

Abstract

Sodium dodecyl sulfate (SDS)·1/8 hydrate (NaC12H25SO4·1/8H2O) crystals were successfully produced by evaporation, antisolvent addition, cooling crystallization, and isothermal aging in a common stirred tank. A clear 33.3 wt % SDS aqueous solution was concentrated by evaporation to a 60 wt % coagel consisting of numerous SDS hydrates and water. The coagel was transformed to a clear solution when two times the volume of acetone relative to the water remaining were added. By this fluid property, a controlled crystallization was made possible in a homogeneous solution. Moreover, acetone with a water-to-acetone volume ratio of 1:15 was then added as an antisolvent to induce crystallization of SDS·1/8 hydrate by cubic addition. Finally, cooling crystallization and isothermal aging were carried out to further increase the yields and gave monodispersed particle size. The stability test showed that the produced SDS·1/8 hydrate could be stored at various relative humidity environments for at least 5 days.
INTRODUCTION
To better engineer the crystal attributes for manufacturability in downstream processes, solution crystallization can be induced by supersaturation through three well-known approaches: evaporation, antisolvent addition, and cooling. Evaporation is one of the convenient ways to concentrate the solution. As the concentration of the solution keeps increasing, the supersaturation will induce crystallization of the solute. Consequently, evaporative crystallization is often combined with other separation processes to reduce equipment requirement.1 Despite the operational advantages of evaporative crystallization, there is a caveat. By employing evaporative crystallization, crystal attributes, including polymorphism, crystallinity, and particle size distribution (PSD), are hard to achieve reproducibly upon scaling up.2,3 The crystallization process is also easily influenced by the uncontrollably grown crystals acting as seeds having inconsistent PSD due to the descent of the mother liquid level. Antisolvent crystallization can be operated at the isothermal condition, which is of paramount importance for heat-sensitive substances aside from convenience and economical aspects. The desired PSD may be achieved by controlling the addition position and rate of the antisolvent.4,5 However, the binary solvent mixture must be subsequently separated to recover and recycle one or both solvents, and the capacity of the vessel is taken up by the additional volume of the antisolvent. On the other hand, cooling crystallization is a popular technique for the production of high-value chemicals.6 Temperature control is a critical factor for determining the crystallization rate by cooling crystallization. A low crystallization rate can be achieved by a slow cooling rate, which gives a relatively large metastable zone width (MSZW) beyond the solubility. Accordingly, a high degree of supersaturation is required to induce crystallization. Slow cooling could increase the product purity because of fewer lattice defects and reduced crystal agglomeration.
Sodium dodecyl sulfate (SDS) is the most common sulfate surfactant and a key component of many domestic cleaning, personal hygiene, cosmetic, pharmaceutical, and food products, as well as of industrial and commercial cleaning and product formulations. SDS is prepared by first reacting dodecyl alcohol (dodecanol) with concentrated sulfuric acid (or with sulfur trioxide gas, oleum, or chlorosulfuric acid)7 and then neutralizing dodecyl sulfate with sodium hydroxide aqueous solution. Since water is generated as a byproduct8 and SDS is highly soluble in water,9 the result is an SDS aqueous solution. Based on our previous experiences, foaming could occur at boiling upon evaporation in a stirred tank, and phases such as micelles, liquid crystals, and coagels would form as the concentration of SDS aqueous solution increases at a given temperature, complicating the nucleation step control. Moreover, SDS has several hydrates other than the anhydrous form,10 such as 1/8 hydrate,11 hemihydrate,12 monohydrate,13 and dihydrate.14 An additional form, called the “α phase”, nearly identical to 1/8 hydrate, has also been reported.15,16 The α phase, produced by slow evaporation of SDS from a 95% ethanol aqueous solution, has a monoclinic Cc or C2/c structure.17 However, the single-crystal structure of the dihydrate SDS solid has not been established, and various kinds of SDS hydrates have only been prepared and analyzed in a small scale by growing the solids in chloroform-methanol cosolvents,11 water through cooling,18,19 or ethanolic aqueous solution by slow evaporation from a saturated solution at a constant temperature.15 These methods can only be utilized for crystallography and are inappropriate for mass production.
Interestingly, the formation of hydrates is affected by the solvent composition, temperature, pressure, and relative humidity (RH) range.20,21 Hydrates can be described in terms of their stoichiometry relative to the host molecule, and they are often classified as stoichiometric or nonstoichiometric. Stoichiometric hydrates have a well-defined water content, and the crystal structure is clearly different from that of other solid forms.22 In comparison to the stoichiometric hydrate, nonstoichiometric hydrates can vary in water content without distinguishable changes in the crystal structure because of the existence of the so-called channel (open structural voids).23 Upon dehydration, water in the channel can be removed without substantially affecting the crystalline structure. The void spaces due to dehydration can also be filled reversibly while still retaining the initial crystalline structure. Various hydrate solid forms possess different properties of thermodynamics, kinetics, and surfaces,24 which are especially important for active pharmaceutical ingredients. In general, knowledge of the water content of hydration is necessary for determining the equivalent weight and dosage amount in formulation.
Commercially, SDS solids are produced by spray-drying.25 Water in the sprayed droplets of the SDS aqueous solution is rapidly evaporated off with a hot gas, and fine powders are made instantly. However, the hydrate solid forms in the fine powders are not well controlled, and the resultant fine powders need to be enlarged for ease of handling into cylindrical granules by an additional step of extrusion.26 Although it was shown that cooling crystallization of the 20 wt % SDS–water micellar solution could give a much larger particle size, this single-solvent process by linear cooling was not robust enough because the production of either SDS monohydrate platelets or SDS hemihydrate needles was sensitive to the cooling ramp and the final temperature.19 Interestingly, commercial SDS solids are sold as a mixture of hydrate forms rather than a specific hydrate. The large-scale production of specific SDS hydrate solids has never been reported in the literature. The complicated evaporation process during spray-drying may easily make the quality of its crystallization product go out of control. The instability of the hydrate forms also makes long-time storage difficult. Nevertheless, a mixture of crystal forms was unfavorable at all times for manufacturing or practical applications due to its inconsistent physical and chemical properties. Stability, molecular weight, dissolution rate, hardness, toughness, and morphology of SDS particles would be greatly affected as the hydrate structure varies. High purity should be pursued not only in chemical structures but also in the crystal form.
