, 131. Thus, liver injury upon chronic ingestion of alcohol (EtOH), or intake of aromatic anti-convulsants [carbamazepine (CBZ), phenytoin and phenobarbital], anesthetics (halothane), anti-tubercular drugs (isoniazid; INH), diuretics (tienilic acid), antihypertensives (dihydralazine), etc.180, 181, 182, 183, 184, 185, 186, 187, 188, 189, 190, 191, 192 are associated with circulating immunoreactive P450 peptide autoantibodies. Less clear is whether such P450 autoantibodies are the pathogenic cause of DILI, or the consequence of DILI-induced immunoproteasome. Nonetheless, once generated, they would be instrumental in further aggravating the severity of the autoimmune pathology. Several normally ER-anchored hepatic P450s are transported to the outer hepatic plasma membrane where they would readily react with circulating P450 autoantibodies with consequent hepatic parenchymal cytotoxicity and subsequent parenchymal cell death181, 188, 189.
In patients with severe INH-induced liver injury, not only serum autoantibodies to CYP2E1, CYP3A4 and CYP2C9 (the three major P450s that metabolize and/or interact with INH), but also serum antibodies to INH-adducted CYP2E1 and other INH-adducted non-P450 proteins are detected192. Interestingly, the antibodies detected against INH-adducted P450s and other proteins were generated against the parent isonicotinic acyl-moiety and not to its previously implicated reactive acetylhydrazine metabolite193. Similar serum antibodies to drug-modified and/or unmodified P450s and other native cellular proteins have also been detected in idiosyncratic liver injury upon halothane-exposure (anti-trifluoroacetyl-CYP2E1), aromatic anti-convulsant (anti-CYP3A), dihydralazine (anti-CYP1A2), tienilic acid (anti-tienilic acid-CYP2C9) and/or chronic alcohol intake (anti-acetaldehyde-CYP2E1 and anti-hydroxyethyl-CYP2E1)182, 183, 184, 185, 186, 187, 188, 189, 190, 191, 192. Anti-P450 autoantibodies (anti-liver kidney microsomal (LKM) Type I) targeting CYP2D6 are also detected in chronic autoimmune hepatitis and hepatitis C viral (HCV) infections194, 195, anti-CYP11A1 in type II autoimmune hepatic polyendocrine syndrome196, and anti-CYP17 and anti-CYP21 in Addison's disease197, 198. These observations underscore an etiological role of inflammatory and/or infectious conditions in immunoproteasome induction and subsequent P450 autoantibody pathogenesis.
We find it intriguing that serum antibodies to one of the CYP3A peptides detected in clinical CBZ-elicited acute hypersensitivity reactions187, corresponds to the same CYP3A4 helix K-region that is heme-modified upon cumene-hydroperoxide-elicited peroxidative CYP3A4 heme-fragmentation199. This suggests not only that peptides derived from hydrophobic P450-active sites may optimally fulfill the target pre-requisites for MHC class I antigenic presentation, but also that such active-site regions being strategically poised for attack by P450-generated ROS would be the primary targets for peptide oxidation and subsequent immunoproteasomal degradation.
3.2.2. Pathogenic implications of hepatic P450 ERAD-disruption through global Chip-ablation: CYP2E1-functional stabilization leading to overt NASH
Functional stabilization of hepatic CYPs 2E1 and 3A in Chip-KO mice (Fig. 3) was previously shown to elicit an age-dependent enhancement of oxidative stress as monitored with hepatic 15-F21-isoprostane and malondialdehyde (MDA) levels as indices of enhanced lipid peroxidation139. This enhancement was largely abolished by specific diagnostic functional inhibitors of these P450s139, with the residual amount being further quenched by Mito-TEMPO200, a mitochondria-targeted antioxidant probe that scavenges superoxide/alkyl radicals produced by various intramitochondrial oxidative processes, including those plausibly generated by intramitochondrial species of these P450s139. Such age-dependent enhancement of oxidative stress was also underscored by progressively increased levels of immunofluorescent 4-hydroxynonenal (HNE)-conjugated proteins in Chip-KO hepatocytes relative to age-matched WT-hepatocytes139. In parallel, starting at 2-months of age, Chip-KO hepatocytes exhibited oxidative stress-induced activation of the pathogenic ASK1-MKK4-JNK1-c-Jun-ATF2 and ASK1-MKK4-JNK1-c-Jun-AP-1 signaling pathways (Fig. 4), which were not only found to be CYP2E1-dependent, but also to remain persistently activated throughout the observation period, i.e., first 12 months of age139. Concurrently, as early as 2 months of age, the hepatic insulin-signaling pathway became mildly impaired as judged by the pJNK-mediated IRS1-S307-phosphorylation and the correspondingly attenuated IRS1-activation (via Y895-phosphorylation), coupled with ROS-mediated inactivation of Akt139 (Fig. 4). This early insulin-signaling impairment resulted in lowered insulin-induced hepatic Insig-1 and Insig-2 protein expression in Chip-KO relative to WT-controls (Fig. 5). The elevated hepatic SREBP-1c-protein levels upon Chip-KO relative to WT, coupled with the lowered ER-bound Insig levels (Fig. 5), most likely contributed to the increased ER-release of precursor SCAP:SREBP-1c-complex. Subsequent Golgi proteolytic processing and nuclear import of the transcriptionally active SREBP-1c resulted in the feed-forward autoregulation of Srebp-1c and upregulation of its target lipogenic genes (Fas, Scd-1, and Acc1), as documented previously139. Concomitantly, NF-κB-activation along with its transcriptional activation of inflammatory cytokine (Tnfα, Il-6, Mcp-1 and Il-β1) expression was also noted in Chip-KO livers139. However, in spite of all these early pathognomonic indices of NASH, corresponding histological analyses of 2-, and 4-month-old Chip-KO mouse livers versus age-matched WT mouse livers showed no major alterations in Hematoxylin and eosin (H&E)-based histology and no inflammatory signs139. Oil red O-staining showed only a progressive increase in hepatic microvesicular fat accumulation, albeit uncharacteristic of NASH, in 2- and 4-month-old Chip-KO animals versus corresponding WT-controls139.
Figure 3.

CYP2E1 and CYP3A levels in cultured hepatocytes. Primary cultured Chip-WT, Gp78-WT, Chip-KO and Gp78-KO mouse hepatocytes after initial attachment and 48 h-culture were treated with INH (1 mmol/L; CYP2E1) or Dex (10 μmol/L; CYP3A) for 3 days. Lysate proteins (5 μg) were subjected to immunoblotting as detailed previously140. Each lane in panels A & B corresponds to an individual cell culture. In panel C, fructose (20 mmol/L) was also included in some cell cultures. For detailed methodology see Supporting Information. These data are unpublished findings by Kwon & Correia, 2019.
Figure 4.

Concurrent activation and inactivation of three critical intersecting cellular signaling pathways in Chip-KO mouse hepatocytes: (i) Activation of the injurious CYP2E1-ROS-JNK-signaling pathway resulting in activation of ROS-elicited ASK1-JNK1-c-Jun- and ASK1-JNK1-ATF2-signaling pathways with consequent nuclear pc-Jun-pATF2-heterodimerization and the transcriptional up-regulation of pro-inflammatory factors/cytokines, and proapoptotic Ache-expression. The CYP2E1-dependent steps inhibited by its diagnostic inhibitor 4-methylpyrazole (4MP) are indicated by red stop-lines. (ii) Suppressed insulin-signaling via activated pJNK-1 phosphorylation of insulin receptor substrate 1 (IRS-1) at Ser307, thereby impairing insulin–insulin receptor (InsR)-mediated IRS-1 activation via Tyr895-phosphorylation and ushering insulin resistance. Furthermore, CYP2E1-generated ROS would also inactivate Akt via oxidation, thereby impairing AktS473-phosphorylation required for effective insulin-signaling. (iii) Beneficial activation of the adiponectin-AMPK-FOXO signaling pathways by virtue of liver kinase B1 (LKB1) and AMP-kinase (AMPK1α) being CHIP-substrates that are stabilized upon Chip-KO and CYP2E1-mediated oxidative stress that activates AMPK1α leading to the activation via C-terminal phosphorylation of the FOXO transcriptional factors. Intranuclear import of phosphorylated FOXOs and subsequent upregulation of energy, lipid metabolism, lipophagic, autophagic and oxidative stress resistance genes and elevation of hepatic adiponectin receptors AdipoR1/AdipoR2 genes206 that would enhance hepatic adiponectin signaling thus further feed-forwarding the ROS-counteracting adiponectin-AMPK-FOXO signaling, at the least during the initial 2–4 months of age. Additionally, enhanced adiponectin-AMPK-signaling also increases the activity of its diagnostic probe, acetyl CoA carboxylase 2 (ACC2), thereby attenuating malonyl CoA production, derepressing carnitine palmitoyltransferase 1 activity, and stimulating mitochondrial FA uptake and β-oxidation. With age progression (>9 months), the persistent oxidative stress, JNK1 activation, ATF2-mediated mitochondrial disruption, inflammation and central venous congestion trigger nuclear pyknosis and hepatocyte ballooning causing the adiponectin-AMPK-FOXO signaling to wane and vanish altogether within 12 months of age, thus enabling the manifestation of overt macrovesicular steatosis and other NASH-hallmarks.
Figure 5.

Relative levels of Insig 1 and Insig 2 and SREBP-1c proteins in INH-treated Chip-KO and Gp78-KO mouse hepatocytes and corresponding WT-controls: Hepatocytes were cultured for 48 h and then treated with INH for 3 days (as in Fig. 3). Lysate proteins (5 or 15 μg for Insig or SREBP-1c, respectively) were subjected to immunoblotting analyses with corresponding antibodies. P, precursor, N, nuclear SREBP-1c species identified on the basis of their corresponding molecular weights. For detailed methodology see Supporting Information. These data are unpublished findings by Kwon & Correia, 2019.
A PathScan Intracellular Signaling array provided an important clue to this apparent resistance of Chip-KO livers to NASH at this stage139. It revealed a two-fold activation of the energy sensor, AMP-protein kinase (AMPKα1) in these 2-month-old Chip-KO livers relative to corresponding age-matched controls that was also CYP2E1-dependent139. Upon further probing, the CHIP-substrate AMPK-activating kinase LKB1201 was indeed verified to be activated139. Moreover, AMPKα1 was itself discovered to be a CHIP-substrate, being stabilized and thus further activated upon Chip-ablation139. Hepatic AMPKα1 activation in turn phosphorylated the transcriptional factors FOXO1 and FOXO3202, 203, 204, 205, leading to the enhanced intranuclear import of their transcriptionally active phosphorylated species139. This not only protected the FOXO proteins from proteasomal degradation, but also triggered the transcriptional upregulation of their target genes (Fig. 4): (i) hepatic adiponectin receptors AdipoR1/R2206; (ii) oxidative-stress resistance [Mn-superoxide dismutase (Sod1), catalase (Cat), peroxisome-proliferator-activated receptor-γ-coactivator (Pgc)-1α and its target, acetyl CoA-oxidase (Acox-1)207, 208, 209, 210, 211, 212, 213]; and (iii) lipophagic (Atg14) and lipoprotein lipase (Lpl) genes139, 214, 215. Additionally, AMPKα1 activation most likely would not only promote mitochondrial fatty acid (FA) β-oxidation through ACC2-phosphorylation139, 208, 209, 210, 211, 212, 213, but also blunt SREB-1c-inducd lipogenesis through AMPK-mediated Insig protein phosphorylation that protects them from gp78-mediated ERAD216 (see below). This concerted upregulation of the hepatic adiponectin-LKB1-AMPK-FOXO1/3 signaling cascade (Fig. 4) and its beneficial anti-lipogenic and anti-oxidative stress responses coupled with pro-lipophagic effects and enhanced FA catabolism, thus was capable of effectively counteracting the concurrent pathognomonic NASH features217, 218, 219, 220, 221 prevalent in the 2- and 4-month-old Chip-KO livers139.
