

Abstract
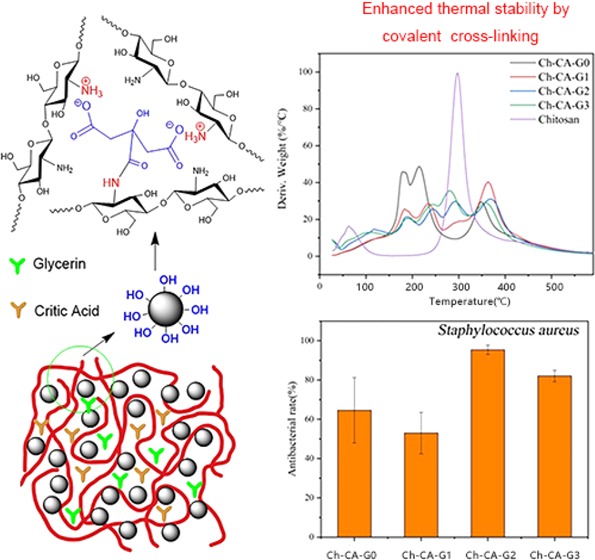
Chitosan–citric biomembranes Ch-CA-Gx (x = 0–3) were prepared by a simple cross-linking. The dependence of mechanical property, water-resisting capacity, microstructural characteristic, oxygen barrier ability, and thermal properties of membranes on the content of glycerin was investigated. The results revealed that vacuum drying at 80 °C can lead to low-yield amidation and the Maillard reaction, thus affecting the thermal stability and water resistance of biomembranes. Owing to the ionic cross-linking and amidation, the chitosan–citrate complex showed weaker compatibility when the glycerin content increased, thereby leading to discontinuity of microstructure in the Ch-CA-Gx (x = 1–3) membranes, which was in line with the weaker mechanical properties and water-resisting abilities of membranes, compared to Ch-CA-G0. Chitosan membranes showed interestingly high oxygen barrier capabilities under 40 and 80% relative humidity (RH) conditions, probably attributed to the increased diffusion length arising from the hydrogen-bonding, ionic, and covalent cross-linking. The oxygen transmission rates of Ch-CA-Gx were below 0.1 cm3 m–2 day–1 at 40% RH. The Ch-CA-Gx membranes showed a good elasticity assigned to the reversibly cross-linked structure. The membranes presented strong antibacterial activities against Staphylococcus aureus and Escherichia coli bacteria, probably owing to the citric acids. The results demonstrated that these materials have potential applications as membranes or protecting coatings for food packaging and successful cross-linking by means of amidation, and the Maillard reaction under the condition of vacuum drying can be probably applied as a green and alternative method for the fabrication of mechanically tough and antibacterial membranes, fibers, and gels.
1. Introduction
Chitosan (Ch) has been intensively investigated in the fields of biomedicines, elastic hydrogels, water treatment, biomembranes, and active food packaging, owing to the nontoxicity, biodegradation, biocompatibility, and the inherent antibacterial ability against the pathogenic and spoilage bacteria.1−4 Antibacterial packaging materials doped by chitosan have shown excellent thermosealability, microwavability, optical properties, and antimicrobial activities against pathogenic and spoilage bacteria.5,6 However, the poor oxygen barrier property and weak water-resisting ability limited the composite films consisting of chitosan as host material in the development of commercial products as coated layer over foods or packaging materials.7,8 The water- and oxygen-barrier characteristics of packaging membrane were influenced by many factors such as the macromolecular structure, morphology, additives, cross-linked network, and so on. Recently, cross-linking by means of covalent and salt bonds has been considered as an effective method to enhance the water-resisting capacities of chitosan-based films.9−12 Glutaraldehyde, genipin, and tripolyphosphate are generally used as cross-linkers.13
Recently, citric acid (CA) as ionic cross-linker has been widely explored because of the excellent biocompatibility in membranes, fibers, and micro/nanogels.14−16 Citric acid, which is used as a food flavoring agent, presented excellent antimicrobial and antioxidant properties. By means of thermocompression molding, chitosan biomembranes with high strength and antibacterial ability can be successfully fabricated by utilizing citric acid as a biodegradable plasticizer.17 Antibacterial membranes using chitosan and cholinium citrate as materials were successfully prepared by Galvis-Sánchez et al.18 Möller et al. fabricated a series of antimicrobial chitosan–citric membranes by solution casting methods, utilizing NaH2PO4 as amidation catalyst.7 Guerrero et al. reported sustainable chitosan films through thermocompression molding and utilization of citric acid as a covalent linker.19 However, covalent cross-linking by means of amidation was mainly assigned to thermocompression at high temperature and chemical catalyst in the above-mentioned film preparation, and the investigation of the catalyst-free amidation reaction is still lacking in the cross-linked chitosan–citric film preparation. Cui et al. reported an effective and catalyst-free modification of chitosan films by means of citric acid for increased water-resisting capacity in high-temperature drying.20 Nowakowska et al. demonstrated that water-soluble chitosan-Rose Bengal photosensitizers can be successfully synthesized by means of catalyst-free amidation reaction between amino and carboxyl groups under the conditions of 80 °C and vacuum. Catalyst-free amidation reaction provided a green and alternative method for cross-linked chitosan films, fibers, and hydrogels using citric acid as cross-linker.21,22 The different processing methods for cross-linked films can probably endow them with versatile physical properties in the aspects of mechanical properties, water-resisting capabilities, and oxygen barrier characteristics.
For this purpose, the aims of the present research are (i) to explore practicability of the temperature-dependent and catalyst-free amidation in vacuum for the preparation of cross-linked chitosan–citric film; (ii) to investigate the dependence of microstructure, mechanical properties, water vapor permeability (WVP), thermal stability, and oxygen barrier ability of chitosan–citric membranes on the content of glycerin (G); and (iii) to assess the antibacterial capabilities of chitosan–citric membranes against Staphylococcus aureus and Escherichia coli bacteria for potential packaging applications.
2. Results and Discussion
2.1. Structural Characterization of Ch-CA-Gx Membranes
The interactions between the citric acids and the primary amine groups of chitosan readily caused the formation of chitosan–citrate complex.14 The general procedure to fabricate chitosan–citric membranes Ch-CA-Gx (x = 0–3) is described in Figure 1. As reported in some previous works, acetic acid was frequently utilized as a solubilizer for chitosan through ammonium acetate in film preparation using citric acid as cross-linker.7 However, ammonium acetate and amides attributed to the dehydration of ammonium acetate in film preparation under the condition of thermocompression at high temperature or chemical catalyst can probably reduce the possibility of ammonium citrate, thus leading to lower ionic cross-linking and amidation. Therefore, citric acid was utilized as the solubilizer and cross-linker in the absence of acetic acid in our work, and the chitosan powder can be dissolved thoroughly in water at the ratio of 2.5/4 (weight ratio, w/w). A vacuum drying oven was set at temperatures of 100, 80, 60, and 40 °C to evaluate the temperature dependence of amidation and the Maillard reaction. To further confirm that citric acids were grafted to the chitosan chain by means of the amide bonds, the model polymer was synthesized for comparison with Ch-CA-Gx films. The model polymer was obtained as a white solid, which was very well soluble in 1% acetic acid aqueous solution, and the synthesis route is shown in Figure 2.
Figure 1.