Although crystallization and phase transformation among different SDS hydrates at various temperatures have been studied,17−19,27,28 the systems were constrained in an aqueous solution and the drying behaviors of the hydrate crystals have not been investigated. The SDS·1/8 hydrate was believed to be stable in air.28 Therefore, the aim of this paper is to develop a robust crystallization protocol in a common stirred tank for readily making large particles of a stable SDS·1/8 hydrate solid form from an SDS aqueous solution reproducibly. The crystallization of the SDS·1/8 hydrate would be broken down into four steps: evaporation, antisolvent addition, cooling, and isothermal aging. Notably, the crystallization know-how learned from SDS may also be applied to some other highly water-soluble zwitterionic sulfonic acid biological buffering agents, which are also synthesized in aqueous media.29
Results and Discussion
Construction of Form Space
Solvents play an important role in controlling yield, purity, solubility, polymorphism, and crystal habit and in selecting the appropriate crystallization process. Choosing a suitable solvent for dissolution and crystallization is essential. Form space30 is a useful table to indicate rapidly whether a solvent is good or bad in terms of its solubility power and show clearly the mutual relationship between any two solvents. A solvent is classified as a pure good solvent when the solubility value of SDS in that solvent at 25 °C is higher than or equal to 5 mg/mL; otherwise, it was considered a pure bad solvent in our present study. Actually, the criterion of 5 mg/mL can be changed to 1 mg/mL or less, 10 mg/mL or more, at 25 °C depending on the volume of the crystallizer, the process, and the nature of the final product. According to the results summarized in the form space (Table 1), SDS had five pure good solvents: N,N-dimethylformamide (DMF), ethanol, dimethyl sulfoxide (DMSO), methanol, and water, as represented by the yellow grids, and 18 pure bad solvents: n-heptane, xylene, p-xylene, ethyl acetate, toluene, methyl tert-butyl ether (MTBE), benzene, methyl ethyl ketone (MEK), chloroform, tetrahydrofuran (THF), N,N-dimethylaniline (DMA), acetone, 1,4-dioxane, nitrobenzene, n-butyl alcohol, isopropyl alcohol (IPA), benzyl alcohol, and acetonitrile, as marked by the red grids. A good solvent is commonly used as a medium for solution crystallization. Its high solubility power can provide a sensible ratio between the solvent and solute for efficient utilization of volume capacity of a crystallizer to give a relatively high yield of product. It should be noted that it is insufficient to select the solvent candidate for crystallization merely by the form space. Other factors, such as the slope of the solubility vs. temperature curve, chemical reactivity, toxicity, and cost, would have to be taken into account. By integrating the information above, a solvent database is constructed for selecting a suitable solvent and designing a crystallization process. For example, the good solvent that displays a high-temperature-dependent solubility behavior to the desired product can be employed for cooling crystallization due to the high theoretical yield.21
Table 1. Form Space of SDS at 25 °C and 1 atm.

The form space in Table 1 is diagonally symmetrical. There are 10 good cosolvent pairs represented by the blue grids, 72 antisolvent pairs designated by the green grids, and 20 immiscible pairs indicated by the gray grids. The bad solvent miscible with the good solvent can play the role of either an antisolvent for crystallization or a rinsing solvent for filter cake washing. An antisolvent is usually used for (1) inducing crystallization under isothermal condition, (2) combining with cooling or evaporative crystallization to further enhance the yields, and (3) modifying the solubility of the solute by other reasons.31 In general, a pure bad solvent, a bad cosolvent system, and an immiscible solvent system are rarely considered for crystallization. As a result, the criterion can be set to less than 5 mg/mL at 25 °C if the good solvents are so few in number that it is difficult to find a suitable solvent medium.
Since water is the reaction medium, a byproduct for SDS synthesis in the final step, and environmentally benign, it is chosen as the good solvent employed out of the five good solvents from Table 1 in the preparation of the SDS·1/8 hydrate to simulate the real situation after reaction, whereas acetone was chosen as the antisolvent out of the 18 bad solvents from Table 1 because of its low cost, slight toxicity, and water miscibility.32
Determination of the End Point for Evaporation
Since SDS was highly water-soluble, it was difficult to get a high yield by using the single solution crystallization method. The phase diagram of the SDS–water binary system also exhibited that the SDS·1/8 hydrate was generated in a narrow region having a very low water content.14 Evaporation was used first to enhance the concentration of the SDS solution for preparing the SDS·1/8 hydrate. However, the high viscosity caused by the high concentration of SDS led to poor mixing. To overcome the unwanted solution thickening, an antisolvent was introduced after evaporation. As a result, the end point of evaporation had to be determined beforehand. At first, evaporation was applied to concentrate the clear, free-flowing 33.3 wt % SDS aqueous solution, which was close to saturation.14 The photo images taken after evaporation for SDS–water systems having final compositions of 55, 60, 65, and 70 wt % with different addition volumes of acetone based on acetone-to-water volume ratios of 0:1, 2:1, 3:1, and 5:1 relative to the volumes of water remaining in the vials of 2.45, 2.00, 1.62, and 1.29 mL, respectively, at 25 °C are summarized in Table 2. It is clearly seen that the coagel was formed on the SDS aqueous solution surface for all SDS–water systems at 55, 60, 65, and 70 wt %. The opaque and inhomogeneous coagel phase comprised water and SDS hydrates.14,33 The micellar solution phase, liquid crystalline phase, coagel, and hydrate were illustrated in the phase diagram by Kékicheff et al.14 Given the concentration of SDS between 37 and 87 wt % at 25 °C, the coagel phase will develop in water, which agrees with our experimental observation. It is well known that good mixing is critical for the crystallization process. Uniform crystal quality and narrow PSD could then be achieved in the common stirred tank during scale-up upon efficient momentum, heat, and mass transfer. Any presence of the tacky coagel could hinder the free flow of the SDS solution and make the mixing very difficult.
Table 2. Phase behaviors of the SDS aqueous solution at the end of evaporation and after acetone addition.

Interestingly, the already-formed coagel phase dissolved and turned into a clear, free-flowing SDS solution when two times the volumes of acetone 4.9 and 4.0 mL relative to the water remaining in the vials of 2.45 and 2 mL were added into the SDS coagel in vial nos. 1 and 4, respectively. The clear, free-flowing SDS solution became turbid when the volume ratios of water to acetone were adjusted to 1:3 and 1:5 in vial nos. 2–3 and 5–6, respectively (Table 2). When the SDS aqueous solutions were concentrated to 65 or 70 wt %, the coagel phases did not disappear even when two times the volume of acetone of 3.24 and 2.58 mL relative to the water remaining in the vials of 1.62 and 1.29 mL were added in vial nos. 7 and 10, respectively. Vial nos. 8–9 and 11–12 illustrated that more and more solids precipitated out as the volume of acetone was increased. Therefore, a 60 wt % SDS–water system was selected to be the end point for the evaporation step, and two times the volume of acetone relative to the residual volume of water would need to be added to achieve a homogeneous phase for carrying out the controllable crystallization process.
Determination of the Final Composition of the Water–Acetone Cosolvent for Preparing the SDS·1/8 Hydrate
Although the hydrate form and micelle structure were investigated a lot in the literature, most of the cases were constrained in the SDS–water binary system. The crystallization and phase transition of the SDS hydrate in the SDS–water–acetone ternary system had never been reported. The composition of the antisolvent was also a significant factor for crystallization; therefore, the final volume ratio of water to acetone for the production of SDS·1/8 hydrate solids at 5 °C was determined in our present study. Figure 1 displays the powder X-ray diffraction (PXRD) patterns for the commercial SDS cylindrical granules and the commercial SDS cylindrical granules aged in water–acetone cosolvents with water-to-acetone volume ratios of 1:10, 1:17, and 1:25. The PXRD patterns of the commercial SDS powders and the aged SDS cylindrical granules looked nearly the same. However, the thermogravimetric analysis (TGA) scans of the commercial SDS cylindrical granules and the aged SDS powders in Figure 2 showed otherwise. All SDS samples exhibited two steps in weight loss regardless of the composition of the aging solvent used. The slight weight loss before 120 °C resulted from dehydration (inset in Figure 2), and the significant weight loss from 200 to 325 °C can be attributed to the decomposition of the SDS molecule. The weight loss from dehydration for each sample is listed in Table 3. The commercial SDS cylindrical granules had a weight loss of 1.64 wt %, and all aged SDS commercial cylinder granules had a weight loss of 0.74–0.75 wt % before 120 °C.