However, at 9 months of age, further aggravated hepatic insulin-resistance markedly reduced both hepatic Insig-1 and Insig-2 gene expression, with correspondingly enhanced SREB-1c-transcriptional upregulation of lipogenic genes and hepatic triglyceride (TG) accumulation139. This together with the sustained JNK1-mediated oxidative liver injury and further magnified NF-κB-activation and inflammatory cytokine expression cumulatively led to hepatocyte dropout139. Accordingly, Chip-KO livers but not the WT-controls exhibited hepatocyte ballooning and apoptotic cells with pyknotic nuclei causing the salutary hepatic adiponectin-LKB1-AMPK-FOXO-1/3 signaling activation begin to wane at 9 months of age, and then with progressively worsened hepatocyte ballooning and enhanced cytotoxicity, to disappear altogether by 12 months of age139. Not surprisingly, during this 9–12-month period, the earlier observed microvesicular hepatic steatosis yielded to the full-blown macrovesicular steatosis, characteristic of NASH139, 217, 218, 219, 220, 221. It is worth noting that such NASH induction in Chip-KO mouse livers did not require any specialized high fat/high sugar-enriched dietary regimens222, 223, as the mice were fed a standard laboratory chow-diet throughout their lifetime.
3.2.3. Hepatic P450 ERAD-disruption through liver-specific Gp78/Amfr-ablation: CYP2E1-functional stabilization but no overt NASH?
Mouse hepatic CYPs 2E1 and 3A are also functionally stabilized upon liver-specific Gp78-ablation (Fig. 3). The magnitude of CYP2E1 and CYP3A functional stabilization is comparable in Gp78-KO and Chip-KO mouse livers139, 140. However, in marked contrast to cultured Chip-KO hepatocytes, Gp78-KO hepatocytes upon culture retained much higher basal CYP2E1 levels (comparable to the INH-induced levels in cultured Chip-KO hepatocytes), which were further increased upon INH-induction (Fig. 3C). Yet, in contrast to the previous findings in age-matched INH-treated Chip-KO hepatocytes139, this was not associated with enhanced oxidative stress as indicated by the lack of substantially elevated hepatic MDA-levels, or even hepatic TG-accumulation in Gp78-KO hepatocytes relative to WT-controls at 2-months of age (Fig. 6). Additionally, in the course of defining the liver specific Gp78-KO mouse phenotype, preliminary SILAC-like (SILL) proteomic analyses of unlabeled Gp78-KO and stable isotope-labeled WT age/strain-matched livers revealed that in addition to the stabilization of several hepatic P450s (including CYP2E1 and CYPs 3A), several cytoplasmic enzymes in the fructose metabolic pathway were increased in Gp78-KO-liver [i.e., ketohexokinase (500%); 3-phosphoglycerokinase (200%–300%); phosphoglyceromutase (177%–250%); Supporting Information Table S1]. This increase would normally enhance the fructose-flux into hepatic FA syntheses and TG generation (Figure 7, Figure 8)224, 225, 226, 227. However, even upon exposure to a lipogenic high fructose (20 mmol/L)-medium that triggers significant FA- and TG-accumulation and cytotoxicity in WT-hepatocytes (Fig. 6A), Gp78-KO hepatocytes consistently exhibited significantly lesser susceptibility to hepatic FA- and TG-accumulation than corresponding WT-controls (Fig. 6A). These relative differences in FA- and TG-accumulation were maintained even upon further INH-mediated induction of CYP2E1 content (Fig. 6B). Furthermore, only minor relative increases in MDA levels were observed, indicative of a mild oxidative stress response, despite the relatively elevated basal CYP2E1 content in Gp78-KO hepatocytes, and its further elevation upon INH-induction (Fig. 6B).
Figure 6.

Relative cytotoxicity, oxidative stress and hepatic FA- and TG-accumulation in cultured WT and Gp78-KO mouse hepatocytes. (A) Hepatocytes from WT and Gp78-KO mice were cultured for 48 h with no P450 inducers (untreated). Hepatocytes were then treated with or without fructose (10 or 20 mmol/L) in the culture medium for the next 3 days. After 5 days of culture, their cytotoxicity was monitored in the culture medium by the Toxilight™ bioassay kit (Cat No. LT07-217, Lonza, Basel, Switzerland), as described in Ref. 19. Hepatic TG- and free FA-levels were determined by the TG colorimetric assay kit (Cat. No. 10010303, Cayman Chemical, Ann Arbor, MI, USA) and free FA quantification kit (Cat. No. MAK044-1KT, Sigma–Aldrich, St. Louis, MO, USA), respectively. (B) Hepatocytes from WT and Gp78-KO mice were cultured with INH treatment as described above. Hepatic TG- and free FA-levels were determined as in (A). MDA levels were monitored as oxidative stress indices by the TBARS assay kit (Cat. No. 10009055, Cayman Chemical). Some hepatocyte cultures were also treated with fructose at 20 mmol/L in the culture medium for the last 3 days of a 5 day-culture. To determine the functional role of CYP2E1, 4-MP (5 mmol/L) was also included along with INH for 2 h before harvest. Values are mean ± SD of three individual cultures. Statistical significance *P < 0.05, **P < 0.01, and ***P < 0.001 was determined relative to corresponding WT-values by the Student's t-test. For detailed methodology see Supporting Information. These data are unpublished findings by Kwon & Correia, 2019.
Figure 7.

Hepatic metabolism of circulating fructose into lipogenic pathways. Upon fructose uptake largely via the hepatic GLUT2 transporter, it is converted largely to fructose-1-P by ketohexokinase (KHK) and to a lesser extent to fructose-6-P by hexokinase (HK). Fructose-6-P is then phosphorylated to fructose-1,6-BiP by phosphofructose kinase (PFK). Fructose-1,6-BiP is then in turn converted by aldolases A & C to glyceraldehyde-3-P (GA-3-P), which via sequential enzymatic steps catalyzed by GA-3-P dehydrogenase, 3-phosphoglycerate kinase 1/2 complex, glyceromutase, and pyruvate kinase is then converted to pyruvate. Pyruvate is taken up into the mitochondria via the mitochondrial pyruvate carrier and fed into the Kreb's/Tricarboxylic acid (TCA) cycle. Fructose-1-P on the other hand, is converted to dihydroxyacetone (DHAP) and to GA via aldolases B and C. Both GA and DHAP can then be converted to glycerol-3-P (G-3-P), the precursor of triglyceride (TG) synthesis. Citrate derived from pyruvate in the TCA cycle is converted by ATP-citrate lyase/synthase (ACLY) to acetyl CoA, which is in turn converted into malonyl CoA by acetyl CoA carboxylase 1 (ACC1), and thence into fatty acid (FA; palmitic acid) synthesis catalyzed by FA-synthase (FAS). Malonyl CoA inhibits FA import into the mitochondria by blocking carnitine palmitoyl-transferase (CPT1a) thereby blocking FA-β-oxidation (β-Oxid). Palmitic acid (C16:O) can be elongated to stearic acid (C18:O), whereas these FAs can be desaturated to palmitoleic (C16:1) and oleic (C18:1) acids, respectively, via stearoyl CoA desaturase (SCD1). Thus, hepatic uptake of circulating fructose leads to increased hepatic FA- and TG-accumulation, under normal conditions. Although glucose uptake shares some of the same metabolic intermediates, a relevant difference is that ATP, citrate and acidic pH generated via the TCA cycle exert a critical allosteric block at the rate-limiting enzyme PHK, but not of KHK238. This would limit glucose or fructose-derived fructose-6-P from further contributing to the FA- and TG-accumulation. By contrast, without such a block, fructose could proceed unimpeded via fructose-1-P to keep contributing to hepatic FA- and TG-syntheses and accumulation as TG-macrovesicles that are normally packaged along with apolipoprotein B 100 (ApoB 100) and exported as VLDL/LDL. Glycerol-3-P acetyltransferase (GPAT); acylglycerol-3-P acetyltransferase AGPAT; elongation of very long fatty acids protein 6 (ELOV6); lipid phosphatidate phosphatase LIPIN1; diacylglycerol-O-acyltransferase, DGAT2; very low-density lipoprotein, (VLDL). Scheme adapted from Park et al.241 and Softic et al.242.
Figure 8.

Relative lipid accumulation in cultured hepatocytes from WT and Gp78-KO hepatocytes. Primary cultured hepatocytes were treated with or without fructose (10 or 20 mmol/L) for the last 3 days of a 5-day culture. Cells were fixed with 10% neutral buffered formalin. Lipids (red) and nuclei (blue) were stained with Oil red O and hematoxylin, respectively. For detailed methodology see Supporting Information. These data are unpublished findings by Kwon & Correia, 2019.
Several converging factors, we believe, synergistically contribute to the observed striking differences in CYP2E1-elicited lipogenic potential in Chip-KO versus Gp78-KO hepatocytes: (i) The differential effects of each E3-KO on hepatic Insig and SREBP-1c protein levels: upon INH-induction of CYP2E1, hepatic Insig 1 and Insig 2 proteins decline in Chip-KO hepatocytes due to the progressive hepatic insulin resistance139, whereas these proteins being bona fide gp78-substrates are markedly stabilized in Gp78-KO hepatocytes (Fig. 5). Moreover, SREBP-1c levels are greatly elevated in Chip-KO relative to WT-controlsf (Fig. 5), thus creating an imbalance in the Insig-dependent SREBP-1c-ER-retention capacity. Because of this, SREBP-1c is increasingly unleashed from the ER and transcriptionally activated to its nuclear form in the Chip-KO hepatocytes (Fig. 5) for its consequent transcriptional upregulation of target lipogenic genes139. By contrast, upon Gp78-KO, these Insig proteins are functionally stabilized leading to their greater ER-retention of the SCAP:SREBP-1c complex, with corresponding suppression of the prolipogenic SREBP-1c transcriptional activation (Fig. 9A). Furthermore, because the LKB1-AMPK-FOXO-signaling cascade is also activated upon Gp78-KO (Fig. 10), it would further augment this anti-lipogenic scenario through enhanced AMPK-mediated SREBP-1c S372-phosphorylation that reportedly blocks its proteolytic processing to a transcriptionally active species228. Thus, in contrast to Chip-KO mice, despite CYP2E1 functional stabilization, the Gp78-KO mice exhibit enhanced energy expenditure relative to WT controls, remain leaner and do not develop NASH, even upon aging and/or a high fat-high sucrose obesity-inducing diet229. (ii) Enhanced extrahepatic FA/TG secretion due to an ∼200% stabilization of apolipoprotein B 100 (Table S1), a documented gp78 substrate involved in hepatic LDL/VLDL-export230. (iii) Concomitant elevation of mitochondrial pyruvate carrier (567%; Table S1), diverting the fructose-derived pyruvate into the mitochondrial tricarboxylic cycle. (iv) Activation of hepatic adiponectin-LKB1-AMPK-FOXO1/3 signaling cascade with consequently enhanced PGC1 α-upregulation of energy metabolism as well as ACC2-stimulated mitochondrial FA β-oxidation211, 212, 213.