Preparation process of cross-linked Ch-CA-Gx membranes.
Figure 2.

Synthesized route of model polymer using 3-ethylcarbodiimide hydrochloride (EDC) as catalyst.
The bands assigned to the N–H bending vibrations of unprotonated amines, amide II, and amide I for chitosan powder peaked at around 1573, 1550, and 1648 cm–1, respectively, as reported by Lawrie et al.23 The Ch-CA-Gx spectrum showed a broad band peaked at around 3400 cm–1 attributed to the stretching vibrations of O–H and N–H, and two small bands centered at 2941 and 2878 cm–1 assigned to C–H stretching vibration, as shown in Figure 3. Before vacuum drying at 80 °C, ionically cross-linked Ch-CA-Gx membranes represented the bands centered at around 1615 and 1512 cm–1, which are mainly attributed to the antisymmetric −NH3+ deformation and the symmetric −NH3+ deformation, respectively, as suggested by Lawrie et al.23 After vacuum drying at 80 °C, the amide I band of chitosan powder without chemical modification at the peak of 1648 cm–1 disappeared in the Ch-CA-Gx spectrum, and the bending vibrations of N–H corresponding to unprotonated primary amines at around 1575 cm–1 were very obvious in the membranes. The shoulder at around 1534 cm–1 probably corresponded to the overlapping band between N–H bending vibrations of amide II and the symmetric deformation of −NH3+. The shoulder at 1623 cm–1 was arising from the bands for N–H bending vibrations of amide I and the antisymmetric −NH3+ deformation. In addition, the small bands peaking at 1374 cm–1 assigned to the C–N of amide III were observed. The C=O stretching characteristic bands with the peak at around 1700 cm–1 corresponding to citric acid were also observed in the Ch-CA-Gx films. The spectrum indicated that no ester bonds peaked at around 1730 cm–1. The 13C NMR measurements of chitosan–citric films showed no signals corresponding to the carbonyl groups of amide. In this work, Fourier transform infrared (FTIR) spectroscopy failed to clearly identify the possible amidation between citric acid and chitosan, owing to the spectrum overlapping between the bands corresponding to the −NH3+ deformation and amide. However, X-ray photoelectron spectroscopy (XPS) N 1s narrow scans of chitosan–citric membranes can probably be utilized to evaluate the degree of amide bonds.23 From the XPS data of films shown in Figure 4, the amidation extent of amine groups after the deduction of the degree of acetylation in raw material chitosan (around 9%) was about 5%, indicating the occurrence of the amidation reaction in Ch-CA-G0 and Ch-CA-G1 under vacuum at 80 °C in low yield.
Figure 3.

Infrarred spectra of the ionically cross-linked Ch-CA-Gx (a) and covalently cross-linked Ch-CA-Gx membranes (b) by means of vacuum drying at 80 °C.
Figure 4.

Curve fit of XPS N 1s narrow scans of chitosan powder (a), Ch-CA-G0 (b), Ch-CA-G1 (c), and model polymer (d).
The 1H NMR spectra of model polymer showed the signals of citric acid in the range of 2.4–2.9 ppm, evidencing that CA was ionically or covalently attached to the chitosan chains, as shown in Figure 5. XPS data showed about 19% acetylation degree of amine groups, evidencing about 10% amidation. From the spectra of model polymer, two peaks at around 1573 and 1530 cm–1 were observed, which was closely consistent with those of Ch-CA-Gx cross-linked membranes. The spectrum of chitosan–citric membranes after extraction in methanol was similar to that of the model polymer, indicating the existence of amide bonds.24 The C=O stretching band with a peak at around 1715 cm–1 corresponding to citric acid and the band centered at about 1380 cm–1 corresponding to the C–N of amide III in chitosan–citric membranes were observed after extraction.
Figure 5.

FTIR spectrum of Ch-CA-Gx after extracted in methanol (a) and 1H NMR spectrum of model polymer in D2O containing 1% CD3COOD (b).
The transition from colorless transparent to light brown membrane originated from the temperature-dependent nonenzymatic Maillard reactions has been reported by many researchers.18Ch-CA-Gx films will gradually change from colorless to light brown when the temperature increased from 60 to 80 °C, then gradually to dark brown at 100 °C, indicating the occurrence of the nonenzymatic Maillard reactions. The ionic cross-linking by means of ammonium salt, together with covalent cross-linking assigned to amidation and nonenzymatic Maillard reactions, can probably enhance the water-resisting capabilities of Ch-CA-Gx films.
2.2. Thermogravimetric Behaviors
The thermogravimetric behaviors of Ch-CA-Gx membranes were affected by plasticization and cross-linking, as shown in Figure 6. Normally, a weight loss of the chitosan powder without chemical modification started at around 250 °C because of the decomposition of crystallized structures and the maximum rate of thermal decomposition occurred at around 300 °C.17,18 The major peak for derivative weight loss at around 300 °C corresponding to the decomposition of crystalline structures nearly disappeared in the Ch-CA-G0 membrane. The interaction between citric acids and amino groups of chitosan can disrupt the crystalline structure in chitosan. Three major peaks were observed for Ch-CA-G0 at around 178, 216, and 366 °C in Figure 6, which probably corresponded to the decomposition of citric acid and amorphous and cross-linked chitosan, respectively.18 However, with the increase in content of glycerol in Ch-CA-Gx (x = 1–3), the major peak at around 300 °C assigned to the degradation of crystalline structures reappeared, probably reflecting the nonignorable effect of glycerol on cleavage of chitosan crystalline structures in the presence of citric acid. The weight loss at around 210–250 °C was associated with the weight loss of glycerol.24 The broad band in the range of 210–270 °C in Ch-CA-Gx (x = 1–3) was probably the overlapping band between decomposition of glycerol and amorphous chitosan. The cross-linking improved the heat-resisting capability of the chitosan, thus leading to around 50 °C higher maximum temperature of thermal decomposition for Ch-CA-Gx (x = 0–3), in comparison to the original chitosan powder.
Figure 6.

