

Abstract
Aim
The current study aimed to examine miR‐145's contribution to thoracic aortic dissection (AD) development by modulating the biological functions of vascular smooth muscle cells (VSMCs).
Methods
The concentration of circulating miR‐145 was determined in patients with AD and healthy controls using quantitative polymerase chain reaction (qPCR). Aortic specimens were obtained from both individuals with Stanford type A AD undergoing surgical treatment and deceased organ donors (serving as controls) whose causes of death were nonvascular diseases. Then, qPCR and fluorescence in situ hybridization were applied to assess miR‐145 amounts and location, respectively. Furthermore, qPCR and immunoblot were employed to determine SMAD3 (the target gene of miR‐145, involved in the TGF‐β pathway) amounts at the gene and protein levels, respectively. Moreover, in vitro transfection of VSMCs with miR‐145 mimics or inhibitors was conducted. Finally, the 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay, Transwell assay and flow cytometry were employed for detecting VSMC proliferation, migration, and apoptosis, respectively.
Results
The amounts of miR‐145 in plasma and aortic specimens were markedly reduced in the AD group in comparison with control values (P < .05). miR‐145 was mostly located in VSMCs. Proliferation and apoptosis of VSMCs were significantly induced in vitro by the downregulation of miR‐145. Also, miR‐145 modulated SMAD3 expression.
Conclusions
miR‐145 was found to be downregulated in patients with AD, which induced the proliferation, migration, and apoptosis of VSMCs by targeting SMAD3. This suggested the involvement of miR‐145 in the pathogenesis of AD.
Keywords: aortic dissection, miR‐145, proliferation, SMAD3, vascular smooth muscle cell
1. INTRODUCTION
Thoracic aortic dissection (AD) is a fatal disease, including approximately 3 cases per 100 000 individuals per year. Nearly 40% of patients with AD do not have sufficient time to reach a hospital and die immediately. The molecular mechanism underlying AD is not well understood. Hence, its prevention is not possible pharmacologically.1, 2, 3, 4 Thoracic AD is associated with laceration and degeneration of aortic media.5 Vascular smooth muscle cells (VSMCs) constitute the major cells in the aortic media layer. They are crucial in maintaining aortic wall homeostasis.1, 6, 7 Vascular smooth muscle cells undergo apoptosis and abnormal proliferation in vascular diseases.8, 9
MicroRNAs (miRNAs) represent highly conserved short noncoding RNAs. They primarily bind to the 3′‐untranslated regions (3′‐UTRs) of mRNAs, thus serving as posttranscriptional regulators. They are closely associated with diverse cardiovascular diseases. In 2009, a series of studies emphasized the importance of miR143/145 cluster in the phenotypic modulation of smooth muscle cells.10, 11, 12, 13, 14 The findings demonstrated a reduction in the miR143/145 levels in proliferative conditions. Ning et al showed that miR‐145 modulated the expression of TGF‐β receptor II (TGFBR2) and downstream matrix genes in smooth muscle cells.15 However, the causal mechanism of miR‐145 in developing vascular diseases, especially AD, is yet to be identified and has gained immense attention of researchers.
The current work aimed to confirm miR‐145 downregulation in serum and aortic specimens from patients with AD. The study revealed that miR‐145 contributed to the proliferation and apoptosis of VSMCs. Furthermore, the expression of SMAD3 was modulated by transfection with miR‐145 inhibitors or mimics in vitro, indicating the involvement of miR‐145 in the development of AD through targeting SMAD3. Moreover, circulating miR‐145 amounts were remarkably reduced in the AD group in comparison with the non‐AD hypertensive control group. These findings might provide insights into the role of miRNAs in regulating the function of VSMC in AD at the molecular level.
2. MATERIALS AND METHODS
2.1. Recruitment of participants and collection of samples
This study was performed on Stanford type A AD cases (total of 80) and healthy control patients (total of 71) After 12‐14 hours of fasting, 5‐10 mL of whole venous blood was extracted from the elbow vein and centrifuged (3000 g, 10 minutes; ambient conditions). The resulting plasma was kept at −80°C until further use. Aortic samples were collected from 6 patients undergoing surgical treatment. The exclusion criteria were as follows: (a) aortic aneurysm, congenital bicuspid aortic valves, Ehlers–Danlos syndrome, Loeys–Dietz syndrome, Marfan syndrome, traumatic dissection, Turner syndrome, or other connective tissue disorders; and (b) age <18 years. Normal aortic samples were obtained from four deceased organ donors whose cause of death was nonvascular diseases. The samples were collected within 30 minutes following excision. The tissue specimens were rinsed with precooled saline for at least 5 times, followed by the quick removal of thrombus and adventitia. The specimens were cut into 2‐mm‐thick pieces and snap frozen in liquid nitrogen for further use. The time taken to complete this whole procedure was 10 minutes. No additional samples were collected. Table 1 presents the clinical and demographic characteristics of participants.
Table 1.