Figure 1.

PXRD patterns of (a) commercial SDS cylindrical granules and commercial SDS cylindrical granules aged in a cosolvent with volume ratios of water to acetone of (b) 1:10, (c) 1:17, and (d) 1:25 at 5 °C for 8 h.
Figure 2.

TGA scans for commercial SDS cylindrical granules before and after aging in a cosolvent with volume ratios of water to acetone of 1:10, 1:17, and 1:25 at 5 °C for 8 h.
Table 3. Weight Loss of Commercial SDS Cylindrical Granules Before and After Aging in a Cosolvent with Different Volume Ratios of Water to Acetone by Heating from 40 to 150 °C by TGA.
| SDS samples | weight loss (%) |
|---|---|
| commercial SDS powder | 1.64 |
| aged in 1:10 cosolvent | 0.74 |
| aged in 1:17 cosolvent | 0.75 |
| aged in 1:25 cosolvent | 0.75 |
The three SDS hydrate forms of 1/8 hydrate, hemihydrate, and monohydrate gave the theoretical weight fractions of water of 0.77, 2.95, and 5.58 wt %, respectively. According to the definition of hydrate, the host molecule and the water molecule have a definite stoichiometry within the crystal lattice. The weight loss arising from dehydration of a pure SDS hydrate should correspond to the weight percent of water originally present in the SDS hydrate. A weight loss of 0.77% was predicted for the SDS·1/8 hydrate crystal during dehydration. Obviously, all of the aged SDS powders were SDS·1/8 hydrates because of the weight loss of close to 0.77% as determined by TGA, whereas the commercial SDS was a mixture of hydrates composed primarily of SDS·1/8 hydrate and a small amount of hemihydrate and/or monohydrate. Figure 3 shows that the differential scanning calorimetry (DSC) scans of the commercial SDS cylindrical granules and the aged SDS powders exhibited more or less the same dehydration endotherm around 103 °C and melting point around 196 °C, indicating that all dehydrated samples have the same crystal structure.
Figure 3.

DSC scans of (a) commercial SDS cylindrical granules and commercial SDS cylindrical granules aged in a cosolvent with volume ratios of water to acetone of (b) 1:10, (c) 1:17, and (d) 1:25 at 5 °C for 8 h.
In summary, the SDS·1/8 hydrate could be produced by aging the commercial SDS cylinder granules in the water–acetone cosolvents with the water-to-acetone volume ratios ranging from 1:10 to 1:25. XRD and TGA could discriminate a specific hydrate form from the others or their mixtures. From the standpoint of optimizing the product yield from solution crystallization, seemingly, the more antisolvent added, the better. The water-to-acetone volume ratio of 1:25 would have been the best choice. However, the large volume of antisolvent added would take up the crystallizer volume, which could have been fed by more SDS aqueous solution. Consequently, we decided to operate at the water-to-acetone volume ratio of 1:17, which was between 1:10 and 1:25; this would also give an operation cushion for unexpected volume fluctuation during scale-up.
Crystallization Process of the SDS·1/8 Hydrate
Figure 4 illustrates the temperature versus time profile for the crystallization process of SDS·1/8 hydrate crystals. First, the 33.3 wt % SDS saturated solution was heated from 25 to 70 °C and evaporated at a constant temperature of 70 °C. Considering the low boiling point of acetone, acetone was added as the temperature was decreased to 25 °C. The cooling ramp was about 1 °C/min, which was controlled by the water bath. The large amount of acetone would give a high degree of supersaturation for SDS and induce crystallization. Then, the resulting SDS suspension was further cooled to 5 °C with a cooling ramp of 0.66 °C/min and further aged isothermally for 8 h. The composition of SDS in wt % in the glass stirred tank was deduced by weighing the evaporated water condensed and collected in the round-bottomed receiver every 30 min during evaporation, and the temperature profile was monitored for the whole 13 h-long crystallization process.
Figure 4.
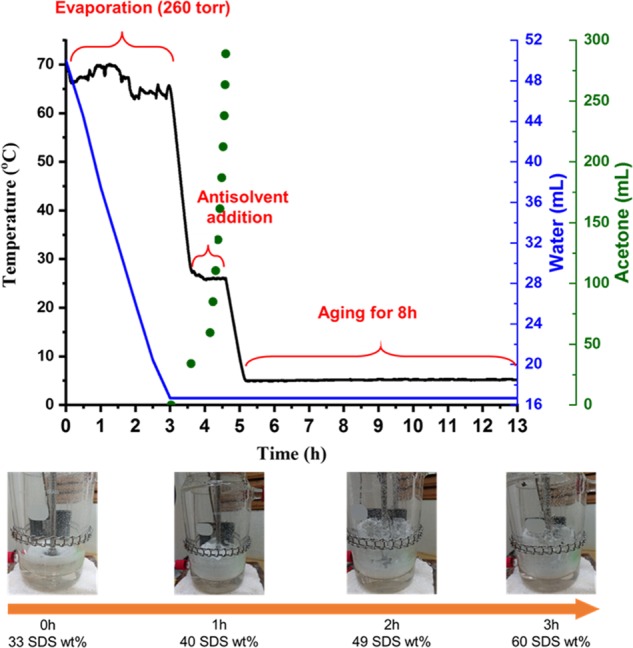
Temperature, volume of water, and volume of acetone vs. time profile of the crystallization process of the SDS·1/8 hydrate.
To speed up the evaporation process, the 33.3 wt % SDS aqueous solution was heated to 70 °C under agitation of 200 rpm at 260 torr. Because the saturated vapor pressure of water at 70 °C is 235 torr, to avoid solution boiling and minimize foaming, the operating pressure of 260 torr was set above 235 torr. Intensive foaming was troublesome since the SDS soap bubbles could move up the stirred tank and go down to the receiver. The same problem would occur if the agitation rate was too high and a lot of air was blown through the solution. The 33.3 wt % SDS aqueous solution was quite viscous in the beginning, and then slight foaming occurred at 36 wt % in 0.5 h due to the decrease in water surface tension. As the concentration went up to 44 wt %, the SDS–water system became more and more tacky, and finally the coagel was formed. By the end of 3 h, the 60 wt % SDS–water system was cooled to 25 °C, and the pressure was recovered to 760 torr. The temperature fluctuation between 0 and 3 h in Figure 4 was caused by water evaporation. Evaporation is an endothermic process, and the temperature of the SDS solution was reduced when water was vaporized. However, the vacuum was temporarily broken from time to time for weighing the water inside the receiver while the hot water jacket still supplied heat to the SDS solution; the temperature of the SDS solution was then increased. As a consequence, the periodic vacuum shutdown had led to the noisy temperature profile during evaporation.