Thermogravimetric graphs of the cross-linked membranes Ch-CA-Gx and chitosan powder: (a) weight percentage and (b) derivative weight.
2.3. Mechanical Properties of the Cross-Linked Films
Table 1 shows the tensile strength (TS) and elongation (E) of the cross-linked films. With the increase of glycerol concentration, the TS and E decreased accordingly, and the films became more extensible. The blend of G caused the decrease in mechanical properties, probably due to the weakened hydrogen-bonding interaction within the polysaccharide network. Concerning the Ch-CA-G0 film, the cross-linking structure assigned to the stronger hydrogen bonding caused extensive interchain bonding and limited flexibility of the film in the absence of glycerin.25−28
Table 1. Mechanical Properties (TS, Tensile Strength; E, Elongation at Break), Water Vapor Permeability (WVP), and Solubility Degree (S) of the Ch-CA-Gx Films Prepared by the in Situ Cross-Linking Strategy.
| samples | TS (MPa) | E (%) | WVP (g m–2 day–1) | S (%) |
|---|---|---|---|---|
| Ch-CA-G0 | 58.39 ± 15.79 | 32.49 ± 2.80 | 28.57 ± 4.64 | 45.85 ± 2.21 |
| Ch-CA-G1 | 5.50 ± 0.30 | 52.58 ± 3.37 | 35.87 ± 13.67 | 52.65 ± 6.86 |
| Ch-CA-G2 | 2.25 ± 0.52 | 86.73 ± 12.71 | 78.33 ± 18.03 | 47.18 ± 6.07 |
| Ch-CA-G3 | 1.29 ± 0.17 | 151.87 ± 6.56 | 160.32 ± 45.51 | 55.24 ± 4.42 |
The effect of frequency on the storage modulus (G′) and loss modulus (G″) of the membranes in the range of 0.01–100 Hz is shown in Figure 7. The elastic behavior at low frequency (G′ > G″) and viscous behavior (G′ < G″) at high frequency can be observed in all samples, evidencing the occurrence of a reversibly cross-linked network in membranes.29 Owing to the absence of glycerin in Ch-CA-G0, the values of G′ and G″ increased obviously in comparison to Ch-CA-Gx (x = 1–3), reaching the maximum values of about 2 MPa (G′) and 1 MPa (G″), respectively, and the crossover point of G′ and G″ located at lower frequency in Ch-CA-G0. The G′ and G″ values declined significantly when the strain increased, as shown in Figure 7b. The strain-induced collapse was possibly assigned to the breaking of physically or covalently cross-linked points of the membranes. The G′ and G″ of the membranes decreased with increasing content of glycerin, probably owing to the enhanced plasticization effect, which is consistent with the change trend of the mechanical properties as the glycerin loading increased.27 The plateaus G′ of the membranes Ch-CA-Gx (x = 0, 1, 2, and 3) are 4, 0.4, 0.2, and 0.05 MPa, respectively. When the materials were loaded, mechanical responses of soft elasticity were observed. Figure 8 shows the shape recovery process of Ch-CA-G3. The original shape of membrane with a 9.5 cm length was stretched to 14 cm (50% strain) at 23 °C, and the length of the sample recovered instantly to 12.2 cm after removing the stress. After 48 h, the length of the membrane recovered to 9.7 cm (2% strain). Although the efficiency of elastic recovery is time dependent for all samples, the recovery efficiency was around 100% under the condition of one stretch. The cross-linked network arising from a large number of hydrogen bonds and covalently cross-linked bonds was responsible for the maintenance of the original shape in the stretched films.
Figure 7.

Rheological data of the membranes Ch-CA-Gx (x = 0–3) in frequency sweeps (a) and strain sweeps (b) at 60 °C.
Figure 8.

Shape memory behavior of Ch-CA-G3 (a); the original shape of the membrane (length = 9.5 cm) (b); membrane after removing the stress upon (length = 12 cm) (c); and the shape of the membrane after 48 h (length = 9.7 cm) (d).
2.4. Water Vapor Permeability and Solubility Degree
The water vapor permeability (WVP) and solubility degree (S) of the films were considered as important indexes in cross-linking degree and water resistance.30,31 The WVP of Ch-CA-Gx membranes increased with the loading concentration of glycerin. Increased G content in Ch-CA-Gx membranes caused a significant increase in WVP, probably due to the stronger hydrophilicity of G facilitating water migration through the membranes.7 The WVP value was 28.57, 35.87, 78.33, and 160.32 g m–2 day–1 for Ch-CA-Gx (x = 0–3), respectively. According to previous reports,32 the chitosan film and the cross-linked chitosan membranes using glutaraldehyde and genipin as cross-linkers showed WVP values of around 835, 724, and 684 g m–2 day–1, respectively, using the ASTM method at 25 °C and 50% RH, which are higher than the values of the Ch-CA-Gx composite membranes. The water migration in packaging membrane is closely associated with the hydrophilic and hydrophobic capacities of material and the microscopic paths for the water migration. Compared to Ch-CA-G0, the higher WVP behavior of Ch-CA-Gx (x = 1–3) could be assigned to the modified hydrophilic character of the film as well as the tortuous micropath for the water migration in the presence of glycerin.33
Figure 9 shows the effect of dehydrated temperature on the solubility of the Ch-CA-G0. The membranes dehydrated in 40 and 60 °C under vacuum completely dissolved in water, but the membrane fabricated at or above 80 °C retained about 45% weight after soaking in water. These results indicated that the inter- and intramolecular interactions by means of amidation and the Maillard reaction were improved under vacuum drying at high temperatures, further affecting the thermal stability and tensile strength of films, as suggested by Coma et al.7 Water solubilities of composite membranes are shown in Table 1. The solubility degree of composite membrane was 45.85, 52.65, 47.18, and 55.24 for Ch-CA-Gx (x = 0, 1, 2 and 3), respectively.
Figure 9.