Clinico‐demographic features of tissue sample donors
|
Non‐AD control group (N = 4) |
AD group (N = 6) |
Total (N = 10) |
P‐value | |
|---|---|---|---|---|
| Gender (male), n (%) | 3 (75.0) | 5 (83.3) | 8 (80.0) | .747 |
| Age (year), mean (SD) | 46.75 (7.50) | 52.67 (10.50) | 50.30 (9.45) | .362 |
| Hypertension, n (%) | 0 (0.0) | 6 (100.0) | 6 (60.0) | .002 |
| Dyslipidemia, n (%) | 1 (25.0) | 0 (0.0) | 1 (10.0) | .197 |
| Diabetes, n (%) | 0 (0.0) | 1 (16.7) | 1 (10.0) | .389 |
| Smoking, n (%) | 0 (0.0) | 2 (33.3) | 2 (20.0) | .197 |
| Drinking, n (%) | 0 (0.0) | 1 (16.7) | 1 (10.0) | .389 |
| AST (U/L), mean (SD) | 71.00 (14.18) | 43.67 (26.82) | 52.78 (26.20) | .150 |
| ALT (U/L), mean (SD) | 32.75 (17.15) | 32.17 (21.00) | 32.40 (18.52) | .964 |
| Cr(mmol/L), mean (SD) | 157.24 (102.26) | 106.05 (25.27) | 126.52 (67.38) | .262 |
This study complied with the Declaration of Helsinki and had approval from the Medical Ethics Committee of Guangdong Provincial People's Hospital. Each participant signed an informed consent form before the initiation of the study.
2.2. Hematoxylin and eosin staining
The aortic samples, after fixation with 10% formalin (procured from Nanjing Jiancheng Bioengineering Institute, Nanjing, China) for 24 hours followed by dehydration in ethyl alcohol series, underwent xylene clearing and deparaffinization. The samples were then submitted to paraffin embedding to produce 4 μm‐thick sections. Upon hematoxylin staining (5 minutes) and differentiation in acid alcohol (1%), the specimens were subsequently stained with eosin (3 minutes), dehydrated with ethyl alcohol series, and washed twice with xylene (10 minutes). Gum Arabic was employed for mounting before observation under an optical microscope (CX‐31 optical microscope).
2.3. Fluorescence in situ hybridization
After fixation with 4% formaldehyde (10 minutes) and three phosphate‐buffered saline (PBS) washes (5 minutes), aortic specimens underwent permeabilization with 0.5% Triton X‐100 in PBS (10 minutes) at 4°C and further washing with PBS 3 times (5 minutes). The FITC‐labeled miR‐145 probes were incubated with prehybridization buffer (1:100) at 88°C (5 minutes) and 4°C (3 minutes) in a polymerase chain reaction (PCR; Bio‐Rad) instrument. miR‐145 probes labeled with FITC were used to incubate the specimens at 37°C overnight. Subsequently, the specimens were washed (2 × SSC 6 times for 3 minutes) and then stained with DAPI. Following these, the specimens were washed again (2 × SSC 3 times for 5 minutes) and scanned under a laser scanning confocal microscope (Leica TCS‐SP2) as described previously.16
2.4. Isolation of RNA and quantitative reverse transcription–PCR
TRIzol (Invitrogen) was employed for total RNA (comprising small RNAs) extraction from the aortic tissue or cultured VSMCs. The Geneseed II First Strand cDNA Synthesis Kit purchased from Geneseed Biotech was applied to reverse transcribe the RNA thus obtained into complementary DNA (cDNA) following the manufacturer's protocols.9 Then, reverse transcription (500 ng of total RNA) was carried out with PrimeScript reverse transcription (RT) reagent kit obtained from TaKaRa. Next, quantitative PCR (qPCR) was performed with SYBR Green Master Mix (Geneseed Biotech) on an ABI 7500 real‐time PCR system (ABI) as described previously.17, 18
The Primer 5.0 software was used to construct primers in line with gene sequences in GenBank. The primers were validated using Oligo 7 and NCBI BLAST (Molecular Biology Insights) (Table 2). Each step involved the use of negative controls with no template. Further, 20 μL of SYBR Green reaction mixture comprised a template (2 μL of cDNA) and primers (10 μmol/L of each). The RT reaction comprised the following steps: 25°C for 10 minutes, 42°C for 15 minutes, and 85°C for 5 minutes; incubation at 95°C (5 minutes); and 40 cycles at 95°C (10 seconds), 60°C (34 seconds) and 60°C (60 seconds). Next, the heat dissociation protocol was applied to the samples, and qRT‐PCR products were assessed with SYBR Green. Whether qRT‐PCR was reliable was evaluated by melting curve analysis using β‐actin as a reference gene. Relative miRNA expression was determined by the 2−△Cq method. This was followed by calculating the mean Cq values and standard deviations in duplicate assays.
Table 2.
Sequences of primers
| Sequence (5′–3′) | |
|---|---|
| SMAD3 | F‐1: AGAAGGCTGGGGCTCATTTG |
| SMAD3 | RT‐1: CCTCTTCCGATGTGTCTCCG |
| Product length: 115 bp | |
| miR145‐5p | F‐1: ATGGTTCGTGGGGTCCAGTTTTCCA |
| miR145‐5p | RT‐1: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCAGGTATTC |
| Com R: GTGCAGGGTCCGAGGT | |
| Product length: 68 bp |
2.5. Western immunoblotting
Aortic tissues or VSMCs were lysed using RIPA lysis buffer containing protease inhibitors and Western blotting along with quantification of images were performed as described previously.18 RIPA buffer (100 mg/1000 mL) was used to extract total protein from aortic specimens. Phosphate buffered Saline and RIPA buffer (100 μL per well) (Cell Signaling Technology) were used to wash VSMCs 3 times. A cell lifter obtained from Corning Costar was employed for lysing the specimens, with protein concentrations adjusted to 1.0 mg/mL. The lysate (20 μL, or 20 μg) was separated by 6%‐12% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) and bands were electro‐transferred onto a polyvinylidene difluoride membrane. Nonfat milk (5%) (Becton Dickinson) was used to block the membranes, followed by washing, probing with rat anti‐human SMAD3 (1:1000; Abcam) and anti‐GAPDH (1:500; Abcam) primary antibodies, and incubation with a horseradish peroxidase (HRP) conjugated goat anti‐rat secondary antibodies (1:2000; Sigma) for 60 minutes. Further, an enhanced chemiluminescence kit was used to wash and expose the membrane following the manufacturer's protocols (Thermo Fisher). The immunoblots obtained from three rounds of Western blot analyses were assessed on a flatbed scanner, and the intensities were calculated with Image‐Pro Plus (v6.0; Media Cybernetics).
