

To ensure US patients realize the public health benefit of a robust, competitive market for biosimilar products, the US Food and Drug Administration (FDA) is focused on improving the efficiency of biosimilar development and approvals. As comparative clinical studies can be costly and time consuming, the FDA is currently conducting research to inform the agency's thinking on critical aspects of the use of pharmacodynamic (PD) biomarkers to demonstrate biosimilarity, which can either streamline or negate the need for comparative clinical studies.
The Opportunity for Clinical Pharmacology in Biosimilar Development
A biosimilar is a biological product that is highly similar to and has no clinically meaningful differences from an existing FDA‐approved reference product. The enactment of the Biologics Price Competition and Innovation Act of 2009 added the 351(k) Biologics License Application pathway to the Public Health Service Act, which established an abbreviated approval pathway for the licensure of biosimilar and interchangeable biological products. Biosimilar sponsors are encouraged to use a stepwise approach to demonstrate biosimilarity, beginning with the establishment of an analytically “highly similar” foundation from comparative analytical studies (product quality), followed by animal studies and clinical studies assessing safety/immunogenicity, pharmacokinetic (PK) similarity and, when appropriate, PD similarity. Additional comparative clinical study(ies) may be necessary to address residual uncertainties that remain to support a demonstration that there are no clinically meaningful differences between the proposed biosimilar and reference product in terms of the safety, purity, and potency of the product.1
As noted above and outlined in FDA guidance documents, biosimilars may be approved based on PK and PD biomarker data without a comparative clinical study with efficacy end point(s).2 Reliance on PK and PD biomarker data allows for shorter and less costly clinical studies than can often be conducted in healthy participants. Evaluation of PK and PD similarity may have an additional advantage of being more sensitive than clinical efficacy end point(s) in detecting differences between a biosimilar and reference product, should differences exist. As an example, a quantitative analysis of clinical data from a comparison of filgrastim products showed that the PD biomarker, area under effect‐time curve of absolute neutrophil count, is in fact a more sensitive end point than the clinical efficacy end point of duration of severe neutropenia.3
While PK similarity has been evaluated in every FDA‐approved biosimilar to date, as of July 3, 2019, five of 21 approved biosimilars have included PD similarity data to support the demonstration that there were no clinically meaningful differences between the proposed biosimilar product and the reference product (i.e., filgrastim‐aafi, filgrastim‐sndz, pegfilgrastim‐jmdb, pegfilgrastim‐cbqv, and epoetin alfa‐epbx). Notably, these products have established sensitive dose‐dependent PD biomarkers (absolute neutrophil count for filgrastim and pegfilgrastim; CD34+ cells for filgrastim; reticulocyte count and hemoglobin level for epoetin alfa) that are either closely associated with or are a surrogate for clinical outcome(s). Under circumstances where an established and sensitive PD biomarker does not exist, there is an opportunity to explore and identify new PD biomarkers to facilitate the use of PD biomarker data in clinical pharmacology studies. In turn, this will expand the number of biosimilar products for which comparative clinical studies with efficacy end point(s) may not be needed to demonstrate biosimilarity.
Considerations for PD Biomarker Assessment and PK/PD Similarity Study Design
Criteria for PD biomarkers intended to support a demonstration of biosimilarity are inherently different from criteria for surrogate biomarkers used to support new drug approvals.4 Biosimilar development programs use biomarkers from PD similarity studies to address residual uncertainties and/or detect clinically meaningful differences between a proposed biosimilar and the reference product. As the purpose is to confirm similarity instead of establishing patient benefit, a correlation between the PD biomarker and clinical outcomes, while beneficial, is not a requirement. Furthermore, PD biomarkers that reflect mechanism(s) of action(s) of the biological product have the potential to be more sensitive end points for detecting clinically meaningful differences between two products. This provides opportunities for biomarkers that were used as secondary and exploratory end points in new drug development programs to play important roles in biosimilar programs. The FDA has described five characteristics for PD biomarkers (Figure 1) in guidances to assist sponsors planning to use PD biomarkers as a component of a biosimilar development program.1, 2
Figure 1.
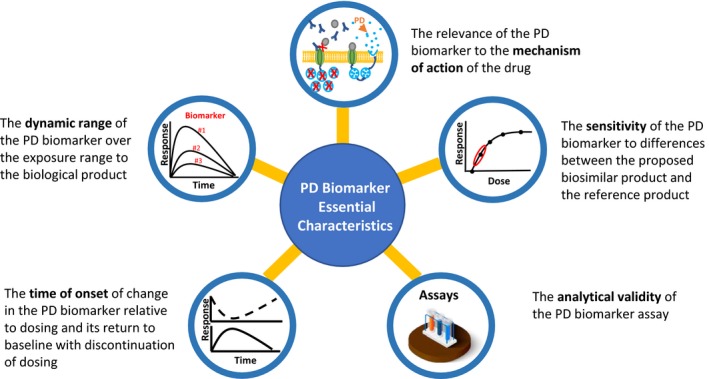
Five essential characteristics of PD (pharmacodynamic) biomarker for biosimilars. Assays image © sidmay ‐ https://stock.adobe.com.