The volume of acetone was determined as 289 mL, which was 17 times the volume of residual water from the previous evaporation experiment. However, if the total volume of acetone was poured in all at once, the already-formed SDS coagel and the newly generated crystals from antisolvent crystallization would coexist. This would make the crystallization process go out of control, resulting in the inconsistency of yield, purity, crystal habit, and PSD of the final SDS hydrate product. The uncontrollable crystallization process was disadvantageous in batch-to-batch reproducibility and scale-up performance. It should be noticed that the particular proportion of acetone to water was found in the end-point determination experiment for the evaporation process (Table 2). The tacky SDS coagel disappeared and became clear upon the introduction of acetone, whose volume was two times the volume of the remaining water. It was much better to divide the addition of acetone into two portions. After adding the first portion of 34 mL of acetone, which was twofold that of 17 mL of the remaining water, at 25 °C, the coagel lumps dissolved and turned into a clear, free-flowing tertiary SDS–water–acetone solution. The drawback brought about by evaporative crystallization could be overcome by this redissolution. The second portion of 255 mL of acetone was then fed by cubic addition at 25 °C. The addition strategy of the antisolvent is an important factor in antisolvent crystallization. Linear addition of the antisolvent is commonly used in antisolvent crystallization. It causes a high level of supersaturation initially but decreases gradually with time. In other words, the level of supersaturation varied with time. The other strategy was nonlinear addition; the addition rate of the antisolvent was increased with time to offer a roughly constant change of the supersaturation level.34 This method could minimize the nucleation rate, so that the promoted concentration was controlled within the metastable zone, leading to a growth-dominant process. Cubic addition is one of the examples of nonlinear addition and has been utilized in our present process. Finally, the SDS slurry at 25 °C was cooled to 5 °C to produce more crystals and aged in the cosolvent comprising 17 mL of water and 289 mL of acetone with the water-to-acetone volume ratio of 1:17 at 5 °C for 8 h. The SDS·1/8 hydrate crystals became larger, and the PSD was narrowed by Ostwald ripening.
The crystal habit and stoichiometry of the produced SDS crystals were monitored by optical microscopy (OM) and TGA after the introduction of acetone by the cubic addition method, respectively, as demonstrated in Table 4. The SDS crystals started to nucleate and grow immediately once acetone was fed. Thin and flaky plates of produced SDS crystals18 grew gradually with time from 50 μm after the addition of the first vial of 25.5 mL of acetone at t = 0 min to about 100–150 μm at the end upon the addition of the tenth vial of 25.5 mL of acetone at t = 27. 0 min. Remarkably, the weight loss of the produced SDS crystals harvested at t = 0 min had already reached 0.77 wt %, which matched well with the theoretical amount of water in the SDS·1/8 hydrate. The same TGA weight loss of 0.77 wt % was maintained for the produced SDS crystals collected after the addition of the tenth vial of 25.5 mL of acetone at t = 27.0 min. These results prove that antisolvent crystallization could produce SDS·1/8 hydrate crystals that are stable in the water–acetone cosolvent system. The produced SDS·1/8 hydrate crystals more or less stayed at 150 μm and became monodispersed after aging for 4 and 8 h at 5 °C (Table 4). The final yield of the SDS·1/8 hydrate crystals was about 83%. Apparently, the SDS crystal had a tendency to become SDS·1/8 hydrate in a water–acetone cosolvent system at both 5 and 25 °C. Changing the temperature for acetone addition from 25 to 5 °C might be feasible. However, the substantial temperature drop would lead to a large solubility difference and a high level of supersaturation. The number of nuclei explodes suddenly, which was disadvantageous for PSD control. In addition, the poor mixing resulting from the coagel phase should be overcome first, or else, the heat exchange in the crystallizer would be hindered. For these given reasons, cooling was done after antisolvent addition even though it was possible to harvest the SDS·1/8 hydrate if those two steps were reversed. The second cooling step from 25 to 5 °C not only enhanced the yield of SDS·1/8 hydrate crystals but also facilitated the growth of SDS·1/8 hydrate crystals.
Table 4. Optical Images and Weight Fractions of Water of the Produced SDS Crystals at Different Statuses Determined by TGA.

The assignments of Fourier transform infrared (FTIR) spectroscopy bands for SDS·1/8 hydrate crystals made from the crystallization process are shown and compared in Figure 5: the 3470 cm–1 band was assigned to −OH stretching; the 2957, 2873, 2848, and 1468 cm–1 peaks corresponded to −CH2 stretching and bending modes, which were characteristic of a polar environment; the 1213 cm–1 peak was assigned to skeletal vibration involving the bridge S–O stretch; the 1083 cm–1 peak was attributed to C–C band stretching; and the 837 and 592 cm-1 peaks were attributed to asymmetric C–H bending of the CH2 group.35 The PXRD pattern of our produced SDS·1/8 hydrate crystals and the theoretical PXRD patterns of the SDS anhydrous form, the SDS·1/8 hydrate, the SDS hemihydrate, and the SDS monohydrate are shown in Figure 6. The PXRD pattern of the produced SDS·1/8 hydrate crystals matched very well with the theoretical one of the SDS·1/8 hydrate. The GC chromatograms in Figure S1 of the Supporting Information (SI) indicated that no acetone remained in our produced SDS·1/8 hydrate crystals.
Figure 5.

FTIR spectrum of the produced SDS·1/8 hydrate crystals.
Figure 6.

PXRD pattern of (a) the produced SDS·1/8 hydrate, and theoretical patterns of (b) anhydrous SDS from CCDC (database identifier: VECYOR01), (c) SDS·1/8 hydrate from CCDC (database identifier: DODSUL), (d) SDS hemihydrate from CCDC (database identifier: COYGEC10), and (e) SDS monohydrate from CCDC (database identifier: ZZZMAI01).
Moisture Stability Test
The hydrate forms could transform mutually with the change of RH, temperature, and pressure.36 It was difficult to evaluate the stability among the various SDS hydrate forms under solvent-free and ambient conditions because the dry powders were not obtainable. Nevertheless, our produced SDS·1/8 hydrate could be stable upon oven-drying at 40 °C in air. To further determine the influence of humidity on the SDS·1/8 hydrate, the moisture stability test for our produced SDS·1/8 hydrate crystals was performed in the present study. Our produced SDS·1/8 hydrate crystals were stored in three different humidity conditions of 25, 52, and 75% RH at 25 °C for 5 days. The TGA scans in Figure 7 demonstrate that SDS·1/8 hydrate crystals subjected to three different %RH values exhibited the same weight loss of around 0.75 wt %. The PXRD patterns in Figure 8 also indicated that all of our produced SDS·1/8 hydrate samples exposed to the three different %RH values looked identical to the PXRD pattern of the original SDS·1/8 hydrate crystals. The wide range of %RH illustrated that the water content of the SDS·1/8 hydrate was not altered by moisture. It was reasonable to deduce that the SDS·1/8 hydrate was more stable than the other hydrate forms at various %RH, 25 °C, and 1 atm.