2.6. Luciferase assay
PCR was used to magnify a specific sequence of SMAD3 gene—3′‐untranslated region (UTR)—harboring miR‐145‐binding sites. For the cloning of the magnified sequence, the pMIR‐REPORT luciferase vector (psiCHECK2‐SMAD3‐wild‐type) was used. The binding site in the aforementioned region of SMAD3 was mutated and inserted into the plasmid psiCHECK2‐SMAD3‐mutant. Lipofectamine 2000 obtained from Invitrogen (Thermo Fisher Scientific) was applied to transfect 293T cells with miR‐145 mimics and luciferase plasmid psiCHECK2‐SMAD3 (wild‐type/mutant, WT/Mut) as directed by the manufacturer. The reporter assay was performed as described previously.19
The cells were cultivated for 48 hours, harvested, and lysed. TransDetect Double‐Luciferase Reporter Assay Kit obtained from TransGen Biotech was applied to assess and standardize the firefly luciferase activity to Renilla luciferase activity. The whole procedure was repeated thrice.
2.7. Culture and transfection of cells
Primary human arterial smooth muscle cells (HASMCs) isolated from aortic tissues were procured from Geneseed Biotech. They were cultivated in smooth muscle cell medium (Cat. No. 1101; ScienCell) containing fetal bovine serum (FBS; 10%), streptomycin (100 μg/mL), and penicillin (100 U/mL) (Invitrogen) at 37°C in a humid environment with 5% CO2. Passage 3‐6 cells were assessed in subsequent experiments. miR‐145 mimics (50nM) or inhibitors (50 nmol/L) (Geneseed Biotech) were employed to transfect HASMCs for 48 hours with Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. All transfections and assays on HASMCs were conducted in low serum medium (1% FBS).
2.8. Transwell HASMC migration assays
Human arterial smooth muscle cells at 2 × 104 cells/well were seeded in 24‐well plates. After overnight incubation of HASMCs, miR‐145 mimics or inhibitors were used to transfect the cells for 24 hours. Trypsin (2.5%) was used to detach the cells, which were resuspended serum‐free medium at 106/mL. Further, 200 μL of cell suspension and 600 μL of growth medium were placed in the upper and lower chambers of the Transwell and incubated at 37°C for 48 hours. For the migration assay, the 8.0 μm pore filter of the Transwell chamber underwent pre‐treatment with 50 μL Matrigel obtained from BD Biosciences. The cells migrating to the lower side underwent fixation with 10% formalin and crystal violet (0.5%) staining. The cell count was determined for five randomly selected high‐power fields (×200) by light microscopy.
2.9. Cell proliferation assay
Cell Counting Kit‐8 (CCK‐8; Dojindo) was employed for assessing cell proliferation as directed by the manufacturer. The cells were plated in 96‐well plates in M199 medium containing 1% FBS at 5 × 103/well followed by incubation for 24 hours. Further, the cells were treated with adenovirus at the MOI of 120. After every 24 hours, the medium was refreshed. After 0, 12, 24, 36, and 48 hours of transfection, respectively, 10 μL CCK‐8 reagent was supplemented to individual wells, for further incubation at 37°C for 4 hours. Optical density was obtained on a plate reader at 450 nm. Vascular smooth muscle cells serving as negative controls were transfected with adenovirus lacking the miR‐145‐interfering effect. Colonies were counted and analyzed using the Prism software (version 6.00).20
2.10. 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay for HASMC viability
A total of 5 × 104 HASMCs per well were plated in 96‐well plates. They were grown the whole night and transfected with miR‐145 mimics or inhibitors with Lipofectamine 2000 (Invitrogen) for 24 hours following the manufacturer's protocols. Then, cells were cultured for various time points (24, 48, and 72 hours) after replacing the growth medium. MTT (Beyotime) was supplemented at the end of each experiment for 4 hours, following the manufacturer's instructions, followed by the replacement of the cell culture medium with 150 μL dimethyl sulfoxide. Upon thorough shaking, optical density at 490 nm was assessed on a microplate reader (Bio‐Rad 680). The data thus obtained were used to plot cell growth curves. The whole procedure was carried out in three sets and repeated at least 3 times.
2.11. Apoptosis assay by flow cytometry
The adhering HASMCs were trypsinized and submitted to staining with Annexin V‐FITC and propidium iodide (Annexin V–FITC Apoptosis Detection Kit), and screened on a FACScan flow cytometer (DB Biosciences). The experiment was conducted in triplicate. The CellQuest software (BD Biosciences) was used to determine the apoptotic rate.
2.12. Statistical analysis
Data are mean ± standard deviation (SD) and were assessed with SPSS 17.0 (SPSS). Independent‐samples t test and the Kolmogorov–Smirnov test were used for intergroup comparison and normality assessment, respectively. One‐way analysis of variance (ANOVA) was employed to evaluate differences in continuous numerical parameters with normal distribution between groups. Post‐hoc analyses were carried out by the Tukey's test. P < .05 indicated statistical significance.