Effect of dehydrated temperature on the solubility degree of Ch-CA-G0 (a) and the photo of the Ch-CA-G0 membrane (b).
Swelling degree as high as 2000% has been reported for the pure chitosan in an acidic environment. In comparison, chitosan microspheres grafted by poly(acrylamide) or poly(acrylic acid) and cross-linked by means of glutaraldheyde and tripolyphosphate exhibited swelling degree as low as 50%.12 The swelling degree was 18.77, 35.23, 83.87, and 218.07% for Ch-CA-Gx (x = 0, 1, 2, and 3), respectively. This obvious difference was attributed to the weaker intermolecular interactions with the increase of G concentration, which was in line with the increased water vapor permeability when glycerin content increased. The model polymer was completely soluble in 1% acetic acid aqueous solution, but the Ch-CA-Gx membranes retained around 40% weight after 1 week soaking in 1% acetic acid aqueous solution, further evidencing that the presence of cross-linked bonds improved the water resistance of membrane matrix.
2.5. Film Morphology and Structure
The scanning electron microscopy (SEM) images of membrane surfaces are compared in Figure 10. When the percentage of glycerol increased in Ch-CA-Gx membranes, the distinctive morphological differences were identified, indicating the non-negligible effect of the glycerol content on microstructure. The nanoscale phase separation was clearly observed in Ch-CA-G2 and Ch-CA-G3 samples. The average roughness of surface in Ch-CA-Gx membranes (x = 1, 4.2 nm; x = 2, 8.5 nm; and x = 3, 16.9 nm) increased with increasing glycerin content, as shown in Figure 11.
Figure 10.

SEM images of Ch-CA-G0 (a1, a2), Ch-CA-G1 (b1, b2), Ch-CA-G2 (c1, c2), and Ch-CA-G3 (d1, d2) surface.
Figure 11.

Atomic force microscopy (AFM) micrographs (80 × 80 μm2) of the surface of Ch-CA-G0 (a), Ch-CA-G1 (b), Ch-CA-G2 (c), and Ch-CA-G3 (d) membrane.
Citric acid was expected to form ionically cross-linked chitosan–citrate complex. The particular arrangement of chitosan chains can occur during vacuum drying, where the hydrophobic polysaccharide skeleton oriented toward the surface and the hydrophilic −NH3+ COO– groups oriented inward in the chitosan–citrate complex.7 Compared to the cationic chitosan acetate, the relatively hydrophobic and cross-linked chitosan–citrate complex was considered to have worse compatibility with glycerin, thus leading to the discontinuity of membrane and obvious nanostructures in SEM. The more serious phase separation with increased glycerin content was in line with the change trend of the weaker mechanical and water-resisting properties of membranes when glycerin content increased.
To further visualize the microstructure, the Ch-CA-G0 sample was immersed in water for 48 h. The sample got swollen and the unbound citric acids got dissolved in water during the immersion. Nanoscale chitosan–citrate particles consisting of cross-linked chitosan chains were clearly observed on the surface and cross section part of membrane, as shown in Figure 12. In addition, the freeze-died Ch-CA-G1 and Ch-CA-G3 samples after soaking in deionized water for 48 h showed the obvious occurrence of nanoscale chitosan–citrate particles. Recent studies associated with the micro- and nanoparticles of chitosan concentrated on the application of the sodium tripolyphosphate and sodium sulfate as cross-linkers.34 However, few studies have explored the CA application in fabricating micro- or nanostructure of chitosan in the fields of membranes, fibers, and gels. Figure 13 shows the X-ray diffraction (XRD) patterns of the Ch-CA-Gx membranes. The diffraction peak at about 19.6° corresponding to the characteristic diffraction of crystallized chitosan without chemical modification became very broad, indicating a decrease of crystallinity. The results confirmed that the citric acid and glycerin can efficiently reduce the crystallinity of chitosan–citric membranes.24
Figure 12.

SEM images corresponding to the surface (a) and cross section (b) of Ch-CA-G0. SEM images of freeze dying Ch-CA-G1 (c) and Ch-CA-G3 (d) surface after immersion in water.
Figure 13.
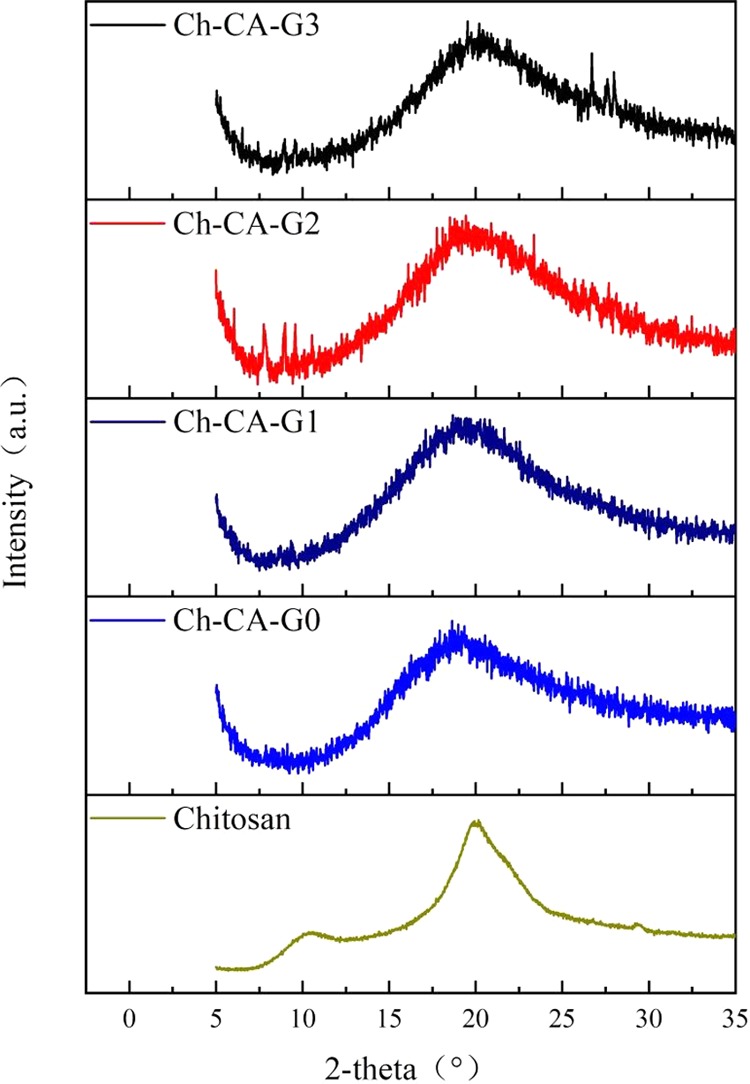
XRD patterns of Ch-CA-Gx (x = 0–3) membranes and chitosan powder without chemical modification.
2.6. Oxygen Barrier Characteristics
The oxygen permeability (OP) of cross-linked membranes was evaluated under 40 and 80% relative humidity, as shown in Table 2. The oxygen permeability of Ch-CA-G0 at 40% RH was 23.73 cm3 μm m–2 day–1, which slightly increased to 28.80 cm3 μm m–2 day–1 at 80% RH. In addition, the oxygen transmission rates (OTRs) for Ch-CA-G0 in different RH conditions showed similar values (∼0.07 cm3 m–2 day–1), indicating that the cross-linked structure of unplasticized Ch-CA-G0 can reduce the moisture sensitivity. However, the plasticized Ch-CA-Gx (x = 1–3) presented obvious moisture sensitivity with increase of glycerin content. The OTR value of Ch-CA-G1 increased from the value below detection limit to 0.07 cm3 m–2 day–1 when the RH raised from 40 to 80%. The OTR value of Ch-CA-G3 at 80% RH (∼0.79 cm3 m–2 day–1) showed a significant improvement relative to the value at 40% RH (∼0.07 cm3 m–2 day–1). The results were consistent with the dependence of WVP, swelling degree, and mechanical properties on the glycerin extent. For Ch-CA-Gx membranes, their OTR results at 40% RH were comparable to those of some high oxygen barrier membranes based on synthetic polymers, such as polyamide (PA) and poly(vinylidene chloride) (PVDC) (around 40 and 26.5 cm3 m–2 day–1 at 23 °C and 0% RH in 25 μm thickness, respectively). High oxygen barrier capacities of membranes were probably assigned to the strong attractive interactions occurring through hydrogen-bonding, ionic, and covalent cross-linking, thus increasing the diffusion length for oxygen,35−40 as shown in Figure 14. Xiang et al. successfully made a stretchable gas barrier thin film by means of hydrogen-bonding assemblies between poly(ethylene oxide) and tannic acid.41 The oxygen barrier and elasticity properties for the application in food packaging and protecting coating can also be improved through CA-involved hydrogen-bonding, ionic, and covalent cross-linking in the polymer matrix.
Table 2. Oxygen Transmission Rate (OTR, cm3 m–2 day–1) and Oxygen Permeability (OP, cm3 μm m–2 day–1) of Ch-CA-Gx Membranes under the Conditions of 40 and 80% Relative Humidity, and 100% Oxygen at 23 °C.
| samples | OTR at 40% RH | OPb at 40% RH | OTR at 80% RH | OPb at 80% RH |
|---|---|---|---|---|
| Ch-CA-G0 | 0.07 | 23.73 | 0.07 | 28.80 |
| Ch-CA-G1 | a | a | 0.07 | 505.48 |
| Ch-CA-G2 | a | a | 1.03 | 545.95 |
| Ch-CA-G3 | 0.02 | 12.48 | 0.79 | 453.24 |
The OTR values of these materials were below the detection limit.
The OP value is equal to the value (OTR value × film thickness).
Figure 14.