Figure 9.

Lipogenic enzymes and insulin-signaling in WT and Gp78-KO mouse hepatocytes upon fructose treatment. Relative levels of hepatic ACC1, FAS and SCD-1 enzymes in the FA-synthetic pathway (A) and Akt-S473/T308 phosphorylation (B) were monitored by immunoblotting analyses. Akt-S473/T308 activation also leads to N-terminal phosphorylation of the FOXO transcription factors, indicative of normal hepatic insulin-signaling function. Hepatocytes were cultured as described above with fructose treatment (20 mmol/L) for the last 3 days of a 5-day culture. Values shown are from two independent cultures. GAPDH immunoreactivity was included as the loading control in panels A & B. For detailed methodology see Supporting Information. These data are unpublished findings by Kwon & Correia, 2019.
Figure 10.
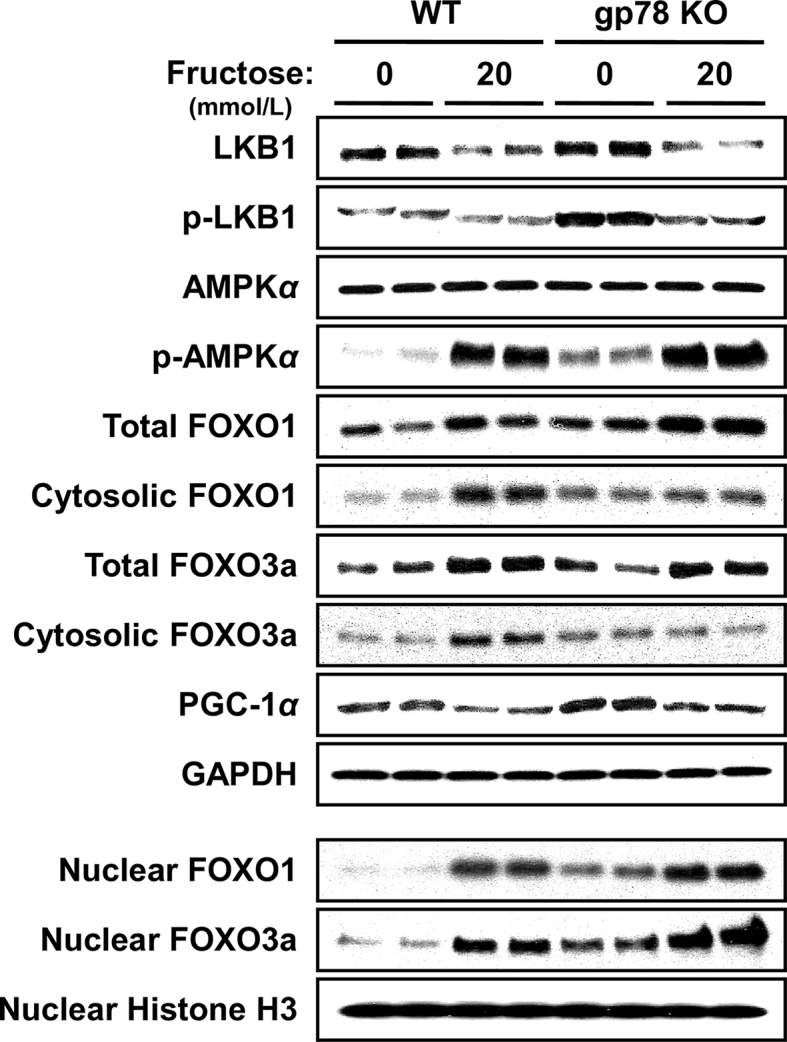
LKB1-AMPK1α-FOXO 1/3-signaling in hepatocytes from WT and Gp78-KO mouse hepatocytes upon fructose treatment. Hepatocytes were cultured as described above with or without fructose treatment (20 mmol/L) for the last 3 days of a 5-day culture. The various proteins were monitored by immunoblotting analyses as previously detailed139. GAPDH immunoreactivity was included as the loading control. Cells were fractionated into cytosolic and nuclear subfractions for FOXO-protein immunoblotting determinations, as previously detailed139. For detailed methodology see Supporting Information. These data are unpublished findings by Kwon & Correia, 2019.
Intriguingly, in spite of the comparable CYP2E1 content upon INH induction, hepatic MDA-levels were only minimally increased in Gp78-KO hepatocytes (Fig. 6B), in contrast to the marked MDA elevation observed in Chip-KO hepatocytes139. Such a very mild oxidative stress response in Gp78-KO precluded not only JNK-1 activation (our preliminary findings), but also oxidative derangement of insulin-signaling (Fig. 9B). This finding strongly suggests that hepatic CYP2E1-dependent ROS-elicited lipid peroxidation with consequent JNK1 activation and insulin resistance occur only when hepatic lipid is markedly elevated and available for peroxidation.
In summary, in concurrence with previous reports of Han et al.216, Li et al.228 and Jiang et al.229, we conclude that a major determinant of the differential responses observed upon comparable CYP2E1-functional stabilization in Chip-KO and Gp78-KO mouse livers is the ER-stability of the Insig-SCAP-SREBP-1c complex. Thus, liver-specific gp78-ablation coupled with the enhanced AMPK1α-phosphorylation of the Insig proteins protects them from ERAD216, resulting in their ER-stabilization (Fig. 5), and corresponding ER-sequestration of the SCAP:SREBP-1c complex216. However, this antilipogenic/NASH-countering response in Gp78-KO livers is plausibly magnified by the synergistic apolipoprotein B100-mediated enhanced hepatic TG export (Table S1) coupled with the activation of the adiponectin-LKB1-AMPK-FOXO1/3 signaling cascade (Fig. 10) and consequent PGC1α-upregulated energy metabolism and ACC2-mediated enhanced mitochondrial FA β-oxidation. Intriguingly, in marked contrast to the liver-specific Gp78-KO, global Gp78-ablation reportedly results in NASH and hepatocellular carcinoma231.
On the other hand, in Chip-KO mice, although the adiponectin-AMPK1α-FOXO1/3 signaling initially protects the livers (Fig. 3), this beneficial signaling is suppressed as progressively augmented CYP2E1-dependent oxidative injury and sustained JNK-signaling activation trigger hepatic injury and hepatocyte dropout with age139. This, coupled with the down regulation of the hepatic Insig proteins upon aggravated insulin resistance along with the concurrent elevation of SREBP-1c levels (Fig. 5), unleashed the transcriptional activation of SREBP-1c-induced hepatic lipogenesis that caused full-blown NASH within 12 months of age even in the absence of dietary obesity139.
4. Plausible therapeutic and pathophysiological relevance of altered P450 ERAD/ALD?
Regrettably, our foregoing discussion of the therapeutic and pathophysiological relevance of P450 ERAD has focused largely on their UPD pathway due both to the paucity and the complexity of the documented evidence in the literature on the physiological relevance of altered P450 ALD that preclude definitive conclusions of its clinical relevance. Presumably, its therapeutic implications should be very similar to those of altered P450 UPD, albeit largely dictated by the particular ALD-targeted P450 isoforms that are affected. Thus, in principle, ALD-targeted CYP2E1 given its critical role in acetaminophen-mediated hepatotoxicity and contribution to alcoholic liver disease could be a prime candidate. However, while the protective effects of rapamycin-induced ALD and the worsening effects of chloroquine-suppressed ALD on acetaminophen-elicited hepatotoxicity seemingly reflect an inherent role of CYP2E1 in acetaminophen bioactivation, they are exerted at a stage beyond CYP2E1-dependent metabolic activation232, 233, 234, and thus, quite independent of corresponding effects on CYP2E1-proteolytic stability. Furthermore, the genetic ALD-deficiency model (liver-specific Atg5-KO) reveals a very different picture. Thus, in contrast to chloroquine-suppressed ALD exacerbating acetaminophen-elicited liver injury232, liver-specific Atg5-KO mice are protected from liver injury triggered by a toxic 500 mg/kg acetaminophen dose234. This apparent discrepancy is due not only to reduced CYP2E1 content but also a persistent, compensatory activation of NRF2 that triggers the transcriptional activation of drug detoxification enzymes and GSH synthesis, as well as greater hepatocyte proliferation in the liver-specific Atg5-KO mice234. All these responses synergistically contribute towards the observed resistance of the liver-specific Atg5-KO mice to acetaminophen-elicited hepatotoxicity234. Interestingly, CYP2E1-stabilization upon chemically-induced autophagic disruption in HepG2 cells promotes, as expected, CYP2E1-dependent oxidative stress, mitochondrial dysfunction, JNK and p38 MAPK-activation, and consequent cytotoxicity235. However, given the compensatory features of genetically disrupted ALD, it is unclear whether similar responses would be observed in vivo in ALD-deficient mice. Thus, although clinical lysosomal disorders involving genetic deficiency of lysosomal proteases and/or their defective trafficking, or even defective lysosomal non-enzymatic and/or membrane proteins are indeed known236, 237, it remains to be determined whether these disorders would specifically affect the clinically/pathophysiologically relevant function of P450s that are largely targeted to ALD.
5. Conclusions
Normal ERAD is vital for the regulation of basal P450 homeostasis and physiologic function. Accelerated P450 ERAD by impairing the metabolism of its drug substrates can prolong their pharmacological duration and therapeutic responses, often triggering adverse DDIs. For drugs with narrow therapeutic windows this could result in undesirable drug-induced toxicities. However, P450 ERAD can also be pathophysiologically relevant. Proinflammatory conditions such as acute drug-induced hypersensitivity reactions and hepatitis as well as viral infections that release cytokines such as IFNγ and TNFα induce immunoproteasomal assembly by re-accessorizing the constitutive 26S proteasome: Its 19S cap is replaced by the 11S/PA28α,β activator and its 20S proteolytic core is equipped with alternate specialized proteolytic iβ-subunits responsible for generating MHC class I compatible peptides for antigenic presentation and consequent generation of pathogenic immunoreactive serum P450 autoantibodies associated with these clinical conditions.