3. RESULTS
3.1. miR‐145 and SMAD3 were largely located in vascular smooth muscle
Vascular smooth muscle cells were less abundant but continuously and normally arranged and medial laceration was observed in aortic specimens from the non‐AD control group (Figure S1). Next, the localization and expression of SMAD3 and miR‐145 in aortic tissue were examined using immunochemical staining and fluorescence in situ hybridization (FISH), respectively. The findings showed that SMAD3 was mainly located in VSMCs (Figure 1A), while miR‐145 was abundantly distributed in the cytoplasm of VSMCs (Figure 1B and Figure S2).
Figure 1.
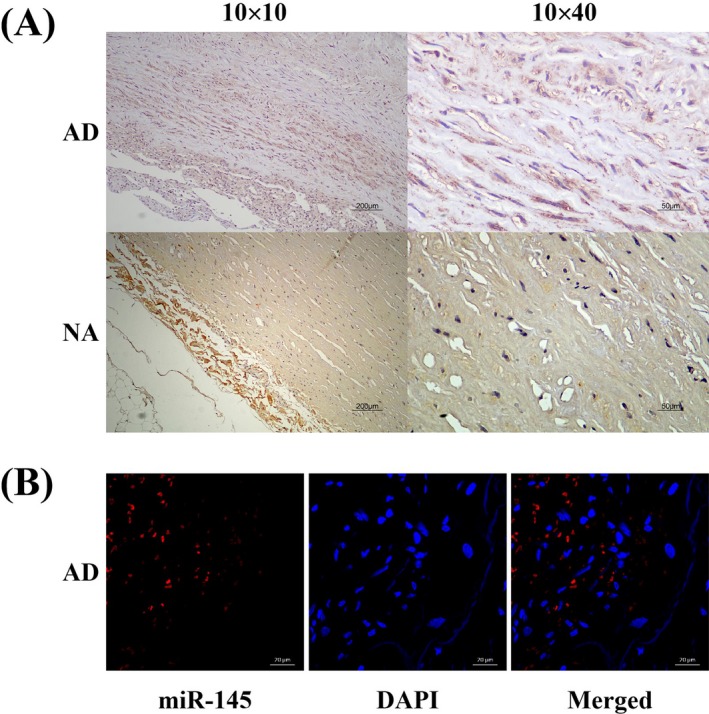
Location of SMAD3 and miR‐145 in human aortic tissues. A, Immunochemistry staining for SMAD3 location in both dissected and nondissected aortic tissues. SMAD3 was mainly located in VSMCs. B, FISH to detect miR‐145 location in dissected aortic tissues (scale bar = 50 μm). DAPI (blue) was used to counterstain the nucleus. The merged images revealed that miR‐145 was located mainly in the cytoplasm of VSMCs
3.2. Specimens of dissected and normal aortic tissues exhibiting differential expression of miR‐145 and SMAD3
SMAD3 protein amounts in the human aorta were detected using the Western blot analysis. Figure 2A,B show the significantly increased expression level of SMAD3 in AD samples than in NA specimens. The mRNA amounts of miR‐145 and SMAD3 in AD and their matched NA samples were determined using qRT‐PCR to assess miR‐145 expression (Figure 2C,D) and SMAD3 in the human aorta, which revealed marked downregulation of miR‐145 and significant SMAD3 upregulation in the AD group. The results suggested the AD group had increased expression levels of SMAD3 in comparison with the non‐AD control group, as opposed to miR‐145.
Figure 2.
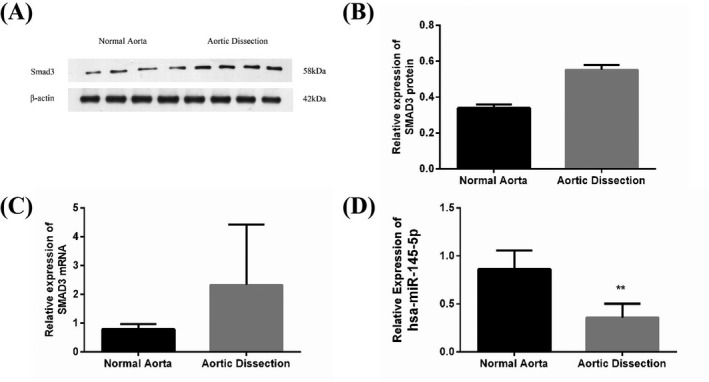
Expression of SMAD3 and miR‐145 in human aortic samples in the non‐AD control and AD groups, respectively. A and B, Immunoblot demonstrated the amounts of the SMAD3 protein in human aortic samples (n = 4 per group). The AD group showed significantly increased SMAD3 amounts (P < .01). C, qPCR demonstrated the expression of SMAD3 mRNA in human aortic samples (n = 6 and 4 for the AD and non‐AD control groups, respectively). The expression of SMAD3 mRNA significantly increased in the AD group (P < .01). D, qPCR was employed to assess miR‐145 amounts in human aortic samples (n = 6 and 4 for the AD and non‐AD control groups, respectively). miR‐145 amounts were markedly downregulated in the AD group (P < .05)
3.3. Overexpressed miR‐145 promoted the proliferation, migration, and apoptosis of HASMCs
Human arterial smooth muscle cells underwent transfection with miR‐145 mimics or inhibitors to investigate the involvement of miR‐145 in regulating the biological functions of VSMCs (Figure S3). The MTT assay revealed miR‐145 downregulation in HASMCs increased the proliferation of HASMCS, while its upregulation significantly suppressed cell proliferation (Figure 3A). The involvement of miR‐145 in the migration of HASMCs was determined using the Transwell test. miR‐145 mimics markedly promoted the migratory response of cultured HASMCs. However, cell migration was not affected by miR‐145 downregulation (Figure 3B,D). Apoptosis in HASMCs was obviously affected upon transfection with miR‐145 mimics, as detected by flow cytometry (Figure 3C,E). These in vitro observations revealed the participation of miR‐145 in HASMC proliferation, migration, and apoptosis.