Figure 7.

TGA scans of the produced SDS·1/8 hydrate crystals subjected to (a) 25%, (b) 52%, and (c) 75% RH at 25 °C for 5 days.
Figure 8.

PXRD patterns of (a) our produced SDS·1/8 hydrate and (b) our produced SDS 1/8 hydrate subjected to (b) 25%, (c) 52%, and (d) 75% RH at 25 °C for 5 days.
Moreover, the SDS powders were dehydrated to evaluate the stability of anhydrous SDS. The commercial SDS sample was first dehydrated by heating in TGA from 40 to 150 °C and then holding isothermally for 5 min under a nitrogen purge. The weight loss of the samples was followed. The TGA dehydrated samples were then characterized immediately by PXRD after the temperature was returned to room temperature to minimize the contact time with air. In Figure 9a,b, the first and second scans of the commercial SDS sample showed weight losses of 1.45 and 0.76 wt %, respectively, indicating that the commercial SDS sample was a mixture of hydrates initially. The SDS sample was then recovered to the mixture of hydrates even though it had been dehydrated previously. Right after the first dehydration for the TGA dehydrated commercial SDS sample (Figure 10a), the diffraction pattern exhibited diffraction peaks of SDS hemihydrate with characteristic 2θ = 5.6 and 8.5° (Figure 10d). The dehydrated SDS crystals, presumably the anhydrous form, were instable upon air contact and showed the tendency of rehydration again. It implied that the SDS·1/8 hydrate was more stable than anhydrous SDS at 25 °C in air. Therefore, the production of SDS·1/8 hydrate was favorable not only for the reproducibility of SDS structural purity and equivalent weight determination but also for the storage quality of SDS.
Figure 9.

TGA scans of commercial SDS cylinder granules at the (a) first TGA heating and (b) second TGA heating.
Figure 10.

PXRD patterns of (a) commercial SDS cylinder granules after the first heating, (b) anhydrous SDS from CCDC (database identifier: VECYOR01), (c) SDS·1/8 hydrate from CCDC (database identifier: DODSUL), (d) SDS hemihydrate from CCDC (database identifier: COYGEC10), and (e) SDS monohydrate from CCDC (database identifier: ZZZMAI01).
Conclusions
To date, SDS has commonly been applied in the areas of detergents, excipients, cosmetics,37 and gas hydration.38 The water present in the crystal lattice of the host molecule was critical for influencing the physical and chemical properties of the solid state such as morphology, hardness, dissolution rate, and toughness, leading to inconsistent quality of the final products. Therefore, it is paramount to make the pure stable hydrate in a large scale reproducibly. In the present study, the form space of SDS has been constructed, which can be utilized as an important reference for determining the solvents used in synthesis, crystallization, and separation processes. SDS·1/8 hydrate crystals were successfully produced by evaporation, antisolvent addition, and cooling in a common stirred tank. A clear, free-flowing 33.3 wt % SDS aqueous solution was concentrated by evaporation to a 60 wt % SDS–water binary system containing the tacky coagel, which was determined as the end point. Acetone that was two times the volume relative to the water remaining was added to dissolve the coagel. The newly formed tertiary SDS–water–acetone system had turned clear again. By this property, a controlled crystallization was made in a homogeneous solution. Acetone was then added to the system at 25 °C until the water-to-acetone volume ratio in the stirred tank had reached 1:17 as an antisolvent to induce crystallization by cubic addition. The cubic addition strategy of acetone and isothermal aging can not only enlarge the crystal size of SDS but also give a narrow PSD. The produced platelike SDS·1/8 hydrate crystals were verified by TGA and PXRD and had a size of 150 μm as characterized by OM. In summary, a robust and reproducible protocol has been developed by combining evaporation, antisolvent addition, and cooling crystallization in a common stirred tank for readily controlling the particle size and the hydration stoichiometry of a stable form of SDS hydrates grown from an SDS aqueous solution. The moisture stability test showed that commercial SDS cylindrical granules became a mixture of SDS hydrates rapidly again after dehydration. Moreover, the produced SDS·1/8 hydrate could be stored at a relatively high %RH environment at 25 °C for at least 5 days.
Materials and Methods
Materials
SDS cylindrical granules (≥99% purity, CMC, 8.2 mM at 25 °C; Lot, STBG9926) were purchased from Sigma-Aldrich (China).
Solvents
n-Heptane (99% purity; Lot, EA8351), toluene (≥99.5% purity; Lot, ETA140403), chloroform (99.99% purity; Lot, E554180), THF (99% purity; Lot, E704198), ethanol (99.5% purity; Lot, 262611), acetone (99.5% purity; Lot, EHAD105), and DMSO (99.8% purity; Lot, EA3M741) were purchased from Echo Chemical Co. Ltd. (Taiwan). Xylene (98.5% purity; Lot, E03B34) and 1,4-dioxane (98% purity; Lot, SP-34332R) were purchased from Avantor Performance Materials Co. Ltd. p-Xylene (99% purity, Lot, 802551), ethyl acetate (99.5% purity; Lot, 711912), MTBE (99.9% purity; Lot, 1005355), benzene (99% purity; Lot, 310008), MEK (99.6% purity; Lot, SMEI40421), n-butyl alcohol (99.4% purity; Lot, 205027), IPA (99.8% purity; Lot, 14030108), benzyl alcohol (99.8% purity; Lot, 70950), acetonitrile (99.96% purity; Lot, 14050419), DMF (99.8% purity; Lot, 912313), and methanol (99.9% purity; Lot, 14050368) were purchased from TEDIA company. DMA (99% purity; Lot, A0213203001), and nitrobenzene (99% purity; Lot, A0282673) were purchased from Acros Organics company. Reversible osmosis water was clarified by a water purification system (model Milli-RO Plus) bought from Millipore (Billerica, MA).
Construction of Form Space
Form space consisted of six parts: pure good solvents, pure bad solvents, good cosolvents, bad cosolvents, antisolvent (i.e., good-solvent–bad-solvent pairs), and immiscible pairs. Twenty-three common solvents, n-heptane, xylene, p-xylene, ethyl acetate, toluene, MTBE, benzene, MEK, chloroform, THF, DMA, acetone, 1,4-dioxane, nitrobenzene, n-butyl alcohol, IPA, benzyl alcohol, acetonitrile, DMF, ethanol, DMSO, methanol, and water, were used for the construction of form space of SDS at 25 °C and 1 atm. The solvents were classified as pure good solvents, pure bad solvents, good cosolvents, bad cosolvents, antisolvents, and immiscible pairs in accordance with their solubility to SDS. These six parts would be displayed as grids by different colors and formed a 23 × 23 table. Solvents that gave the solubility value of SDS larger than 5 mg per mL were defined as pure good solvents, or else as pure bad solvents. The pure solvent system was represented as diagonal grids in the form space. The binary solvent systems mixed by two good solvents, two bad solvents, and one good and one bad solvent were classified as good cosolvents, bad cosolvents, and Antisolvent-good-solvent system, respectively. The immiscible solvent pair indicated that the solvent mixture was immiscible, for example, water and n-heptane.