Diagram of the intramolecular hydrogen-bonding interaction.
2.7. Antimicrobial Activity
To explore the potential antimicrobial activity of membranes, antibacterial measurements against E. coli and S. aureus bacteria were assessed by using a reference method.33 As illustrated in Figure 15, complete inhibition of the growth of E. coli and S. aureus bacterial can be observed. The polycationic chitosan and citric acids were probably the bioactive compounds assigned to the antibacterial abilities of the films. Considering the large number of citric acids used in the membranes, it was necessary to evaluate the bioactive properties of membranes after removing citric acids which were uninvolved in ionic and covalent cross-linking. The unbound citric acids can be removed by immersing membranes in water for 2 weeks. Toward S. aureus, the biocide activities of membranes declined after soaking in water, evidencing that the antibacterial activity from membranes could partially be attributed to the free citric acids, as shown in Figure 16.7 Colony counting showed that the killing efficiency of Ch-CA-Gx (x = 0, 1, 2, 3) membranes was approximately 65, 51, 95, and 82%, respectively.
Figure 15.

Inhibitory effect of Ch-CA-Gx membranes (a: Ch-CA-G0; b: Ch-CA-G1; c: Ch-CA-G2; d: Ch-CA-G3) against S. aureus (left) and E. coli (right).
Figure 16.
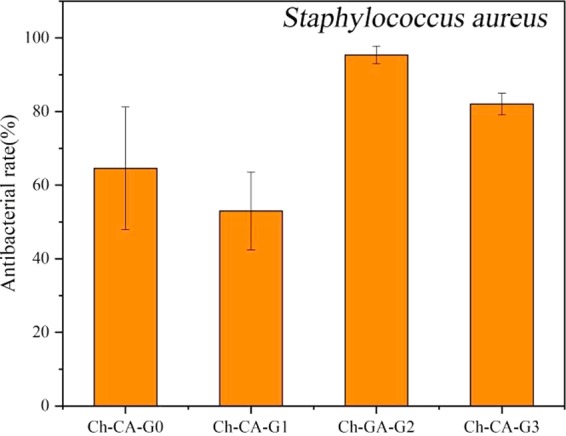
Biocidal effect of Ch-CA-Gx (x = 0–3) films against S. aureus after removing unbound citric acids.
3. Conclusions
Chitosan–citric biomembranes have been successfully prepared by a simple cross-linked strategy under the condition of vacuum drying at 80 °C. The successful cross-linking by means of amidation and the Maillard reaction under the condition of vacuum drying can be probably applied as a green and potential method for the fabrication of mechanically tough membranes, fibers, and gels. The results demonstrated that the mechanical properties, water resistance, microstructure, and oxygen barrier capabilities of membranes showed strong dependence on the content of glycerin. The vacuum drying at 80 °C can facilitate amidation and the Maillard reaction in the membrane matrix, thus further improving their water resistance. Chitosan membranes showed an interestingly high oxygen barrier capability under 40 and 80% RH conditions, probably attributed to the cross-linked structure formed by the strong interactions occurring through hydrogen-bonding and covalent cross-linking. The OTR values of Ch-CA-Gx were below 0.1 cm3 m–2 day–1 under 40% RH. The Ch-CA-Gx membranes exhibited good elasticity, indicating the existence of the reversibly cross-linked network. The membranes presented effective antimicrobial activities against E. coli and S. aureus bacteria. The results demonstrated the potential application of these cross-linked materials as membranes or coatings for food packaging.
4. Materials and Methods
4.1. Materials
The commercial chitosan (Mw, 176 kDa) was purchased from Sinopharm Chemical Reagent Co., Ltd. The deacetylation degree of chitosan powder at 91% was determined by 1H NMR and XPS methods. Glycerin with a purity of 99.0% (Beijing Huayitai Biotechnology Co., Ltd.), citric acid with a purity of 99.5% (J&K Scientific Co., Ltd.), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, J&K Scientific) were utilized without purification. S. aureus and E. coli used for the antibacterial experiments were obtained from the School of Food Science and Technology in Beijing University of Agriculture.
4.2. Preparation of the Ch-CA-Gx Membranes
The abbreviation Ch-CA-Gx (x = the volume of glycerin) was adopted for the chitosan–citric films. As shown in Figure 1, the general procedure for the fabrication of Ch-CA-G1 membrane is as follows: citric acid (4.0 g) and glycerin (1 mL) were dissolved in deionized water (100 mL). After thorough mixing, chitosan (2.5 g) was added to the mixed solution at 40 °C for 4 h to ensure the dissolving of chitosan. Then, the mixture was poured into a glass plate for film casting and dried at 25 °C for 2 days. The dried film was placed in a vacuum-drying oven for 12 h under the conditions of 80 °C and vacuum degree of 0.1 atm to obtain Ch-CA-G1 film. The glycerol weight corresponded to 50% of the weight of chitosan. The chitosan/citric acid ratio stayed the same for all membranes at 2.5/4 (weight ratio, w/w). The procedures for preparing other membranes were the same as described for Ch-CA-G1. The glycerol weight corresponded to 50, 100, and 150% of the weight of chitosan for Ch-CA-G1, Ch-CA-G2, and Ch-CA-G3. The temperature of the vacuum-drying oven was also set at 100, 80, 60, and 40 °C to evaluate the temperature dependence of amidation and the Maillard reaction. The Ch-CA-Gx (x = 0–3) membranes were kept at 25 °C and 50% RH for 72 h before their characterization.
4.3. Synthesis of the Model Polymer