Figure 3.
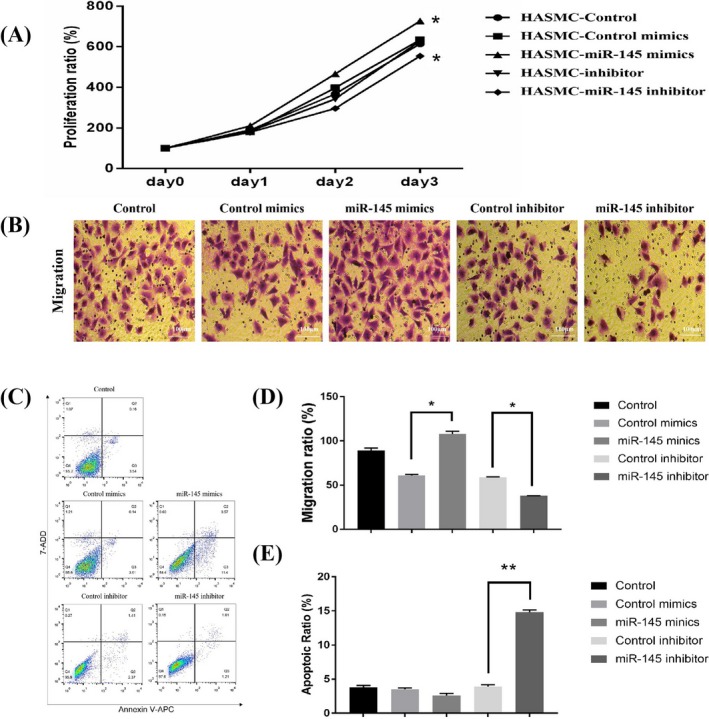
In vitro transfection with miR‐145 mimics or inhibitors strongly induced the proliferation, migration, and apoptosis of HASMCs. A, Proliferation curves of HASMCs transfected with control, miR‐145 mimics, mimics control, inhibitors, and inhibitor control. The proliferation of HASMCs was found to be significantly suppressed after 72‐h treatment with miR‐145 inhibitors. In contrast, miR‐145 mimics promoted the proliferation of HASMCs compared with miR‐145 inhibitor control (n = 5 per group). B and D, The Transwell test results showed that HASMC migration in the miR‐145 inhibitor group was stunted. However, the migration markedly increased in the miR‐145 mimics group. Data were expressed as the number of migrating cells per high‐power field (n = 5/group). C and E, transfection with miR‐145 mimics promoted the apoptosis of HASMCs, as shown by flow cytometric analysis (n = 5/group). D and E, Data are mean ± standard deviation. All assays were conducted in three sets
3.4. miR‐145 regulated the expression of SMAD3
The targets of miR‐145 were predicted using TargetScan and miRanda, aiming to determine the effects of miR‐145 on HASMC proliferation, migration, and apoptosis. The data demonstrated that one of the predicted target genes for miR‐145 was SMAD3. Two putative binding sites were confirmed at 56‐69 nt and 1397‐1403 nt. They were highly conserved across species (Figure 4A). Hence, the WT and Mut 3′‐UTRs of SMAD3 were cloned into a luciferase vector, respectively.
Figure 4.
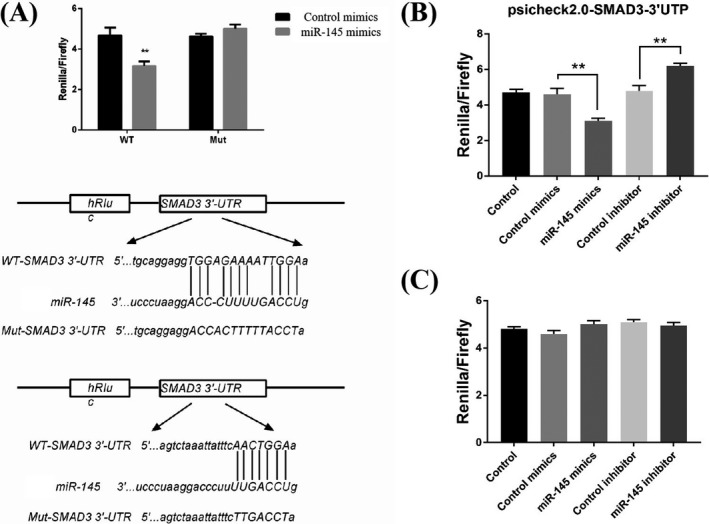
Direct target of SMAD3 by miR‐145. A, Prediction of the sequence alignment of miR‐145 with 3¢‐UTR of SMAD3using TargetScan. B, Decreased luciferase activity due to the upregulated expression of miR‐145, as indicated by luciferase assay results for 293T cells. C, Upregulation of the expression of miR‐145 did not decrease the luciferase activity, as indicated by luciferase assay results for 293T cells with Mut 3¢‐UTR of SMAD3
The predictions were confirmed using the luciferase reporter assay. The WT and Mut 3′‐UTRs of SMAD3 luciferase reporter vectors were developed. Further, miR‐145 mimics and WT/Mut luciferase vector were used to cotransfect 293T cells, revealing a remarkable impairment of the relative luciferase activity (Figure 4B). However, the activity was not altered in 293T cells transfected with Mut luciferase vector (Figure 4C). The findings indicated miR‐145 directly bound to the 3′‐UTR of SMAD3.