Additional PD study considerations include the selection of study design (single dose vs. multiple dose, parallel vs. crossover), study population (healthy participants vs. patient population), and study dose. If the dose–response relationship and the sensitivity of a PD biomarker are not well defined, sponsors may need to perform a pilot study evaluating various dose levels in order to obtain this information. Additionally, if different dose levels are studied (e.g., doses at, above, and below the therapeutic‐approved dose), model‐based assessments can then be applied to aid in understanding the dynamic range of the PD biomarker, ultimately informing the subsequent PD similarity study design.
Current FDA Action on PD Biomarker Assessment for Biosimilar Development
To support the development of biosimilars and to increase scientific and regulatory clarity for the biosimilar development community, among other things, the FDA released its Biosimilars Action Plan in July 2018,5 which included the key action to create “information resources and development tools that can assist biosimilar sponsors in developing high quality biosimilar and interchangeable products using state‐of‐the‐art techniques.” Under the Biosimilars Action Plan, the FDA plans to develop a framework to support the use of PD similarity studies in biosimilar development. This framework initiative aims to outline steps to follow, including the necessary data to demonstrate suitability of PD biomarker(s), the study design (e.g., doses, study population), and other elements for PD similarity studies to support biosimilar development.
The ultimate goal of this initiative is to inform evidentiary strategies for the identification of PD biomarkers and explore study design characteristics for PD similarity studies through a range of activities. One activity involves collecting and summarizing existing information on potential PD biomarkers for different products. This information will also assist FDA staff in advising sponsors on their biosimilar development programs. In addition, in silico models and simulations to correlate PK and PD responses with measures of clinical performance will also be explored to increase confidence in relying on PD biomarkers for biosimilar determination.
Finally, pilot clinical studies with biologics selected from three scenarios are being planned. These scenarios include:
PD biomarker used as a surrogate end point for the reference product
Known PD biomarker tied to the mechanism of action but not used as a surrogate end point for the reference product
No existing well‐characterized mechanism of action and/or PD biomarker(s) for the reference product
These planned pilot studies will collect intensive PK and PD biomarker data at different dose levels, allowing for the evaluation of model‐based approaches for analyzing data and inform future clinical pharmacology PK and PD similarity studies. These studies also offer an opportunity to investigate the utility of new technologies for the identification of exploratory PD biomarkers. As discussed in FDA guidance,3 the PD biomarker(s) used to measure PD response can be a single biomarker or a composite of more than one relevant PD biomarker that effectively demonstrates the characteristics of the product's target effects. Using broader panels of PD biomarkers (e.g., by measuring features of the transcriptome, proteome, and epigenome) capturing multiple pharmacological effects of the product may be of additional value. Applying these new and emerging technologies will enhance PD biomarker identification and inform the analytical strategy in new drug development programs and will begin to have uptake in biosimilar development programs moving into the future.
Summary
Development and approval of biosimilars is critical for enhancing the availability of affordable, safe, and effective treatment options for patients. Utilization of PD biomarkers can help streamline clinical programs for biosimilar development as the current process can be costly and time consuming. While PD biomarkers have not been prominently used across biosimilar approvals to date, there is ample opportunity to utilize such information alongside or in place of comparative clinical studies with efficacy end point(s) moving forward. PD biomarkers have been successfully utilized as primary end points in PD similarity studies when the PD biomarker was a surrogate end point or a direct measure of clinical outcome. Extending such application beyond surrogate end point PD biomarkers will require some or all the following aspects to be explored: (i) investment in evaluating and synthesizing available information in the literature, (ii) conducting pilot studies, (iii) completing model‐based assessments using available data, and (iv) adopting other novel or emerging technologies, among others. Biosimilar development presents an opportunity for the clinical pharmacology discipline to advance the science around identification and application of PD biomarkers as primary end points in PD similarity studies. The impact on public health is such that the use of PD similarity data in biosimilar programs is a clear means for bringing more affordable, safe, and effective treatments to patients faster.
Funding
This work is supported by the US Food and Drug Administration.
Conflict of Interest
As an Associate Editor for Clinical Pharmacology & Therapeutics, David G. Strauss was not involved in the review or decision process for this paper. All authors declared no competing interests for this work.
Disclaimer
This article reflects the views of the authors and should not be construed to represent the FDA's or Booz Allen Hamilton's views or policies.
References
- 1. US Food and Drug Administration . FDA guidance: scientific considerations in demonstrating biosimilarity to a reference product <https://www.fda.gov/media/82647/download> (2015). Accessed April 15, 2019.
- 2. US Food and Drug Administration . FDA guidance: clinical pharmacology data to support a demonstration of biosimilarity to a reference product <https://www.fda.gov/media/88622/download> (2016). Accessed April 15, 2019.
- 3. Li, L. et al Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for G‐CSF in breast cancer patients. Clin. Pharmacol. Ther. 104, 742–748 (2018). [DOI] [PubMed] [Google Scholar]
- 4. US Food and Drug Administration . FDA draft guidance: biomarker qualification: evidentiary framework: guidance for industry and FDA staff. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM628118.pdf> (2018). Accessed May 10, 2019
- 5. US Food and Drug Administration . Biosimilars action plan: balancing innovation and competition <https://www.fda.gov/media/114574/download> (2018). Accessed May 10, 2019.