Citric acid can be successfully attached to the chitosan backbone to fabricate the model polymer, according to the reference method.42 EDC (4.0 g) dissolved in 50 mL of solution containing deionized water and ethanol (1:1, v/v) was slowly dropped to the deionized water solution (50 mL) containing chitosan (2.5 g) and citric acid (4.0 g). The reaction mixture was stirred with pH value maintained at 5.0 at room temperature. After 12 h, the solution was dialyzed (molecular weight cutoff: 3500, union carbide) in deionized water (pH 5.0) for 2 days and in deionized water for 4 h to clean out unbound CA. The dialysis ended until no CA residue was detected by means of the electrospray ionization mass spectrometry (ESI-MS) spectrum. The degree of amidation at about 10% was determined by XPS and 1H NMR.
4.4. Instrumentation and Methods
1H NMR spectroscopy was performed on a Bruker (300 M) spectrometer. Sample was dissolved in D2O solution containing 1% CD3COOD. Fourier transform infrared (FTIR) spectra were acquired by means of a Nicolet 6700 spectroscopy analyzer in the scanning range of 4000–400 cm–1. X-ray photoelectron spectroscopy (XPS) survey was performed by a Thermo Fischer XPS spectrometer (ESCALAB 250Xi) with a monochromatic Al Kα X-ray source operating at 12.5 kV and 16 mA. Thermogravimetric analysis (TGA) was collected from a TGA (SDT Q600 V20.9 Build 20) thermogravimetric analyzer at a 20 °C min–1 heating rate in the range of 20–600 °C. The surface images of the membranes were recorded on an atomic force microscope (Oxford Cypher VRS, U.K.). Wide-angle X-ray diffraction (WXRD) measurements were carried out by means of a Rigaku RINT-2200V diffractometer at 20 °C and in reflection mode, and the intensity profiles of diffraction were collected in the range of 5–40°. Scanning electron microscopy (SEM) was applied to investigate the morphologies of surface and cross section of dry films by means of scanning electron microscopy (JEOL Tsm-6700F, JP). Oxygen transmission rate (OTR) testing was carried out by means of the Oxtran 2/21 ML apparatus at 23 °C and 40 or 80% RH. The mechanical properties were investigated by means of a texture analyzer (MTS Criterion model C43.104). A 500 mm min–1 strain rate was used in all experiments, and data were collected in triplicate for each sample. A micrometer was utilized to assess membrane thickness with an accuracy of 1 μm, and the mean thickness was determined by 10 measurements for each sample. Viscoelastic measurements were carried out by means of a Physica MCR302 (Anton Paar) rotary rheometer. The frequency sweeps in viscoelastic analysis were carried out in the range of 0.1–100 rad s–1 at 60 °C with 0.1% elongation. Water vapor permeability (WVP) tests were carried out by W303 water vapor transmission rate tester (GBPI Packaging Test Instrument Co., China). The film samples with the diameter of 80 mm were sealed on a water-containing permeation cell. The cell was placed on a balance in a cavity equipped with a desiccator facilitating water absorbance. The cell was weighed at different time intervals, and the WVP value (g m–2 day–1) was evaluated by the equation of WVP = Δm/AΔt, where Δm is the weight reduction (g) and A is the exposed area (m2) for the time Δt (day) at 37 °C. Swelling measurements were carried out in water. Swollen hydrogels were weighed after they were taken out from the water and wiped dry. The swelling percentage was calculated according to the following equation: DS = [(Weq – Wd)/Wd] × 100%, where Wd is the initial weight of the dried membrane and Weq is the saturated weight of membranes. The solubility was determined by comparison between the residual weight (W1) of membranes after soaking in water for 48 h and starting weight (W2) of membranes according to the equation S = [(W1 – W2)/W2] × 100%. All experiments were conducted in triplicate at room temperature.
4.5. Antibacterial Assessment
Both sides of the Ch-CA-Gx (x = 0–3) films were respectively sterilized under UV for 15 min. The membrane was cut into round films with a diameter of 14 mm, and round films were placed at the bottom of each well in a 24-well plate. Bacterial fluid (50 μL) was inoculated on the surface of membranes (105 CFU mL–1, CFU: colony-forming unit). The bacterial fluid cultured in the well in the absence of chitosan–citric membrane was considered as a control sample. Then, the plate was placed in a constant-temperature incubator at 37 °C for 4 h. The membranes with bacterial inoculated were washed using phosphate-buffered saline (PBS, 1 mL, pH 7.4) under the condition of ultrasonic treatment for 75 s. A bacterial solution (50 μL) was incubated for 24 h at 37 °C by Lysogeny broth (LB) agar plates. Viable counts were estimated by the plate count technique in colony-forming units (CFU mL–1), and all antibacterial experiments were carried out in triplicate. The antibacterial rate (I) was evaluated by the following equation: I (%) = [(N1 – N2)/N1] × 100%, where N1 is the colony count (CFU mL–1) in control experiment and N2 is the colony count in the presence of membranes (CFU mL–1).
Acknowledgments
This work was funded by the National Science Foundation of China (Nos. 21772014 and 21372029) and Beijing Laboratory of Food Quality and Safety.
Author Contributions
§ L.Z. and X.Z. are co-first authors.