Human arterial smooth muscle cells underwent transfection with miR‐145 mimics and inhibitors to confirm the direct regulatory activity of miR‐145 on SMAD3. qPCR (Figure 5A) and immunoblot (Figure 5B,C) were employed to determine SMAD3 mRNA and protein levels, respectively, indicating a reduction in the expression of SMAD3 with the overexpression of miR‐145 in HASMCs. However, SMAD3 mRNA and protein amounts in HASMCs were elevated by miR‐145 inhibitor.
Figure 5.
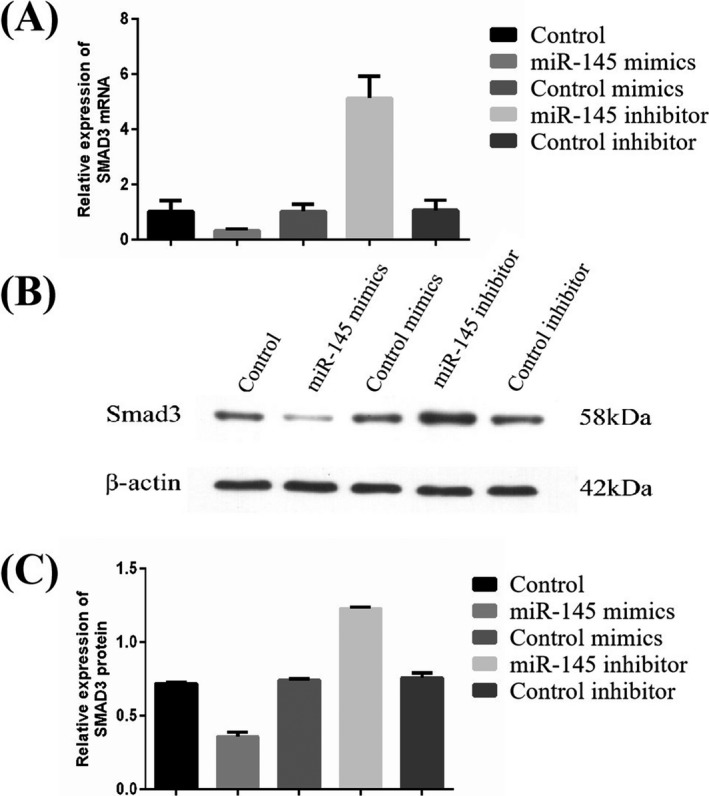
Suppression of SMAD3 expression in HASMCs by miR‐145. The expression of SMAD3 in HASMCs was inhibited by miR‐145, as confirmed by qPCR (A) and immunoblot (B and C) results
3.5. Circulating miR‐145 amounts are reduced in the AD population
Circulating miR‐145 amounts were determined in 80 AD cases and 71 non‐AD controls, whose clinic‐demographic features demonstrated marked differences between the two groups in gender (66.2% and 94.4%), age (65.12% and 74.4%), smoking rate(18.8% vs 46.5%), as well as incidence of hypertension (75.0% vs 85.9%), dyslipidemia (97.5% vs 18.8%), diabetes (37.5% vs 8.5%) (P < .001, respectively) (Table 3). Notably, circulating miRNA‐145 amounts were significantly lower in the AD group in comparison with the non‐AD control group, as expected (Figure 6).
Table 3.
Clinico‐demographic features of blood sample donors
|
Non‐AD group (N = 71) |
AD group (N = 80) |
P‐value | |
|---|---|---|---|
| Gender (male), n (%) | 67 (94.4) | 53 (66.2) | <.001 |
| Age (y), mean (SD) | 74.41 (3.53) | 65.12 (10.75) | <.001 |
| Hypertension, n (%) | 61 (85.9) | 60 (75.0) | <.001 |
| Dyslipidemia, n (%) | 13 (18.8) | 78 (97.5) | <.001 |
| Diabetes, n (%) | 6 (8.5) | 30 (37.5) | <.001 |
| Smoking, n (%) | 33 (46.5) | 15 (18.8) | <.001 |
| Drinking, n (%) | 2 (2.8) | 4 (5.0) | .493 |
| AST (U/L), mean (SD) | 73.14 (393.26) | 30.50 (31.43) | .338 |
| ALT (U/L), mean (SD) | 56.32 (270.22) | 35.67 (54.53) | .510 |
| Cr (mmol/L), mean (SD) | 123.37 (123.13) | 91.46 (51.74) | .036 |
Figure 6.