On the other hand, disruption of hepatic P450 ERAD via UPD or ALD, resulting in functional stabilization of P450s can have grave therapeutic consequences either through curtailing the pharmacological response of a drug, increasing its adverse effects through bioactivation to toxic metabolites, and/or triggering clinically significant DDIs similar to those observed with P450-inducers. However, such P450 functional stabilization can also be therapeutically harnessed into increased bioactivation of a chemotherapeutic prodrug, particularly one with an otherwise intrinsically low therapeutic index. Furthermore, under certain conspiring inopportune circumstances that cannot be effectively counteracted, disruption of P450 ERAD with subsequent P450 functional stabilization can also have pathological consequences. Thus, CYP2E1 functional stabilization upon Chip-KO results in hepatic oxidative stress, JNK1-activation, insulin resistance, FA- and TG-accumulation and liver injury that contribute to NASH139. By contrast, despite comparable CYP2E1 functional stabilization, no significant hepatic FA- and TG-accumulation is observed upon Gp78-KO due to the ER-stabilization of the Insig proteins that prevents SREBP-1c-elicited transcriptional activation of hepatic lipogenesis. Thus, incipient P450-elicited oxidative stress and JNK1-activation coupled with a high intrahepatic lipogenic potential and sustained insulin resistance are the single most critical determinants of pathogenic NASH manifestations of disrupted P450 ERAD. The striking resemblance of these inherent features to those contributing to the currently explosive epidemic of type II diabetes/dietary obesity-induced nonalcoholic fatty liver disease (NAFLD)/NASH217, 218, 219, 220, 221 is inescapable. The documented beneficial role of the adiponectin-LKB1-AMPK-FOXO1/3 signaling activation in suppressing early NASH development in Chip-KO mice139 coupled with the recently documented AMPK1α-mediated anti-lipogenic effect via its ER-stabilization of the Insig proteins216 and impaired SREBP-1c transcriptional processing228, once again reinforce the concept that pharmacological activation of hepatic AMPK1α may indeed be a therapeutically beneficial NAFLD/NASH-counteractive strategy139, 238, 239.
Acknowledgments
We thank Mr. Chris Her for liver cell isolation at the UCSF Liver Center Core on Cell & Tissue Biology, supported by NIDDK Center Grant DK26743. This work was supported by NIH Grants GM44037 and DK26506 (USA) to Maria Almira Correia.
Author contributions
Dr. Kwon carried out all the experiments, designed some of the studies, wrote the methods and prepared many of the figures and figure legends, and completed the proteomic analyses. Dr. Kim generated the Gp78-KO mouse and carried out most of the studies in Chip-KO, except those in Figure 3, Figure 5. Dr. Correia conceived the manuscript, designed the studies and wrote most of the discussion and prepared some of the schemes/figures and Table S1.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
IL-1-elicited degradation of NO-inactivated CYP2B proteins in rat hepatocytes occurs via immunoproteasomal LMP2 subunit but not LMP7 subunit, as determined with selective inhibitors of each proteolytic subunit. Thus, it is conceivable that oxidized and/or drug-modified P450s could similarly be immunoproteasomal targets240.
This novel finding suggests that SREBP-1c is a CHIP-target.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.11.002.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Guengerich F.P. Human cytochrome P450 enzymes. In: Ortiz de Montellano P., editor. Cytochrome P450: structure, mechanism and biochemistry. Springer International Publishing; Heidelberg: 2015. pp. 523–785. [Google Scholar]
- 2.Correia M.A. Drug biotransformation. In: Katzung B.G., editor. Basic and clinical pharmacology. McGraw Hill & Lange; 2018. pp. 56–73. [Google Scholar]
- 3.Gonzalez F.J. The molecular biology of cytochrome P450s. Pharmacol Rev. 1988;40:243–288. [PubMed] [Google Scholar]
- 4.Gonzalez F.J., Liu S.Y., Yano M. Regulation of cytochrome P450 genes: molecular mechanisms. Pharmacogenetics. 1993;3:51–57. doi: 10.1097/00008571-199302000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Gotoh S., Ohno M., Yoshinari K., Negishi M., Kawajiri K. Nuclear receptor-mediated regulation of cytochrome P450 genes. In: Ortiz de Montellano P., editor. Cytochrome P450: structure, mechanism and biochemistry. Springer International Publishing; Heidelberg: 2015. pp. 787–812. [Google Scholar]
- 6.Watkins P.B., Wrighton S.A., Schuetz E.G., Maurel P., Guzelian P.S. Macrolide antibiotics inhibit the degradation of the glucocorticoid-responsive cytochrome P-450p in rat hepatocytes in vivo and in primary monolayer culture. J Biol Chem. 1986;261:6264–6271. [PubMed] [Google Scholar]
- 7.Song B.J., Veech R.L., Park S.S., Gelboin H.V., Gonzalez F.J. Induction of rat hepatic N-nitrosodimethylamine demethylase by acetone is due to protein stabilization. J Biol Chem. 1989;264:3568–3572. [PubMed] [Google Scholar]
- 8.Roberts B.J., Shoaf S.E., Jeong K.S., Song B.J. Induction of CYP2E1 in liver, kidney, brain and intestine during chronic ethanol administration and withdrawal: evidence that CYP2E1 possesses a rapid phase half-life of 6 hours or less. Biochem Biophys Res Commun. 1994;205:1064–1071. doi: 10.1006/bbrc.1994.2774. [DOI] [PubMed] [Google Scholar]
- 9.Correia M.A., Davoll S.H., Wrighton S.A., Thomas P.E. Degradation of rat liver cytochromes P450 3A after their inactivation by 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine: characterization of the proteolytic system. Arch Biochem Biophys. 1992;297:228–238. doi: 10.1016/0003-9861(92)90666-k. [DOI] [PubMed] [Google Scholar]
- 10.Schmiedlin-Ren P., Edwards D.J., Fitzsimmons M.E., He K., Lown K.S., Woster P.M. Mechanisms of enhanced oral availability of CYP3A4 substrates by grapefruit constituents. Decreased enterocyte CYP3A4 concentration and mechanism-based inactivation by furanocoumarins. Drug Metab Dispos. 1997;25:1228–1233. [PubMed] [Google Scholar]
- 11.Olzmann J.A., Kopito R.R., Christianson J.C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol. 2013;5:a013185. doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christianson J.C., Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Preston G.M., Brodsky J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem J. 2017;474:445–469. doi: 10.1042/BCJ20160582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Correia M.A. Cytochrome P450 turnover. Methods Enzymol. 1991;206:315–325. doi: 10.1016/0076-6879(91)06101-8. [DOI] [PubMed] [Google Scholar]
- 15.Correia M.A. Hepatic cytochrome P450 degradation: mechanistic diversity of the cellular sanitation brigade. Drug Metab Rev. 2003;35:107–143. doi: 10.1081/dmr-120023683. [DOI] [PubMed] [Google Scholar]
- 16.Correia M.A., Sadeghi S., Mundo-Paredes E. Cytochrome P450 ubiquitination: branding for the proteolytic slaughter?. Annu Rev Pharmacol Toxicol. 2005;45:439–464. doi: 10.1146/annurev.pharmtox.45.120403.100127. [DOI] [PubMed] [Google Scholar]
- 17.Kim S.M., Wang Y., Nabavi N., Liu Y., Correia M.A. Hepatic cytochromes P450: structural degrons and barcodes, posttranslational modifications and cellular adapters in the ERAD-endgame. Drug Metab Rev. 2016;48:405–433. doi: 10.1080/03602532.2016.1195403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H.F., Figueiredo Pereira M.E., Correia M.A. Cytochrome P450 3A degradation in isolated rat hepatocytes: 26S proteasome inhibitors as probes. Arch Biochem Biophys. 1999;365:45–53. doi: 10.1006/abbi.1999.1139. [DOI] [PubMed] [Google Scholar]
- 19.Faouzi S., Medzihradszky K.F., Hefner C., Maher J.J., Correia M.A. Characterization of the physiological turnover of native and inactivated cytochromes P450 3A in cultured rat hepatocytes: a role for the cytosolic AAA ATPase p97?. Biochemistry. 2007;46:7793–7803. doi: 10.1021/bi700340n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sohn D.H., Yun Y.P., Park K.S., Veech R.L., Song B.J. Post-translational reduction of cytochrome P450IIE by CCl4, its substrate. Biochem Biophys Res Commun. 1991;179:449–454. doi: 10.1016/0006-291x(91)91391-o. [DOI] [PubMed] [Google Scholar]
- 21.Roberts B.J. Evidence of proteasome-mediated cytochrome P-450 degradation. J Biol Chem. 1997;272:9771–9778. doi: 10.1074/jbc.272.15.9771. [DOI] [PubMed] [Google Scholar]
- 22.Tierney D.J., Haas A.L., Koop D.R. Degradation of cytochrome P450 2E1: selective loss after labilization of the enzyme. Arch Biochem Biophys. 1992;293:9–16. doi: 10.1016/0003-9861(92)90358-4. [DOI] [PubMed] [Google Scholar]
- 23.Lee C.M., Kim B.Y., Li L., Morgan E.T. Nitric oxide-dependent proteasomal degradation of cytochrome P450 2B proteins. J Biol Chem. 2008;283:889–898. doi: 10.1074/jbc.M708821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee C.M., Tripathi S., Morgan E.T. Nitric oxide-regulated proteolysis of human CYP2B6 via the ubiquitin-proteasome system. Free Radic Biol Med. 2017;108:478–486. doi: 10.1016/j.freeradbiomed.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Von Wachenfeldt C., Johnson E.F. Structures of eukaryotic cytochromes P450 enzymes. In: Ortiz de Montellano P., editor. Cytochrome P450: structure, mechanism and biochemistry. Plenum Press; New York: 1995. pp. 183–223. [Google Scholar]
- 26.von Wachenfeldt C., Richardson T.H., Cosme J., Johnson E.F. Microsomal P450 2C3 is expressed as a soluble dimer in Escherichia coli following modification of its N-terminus. Arch Biochem Biophys. 1997;339:107–114. doi: 10.1006/abbi.1996.9859. [DOI] [PubMed] [Google Scholar]
- 27.Zhao J., Zhai B., Gygi S.P., Goldberg A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U S A. 2015;112:15790–15797. doi: 10.1073/pnas.1521919112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahner A., Brodsky J.L. Checkpoints in ER-associated degradation: excuse me, which way to the proteasome?. Trends Cell Biol. 2004;14:474–478. doi: 10.1016/j.tcb.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 29.Taxis C., Hitt R., Park S.H., Deak P.M., Kostova Z., Wolf D.H. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem. 2003;278:35903–35913. doi: 10.1074/jbc.M301080200. [DOI] [PubMed] [Google Scholar]
- 30.Vashist S., Ng D.T. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cederbaum A.I. CYP2E1—biochemical and toxicological aspects and role in alcohol-induced liver injury. Mt Sinai J Med. 2006;73:657–672. [PubMed] [Google Scholar]
- 32.Bardag-Gorce F., French B.A., Nan L., Song H., Nguyen S.K., Yong H. CYP2E1 induced by ethanol causes oxidative stress, proteasome inhibition and cytokeratin aggresome (Mallory body-like) formation. Exp Mol Pathol. 2006;81:191–201. doi: 10.1016/j.yexmp.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Porubsky P.R., Meneely K.M., Scott E.E. Structures of human cytochrome P-450 2E1. Insights into the binding of inhibitors and both small molecular weight and fatty acid substrates. J Biol Chem. 2008;283:33698–33707. doi: 10.1074/jbc.M805999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eliasson E., Mkrtchian S., Halpert J.R., Ingelman-Sundberg M. Substrate-regulated, cAMP-dependent phosphorylation, denaturation, and degradation of glucocorticoid-inducible rat liver cytochrome P450 3A1. J Biol Chem. 1994;269:18378–18383. [PubMed] [Google Scholar]