The authors declare no competing financial interest.
References
- Synowiecki J.; Al-Khateeb N. A. Production, Properties, and Some New Applications of Chitin and Its Derivatives. Crit. Rev. Food Sci. Nutr. 2003, 43, 145–171. 10.1080/10408690390826473. [DOI] [PubMed] [Google Scholar]
- Dash M.; Chiellini F.; Ottenbrite R. M.; Chiellini E. Chitosan-A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. 10.1016/j.progpolymsci.2011.02.001. [DOI] [Google Scholar]
- Dutta P. K.; Tripathi S.; Mehrotra G. K.; Dutta J. Perspectives for chitosan based antimicrobial films in food applications. Food Chem. 2009, 114, 1173–1182. 10.1016/j.foodchem.2008.11.047. [DOI] [Google Scholar]
- Marsh K.; Bugusu B. Food Packaging-Roles, Materials, and Environmental Issues. J. Food Sci. 2007, 72, R39–R55. 10.1111/j.1750-3841.2007.00301.x. [DOI] [PubMed] [Google Scholar]
- Sébastien F.; Stéphane G.; Copinet A.; Coma V. Novel biodegradable films made from chitosan and poly(lactic acid) with antifungal properties against mycotoxinogen strains. Carbohydr. Polym. 2006, 65, 185–193. 10.1016/j.carbpol.2006.01.006. [DOI] [Google Scholar]
- Ma Y.; Zhou T.; Zhao C. Preparation of chitosan–nylon-6 blended membranes containing silver ions as antibacterial materials. Carbohydr. Res. 2008, 343, 230–237. 10.1016/j.carres.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Möller H.; Grelier S.; Pardon P.; Coma V. Antimicrobial and Physicochemical Properties of Chitosan-HPMC-Based Films. J. Agric. Food Chem. 2004, 52, 6585–6591. 10.1021/jf0306690. [DOI] [PubMed] [Google Scholar]
- Clasen C.; Wilhelms T.; Kulicke W. M. Formation and Characterization of Chitosan Membranes. Biomacromolecules 2006, 7, 3210–3222. 10.1021/bm060486x. [DOI] [PubMed] [Google Scholar]
- Sacco P.; Borgogna M.; Travan A.; Marsich E.; Paoletti S.; Asaro F.; Grassi M.; Donati I. Polysaccharide-Based Networks from Homogeneous Chitosan-Tripolyphosphate Hydrogels: Synthesis and Characterization. Biomacromolecules 2014, 15, 3396–3405. 10.1021/bm500909n. [DOI] [PubMed] [Google Scholar]
- Moura M. J.; Faneca H.; Lima M. P.; Gil M. H.; Figueiredo M. M. In Situ Forming Chitosan Hydrogels Prepared via Ionic/Covalent Co-Cross-Linking. Biomacromolecules 2011, 12, 3275–3284. 10.1021/bm200731x. [DOI] [PubMed] [Google Scholar]
- Chaudhary J. P.; Vadodariya N.; Nataraj S. K.; Meena R. Chitosan-Based Aerogel Membrane for Robust Oil-in-Water Emulsion Separation. ACS Appl. Mater. Interfaces 2015, 7, 24957–24962. 10.1021/acsami.5b08705. [DOI] [PubMed] [Google Scholar]
- Kyzas G. Z.; Bikiaris D. N.; Lazaridis N. K. Low-Swelling Chitosan Derivatives as Biosorbents for Basic Dyes. Langmuir 2008, 24, 4791–4799. 10.1021/la7039064. [DOI] [PubMed] [Google Scholar]
- Shen X. P.; Shamshina J. L.; Berton P.; Gurauc G.; Rogers R. D. Hydrogels based on cellulose and chitin: fabrication, properties, and applications. Green Chem. 2016, 18, 53–75. 10.1039/C5GC02396C. [DOI] [Google Scholar]
- Watthanaphanit A.; Supaphol P.; Furuike T.; Tokura S.; Tamura H.; Rujiravanit R. Novel Chitosan-Spotted Alginate Fibers from Wet-Spinning of Alginate Solutions Containing Emulsified Chitosan-Citrate Complex and their Characterization. Biomacromolecules 2009, 10, 320–327. 10.1021/bm801043d. [DOI] [PubMed] [Google Scholar]
- Franklin D. S.; Guhanathan S. Investigation of citric acid–glycerol based pH-sensitive biopolymeric hydrogels for dye removal applications: A green approach. Ecotoxicol. Environ. Saf. 2015, 121, 80–86. 10.1016/j.ecoenv.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Spinella S.; Maiorana A.; Qian Q.; Dawson N. J.; Hepworth V.; McCallum S. A.; Ganesh M.; Singer K. D.; Gross R. A. Concurrent Cellulose Hydrolysis and Esterification to Prepare a Surface-Modified Cellulose Nanocrystal Decorated with Carboxylic Acid Moieties. ACS Sustainable Chem. Eng. 2016, 4, 1538–1550. 10.1021/acssuschemeng.5b01489. [DOI] [Google Scholar]
- Tomé L. C.; Silva N. H. C. S.; Soares H. R.; Coroadinha A. S.; Sadocco P.; Marruchoa I. M.; Freire C. S. R. Bioactive transparent films based on polysaccharides and cholinium carboxylate ionic liquids. Green Chem. 2015, 17, 4291–4299. 10.1039/C5GC00416K. [DOI] [Google Scholar]
- Galvis-Sánchez A. C.; Sousa M. M.; Hilliou L.; Gonçalvesa M. P.; Souza H. K. S. Thermo-compression molding of chitosan with a deep eutectic mixture for biofilms development. Green Chem. 2016, 18, 1571–1580. 10.1039/C5GC02231B. [DOI] [Google Scholar]
- Guerrero P.; Muxika A.; Zarandona I.; de la Caba K. Crosslinking of chitosan films processed by compression molding. Carbohydr. Polym. 2019, 206, 820–826. 10.1016/j.carbpol.2018.11.064. [DOI] [PubMed] [Google Scholar]
- Cui Z.; Beach E. S.; Anastas P. T. Modification of chitosan films with environmentally benign reagents for increased water resistance. Green Chem. Lett. Rev. 2011, 4, 35–40. 10.1080/17518253.2010.500621. [DOI] [Google Scholar]
- Nowakowska M.; Moczek Ł.; Szczubiałka K. Photoactive Modified Chitosan. Biomacromolecules 2008, 9, 1631–1636. 10.1021/bm800141v. [DOI] [PubMed] [Google Scholar]
- Moczek Ł.; Nowakowska M. Novel Water-Soluble Photosensitizers from Chitosan. Biomacromolecules 2007, 8, 433–438. 10.1021/bm060454+. [DOI] [PubMed] [Google Scholar]
- Lawrie G.; Keen I.; Drew B.; Chandler-Temple A.; Rintoul L.; Fredericks P.; Grøndahl L. Interactions between Alginate and Chitosan Biopolymers Characterized Using FTIR and XPS. Biomacromolecules 2007, 8, 2533–2541. 10.1021/bm070014y. [DOI] [PubMed] [Google Scholar]
- Meng Q. K.; Heuzey M. C.; Carreau P. J. Hierarchical Structure and Physicochemical Properties of Plasticized Chitosan. Biomacromolecules 2014, 15, 1216–1224. 10.1021/bm401792u. [DOI] [PubMed] [Google Scholar]
- Ware T. H.; Perry Z. P.; Middleton C. M.; Iacono S. T.; White T. J. Programmable Liquid Crystal Elastomers Prepared by Thiol–Ene Photopolymerization. ACS Macro Lett. 2015, 4, 942–946. 10.1021/acsmacrolett.5b00511. [DOI] [PubMed] [Google Scholar]
- Hao J. K.; Weiss R. A. Mechanically Tough, Thermally Activated Shape Memory Hydrogels. ACS Macro Lett. 2013, 2, 86–89. 10.1021/mz3006389. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Peng Y. H.; Zhang L. Q.; Zhao Y.; Vyzhimov R.; Tan T. W.; Fong H. Investigation of Palm Oil as Green Plasticizer on the Processing and Mechanical Properties of Ethylene Propylene Diene Monomer Rubber. Ind. Eng. Chem. Res. 2016, 55, 2784–2789. 10.1021/acs.iecr.5b04527. [DOI] [Google Scholar]
- Lu H. L.; Wang W. B.; Wang A. Q. Ethanol–NaOH solidification method to intensify chitosan/poly(vinyl alcohol)/attapulgite composite film. RSC Adv. 2015, 5, 17775–17781. 10.1039/C4RA09835H. [DOI] [Google Scholar]
- Lu B. L.; Lin F. C.; Jiang X.; Cheng J. J.; Lu Q. L.; Song J. B.; Chen C.; Huang B. One-Pot Assembly of Microfibrillated Cellulose Reinforced PVA–Borax Hydrogels with Self-Healing and pH-Responsive Properties. ACS Sustainable Chem. Eng. 2017, 5, 948–956. 10.1021/acssuschemeng.6b02279. [DOI] [Google Scholar]
- Yoon S. D. Cross-Linked Potato Starch-Based Blend Films Using Ascorbic Acid as a Plasticizer. J. Agric. Food Chem. 2014, 62, 1755–1764. 10.1021/jf4024855. [DOI] [PubMed] [Google Scholar]
- Liu X. L.; Xia W. S.; Jiang Q. X.; Xu Y. S.; Yu P. P. Synthesis, Characterization, and Antimicrobial Activity of Kojic Acid Grafted Chitosan Oligosaccharide. J. Agric. Food Chem. 2014, 62, 297–303. 10.1021/jf404026f. [DOI] [PubMed] [Google Scholar]
- Singha N. R.; Karmakar M.; Chattopadhyay P. K.; Roy S.; Deb M.; Mondal H.; Mahapatra M.; Dutta A.; Mitra M.; Roy J. S. D. Structures, Properties, and Performances—Relationships of Polymeric Membranes for Pervaporative Desalination. Membranes 2019, 9, 58 10.3390/membranes9050058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi F. L.; Huang C. T.; Liang H. F.; Chen M. C.; Chiu Y. L.; Chen C. H.; Sung H. W. Physicochemical, Antimicrobial, and Cytotoxic Characteristics of a Chitosan Film Cross-Linked by a Naturally Occurring Cross-Linking Agent, Aglycone Geniposidic Acid. J. Agric. Food Chem. 2006, 54, 3290–3296. 10.1021/jf0529868. [DOI] [PubMed] [Google Scholar]
- Ma Z. X.; Garrido-Maestua A.; Jeonga K. C. Application, mode of action, and in vivo activity of chitosan and its micro and nanoparticles as antimicrobial agents: A review. Carbohydr. Polym. 2017, 176, 257–265. 10.1016/j.carbpol.2017.08.082. [DOI] [PubMed] [Google Scholar]
- Laufer G.; Kirkland C.; Cain A. A.; Grunlan J. C. Clay–Chitosan Nanobrick Walls: Completely Renewable Gas Barrier and Flame-Retardant Nanocoatings. ACS Appl. Mater. Interfaces 2012, 4, 1643–1649. 10.1021/am2017915. [DOI] [PubMed] [Google Scholar]
- Way A. E.; Hsu L.; Shanmuganathan K.; Weder C.; Rowan S. J. pH-Responsive Cellulose Nanocrystal Gels and Nanocomposites. ACS Macro Lett. 2012, 1, 1001–1006. 10.1021/mz3003006. [DOI] [PubMed] [Google Scholar]
- Liu A. D.; Walther A.; Ikkala O.; Belova L.; Berglund L. A. Clay Nanopaper with Tough Cellulose Nanofiber Matrix for Fire Retardancy and Gas Barrier Functions. Biomacromolecules 2011, 12, 633–641. 10.1021/bm101296z. [DOI] [PubMed] [Google Scholar]
- Liu C.; Thormann E.; Claesson P. M.; Tyrode E. Surface Grafted Chitosan Gels. Part II. Gel Formation and Characterization. Langmuir 2014, 30, 8878–8888. 10.1021/la501319r. [DOI] [PubMed] [Google Scholar]
- Yang Y. H.; Haile M.; Park Y. T.; Malek F. A.; Grunlan J. C. Super Gas Barrier of All-Polymer Multilayer Thin Films. Macromolecules 2011, 44, 1450–1459. 10.1021/ma1026127. [DOI] [Google Scholar]
- Yang Q. L.; Fukuzumi H. K.; Saito T.; Isogai A.; Zhang L. N. Transparent Cellulose Films with High Gas Barrier Properties Fabricated from Aqueous Alkali/Urea Solutions. Biomacromolecules 2011, 12, 2766–2771. 10.1021/bm200766v. [DOI] [PubMed] [Google Scholar]
- Xiang F. M.; Givens T. M.; Ward S. M.; Grunlan J. C. Elastomeric Polymer Multilayer Thin Film with Sustainable Gas Barrier at High Strain. ACS Appl. Mater. Interfaces 2015, 7, 16148–16151. 10.1021/acsami.5b04500. [DOI] [PubMed] [Google Scholar]
- Rimondino G. N.; Strumia M. C.; Martinelli M. Synthesis and Characterization of Water-Soluble Dendronized Chitosan Using Newkome-Type Dendrons. ACS Sustainable Chem. Eng. 2014, 2, 2582–2587. 10.1021/sc500476k. [DOI] [Google Scholar]