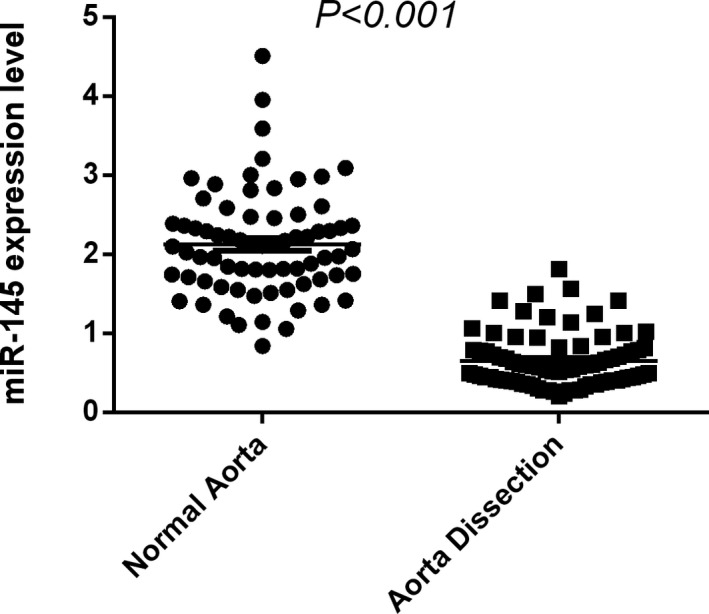
Significantly reduced circulating miR‐145 amounts were found in the AD group compared with the non‐AD control group
4. DISCUSSION
Previous findings indicate a vital role for VSMCs in degenerating the aortic wall, which marks the initiation of pathological remodeling in AD and aneurysms.21 The present study revealed prominent signs of aortic medial laceration in AD and emphasized the participation of VSMCs in the development of AD. Importantly, several miRNAs were involved in regulating VSMC phenotypes. Previous studies indicated a role of miR‐145 in modulating smooth muscle differentiation.22 Smooth muscle cells received miR‐145 from exosomes excreted by endothelial cells.23 Further, miR‐145 was found to have an atheroprotective role.21
However, a few studies identified a function for miR‐145 in AD pathogenesis. Hence, this study intended to determine the pattern of expression of miR‐145 in AD and also its participation in the biological functions of VSMCs. The findings indicated the location of miR‐145 in VSMCs and significant downregulation of its expression levels in AD settings. The study further investigated the significance and potential impact of miR‐145 dysregulation on proliferation, migration, and apoptosis in HASMCs using functional assays. The results showed that miR‐145 depletion induced proliferation and migration in VSMCs. However, the overexpression of miR‐145 using specific mimics suppressed the proliferation and apoptosis of VSMCs. The levels of circulating miRNA‐145 also markedly reduced in patients with AD compared with non‐AD hypertensive controls, further substantiating the association of miR‐145 with AD, and indicating that miRNA‐145 could be novel serum biomarkers for AD.
Abnormal proliferation and migration of VSMCs are the major contributing factors for pathological vascular remodeling through undermining vasculature stability.24 Müller et al and Wang et al demonstrated the more rapid proliferation of VSMCs from dissected aorta compared with VSMCs from normal aortic tissues. Moreover, the expression levels of genes involved in proliferation were elevated.25, 26 On the contrary, VSMCs showed signs of apoptosis in AD settings.27, 28, 29, 30
The above in vitro findings provided valid evidence that miR‐145 knockdown promotes the proliferation, migration, and apoptosis of VSMCs, which implied that miR‐145 is vital in the pathogenesis and development of AD.
Further, the results confirmed SMAD3 as a direct miR‐145 target in VSMCs. SMADs are pivotal elements among the intracellular effectors of TGF‐β1. They regulate various biological activities, including cell proliferation, apoptosis, migration, differentiation, and adhesion.31 Dysregulated TGF‐β signaling is crucial in the pathogenesis of AD.32 Mutations in genes encoding the TGF‐β‐signaling components, including FBN1, TGFBR1, TGFBR2, TGFB2, TGFB3, SMAD2, and SMAD3, have been detected in thoracic AD.33 Of note, SMAD3 mutations are found in families with TAAD in an autosomal dominant manner.34 The findings indicated the dissected aorta had upregulated expression of SMAD3 in comparison with non‐AD tissues. Therefore, in vitro assays were carried out to better elucidate the relationship between miR‐145 and SMAD3 in VSMCs. The findings revealed an altered expression of SMAD3 due to the abnormal expression of miR‐145 in VSMCs, implying a direct regulatory effect of miR‐145 on SMAD3.
This study had several limitations. First, the sample size was relatively small. Therefore, large‐scale studies should be performed on tissue and plasma specimens of AD to further validate the findings. Second, the association of miRNA‐145 with VSMCs in aortic wall degeneration deserves further investigation. Third, the present study shows that SMAD3 is a target of miR‐145, hence in vitro studies will be needed to perform to address whether overexpression or knockdown of SMAD3 will affect the effect of miR‐145 on proliferation, migration, and apoptosis of VSMCs. Finally and notably, the similar expression pattern of miR‐145 has been observed in aortic tissues and circulating‐cell free miRNAs are relatively stable in blood due to the protection by their inclusion in exosomes, therefore, the interaction between miR‐145 and exosomes and the detailed mechanisms remains unclear. All mentioned above limited the role of miR‐145 as a biomarker in the prognosis of AD. These shortcomings should be addressed by performing prospective AD cohort and by using molecular, cell, animal, as well as human approaches.
Overall, the study showed the downregulation of miR‐145 and the upregulation of SMAD3 in AD tissues. The miR‐145 ectopic expression promoted the proliferation and apoptosis of VSMCs. However, miR‐145 suppressed the expression of SMAD3 through targeting the 3′‐UTR of SMAD3. The findings of this study indicated the involvement of miR‐145 in AD pathogenesis through direct targeting of SMAD3, thereby providing novel insights into the effective clinical management of AD.
CONFLICT OF INTEREST
The authors do not have a conflict of interest.
Supporting information
ACKNOWLEDGMENTS
The authors acknowledge the efforts and contributions of all the participating volunteers.
Huang W, Huang C, Ding H, et al. Involvement of miR‐145 in the development of aortic dissection via inducing proliferation, migration, and apoptosis of vascular smooth muscle cells. J Clin Lab Anal. 2020;34:e23028 10.1002/jcla.23028
Wenhui Huang and Cheng Huang contributed equally to this work.
Funding information
This work was financially supported by the National Natural Science Foundation of China (81300230), and High‐level Hospital Construction Project (DFJH201807).