- 35.Liao M., Kang P., Murray B.P., Correia M.A. Cytochrome P450 degradation and its clinical relevance. In: Lu C., Li A.P., editors. Enzyme inhibition in drug discovery & development. John Wiley & Sons; Hoboken: 2010. pp. 363–406. [Google Scholar]
- 36.Gu J., Weng Y., Zhang Q.Y., Cui H., Behr M., Wu L. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem. 2003;278:25895–25901. doi: 10.1074/jbc.M303125200. [DOI] [PubMed] [Google Scholar]
- 37.Henderson C.J., Otto D.M., Carrie D., Magnuson M.A., McLaren A.W., Rosewell I. Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J Biol Chem. 2003;278:13480–13486. doi: 10.1074/jbc.M212087200. [DOI] [PubMed] [Google Scholar]
- 38.Goasduff T., Cederbaum A.I. NADPH-dependent microsomal electron transfer increases degradation of CYP2E1 by the proteasome complex: role of reactive oxygen species. Arch Biochem Biophys. 1999;370:258–270. doi: 10.1006/abbi.1999.1399. [DOI] [PubMed] [Google Scholar]
- 39.Zhukov A., Ingelman-Sundberg M. Relationship between cytochrome P450 catalytic cycling and stability: fast degradation of ethanol-inducible cytochrome P450 2E1 (CYP2E1) in hepatoma cells is abolished by inactivation of its electron donor NADPH-cytochrome P450 reductase. Biochem J. 1999;340:453–458. [PMC free article] [PubMed] [Google Scholar]
- 40.Eliasson E., Mkrtchian S., Ingelman-Sundberg M. Hormone- and substrate-regulated intracellular degradation of cytochrome P450 (2E1) involving MgATP-activated rapid proteolysis in the endoplasmic reticulum membranes. J Biol Chem. 1992;267:15765–15769. [PubMed] [Google Scholar]
- 41.Korsmeyer K.K., Davoll S., Figueiredo-Pereira M.E., Correia M.A. Proteolytic degradation of heme-modified hepatic cytochromes P450: a role for phosphorylation, ubiquitination, and the 26S proteasome?. Arch Biochem Biophys. 1999;365:31–44. doi: 10.1006/abbi.1999.1138. [DOI] [PubMed] [Google Scholar]
- 42.Wang X., Medzihradszky K.F., Maltby D., Correia M.A. Phosphorylation of native and heme-modified CYP3A4 by protein kinase C: a mass spectrometric characterization of the phosphorylated peptides. Biochemistry. 2001;40:11318–11326. doi: 10.1021/bi010690z. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y., Liao M., Hoe N., Acharya P., Deng C., Krutchinsky A.N. A role for protein phosphorylation in cytochrome P450 3A4 ubiquitin-dependent proteasomal degradation. J Biol Chem. 2009;284:5671–5684. doi: 10.1074/jbc.M806104200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y., Guan S., Acharya P., Liu Y., Thirumaran R.K., Brandman R. Multisite phosphorylation of human liver cytochrome P450 3A4 enhances its gp78- and CHIP-mediated ubiquitination: a pivotal role of its Ser-478 residue in the gp78-catalyzed reaction. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Guan S., Acharya P., Koop D.R., Liu Y., Liao M. Ubiquitin-dependent proteasomal degradation of human liver cytochrome P450 2E1: identification of sites targeted for phosphorylation and ubiquitination. J Biol Chem. 2011;286:9443–9456. doi: 10.1074/jbc.M110.176685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y., Kim S.M., Trnka M.J., Liu Y., Burlingame A.L., Correia M.A. Human liver cytochrome P450 3A4 ubiquitination: molecular recognition by UBC7-gp78 autocrine motility factor receptor and UbcH5a-CHIP-Hsc70-Hsp40 E2-E3 ubiquitin ligase complexes. J Biol Chem. 2015;290:3308–3332. doi: 10.1074/jbc.M114.611525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pabarcus M.K., Hoe N., Sadeghi S., Patterson C., Wiertz E., Correia M.A. CYP3A4 ubiquitination by gp78 (the tumor autocrine motility factor receptor, AMFR) and CHIP E3 ligases. Arch Biochem Biophys. 2009;483:66–74. doi: 10.1016/j.abb.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bays N.W., Wilhovsky S.K., Goradia A., Hodgkiss-Harlow K., Hampton R.Y. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elkabetz Y., Shapira I., Rabinovich E., Bar-Nun S. Distinct steps in dislocation of luminal endoplasmic reticulum-associated degradation substrates: roles of endoplasmic reticulum-bound p97/Cdc48p and proteasome. J Biol Chem. 2004;279:3980–3989. doi: 10.1074/jbc.M309938200. [DOI] [PubMed] [Google Scholar]
- 50.Zhong X., Shen Y., Ballar P., Apostolou A., Agami R., Fang S. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem. 2004;279:45676–45684. doi: 10.1074/jbc.M409034200. [DOI] [PubMed] [Google Scholar]
- 51.Richly H., Rape M., Braun S., Rumpf S., Hoege C., Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 52.Ye Y., Shibata Y., Kikkert M., van Voorden S., Wiertz E., Rapoport T.A. Inaugural article: recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A. 2005;102:14132–14138. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liao M., Faouzi S., Karyakin A., Correia M.A. Endoplasmic reticulum-associated degradation of cytochrome P450 CYP3A4 in Saccharomyces cerevisiae: further characterization of cellular participants and structural determinants. Mol Pharmacol. 2006;69:1897–1904. doi: 10.1124/mol.105.021816. [DOI] [PubMed] [Google Scholar]
- 54.Acharya P., Liao M., Engel J.C., Correia M.A. Liver cytochrome P450 3A endoplasmic reticulum-associated degradation: a major role for the p97 AAA ATPase in cytochrome P450 3A extraction into the cytosol. J Biol Chem. 2011;286:3815–3828. doi: 10.1074/jbc.M110.186981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weissman A.M. Regulating protein degradation by ubiquitination. Immunol Today. 1997;18:189–198. doi: 10.1016/s0167-5699(97)84666-x. [DOI] [PubMed] [Google Scholar]
- 56.Weissman A.M. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–178. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 57.Pickart C.M., Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 58.Fang S., Weissman A.M. A field guide to ubiquitylation. Cell Mol Life Sci. 2004;61:1546–1561. doi: 10.1007/s00018-004-4129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pickart C.M., Eddins M.J. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 60.Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009;37:937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 61.Kim P.K., Hailey D.W., Mullen R.T., Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirkin V., McEwan D.G., Novak I., Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 63.Ishikura S., Weissman A.M., Bonifacino J.S. Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J Biol Chem. 2010;285:23916–23924. doi: 10.1074/jbc.M110.127936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang X., Herr R.A., Rabelink M., Hoeben R.C., Wiertz E.J., Hansen T.H. Ube2j2 ubiquitinates hydroxylated amino acids on ER-associated degradation substrates. J Cell Biol. 2009;187:655–668. doi: 10.1083/jcb.200908036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vosper J.M., McDowell G.S., Hindley C.J., Fiore-Heriche C.S., Kucerova R., Horan I. Ubiquitylation on canonical and non-canonical sites targets the transcription factor neurogenin for ubiquitin-mediated proteolysis. J Biol Chem. 2009;284:15458–15468. doi: 10.1074/jbc.M809366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li W., Tu D., Brunger A.T., Ye Y. A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature. 2007;446:333–337. doi: 10.1038/nature05542. [DOI] [PubMed] [Google Scholar]
- 67.Li W., Tu D., Li L., Wollert T., Ghirlando R., Brunger A.T. Mechanistic insights into active site-associated polyubiquitination by the ubiquitin-conjugating enzyme Ube2g2. Proc Natl Acad Sci U S A. 2009;106:3722–3727. doi: 10.1073/pnas.0808564106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kulathu Y., Komander D. Atypical ubiquitylation—the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- 69.Morishima Y., Peng H.M., Lin H.L., Hollenberg P.F., Sunahara R.K., Osawa Y. Regulation of cytochrome P450 2E1 by heat shock protein 90-dependent stabilization and CHIP-dependent proteasomal degradation. Biochemistry. 2005;44:16333–16340. doi: 10.1021/bi0515570. [DOI] [PubMed] [Google Scholar]
- 70.Kim S.M., Acharya P., Engel J.C., Correia M.A. Liver cytochrome P450 3A ubiquitination in vivo by gp78/autocrine motility factor receptor and C-terminus of Hsp70-interacting protein (CHIP) E3 ubiquitin ligases: physiological and pharmacological relevance. J Biol Chem. 2010;285:35866–35877. doi: 10.1074/jbc.M110.167189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ballinger C.A., Connell P., Wu Y., Hu Z., Thompson L.J., Yin L.Y. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–4545. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Connell P., Ballinger C.A., Jiang J., Wu Y., Thompson L.J., Hohfeld J. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 73.Murata S., Minami Y., Minami M., Chiba T., Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2:1133–1138. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang J., Ballinger C.A., Wu Y., Dai Q., Cyr D.M., Hohfeld J. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- 75.Patterson C., Hohfeld J. Molecular chaperones and the ubiquitin-proteasome system. In: Mayer J., Ciechanover A., Rechsteiner M., editors. vol. 2. Wiley-VCH Verlag GmbH & Co KGaA; Weinheim: 2005. pp. 1–30. (Protein degradation). [Google Scholar]
- 76.Pratt W.B., Morishima Y., Peng H.M., Osawa Y. Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp Biol Med. 2010;235:278–289. doi: 10.1258/ebm.2009.009250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nabi I.R., Watanabe H., Silletti S., Raz A. Tumor cell autocrine motility factor receptor. EXS. 1991;59:163–177. doi: 10.1007/978-3-0348-7494-6_11. [DOI] [PubMed] [Google Scholar]
- 78.Fang S., Ferrone M., Yang C., Jensen J.P., Tiwari S., Weissman A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Z., Du S., Fang S. gp78: a multifaceted ubiquitin ligase that integrates a unique protein degradation pathway from the endoplasmic reticulum. Curr Protein Pept Sci. 2012;13:414–424. doi: 10.2174/138920312802430590. [DOI] [PubMed] [Google Scholar]
- 80.Joshi V., Upadhyay A., Kumar A., Mishra A. Gp78 E3 ubiquitin ligase: essential functions and contributions in proteostasis. Front Cell Neurosci. 2017;11:259. doi: 10.3389/fncel.2017.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen B., Mariano J., Tsai Y.C., Chan A.H., Cohen M., Weissman A.M. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci U S A. 2006;103:341–346. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meyer H.H., Shorter J.G., Seemann J., Pappin D., Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. Embo J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dai R.M., Li C.C. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol. 2001;3:740–744. doi: 10.1038/35087056. [DOI] [PubMed] [Google Scholar]
- 84.Ye Y., Meyer H.H., Rapoport T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 85.Jarosch E., Taxis C., Volkwein C., Bordallo J., Finley D., Wolf D.H. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- 86.Bays N.W., Hampton R.Y. Cdc48-Ufd1-Npl4: stuck in the middle with Ub. Curr Biol. 2002;12:R366–R371. doi: 10.1016/s0960-9822(02)00862-x. [DOI] [PubMed] [Google Scholar]
- 87.Tsai B., Ye Y., Rapoport T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3:246–255. doi: 10.1038/nrm780. [DOI] [PubMed] [Google Scholar]
- 88.Rabinovich E., Kerem A., Frohlich K.U., Diamant N., Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ye Y., Meyer H.H., Rapoport T.A. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ye Y., Shibata Y., Yun C., Ron D., Rapoport T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 91.Bar-Nun S. The role of p97/Cdc48p in endoplasmic reticulum-associated degradation: from the immune system to yeast. Curr Top Microbiol Immunol. 2005;300:95–125. doi: 10.1007/3-540-28007-3_5. [DOI] [PubMed] [Google Scholar]
- 92.Elsasser S., Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol. 2005;7:742–749. doi: 10.1038/ncb0805-742. [DOI] [PubMed] [Google Scholar]
- 93.Jentsch S., Rumpf S. Cdc48 (p97): a “molecular gearbox” in the ubiquitin pathway?. Trends Biochem Sci. 2007;32:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 94.Leichner G.S., Avner R., Harats D., Roitelman J. Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol Biol Cell. 2009;20:3330–3341. doi: 10.1091/mbc.E08-09-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lipson C., Alalouf G., Bajorek M., Rabinovich E., Atir-Lande A., Glickman M. A proteasomal ATPase contributes to dislocation of endoplasmic reticulum-associated degradation (ERAD) substrates. J Biol Chem. 2008;283:7166–7175. doi: 10.1074/jbc.M705893200. [DOI] [PubMed] [Google Scholar]
- 96.Ikeda Y., Demartino G.N., Brown M.S., Lee J.N., Goldstein J.L., Ye J. Regulated endoplasmic reticulum-associated degradation of a polytopic protein: p97 recruits proteasomes to Insig-1 before extraction from membranes. J Biol Chem. 2009;284:34889–34900. doi: 10.1074/jbc.M109.044875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morris L.L., Hartman I.Z., Jun D.J., Seemann J., DeBose-Boyd R.A. Sequential actions of the AAA-ATPase valosin-containing protein (VCP)/p97 and the proteasome 19 S regulatory particle in sterol-accelerated, endoplasmic reticulum (ER)-associated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2014;289:19053–19066. doi: 10.1074/jbc.M114.576652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Woodman P.G. p97, a protein coping with multiple identities. J Cell Sci. 2003;116:4283–4290. doi: 10.1242/jcs.00817. [DOI] [PubMed] [Google Scholar]
- 99.Wojcik C., Rowicka M., Kudlicki A., Nowis D., McConnell E., Kujawa M. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol Biol Cell. 2006;17:4606–4618. doi: 10.1091/mbc.E06-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dreveny I., Pye V.E., Beuron F., Briggs L.C., Isaacson R.L., Matthews S.J. p97 and close encounters of every kind: a brief review. Biochem Soc Trans. 2004;32:715–720. doi: 10.1042/BST0320715. [DOI] [PubMed] [Google Scholar]
- 101.Chou T.F., Brown S.J., Minond D., Nordin B.E., Li K., Jones A.C. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A. 2011;108:4834–4839. doi: 10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krick R., Bremer S., Welter E., Schlotterhose P., Muehe Y., Eskelinen E.L. Cdc48/p97 and Shp1/p47 regulate autophagosome biogenesis in concert with ubiquitin-like Atg8. J Cell Biol. 2010;190:965–973. doi: 10.1083/jcb.201002075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huyton T., Pye V.E., Briggs L.C., Flynn T.C., Beuron F., Kondo H. The crystal structure of murine p97/VCP at 3.6A. J Struct Biol. 2003;144:337–348. doi: 10.1016/j.jsb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 104.Isaacson R.L., Pye V.E., Simpson P., Meyer H.H., Zhang X., Freemont P.S. Detailed structural insights into the p97-Npl4-Ufd1 interface. J Biol Chem. 2007;282:21361–21369. doi: 10.1074/jbc.M610069200. [DOI] [PubMed] [Google Scholar]
- 105.Davies J.M., Brunger A.T., Weis W.I. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure. 2008;16:715–726. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 106.Peters J.M., Cejka Z., Harris J.R., Kleinschmidt J.A., Baumeister W. Structural features of the 26 S proteasome complex. J Mol Biol. 1993;234:932–937. doi: 10.1006/jmbi.1993.1646. [DOI] [PubMed] [Google Scholar]
- 107.Walz J., Erdmann A., Kania M., Typke D., Koster A.J., Baumeister W. 26S proteasome structure revealed by three-dimensional electron microscopy. J Struct Biol. 1998;121:19–29. doi: 10.1006/jsbi.1998.3958. [DOI] [PubMed] [Google Scholar]
- 108.Rechsteiner M., Hoffman L., Dubiel W. The multicatalytic and 26 S proteases. J Biol Chem. 1993;268:6065–6068. [PubMed] [Google Scholar]
- 109.Rechsteiner M.C. Ubiquitin-mediated proteolysis: an ideal pathway for systems biology analysis. Adv Exp Med Biol. 2004;547:49–59. doi: 10.1007/978-1-4419-8861-4_5. [DOI] [PubMed] [Google Scholar]
- 110.Pickart C.M., Cohen R.E. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5:177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- 111.Rechsteiner M. The 26S proteasome. In: Mayer J., Ciechanover A., Rechsteiner M., editors. vol. 1. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2005. pp. 220–247. (Protein degradation). [Google Scholar]
- 112.Rechsteiner M., Hill C.P. Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 2005;15:27–33. doi: 10.1016/j.tcb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 113.Bedford L., Paine S., Sheppard P.W., Mayer R.J., Roelofs J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010;20:391–401. doi: 10.1016/j.tcb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park S., Roelofs J., Kim W., Robert J., Schmidt M., Gygi S.P. Hexameric assembly of the proteasomal ATPases is templated through their C termini. Nature. 2009;459:866–870. doi: 10.1038/nature08065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tomko R.J., Jr., Funakoshi M., Schneider K., Wang J., Hochstrasser M. Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: implications for proteasome structure and assembly. Mol Cell. 2010;38:393–403. doi: 10.1016/j.molcel.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ruschak A.M., Religa T.L., Breuer S., Witt S., Kay L.E. The proteasome antechamber maintains substrates in an unfolded state. Nature. 2010;467:868–871. doi: 10.1038/nature09444. [DOI] [PubMed] [Google Scholar]
- 117.Rock K.L., Gramm C., Rothstein L., Clark K., Stein R., Dick L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 118.Fenteany G., Standaert R.F., Lane W.S., Choi S., Corey E.J., Schreiber S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 119.Dick L.R., Cruikshank A.A., Destree A.T., Grenier L., McCormack T.A., Melandri F.D. Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells. J Biol Chem. 1997;272:182–188. doi: 10.1074/jbc.272.1.182. [DOI] [PubMed] [Google Scholar]
- 120.Meng L., Mohan R., Kwok B.H., Elofsson M., Sin N., Crews C.M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci U S A. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baljevic M., Orlowski R.Z. Pharmacodynamics and pharmacokinetics of proteasome inhibitors for the treatment of multiple myeloma. Expert Opin Drug Metab Toxicol. 2019;15:459–473. doi: 10.1080/17425255.2019.1621839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Raynes R., Pomatto L.C., Davies K.J. Degradation of oxidized proteins by the proteasome: distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Mol Aspects Med. 2016;50:41–55. doi: 10.1016/j.mam.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shringarpure R., Grune T., Mehlhase J., Davies K.J. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem. 2003;278:311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 124.Pickering A.M., Davies K.J. Differential roles of proteasome and immunoproteasome regulators Pa28alphabeta, Pa28gamma and Pa200 in the degradation of oxidized proteins. Arch Biochem Biophys. 2012;523:181–190. doi: 10.1016/j.abb.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Michalek M.T., Grant E.P., Gramm C., Goldberg A.L., Rock K.L. A role for the ubiquitin-dependent proteolytic pathway in MHC class I-restricted antigen presentation. Nature. 1993;363:552–554. doi: 10.1038/363552a0. [DOI] [PubMed] [Google Scholar]
- 126.Monaco J.J. Pathways for the processing and presentation of antigens to T cells. J Leukoc Biol. 1995;57:543–547. doi: 10.1002/jlb.57.4.543. [DOI] [PubMed] [Google Scholar]
- 127.Kloetzel P.M. Antigen processing by the proteasome. Nat Rev Mol Cell Biol. 2001;2:179–187. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 128.Kloetzel P.M. Generation of major histocompatibility complex class I antigens: functional interplay between proteasomes and TPPII. Nat Immunol. 2004;5:661–669. doi: 10.1038/ni1090. [DOI] [PubMed] [Google Scholar]
- 129.Monaco J.J., Nandi D. The genetics of proteasomes and antigen processing. Annu Rev Genet. 1995;29:729–754. doi: 10.1146/annurev.ge.29.120195.003501. [DOI] [PubMed] [Google Scholar]
- 130.Kloetzel P.M., Ossendorp F. Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr Opin Immunol. 2004;16:76–81. doi: 10.1016/j.coi.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 131.Kloetzel P.M. The proteasome and MHC class I antigen processing. Biochim Biophys Acta. 2004;1695:225–233. doi: 10.1016/j.bbamcr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 132.Kimura H., Caturegli P., Takahashi M., Suzuki K. New insights into the function of the immunoproteasome in immune and nonimmune cells. J Immunol Res. 2015 doi: 10.1155/2015:541984. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cascio P., Call M., Petre B.M., Walz T., Goldberg A.L. Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 2002;21:2636–2645. doi: 10.1093/emboj/21.11.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kopp F., Dahlmann B., Kuehn L. Reconstitution of hybrid proteasomes from purified PA700-20 S complexes and PA28alphabeta activator: ultrastructure and peptidase activities. J Mol Biol. 2001;313:465–471. doi: 10.1006/jmbi.2001.5063. [DOI] [PubMed] [Google Scholar]
- 135.Wang X., Yen J., Kaiser P., Huang L. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal. 2010;3:ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Grune T., Catalgol B., Licht A., Ermak G., Pickering A.M., Ngo J.K. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic Biol Med. 2011;51:1355–1364. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reinheckel T., Sitte N., Ullrich O., Kuckelkorn U., Davies K.J., Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J. 1998;335:637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Reinheckel T., Ullrich O., Sitte N., Grune T. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch Biochem Biophys. 2000;377:65–68. doi: 10.1006/abbi.2000.1717. [DOI] [PubMed] [Google Scholar]
- 139.Kim S.M., Grenert J.P., Patterson C., Correia M.A. CHIP–/–-mouse liver: adiponectin-AMPK-FOXO-activation overrides CYP2E1-elicited JNK1-activation, delaying onset of NASH: therapeutic implications. Sci Rep. 2016;6:29423. doi: 10.1038/srep29423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kwon D., Kim S.-M., Jacob P., 3rd, Liu Y., Correia M.A. Induction via functional protein stabilization of hepatic cytochromes P450 upon gp78/autocrine motility factor receptor (AMFR) ubiquitin E3-ligase genetic ablation in mice: therapeutic and toxicological relevance. Mol Pharmacol. 2019;96:641–654. doi: 10.1124/mol.119.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Masaki R., Yamamoto A., Tashiro Y. Cytochrome P-450 and NADPH-cytochrome P-450 reductase are degraded in the autolysosomes in rat liver. J Cell Biol. 1987;104:1207–1215. doi: 10.1083/jcb.104.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ronis M.J., Johansson I., Hultenby K., Lagercrantz J., Glaumann H., Ingelman-Sundberg M. Acetone-regulated synthesis and degradation of cytochrome P450E1 and cytochrome P4502B1 in rat liver [corrected] Eur J Biochem. 1991;198:383–389. doi: 10.1111/j.1432-1033.1991.tb16026.x. [DOI] [PubMed] [Google Scholar]
- 143.Murray B.P., Zgoda V.G., Correia M.A. Native CYP2C11: heterologous expression in Saccharomyces cerevisiae reveals a role for vacuolar proteases rather than the proteasome system in the degradation of this endoplasmic reticulum protein. Mol Pharmacol. 2002;61:1146–1153. doi: 10.1124/mol.61.5.1146. [DOI] [PubMed] [Google Scholar]
- 144.Liao M., Zgoda V.G., Murray B.P., Correia M.A. Vacuolar degradation of rat liver CYP2B1 in Saccharomyces cerevisiae: further validation of the yeast model and structural implications for the degradation of mammalian endoplasmic reticulum P450 proteins. Mol Pharmacol. 2005;67:1460–1469. doi: 10.1124/mol.104.009654. [DOI] [PubMed] [Google Scholar]
- 145.Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017;86:193–224. doi: 10.1146/annurev-biochem-061516-044908. [DOI] [PubMed] [Google Scholar]
- 146.Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Klionsky D.J., Codogno P., Cuervo A.M., Deretic V., Elazar Z., Fueyo-Margareto J. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6:438–448. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cuervo A.M. Cell biology. Autophagy's top chef. Science. 2011;332:1392–1393. doi: 10.1126/science.1208607. [DOI] [PubMed] [Google Scholar]
- 150.Bento C.F., Renna M., Ghislat G., Puri C., Ashkenazi A., Vicinanza M. Mammalian autophagy: how does it work?. Annu Rev Biochem. 2016;85:685–713. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- 151.Lamark T., Svenning S., Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61:609–624. doi: 10.1042/EBC20170035. [DOI] [PubMed] [Google Scholar]
- 152.Lamark T., Perander M., Outzen H., Kristiansen K., Overvatn A., Michaelsen E. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- 153.Seibenhener M.L., Babu J.R., Geetha T., Wong H.C., Krishna N.R., Wooten M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 155.Komatsu M., Waguri S., Koike M., Sou Y.S., Ueno T., Hara T. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 156.Lamark T., Kirkin V., Dikic I., Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 157.Linares J.F., Duran A., Yajima T., Pasparakis M., Moscat J., Diaz-Meco M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell. 2013;51:283–296. doi: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Birgisdottir A.B., Lamark T., Johansen T. The LIR motif—crucial for selective autophagy. J Cell Sci. 2013;126:3237–3247. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 159.Dice J.F., Terlecky S.R., Chiang H.L., Olson T.S., Isenman L.D., Short-Russell S.R. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin Cell Biol. 1990;1:449–455. [PubMed] [Google Scholar]
- 160.Cuervo A.M., Dice J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 161.Arndt V., Dick N., Tawo R., Dreiseidler M., Wenzel D., Hesse M. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–148. doi: 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 162.Gamerdinger M., Kaya A.M., Wolfrum U., Clement A.M., Behl C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011;12:149–156. doi: 10.1038/embor.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kruse K.B., Brodsky J.L., McCracken A.A. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yang H., Ni H.M., Guo F., Ding Y., Shi Y.H., Lahiri P. Sequestosome 1/p62 protein is associated with autophagic removal of excess hepatic endoplasmic reticulum in mice. J Biol Chem. 2016;291:18663–18674. doi: 10.1074/jbc.M116.739821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stolz A., Ernst A., Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 166.Khaminets A., Heinrich T., Mari M., Grumati P., Huebner A.K., Akutsu M. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. doi: 10.1038/nature14498. [DOI] [PubMed] [Google Scholar]
- 167.Khaminets A., Behl C., Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 2016;26:6–16. doi: 10.1016/j.tcb.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 168.Chien J.Y., Thummel K.E., Slattery J.T. Pharmacokinetic consequences of induction of CYP2E1 by ligand stabilization. Drug Metab Dispos. 1997;25:1165–1175. [PubMed] [Google Scholar]
- 169.Ghanbari F., Rowland-Yeo K., Bloomer J.C., Clarke S.E., Lennard M.S., Tucker G.T. A critical evaluation of the experimental design of studies of mechanism based enzyme inhibition, with implications for in vitro–in vivo extrapolation. Curr Drug Metab. 2006;7:315–334. doi: 10.2174/138920006776359293. [DOI] [PubMed] [Google Scholar]
- 170.Kalgutkar A.S., Obach R.S., Maurer T.S. Mechanism-based inactivation of cytochrome P450 enzymes: chemical mechanisms, structure–activity relationships and relationship to clinical drug–drug interactions and idiosyncratic adverse drug reactions. Curr Drug Metab. 2007;8:407–447. doi: 10.2174/138920007780866807. [DOI] [PubMed] [Google Scholar]
- 171.Yang J., Liao M., Shou M., Jamei M., Yeo K.R., Tucker G.T. Cytochrome P450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab. 2008;9:384–394. doi: 10.2174/138920008784746382. [DOI] [PubMed] [Google Scholar]
- 172.Fuhr U. Drug interactions with grapefruit juice. Extent, probable mechanism and clinical relevance. Drug Saf. 1998;18:251–272. doi: 10.2165/00002018-199818040-00002. [DOI] [PubMed] [Google Scholar]
- 173.Bailey D.G., Dresser G., Arnold J.M. Grapefruit-medication interactions: forbidden fruit or avoidable consequences?. Can Med Assoc J. 2013;185:309–316. doi: 10.1503/cmaj.120951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bandiera S., Weidlich S., Harth V., Broede P., Ko Y., Friedberg T. Proteasomal degradation of human CYP1B1: effect of the Asn453Ser polymorphism on the post-translational regulation of CYP1B1 expression. Mol Pharmacol. 2005;67:435–443. doi: 10.1124/mol.104.006056. [DOI] [PubMed] [Google Scholar]
- 175.Beaune P., Dansette P.M., Mansuy D., Kiffel L., Finck M., Amar C. Human anti-endoplasmic reticulum autoantibodies appearing in a drug-induced hepatitis are directed against a human liver cytochrome P-450 that hydroxylates the drug. Proc Natl Acad Sci U S A. 1987;84:551–555. doi: 10.1073/pnas.84.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Beaune P., Pessayre D., Dansette P., Mansuy D., Manns M. Autoantibodies against cytochromes P450: role in human diseases. Adv Pharmacol. 1994;30:199–245. doi: 10.1016/s1054-3589(08)60175-1. [DOI] [PubMed] [Google Scholar]
- 177.Uetrecht J.P. New concepts in immunology relevant to idiosyncratic drug reactions: the “danger hypothesis” and innate immune system. Chem Res Toxicol. 1999;12:387–395. doi: 10.1021/tx980249i. [DOI] [PubMed] [Google Scholar]
- 178.Boitier E., Beaune P. Xenobiotic-metabolizing enzymes as autoantigens in human autoimmune disorders. An update. Clin Rev Allergy Immunol. 2000;18:215–239. doi: 10.1385/CRIAI:18:2:215. [DOI] [PubMed] [Google Scholar]
- 179.Uetrecht J. Current trends in drug-induced autoimmunity. Autoimmun Rev. 2005;4:309–314. doi: 10.1016/j.autrev.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 180.Bourdi M., Gautier J.C., Mircheva J., Larrey D., Guillouzo A., Andre C. Anti-liver microsomes autoantibodies and dihydralazine-induced hepatitis: specificity of autoantibodies and inductive capacity of the drug. Mol Pharmacol. 1992;42:280–285. [PubMed] [Google Scholar]
- 181.Loeper J., Descatoire V., Maurice M., Beaune P., Belghiti J., Houssin D. Cytochromes P-450 in human hepatocyte plasma membrane: recognition by several autoantibodies. Gastroenterology. 1993;104:203–216. doi: 10.1016/0016-5085(93)90853-5. [DOI] [PubMed] [Google Scholar]
- 182.Bourdi M., Chen W., Peter R.M., Martin J.L., Buters J.T., Nelson S.D. Human cytochrome P450 2E1 is a major autoantigen associated with halothane hepatitis. Chem Res Toxicol. 1996;9:1159–1166. doi: 10.1021/tx960083q. [DOI] [PubMed] [Google Scholar]
- 183.Eliasson E., Kenna J.G. Cytochrome P450 2E1 is a cell surface autoantigen in halothane hepatitis. Mol Pharmacol. 1996;50:573–582. [PubMed] [Google Scholar]
- 184.Clot P., Albano E., Eliasson E., Tabone M., Arico S., Israel Y. Cytochrome P4502E1 hydroxyethyl radical adducts as the major antigen in autoantibody formation among alcoholics. Gastroenterology. 1996;111:206–216. doi: 10.1053/gast.1996.v111.pm8698201. [DOI] [PubMed] [Google Scholar]
- 185.Clot P., Bellomo G., Tabone M., Arico S., Albano E. Detection of antibodies against proteins modified by hydroxyethyl free radicals in patients with alcoholic cirrhosis. Gastroenterology. 1995;108:201–207. doi: 10.1016/0016-5085(95)90025-x. [DOI] [PubMed] [Google Scholar]
- 186.Lytton S.D., Helander A., Zhang-Gouillon Z.Q., Stokkeland K., Bordone R., Arico S. Autoantibodies against cytochromes P-4502E1 and P-4503A in alcoholics. Mol Pharmacol. 1999;55:223–233. doi: 10.1124/mol.55.2.223. [DOI] [PubMed] [Google Scholar]
- 187.Leeder J.S., Gaedigk A., Lu X., Cook V.A. Epitope mapping studies with human anti-cytochrome P450 3A antibodies. Mol Pharmacol. 1996;49:234–243. [PubMed] [Google Scholar]
- 188.Robin M.A., Le Roy M., Descatoire V., Pessayre D. Plasma membrane cytochromes P450 as neoantigens and autoimmune targets in drug-induced hepatitis. J Hepatol. 1997;26:23–30. doi: 10.1016/s0168-8278(97)82329-x. [DOI] [PubMed] [Google Scholar]
- 189.Robin M.A., Maratrat M., Le Roy M., Le Breton F.P., Bonierbale E., Dansette P. Antigenic targets in tienilic acid hepatitis. Both cytochrome P450 2C11 and 2C11-tienilic acid adducts are transported to the plasma membrane of rat hepatocytes and recognized by human sera. J Clin Invest. 1996;98:1471–1480. doi: 10.1172/JCI118936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lytton S.D., Berg U., Nemeth A., Ingelman-Sundberg M. Autoantibodies against cytochrome P450s in sera of children treated with immunosuppressive drugs. Clin Exp Immunol. 2002;127:293–302. doi: 10.1046/j.1365-2249.2002.01754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Thervet E., Legendre C., Beaune P., Anglicheau D. Cytochrome P450 3A polymorphisms and immunosuppressive drugs. Pharmacogenomics. 2005;6:37–47. doi: 10.1517/14622416.6.1.37. [DOI] [PubMed] [Google Scholar]
- 192.Metushi I.G., Sanders C., Acute Liver Study G., Lee W.M., Uetrecht J. Detection of anti-isoniazid and anti-cytochrome P450 antibodies in patients with isoniazid-induced liver failure. Hepatology. 2014;59:1084–1093. doi: 10.1002/hep.26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Timbrell J.A., Mitchell J.R., Snodgrass W.R., Nelson S.D. Isoniazid hepatoxicity: the relationship between covalent binding and metabolism in vivo. J Pharmacol Exp Ther. 1980;213:364–369. [PubMed] [Google Scholar]
- 194.Manns M.P., Johnson E.F., Griffin K.J., Tan E.M., Sullivan K.F. Major antigen of liver kidney microsomal autoantibodies in idiopathic autoimmune hepatitis is cytochrome P450db1. J Clin Invest. 1989;83:1066–1072. doi: 10.1172/JCI113949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Choudhuri K., Mieli-Vergani G., Vergani D. Cytochrome P4502D6: understanding an autoantigen. Clin Exp Immunol. 1997;108:381–383. doi: 10.1046/j.1365-2249.1997.4131311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Winqvist O., Gustafsson J., Rorsman F., Karlsson F.A., Kampe O. Two different cytochrome P450 enzymes are the adrenal antigens in autoimmune polyendocrine syndrome type I and Addison's disease. J Clin Invest. 1993;92:2377–2385. doi: 10.1172/JCI116843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Winqvist O., Karlsson F.A., Kampe O. 21-Hydroxylase, a major autoantigen in idiopathic Addison's disease. Lancet. 1992;339:1559–1562. doi: 10.1016/0140-6736(92)91829-w. [DOI] [PubMed] [Google Scholar]
- 198.Krohn K., Uibo R., Aavik E., Peterson P., Savilahti K. Identification by molecular cloning of an autoantigen associated with Addison's disease as steroid 17 alpha-hydroxylase. Lancet. 1992;339:770–773. doi: 10.1016/0140-6736(92)91894-e. [DOI] [PubMed] [Google Scholar]
- 199.He K., Bornheim L.M., Falick A.M., Maltby D., Yin H., Correia M.A. Identification of the heme-modified peptides from cumene hydroperoxide-inactivated cytochrome P450 3A4. Biochemistry. 1998;37:17448–17457. doi: 10.1021/bi9808464. [DOI] [PubMed] [Google Scholar]
- 200.Jiang J., Stoyanovsky D.A., Belikova N.A., Tyurina Y.Y., Zhao Q., Tungekar M.A. A mitochondria-targeted triphenylphosphonium-conjugated nitroxide functions as a radioprotector/mitigator. Radiat Res. 2009;172:706–717. doi: 10.1667/RR1729.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gaude H., Aznar N., Delay A., Bres A., Buchet-Poyau K., Caillat C. Molecular chaperone complexes with antagonizing activities regulate stability and activity of the tumor suppressor LKB1. Oncogene. 2012;31:1582–1591. doi: 10.1038/onc.2011.342. [DOI] [PubMed] [Google Scholar]
- 202.Greer E.L., Oskoui P.R., Banko M.R., Maniar J.M., Gygi M.P., Gygi S.P. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 203.Webb A.E., Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39:159–169. doi: 10.1016/j.tibs.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.van der Horst A., Burgering B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–450. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 205.Tikhanovich I., Cox J., Weinman S.A. Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol. 2013;28:125–131. doi: 10.1111/jgh.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tsuchida A., Yamauchi T., Ito Y., Hada Y., Maki T., Takekawa S. Insulin/Foxo1 pathway regulates expression levels of adiponectin receptors and adiponectin sensitivity. J Biol Chem. 2004;279:30817–30822. doi: 10.1074/jbc.M402367200. [DOI] [PubMed] [Google Scholar]
- 207.de Keizer P.L., Burgering B.M., Dansen T.B. Forkhead box O as a sensor, mediator, and regulator of redox signaling. Antioxid Redox Signal. 2011;14:1093–1106. doi: 10.1089/ars.2010.3403. [DOI] [PubMed] [Google Scholar]
- 208.Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Merrill G.F., Kurth E.J., Hardie D.G., Winder W.W. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 210.Yamauchi T., Nio Y., Maki T., Kobayashi M., Takazawa T., Iwabu M. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 211.Jager S., Handschin C., St-Pierre J., Spiegelman B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yamauchi T., Kamon J., Minokoshi Y., Ito Y., Waki H., Uchida S. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 213.Iwabu M., Yamauchi T., Okada-Iwabu M., Sato K., Nakagawa T., Funata M. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca2+ and AMPK/SIRT1. Nature. 2010;464:1313–1319. doi: 10.1038/nature08991. [DOI] [PubMed] [Google Scholar]
- 214.Xiong X., Tao R., DePinho R.A., Dong X.C. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem. 2012;287:39107–39114. doi: 10.1074/jbc.M112.412569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kamei Y., Mizukami J., Miura S., Suzuki M., Takahashi N., Kawada T. A forkhead transcription factor FKHR up-regulates lipoprotein lipase expression in skeletal muscle. FEBS Lett. 2003;536:232–236. doi: 10.1016/s0014-5793(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 216.Han Y., Hu Z., Cui A., Liu Z., Ma F., Xue Y. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat Commun. 2019;10:623. doi: 10.1038/s41467-019-08585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Czaja M.J. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol Metab. 2010;21:707–713. doi: 10.1016/j.tem.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Seki E., Brenner D.A., Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Farrell G.C., van Rooyen D., Gan L., Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Duwaerts C.C., Maher J.J. Mechanisms of liver injury in non-alcoholic steatohepatitis. Curr Hepatol Rep. 2012;13:119–129. doi: 10.1007/s11901-014-0224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Noureddin M., Mato J.M., Lu S.C. Nonalcoholic fatty liver disease: update on pathogenesis, diagnosis, treatment and the role of S-adenosylmethionine. Exp Biol Med. 2015;240:809–820. doi: 10.1177/1535370215579161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ariz U., Mato J.M., Lu S.C., Martinez Chantar M.L. Nonalcoholic steatohepatitis, animal models, and biomarkers: what is new?. Methods Mol Biol. 2010;593:109–136. doi: 10.1007/978-1-60327-194-3_6. [DOI] [PubMed] [Google Scholar]
- 223.Maher J.J. New insights from rodent models of fatty liver disease. Antioxid Redox Signal. 2011;15:535–550. doi: 10.1089/ars.2010.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Dekker M.J., Su Q., Baker C., Rutledge A.C., Adeli K. Fructose: a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2010;299:E685–E694. doi: 10.1152/ajpendo.00283.2010. [DOI] [PubMed] [Google Scholar]
- 225.Ishimoto T., Lanaspa M.A., Rivard C.J., Roncal-Jimenez C.A., Orlicky D.J., Cicerchi C. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology. 2013;58:1632–1643. doi: 10.1002/hep.26594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lanaspa M.A., Ishimoto T., Li N., Cicerchi C., Orlicky D.J., Ruzycki P. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun. 2013;4:2434. doi: 10.1038/ncomms3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Jensen T., Abdelmalek M.F., Sullivan S., Nadeau K.J., Green M., Roncal C. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;6:1063–1075. doi: 10.1016/j.jhep.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Li Y., Xu S., Mihaylova M.M., Zheng B., Hou X., Jiang B. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Liu T.F., Tang J.J., Li P.S., Shen Y., Li J.G., Miao H.H. Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab. 2012;16:213–225. doi: 10.1016/j.cmet.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 230.Liang J.S., Kim T., Fang S., Yamaguchi J., Weissman A.M., Fisher E.A. Overexpression of the tumor autocrine motility factor receptor Gp78, a ubiquitin protein ligase, results in increased ubiquitinylation and decreased secretion of apolipoprotein B100 in HepG2 cells. J Biol Chem. 2003;278:23984–23988. doi: 10.1074/jbc.M302683200. [DOI] [PubMed] [Google Scholar]
- 231.Zhang T., Kho D.H., Wang Y., Harazono Y., Nakajima K., Xie Y. Gp78, an E3 ubiquitin ligase acts as a gatekeeper suppressing nonalcoholic steatohepatitis (NASH) and liver cancer. PLoS One. 2015;10 doi: 10.1371/journal.pone.0118448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ni H.M., Bockus A., Boggess N., Jaeschke H., Ding W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ni H.M., Jaeschke H., Ding W.X. Targeting autophagy for drug-induced hepatotoxicity. Autophagy. 2012;8:709–710. doi: 10.4161/auto.19659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ni H.M., Boggess N., McGill M.R., Lebofsky M., Borude P., Apte U. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci. 2012;127:438–450. doi: 10.1093/toxsci/kfs133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wu D., Cederbaum A.I. Inhibition of autophagy promotes CYP2E1-dependent toxicity in HepG2 cells via elevated oxidative stress, mitochondria dysfunction and activation of p38 and JNK MAPK. Redox Biol. 2013;1:552–565. doi: 10.1016/j.redox.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Platt F.M., Boland B., van der Spoel A.C. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Czaja M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895–1908. doi: 10.1053/j.gastro.2011.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sun H., Lee C.M., Tripathi S., Kim K.B., Morgan E.T. Nitric oxide-dependent CYP2B degradation is potentiated by a cytokine-regulated pathway and utilizes the immunoproteasome subunit LMP2. Biochem J. 2012;445:377–382. doi: 10.1042/BJ20120820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Handa P., Maliken B.D., Nelson J.E., Morgan-Stevenson V., Messner D.J., Dhillon B.K. Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology. 2014;60:133–145. doi: 10.1002/hep.26946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Takaki A., Kawai D., Yamamoto K. Molecular mechanisms and new treatment strategies for non-alcoholic steatohepatitis (NASH) Int J Mol Sci. 2014;15:7352–7379. doi: 10.3390/ijms15057352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Park T.J., Reznick J., Peterson B.L., Blass G., Omerbasic D., Bennett N.C. Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science. 2017;356:307–311. doi: 10.1126/science.aab3896. [DOI] [PubMed] [Google Scholar]
- 242.Softic S., Cohen D.E., Kahn C.R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci. 2016;61:1282–1293. doi: 10.1007/s10620-016-4054-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.