The procedure for constructing the form space was followed.30 About 10–30 mg of SDS powder sample was weighed in 7 mL scintillation vials, and the 23 solvents were added dropwise into the vials individually with intermittent shaking at 25 °C until SDS solids were totally dissolved as observed manually.
Determination of the End Point for Evaporation
This process aimed to simulate and determine the end point for the concentration step of SDS aqueous solution by evaporation. Twelve 100 mL scintillation vials, with each containing 33.3 wt % SDS aqueous solution by dissolving 3 g of commercial SDS cylindrical granules in 6 mL of water at 25 °C, were prepared. All 12 clear 33.3 wt % SDS aqueous solutions were then evaporated under 760 torr at a constant temperature of 70 °C by a water bath until the final compositions of the SDS aqueous solutions of vial nos. 1–3, 4–6, 7–9, and 10–12 had become 55, 60, 65, and 70 wt % SDS–water systems, respectively, which were equal to the water volumes remaining of 2.45, 2.00, 1.62, and 1.29 mL, respectively. All 12 vials were then cooled to 25 °C. Two times the volume of acetone of 4.90, 4.00, 3.24, and 2.58 mL relative to the water volume remaining were introduced into vial nos. 1, 4, 7, and 10 of 55, 60, 65, and 70 wt % SDS–water systems with agitation at 25 °C, respectively. Three times the volume of acetone of 7.35, 6.00, 4.86, and 3.87 mL relative to the water volume remaining were added into vial nos. 2, 5, 8, and 11 of 55, 60, 65, and 70 wt % SDS–water systems with agitation at 25 °C, respectively. Five times the volume of acetone of 12.25, 10.00, 8.10, and 6.45 mL relative to the water volume remaining were added into vial nos. 3, 6, 9, and 12 of 55, 60, 65, and 70 wt % SDS–water systems with agitation at 25 °C, respectively. If a given wt % of the SDS aqueous solution could form a slurry or coagel upon evaporation and be turned into a free-flowing clear solution by acetone addition at the same time, that particular wt % of the SDS aqueous solution and the acetone volume were determined as the end-point conditions for evaporation. The compositions of vial nos. 1–12 and the flowchart are shown in Table S1 and Figure S2 in the SI, respectively.
Determination of the Final Composition of the Water–Acetone Cosolvent for Preparing the SDS·1/8 Hydrate by Aging
First, 0.5 g of commercial SDS cylindrical granules was immersed into a water–acetone cosolvent with different water-to-acetone volume ratios at 25 °C for 8 h. The volume ratios of water to acetone are listed in Table S2 of the SI. After 8 h of aging, the immersed SDS solids were filtered, oven-dried at 40 °C for 8 h, and characterized by PXRD, TGA, and DSC to determine the water-to-acetone ratio, which gave the SDS·1/8 hydrate.
Crystallization Process of the SDS·1/8 Hydrate
Considering the unpredictability in engineering the crystal quality by evaporative crystallization, evaporation at high temperature and under vacuum was only used for concentrating the SDS aqueous solution up to an end point where a thick coagel was formed. A specific organic solvent was then identified and added to the coagel at room temperature at a given volume ratio to turn the binary SDS–water gelation system into a clear ternary SDS–water–solvent solution system for better flowing and mixing. An extra amount of the same organic solvent was then slowly added to the clear SDS aqueous solution as an antisolvent at room temperature at a particular volume ratio of water to solvent for carrying out the antisolvent crystallization step mainly for controlling the SDS hydrate form and its PSD. Cooling crystallization, which goes from room temperature down to a lower temperature at which point it is kept for a while, was employed primarily for maximizing the yield and increasing the monodispersity of the desired hydrate crystals. The experimental setups are demonstrated in Figure S3. Commercial SDS cylindrical granules of weight 25 g were dissolved in 50 mL of water in a 500 mL glass stirred tank at 25 °C and 200 rpm under 760 torr to prepare a 33.3 wt % SDS aqueous solution. The stirred tank had an inner diameter and height of 8.0 and 17 cm, respectively, and was installed with a four-bladed 45° impeller having a diameter of 3.5 cm. The distance between the impeller and the tank bottom was set to 1 cm. The SDS aqueous solution was then evaporated at 70 °C by a water bath and 200 rpm under a partial pressure of 260 torr with minimum foaming until the volume of water remaining decreased to 17 mL. Once the composition of the resulting SDS slurry had reached 60 wt % for about 3 h, the evaporation was stopped, which was the predetermined end point for evaporation. The composition of the SDS solution was monitored and deduced by weighing the water distillate collected in the round-bottomed receiver. The solution was then cooled to 25 °C, and the pressure was returned to 760 torr. Next, 34 mL of acetone, which was two times the volume of 17 mL of water remaining in the 60 wt % concentrated SDS–water system, was introduced into the glass stirred tank at a constant addition rate of 1 mL/s and agitation rate of 200 rpm to make a free-flowing clear solution ready for the next antisolvent crystallization step.
First, 255 mL of acetone was added at slower rates in the beginning and then at higher rates over the time period of 27 min into the solution by a cubic addition method at 25 °C and under an agitation of 200 rpm to crystallize the SDS·1/8 hydrate. Next, 255 mL of acetone, which was 15 times the volume of the 17 mL of water remaining in the concentrated SDS aqueous system, was divided into 10 equal portions of 25.5 mL each. The time intervals for the addition of each portion through the addition funnel into the glass stirred tank were shortened over time at 0, 5.0, 9.5, 13.5, 17.0, 20.0, 22.5, 24.5, 26.0, and 27.0 min. The accumulated volume of acetone addition with time by cubic addition is displayed in Figure S4.
Finally, the SDS–water-containing acetone solution was cooled to 5 °C in 30 min, and the SDS·1/8 hydrate crystals were further aged in the water–acetone cosolvent at 5 °C for 8 h. The SDS·1/8 hydrate crystals were filtered, oven-dried at 40 °C for 8 h, and characterized by OM, FTIR, TGA, and PXRD.
Moisture Stability Test
Three open 20 mL scintillation vials, each containing 0.5 g of SDS·1/8 hydrate crystals were placed separately in three tightly capped 100 mL scintillation vials filled with saturated MgCl2, Mg(NO3)2, and NaCl aqueous solutions at 25 °C, providing 25, 52, and 75% RH conditions, respectively, for 5 days. Afterward, the SDS·1/8 hydrate crystals exposed to different RH conditions were characterized by TGA and PXRD.
The commercial SDS cylindrical granules were dehydrated in TGA by heating to 150 °C at a heating ramp of 10 °C/min, holding isothermally for 5 min, and then cooling back to 40 °C. The dehydrated SDS sample was characterized by PXRD as soon as possible to minimize the time for air contact. Moreover, the dehydrated SDS sample was then characterized by TGA again.
Instruments
OM
The crystal habits and PSD of harvested SDS solids were observed by Olympus BX-51 (Tokyo, Japan) equipped with a digital camera (Hong Kong, China) Moticam 2000. The obtained images were transformed by Motic Image Plus (version 2.0) into digital photographs. Analysis of the photographs was done by ImageJ 1.51 g software equipped with Microscope Measurement Tools v1 plugin.
FTIR
FTIR spectroscopy was utilized to verify the chemical structure of SDS. FTIR spectroscopy was conducted on Perkin Elmer Spectrum One (Norwalk, CT). Each sample was ground with KBr powders with a weight ratio of about 1 to 100 in an agate mortar; then, a hydraulic press was used to turn the powder mixture into a pellet. The pellet was scanned 8 times with a resolution of 2 cm–1 ranging from 4000 to 400 cm–1.
TGA
TGA was employed to determine the dehydration temperature and drying temperature. TGA analysis was conducted by Perkin-Elmer TGA Pyris 1 (Norwalk, CT) to monitor the weight loss of the SDS sample as a function of temperature. The stoichiometric ratios of water to SDS in the hydrates were calculated from the weight loss. Samples of 5–10 mg weight were placed on the open platinum pan suspended in a heating furnace. All samples were heated under a nitrogen atmosphere to avoid oxidization. The heating ramp was 10 °C/min ranging from 40 to 350 °C.
DSC
DSC was used to measure the dehydration temperature, enthalpy of dehydration, solid–solid and solid–liquid transition temperatures, and enthalpies of solid–solid and solid–liquid transitions. Thermal analytical data of samples in perforated aluminum sample pans were collected by PerkinElmer DSC 7 (Shelton, CT) at a temperature scanning rate of 10 °C/min from 40 to 210 °C under a constant nitrogen purge of 99.990%.
PXRD
PXRD was used to determine the crystal forms of the samples by comparing the PXRD patterns of the samples with references obtained from Cambridge Crystallography Data Center (CCDC). PXRD patterns of SDS samples were obtained by Bruker D8 Advance (Karlsruhe, Germany). The source of PXRD was Cu Kα (λ = 1.5418 Å), and the diffractometer was operated at 40 keV and 25 mA passing through a nickel filter. Samples were subjected to PXRD analysis with a sampling width of 0.01° in a continuous mode at a scanning rate of 2°/min over an angular range from 2θ = 5 to 35°.
Acknowledgments
This research was supported by the grant from the Ministry of Science and Technology of Taiwan ROC (MOST 104-2221-E-008-070-MY3 and 107-2221-E-008-037-MY3). We are greatly indebted to Ms. Li Fan Chen for her assistance in TGA and DSC and to Mr. Chin-Chuan Huang for his assistance in PXRD and the technical support in the Precision Instrument Center at National Central University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03067.
Compositions of vial nos. 1–12 in the experiments for end-point determination for evaporation; cosolvent compositions for preparing SDS·1/8 hydrate by aging; flowchart and apparatuses for crystallization experiments of SDS·1/8 hydrate in a common stirred tank; plot for accumulated volume of acetone addition versus time by the cubic addition method; GC chromatograms for the produced SDS·1/8 hydrate (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tung H.-H.; Paul E. L.; Midler M.; McCauley J. A.. Evaporative Crystallization, In Crystallization of Organic Compounds: An Industrial Perspective; John Wiley & Sons: Hoboken, New Jersey, 2009; p 167. [Google Scholar]
- Coquerel G. Crystallization of Molecular Systems from Solution: Phase Diagrams, Supersaturation and Other Basic Concepts. Chem. Soc. Rev. 2014, 43, 2286–2300. 10.1039/C3CS60359H. [DOI] [PubMed] [Google Scholar]
- Lee T.; Hung S. T.; Kuo C. S. Polymorph Farming of Acetaminophen and Sulfathiazole on a Chip. Pharm. Res. 2006, 23, 2542–2555. 10.1007/s11095-006-9078-2. [DOI] [PubMed] [Google Scholar]
- Shariare M.; de Matas M.; York P. Effect of Crystallization Conditions and Feedstock Morphology on the Aerosolization Performance of Micronized Salbutamol Sulphate. Int. J. Pharm. 2011, 415, 62–72. 10.1016/j.ijpharm.2011.05.043. [DOI] [PubMed] [Google Scholar]
- Hu T. T.; Zhao H.; Jiang L. C.; Le Y.; Chen J. F.; Yun J. Engineering Pharmaceutical Fine Particles of Budesonide for Dry Powder Inhalation (DPI). Ind. Eng. Chem. Res. 2008, 47, 9623–9627. 10.1021/ie0715052. [DOI] [Google Scholar]
- Tavare N.Industrial Crystallization: Process Simulation, Analysis and Design; Plenum Publishing: New York, 1995; pp 1–564. [Google Scholar]
- Takei K.; Tsuto K.; Miyamoto S.; Wakatsuki J. Anionic Surfactants: Lauric Products. J. Am. Oil Chem. Soc. 1985, 62, 341–347. 10.1007/BF02541402. [DOI] [Google Scholar]
- Katz D. A.The Science of Soaps and Detergents. 2019, http://www.chymist.com/Soap%20and%20detergent.pdf (accessed July 10, 2019.
- Pollard J. M.; Shi A. J.; Göklen K. E. Solubility and Partitioning Behavior of Surfactants and Additives Used in Bioprocesses. J. Chem. Eng. Data 2006, 51, 230–236. 10.1021/je0503498. [DOI] [Google Scholar]
- Smith L. A.; Hammond R. B.; Roberts K. J.; Machin D.; McLeod G. Determination of the Crystal Structure of Anhydrous Sodium Dodecyl Sulphate Using a Combination of Synchrotron Radiation Powder Diffraction and Molecular Modelling Techniques. J. Mol. Struct. 2000, 554, 173–182. 10.1016/S0022-2860(00)00666-9. [DOI] [Google Scholar]
- Sundell S. The Crystal Structure of Sodium Dodecyl Sulfate. Acta Chem. Scand. 1978, A31, 799–807. 10.3891/acta.chem.scand.31a-0799. [DOI] [Google Scholar]
- Coiro V. M.; Mazza F.; Pochetti G. Crystal Phases Obtained from Aqueous Solutions of Sodium Dodecyl Sulfate. The Structure of a Monoclinic Phase of Sodium Dodecyl Sulfate Hemihydrate. Acta Crystallogr., Sect. C: Struct. Chem. 1986, C42, 991–995. 10.1107/S0108270186093757. [DOI] [Google Scholar]
- Coiro V. M.; Manigrasso M.; Mazza F.; Pochetti G. Structure of a Triclinic Phase of Sodium Dodecyl Sulfate Monohydrate. A Comparison with Other Sodium Dodecyl Sulfate Crystal Phases. Acta Crystallogr., Sect. C: Struct. Chem. 1987, C43, 850–854. 10.1107/S010827018709382X. [DOI] [Google Scholar]
- Kékicheff P.; Grabielle-Madelmont C.; Ollivon M. Phase Diagram of Sodium Sulfate–Water System 1. A Calorimetric Study. J. Colloid Interface Sci. 1989, 131, 112–131. 10.1016/0021-9797(89)90151-3. [DOI] [Google Scholar]
- Rawlings F. F. Jr; Lingafelter E. C. The Alpha Phase of Sodium Dodecyl Sulfate. J. Am. Chem. Soc. 1950, 72, 1852. 10.1021/ja01160a521. [DOI] [Google Scholar]
- Rawlings F. F. Jr; Lingafelter E. C. X-Ray Crystallography of the Sodium n-Alkyl Sulfates. J. Am. Chem. Soc. 1955, 77, 870–872. 10.1021/ja01609a015. [DOI] [Google Scholar]
- Miller R. M.; Poulos A. S.; Robles E. S.; Brooks N. J.; Ces O.; Cabral J. T. Isothermal Crystallization Kinetics of Sodium Dodecyl Sulfate–Water Micellar Solutions. Cryst. Growth Des. 2016, 16, 3379–3388. 10.1021/acs.cgd.6b00353. [DOI] [Google Scholar]
- Smith L. A.; Duncan A.; Thomson G. B.; Roberts K. J.; Machin D.; McLeod G. Crystallization of Sodium dodecyl Sulphate from Aqueous Solution: Phase Identification, Crystal Morphology, Surface Chemistry and Kinetic Interface Roughening. J. Cryst. Growth 2004, 263, 480–490. 10.1016/j.jcrysgro.2003.11.025. [DOI] [Google Scholar]
- Miller R. M.; Ces O.; Brooks N. J.; Robles E. S. J.; Cabral J. T. Crystallization of Sodium Dodecyl Sulfate-Water Micellar Solutions under Linear Cooling. Cryst. Growth Des. 2017, 17, 2428–2437. 10.1021/acs.cgd.6b01841. [DOI] [Google Scholar]
- Otsuka M.; Matsuda Y. Effect of Environmental Humidity on the Transformation Pathway of Nitrofurantoin Modifications During Grinding and the Physicochemical Properties of Ground Products. J. Pharm. Pharmacol 1993, 45, 406–413. 10.1111/j.2042-7158.1993.tb05566.x. [DOI] [PubMed] [Google Scholar]
- Tian F.; Qu H.; Zimmermann A.; Munk T.; Jørgensen A. C.; Rantanen J. Factors Affecting Crystallization of Hydrates. J. Pharm. Pharmacol. 2010, 62, 1534–1546. 10.1111/j.2042-7158.2010.01186.x. [DOI] [PubMed] [Google Scholar]
- Braun D. E.; Griesser U. J. Stoichiometric and Nonstoichiometric Hydrates of Brucine. Cryst. Growth Des. 2016, 16, 6111–6121. 10.1021/acs.cgd.6b01231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt F. G.; Brum J.; Katrincic L. M.; Flach A.; Socha J. M.; Goodman R. M.; Haltiwanger R. C. Physical, Crystallographic, and Spectroscopic Characterization of a Crystalline Pharmaceutical Hydrate: Understanding the Role of Water. Cryst. Growth Des. 2006, 6, 2333–2354. 10.1021/cg060324k. [DOI] [Google Scholar]
- Healy A. M.; Worku Z. A.; Kumar D.; Madi A. M. Pharmaceutical Solvates, Hydrates and Amorphous Forms: A Special Emphasis on Cocrystals. Adv. Drug Delivery Rev. 2017, 117, 25–46. 10.1016/j.addr.2017.03.002. [DOI] [PubMed] [Google Scholar]
- Ramakers H.-P.-E.Alkyl Sulphate Salts. US Patent US5147633A1992.
- Muley S.; Nandgude T.; Poddar S. Extrusion–Spheronization A Promising Pelletization Technique: In-Depth Review. Asian J. Pharm. Sci. 2016, 11, 684–699. 10.1016/j.ajps.2016.08.001. [DOI] [Google Scholar]
- Smith L. A.; Thomson G. B.; Roberts K. J.; Machin D.; McLeod G. Modeling the Crystal Morphology of Alkali-metal Alkyl Surfactants: Sodium and Rubidium Dodecyl Sulfates. Cryst. Growth Des. 2005, 5, 2164–2172. 10.1021/cg058003g. [DOI] [Google Scholar]
- Sperline R. P. Infrared Spectroscopic Study of the Crystalline Phases of Sodium Dodecyl Sulfate. Langmuir 1997, 13, 3715–3726. 10.1021/la9702087. [DOI] [Google Scholar]
- Pugliese P. T.; Pugliese P. M.. Zwitterionic-Fatty Acid Compounds having Anti-Inflammatory Properties. US Patent US6448251B12002.
- Lee T.; Kuo C. S.; Chen Y. H. Solubility, Polymorphism, Crystallinity, and Crystal Habit of Acetaminophen and Ibuprofen by Initial Solvent. Screening. Pharm. Technol. 2006, 30, 72–87. [Google Scholar]
- Wieckhusen D.Development of Batch Crystallizations, In Crystallization: Basic Concepts and Industrial Applications; Beckmann, W. Wiley-VCH: Weinheim: 2013; pp 190–191. [Google Scholar]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use . ICH Q3C: Impurities: Guideline for Residual Solvents, 2016. [Google Scholar]
- Kawai T.; Umemura J.; Takenaka T. Fourier Transform Infrared Study on the Phase Transitions of a Sodium Dodecyl Sulfate-water System. Bull. Inst. Chem. Res., Kyoto Univ. 1983, 61, 314–323. [Google Scholar]
- Tung H. H.; Paul E. L.; Midler M.; McCauley J. A.. Antisolvent Crystallization. In Crystallization of Organic Compounds: An Industrial Perspective; John Wiley & Sons, Inc: Hoboken, New Jersey, 2008; pp 179–182. [Google Scholar]
- Colthup N.; Daly L.; Wiberley S.. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: New York, 1990; Vol. 332, pp 225–228. [Google Scholar]
- Redman-Furey N.; Dicks M.; Bigalow-Kern A.; Cambron R. T.; Lubey G.; Lester C.; Vaughn D. Structural and Analytical Characterization of Three Hydrates and An Anhydrate Form of Risedronate. J. Pharm. Sci. 2005, 94, 893–911. 10.1002/jps.20308. [DOI] [PubMed] [Google Scholar]
- Mészáros R.; Thompson L.; Bos M.; Varga I.; Gilányi T. Interaction of Sodium Dodecyl Sulfate with Polyethyleneimine: Surfactant-induced Polymer Solution Colloid Dispersion Transition. Langmuir 2003, 19, 609–615. 10.1021/la026616e. [DOI] [Google Scholar]
- Pan Z.; Liu Z.; Zhang Z.; Shang L.; Ma S. Effect of Silica Sand Size and Saturation on Methane Hydrate Formation in the Presence of SDS. J. Nat. Gas Sci. Eng. 2018, 56, 266–280. 10.1016/j.jngse.2018.06.018. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.