REFERENCES
- 1. Golledge J, Eagle KA. Acute aortic dissection. Lancet. 2008;372(9632):55‐66. [DOI] [PubMed] [Google Scholar]
- 2. Clouse WD, Hallett JJ, Schaff HV, et al. Acute aortic dissection: population‐based incidence compared with degenerative aortic aneurysm rupture. Mayo Clin Proc. 2004;79(2):176‐180. [DOI] [PubMed] [Google Scholar]
- 3. Meszaros I, Morocz J, Szlavi J, et al. Epidemiology and clinicopathology of aortic dissection. Chest. 2000;117(5):1271‐1278. [DOI] [PubMed] [Google Scholar]
- 4. Kochanek KD, Xu J, Murphy SL, et al. Deaths: final data for 2009. Natl Vital Stat Rep. 2011;60(3):1‐116. [PubMed] [Google Scholar]
- 5. Wu D, Shen YH, Russell L, Coselli JS, LeMaire SA. Molecular mechanisms of thoracic aortic dissection. J Surg Res. 2013;184(2):907‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barbour JR, Spinale FG, Ikonomidis JS. Proteinase systems and thoracic aortic aneurysm progression. J Surg Res. 2007;139(2):292‐307. [DOI] [PubMed] [Google Scholar]
- 7. Wang X, Lemaire SA, Chen L, et al. Increased collagen deposition and elevated expression of connective tissue growth factor in human thoracic aortic dissection. Circulation. 2006;114(1 Suppl):I200‐I205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev. 2013;93(3):1317‐1542. [DOI] [PubMed] [Google Scholar]
- 9. Choi MH, Lee IK, Kim GW, et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435(7040):347‐353. [DOI] [PubMed] [Google Scholar]
- 10. Boettger T, Beetz N, Kostin S, et al. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119(9):2634‐2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng Y, Liu X, Yang J, et al. MicroRNA‐145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105(2):158‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cordes KR, Sheehy NT, White MP, et al. MiR‐145 and miR‐143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460(7256):705‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elia L, Quintavalle M, Zhang J, et al. The knockout of miR‐143 and ‐145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16(12):1590‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xin M, Small EM, Sutherland LB, et al. MicroRNAs miR‐143 and miR‐145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23(18):2166‐2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao N, Koenig SN, Trask AJ, et al. MicroRNA miR145 regulates TGFBR2 expression and matrix synthesis in vascular smooth muscle cells. Circ Res. 2015;116(1):23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar S, Gupta S. Thymosin beta 4 prevents oxidative stress by targeting antioxidant and anti‐apoptotic genes in cardiac fibroblasts. PLoS ONE. 2011;6(10):e26912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li L, Wei C, Kim IK, Janssen‐Heininger Y, Gupta S. Inhibition of nuclear factor‐kappaB in the lungs prevents monocrotaline‐induced pulmonary hypertension in mice. Hypertension. 2014;63(6):1260‐1269. [DOI] [PubMed] [Google Scholar]
- 18. Kumar S, Seqqat R, Chigurupati S, et al. Inhibition of nuclear factor kappaB regresses cardiac hypertrophy by modulating the expression of extracellular matrix and adhesion molecules. Free Radic Biol Med. 2011;50(1):206‐215. [DOI] [PubMed] [Google Scholar]
- 19. Wei C, Kim IK, Kumar S, et al. NF‐kappaB mediated miR‐26a regulation in cardiac fibrosis. J Cell Physiol. 2013;228(7):1433‐1442. [DOI] [PubMed] [Google Scholar]
- 20. Marko TA, Shamsan GA, Edwards EN, et al. Slit‐Robo GTPase‐Activating protein 2 as a metastasis suppressor in osteosarcoma. Sci Rep. 2016;6:39059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu L, Vranckx R, Khau V, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38(3):343‐349. [DOI] [PubMed] [Google Scholar]
- 22. Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA‐145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137(4):647‐658. [DOI] [PubMed] [Google Scholar]
- 23. Du Y, Li J, Xu T, Zhou DD, Zhang L, Wang X. MicroRNA‐145 induces apoptosis of glioma cells by targeting BNIP3 and Notch signaling. Oncotarget. 2017;8(37):61510‐61527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Satoh K, Matoba T, Suzuki J, et al. Cyclophilin a mediates vascular remodeling by promoting inflammation and vascular smooth muscle cell proliferation. Circulation. 2008;117(24):3088‐3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muller BT, Modlich O, Prisack HB, et al. Gene expression profiles in the acutely dissected human aorta. Eur J Vasc Endovasc Surg. 2002;24(4):356‐364. [DOI] [PubMed] [Google Scholar]
- 26. Wang L, Zhang J, Fu W, et al. Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. J Vasc Surg. 2012;56(6):1698‐1709, 1701–1709. [DOI] [PubMed] [Google Scholar]
- 27. Henderson EL, Geng YJ, Sukhova GK, et al. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99(1):96‐104. [DOI] [PubMed] [Google Scholar]
- 28. Liu Y, Sun X, Wu Y, et al. Effects of miRNA‐145 on airway smooth muscle cells function. Mol Cell Biochem. 2015;409(1‐2):135‐143. [DOI] [PubMed] [Google Scholar]
- 29. Du J, Li Q, Shen L, et al. MiR‐145a‐5p promotes myoblast differentiation. Biomed Res Int. 2016;2016:5276271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y, Dong CQ, Peng GY, et al. MicroRNA‐134‐5p regulates media degeneration through inhibiting VSMC phenotypic switch and migration in thoracic aortic dissection. Mol Ther Nucleic Acids. 2019;16:284‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF‐beta signaling. J Cell Biochem. 2007;101(1):9‐33. [DOI] [PubMed] [Google Scholar]
- 32. Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473(7347):308‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early‐onset osteoarthritis. Nat Genet. 2011;43(2):121‐126. [DOI] [PubMed] [Google Scholar]
- 34. Regalado ES, Guo DC, Villamizar C, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109(6):680‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials