

Abstract
The Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have robust scientific and technical collaborations. As a window to the impact of these activities we compared the agencies’ decisions on drug marketing applications. Decisions were compared for 107 new drug applications with a regulatory outcome at both agencies in the period 2014–2016. Further analysis addressed individual applications for which the agencies had differing outcomes in terms of marketing approval, type of approval, and approved indication, including reasons underlying differences. The EMA and the FDA had high concordance (91–98%) in decisions on marketing approvals. Divergence in approval decisions, type of approval, and approved indication were primarily due to differences in agencies’ conclusions about efficacy based on review of the same data or differing clinical data submitted to support the application. This high rate of concordance suggests that engagement and collaboration on regulatory science has a positive impact.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ There have been previous comparisons of regulatory approvals by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), but they have been limited. This is the most comprehensive study to date.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This examination of a 3‐year cohort of marketing applications reviewed by both the EMA and the FDA compared the agencies’ approval/licensing decisions, types of approval, and approved indications, as well as the underlying reasons for different decision reached.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The focus on reasons for differences in decisions is a feature not previously addressed and provides more insight into factors underlying how the agencies approach decisions.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The high concordance of FDA and EMA decisions suggests ongoing efforts to collaborate on regulatory science may be contributing to more global harmonization in the field, including clinical pharmacology and translational science as applied to drug development.
Globalization of drug development has increased the need for harmonization and collaboration among drug regulatory authorities.1 As such, activities and decisions of the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) are often compared, particularly regarding the time it takes to review marketing applications.2, 3, 4 However, understanding differences in regulators’ expectations for drug development and standards for assessment of efficacy and safety is critical to facilitating more global alignment while allowing for unique considerations of regional needs. Examination and comparison of decisions on applications may provide an understanding of how agencies consider and apply regulatory science.5, 6, 7 To date, only a few comparisons of this type for the FDA and the EMA have been conducted, mostly qualitative and in specific therapeutic areas.
The FDA and the EMA are committed to further global alignment of sound regulatory standards in drug development, which ultimately link to assessment of marketing applications. Both participate in and have adopted guidelines of the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH, http://www.ich.org/home.html), an organization whose aim is to facilitate harmonization across national and regional regulatory agencies to ensure safe, effective, and high‐quality drug products.8 Over the past decade, under the auspices of their confidentiality agreements, both agencies have established forums for information sharing and collaboration around many aspects of medicinal drug product development and regulation through standing working groups. These “clusters” bring together technical experts to share information on topics such as plans for manufacturing or clinical site inspections, pediatric drug development, oncology products, pharmacogenomics, biostatistics, rare diseases, and vaccines (http://www.ema.europa.eu/ema/index.jsp?curl=pages/partners_and_networks/general/general_content_000655.jsp&mxml:id=WC0b01ac0580953d9).9 The discussions on basics in regulatory science are a bedrock to facilitating alignment on high impact standards and approaches, even though specific development and decisions about marketing authorization for individual products must be made by the FDA and the EMA within their own legal and regulatory frameworks.
Our objective was to compare EMA and FDA decisions on new drug marketing applications over three calendar years (2014–2016) as a window to the impact of the agencies’ activities in technical collaboration. We next examined applications for which the two agencies had differing outcomes in terms of approval and indication, assessing the scientific and regulatory reasons underlying these differences.
Results
Cohort characteristics
There were 126 and 125 new drug marketing applications for a new active substance, a new chemical entity (NCE), or a therapeutic biologic product submitted to the FDA and the EMA, respectively, and which had an outcome in the period 2014–2016. Of these, 74 applications were identified as having an initial outcome by both agencies during the study period. Also, 33 applications had been submitted to both agencies, with one agency's initial outcome in the study period and the other's outside of the period. In total, the cohort included 107 marketing applications.
The majority (71%) of the drugs in the cohort were for NCEs, while 29% were therapeutic biologics. Therapeutic areas of the applications were led by oncology (25%), as shown in Table 1.
Table 1.
Product characteristics for cohort (2014–2016) of marketing applications reviewed by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA)
| Characteristic |
Applications (N / %) N = 107 |
|---|---|
| Product type | |
| New chemical entity | 76 (71) |
| Therapeutic biologic | 31 (29) |
| Therapeutic area | |
| Oncology | 27 (25) |
| Infectious diseases | 18 (17) |
| Metabolic diseases | 13 (12) |
| Neurology/psychiatry | 10 (9) |
| Hematology | 8 (7) |
| Cardiovascular diseases | 7 (6) |
| Gastroenterology/hepatology | 6 (6) |
| Pulmonology/allergy | 6 (6) |
| Medical imaging/radiation injury | 5 (5) |
| Rheumatology/immunology | 4 (4) |
| Reproductive medicine | 2 (2) |
| Dermatology | 1 (1) |
Comparison of initial outcomes
Overall, 84% of the applications in the cohort were approved on their first submission (“initially approved”) by both the EMA and the FDA, with the EMA having a higher rate of “first review cycle” approval (92%) compared with the FDA (85%).
Initial concordant and discordant regulatory outcomes
FDA and EMA decisions on whether to approve a product for marketing upon first submission and review were concordant (both agencies had the same regulatory outcome) for 92% (98/107) of the applications and discordant for 8% (9/107) (Table 2). Both agencies approved 84% (90/107) of the applications and both had initial negative outcomes (nonapproval or withdrawal) for 4% (4/107). There were four applications that were not approved by the FDA but withdrawn by the applicant prior to the regulatory decision by the EMA. In all four cases, at the time of the withdrawal from the EMA, the applicant was aware that the Committee for Medicinal Products for Human Use (CHMP) had concerns and was of the provisional opinion that the products were not approvable. Table 3 lists the nine drugs for which initial application decisions were discordant.
Table 2.
Initial decisions on cohort marketing applications by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA)
| Initial EMA outcome, N = 107 | ||||
|---|---|---|---|---|
| Approved (98) | Not approved (3) | Withdrawn (6) | ||
|
Initial FDA outcome, N = 107 |
Approved (91) | 90 | 1 | 0 |
| Not approved (14) | 8 | 2 | 4 | |
| Withdrawn (2) | 0 | 0 | 2 | |
Shaded cells indicate concordant outcomes.
Table 3.
Marketing applications with initial discordant outcomes
| Drug | Indication summary | Initial European Medicines Agency (EMA) outcome | Initial US Food and Drug Administration (FDA) outcome |
|---|---|---|---|
| 177 Lu‐Dota0‐Tyr3‐octreotate | Gastroentero‐pancreatic neuroendocrine tumors (GEP‐NETs) | Approved | Not approved |
| Corifollitropin alfa | Controlled Ovarian Stimulation (COS) or the development of multiple follicles in women participating in an Assisted Reproductive Technology (ART) program | Approved | Not approved |
| Cangrelor | Reduction of thrombotic cardiovascular events (MI, stent thrombosis) in patients undergoing percutaneous coronary intervention (PCI) | Approved | Not approved |
| Daclatasvir | Chronic hepatitis C (CHC) infection | Approved | Not approved |
| Empagliflozin | Improvement of glycemic control in Type 2 diabetes mellitus | Approved | Not approved |
| Etelcalcetide | Treatment of secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease on haemodialysis | Approved | Not approved |
| Safinamide mesylate | Fluctuating Parkinson's Disease | Approved | Not approved |
| Sarilumab | Moderate‐severe rheumatoid arthritis | Approved | Not approved |
| Ixazomib | Multiple myeloma | Not approved | Approved |
Resubmissions and reexaminations
As shown in Table 2, 15 applications in the cohort were initially not approved by one or both agencies: 1 application was not approved by the EMA only, 12 by the FDA only, and 2 by both agencies. After initial assessment, 8 applications were resubmitted to the FDA and 3 were the subject of a request for reexamination of the CHMP opinion at the EMA, representing a total of 11 resubmitted or reexamined applications (Table 4). There was no overlap in marketing application authorizations (MAAs) that were resubmitted or reexamined. Table 4 also shows the final regulatory outcomes for the applications that were resubmitted or reexamined. The FDA approved seven of the eight resubmitted applications, and the EMA approved two of the three reexamined applications. Overall, most applications that had a second submission were ultimately approved in both jurisdictions.
Table 4.
Final outcomes for resubmitted (FDA) and reexamined (EMA) marketing applications
| Drug | Indication summary | Final Outcomea | ||
|---|---|---|---|---|
| EMA | FDA | |||
| Applications reexamined by EMA onlyb | Ataluren | Duchenne's Muscular Dystrophy | Approved | Not approved |
| Ixazomib | Multiple myeloma | Approved | Approved | |
| Serelaxin | Symptoms of acute heart failure | Not approved | Not approved | |
| Applications resubmitted to FDA onlyc | 177 Lu‐Dota0‐Tyr3‐octreotate | Gastroentero‐pan‐creatic neuroendocrine tumors | Approved | Approved |
| Cangrelor | Reduction of thrombotic cardiovascular events (MI, stent thrombosis) in patients undergoing percutaneous coronary intervention (PCI) | Approved | Approved | |
| Daclatasvir | Chronic hepatitis C (CHC) infection | Approved | Approved | |
| Empagliflozin | Improvement of glycemic control in Type 2 diabetes mellitus | Approved | Approved | |
| Etelcalcetide | Treatment of secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease on haemodialysis | Approved | Approved | |
| Safinamide mesylate | Fluctuating Parkinson's Disease | Approved | Approved | |
| Sarilumab | Moderate‐severe rheumatoid arthritis | Approved | Approved | |
| Drisapersen | Duchenne's Muscular Dystrophy | Withdrawn | Withdrawn | |
EMA, European Medicines Agency; FDA, US Food and Drug Administration; MI, myocardial infarction.
aIf an application was not resubmitted/reexamined by an agency, the initial outcome was also considered to be the final outcome. bThe three applications reexamined by the EMA were either initially approved by the FDA and therefore were not resubmitted to the FDA (ixasomib) or initially rejected by the FDA and not resubmitted (ataluren and serelaxin). cThe applications resubmitted to the FDA were all initially approved by the EMA, with one exception, and therefore were not reexamined by the EMA.
Final concordant and discordant regulatory outcomes
Further to the above resubmissions and reexaminations of applications, we compared final regulatory outcomes. If an application was not resubmitted/reexamined by an agency, the initial outcome was also considered to be the final outcome. By this accounting, the proportions of applications finally approved by the EMA and the FDA were similar (93% and 92%, respectively). Resubmission/reexamination of initially nonapproved applications increased the concordance between the agencies to 98% (105/107) of applications (Table 5).
Table 5.
Final concordant and discordant marketing application outcomes
| Finala EMA outcome, N = 107 | ||||
|---|---|---|---|---|
| Approved (100) | Not approved (1) | Withdrawn (6) | ||
|
Finala FDA outcome, N = 107 |
Approved (98) | 98 | 0 | 0 |
| Not approved (6) | 2 | 1 | 3 | |
| Withdrawn (3) | 0 | 0 | 3 | |
Shaded cells indicate concordant outcomes.
EMA, European Medicines Agency; FDA, US Food and Drug Administration.
If an application was not resubmitted/reexamined by an agency, the initial outcome was also considered to be the final outcome.
Overall, taking account of the resubmitted and reexamined applications, the EMA and the FDA had final discordant marketing authorization decisions for two drugs: corifollitropin alfa and ataluren.
Concordant approvals with differences in type of marketing authorization
For the 98 applications approved, the FDA and the EMA diverged in the type of marketing authorization for 15 (one agency granted standard approval, the other did not), whereas 76% (74/98) were approved by both agencies as standard approval (Table 6). Nineteen percent (19/98) of the approved applications received accelerated approval from the FDA, and the EMA granted conditional marketing approval to 11% (11/98). These approval mechanisms were employed by both agencies for the same products for only 9% (9/98) of approvals. The EMA also granted approval under “exceptional circumstances” for three applications. The FDA does not have a parallel approval mechanism, but it did approve these same applications as standard approvals.
Table 6.
Final approval outcomes by type of marketing authorization
| EMA marketing approval type, N = 98 | ||||
|---|---|---|---|---|
| Standard (84) | Conditional (11) | Exceptional (3) | ||
| FDA marketing approval type, N = 98 | Standard approval (79) | 74 (76%) | 2 (2%) | 3 (3%) |
| Accelerated approval (19) | 10 (10%) | 9 (9%) | N/A | |
EMA, European Medicines Agency; FDA, US Food and Drug Administration.
The therapeutic areas in which accelerated approval, conditional marketing authorization, or marketing authorization under exceptional circumstances were used most often were oncology and hematology. It was also noted that in these areas, the FDA more commonly granted accelerated approval (12/25 in oncology and 5/8 in hematology) than the EMA granted conditional marketing authorization or authorization under exceptional circumstances (7/25 in oncology and 2/8 in hematology).
Concordant approval decisions with notable differences in the approved indication
Among the 98 applications finally approved by both the EMA and the FDA, 79% (77/98) had the same approved indication, and 21% (21/98) had notable differences in the indication. The proportion of products for which there were differences in the approved indication was similar across therapeutic areas.
Reasons for differing agency outcomes
Altogether, there were 34 drugs with at least one divergent regulatory outcome at the EMA and the FDA among the three types examined: 9 drugs had different initial decisions on marketing approval and, after accounting for resubmission or reexamination of certain applications, 2 drugs had a different final decision on marketing approval. For approved drugs (final approval for purposes of our study), 15 had differences in the type of marketing authorization, and 21 drugs had notable differences in the labeled indications. Eight drugs had differences in both indication and type of marketing authorization. Figure 1 provides an overview of the reasons for initial discordant outcomes in all three categories assessed.
Figure 1.
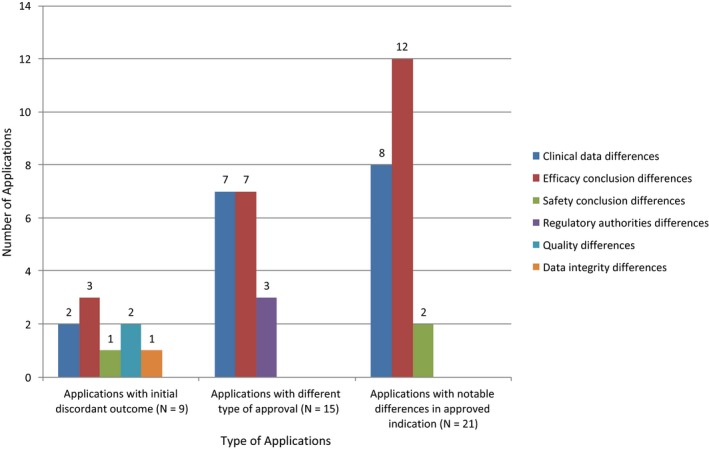
Reasons for initial discordant outcomes, for differences in type of marketing approval and for notable differences in approved indications. Note: Some applications had more than one reason for the difference in outcome; therefore the number of differences is superior to the number of applications in the respective categories.
The most common reason for the nine initial discordant marketing authorization outcomes was a difference between the FDA and the EMA conclusions about efficacy (33%, 3/9 drugs). In all three cases, the agencies reviewed the same clinical efficacy data. Concerns raised included adequacy of the trial design, strength of the findings, or clinical relevance of the efficacy results. Other reasons for the initial discordant outcomes were dissimilarities in the submitted clinical data, differing conclusions about the strength of evidence to support safety, one agency raising concern about the integrity of the data in the application, or an applicant's compliance with good manufacturing practices.
As shown in Figure 1, there were two equally common reasons for agency differences in the type of marketing approval, namely, differing conclusions about efficacy (47%, 7/15) and differences in the submitted clinical data (47%, 7/15). In most cases (5/7 applications), the agencies had dissimilar clinical data because one agency reviewed additional trials (rather than updated data from an initially submitted trial).
Regulatory authority differences explained three cases of the EMA granting approval under exceptional circumstances and the FDA granting standard approval. Unlike the FDA, the EMA can grant marketing authorization for a drug treatment for a disease encountered so rarely that comprehensive evidence cannot be reasonably expected or provided or that it would be contrary to generally accepted principles of medical ethics to collect such information.
With respect to applications with notable differences in the approved indications, the most common reason for this divergent outcome was a difference in the agencies’ conclusions about the strength of evidence to support drug efficacy (52%, 11/21) based on review of the same clinical data, followed by differences in the submitted clinical data (38%, 8/21). The other reason for notable differences in approved indication was diverging conclusions about safety based on review of the same clinical data, namely, different conclusions about the acceptability of the toxicity profile of the drug and therefore how this would be considered in the indication (10%, 2/21).
Postapproval convergence in type of marketing authorization and indications
As shown in Table 6, among the 98 applications that were approved by both the EMA and the FDA, 15% (15/98) had differences in the type of marketing authorization. At the time of our analysis, the marketing authorization for 6 of the 15 had converted to standard approval, with all but one product having the same indication: 5/19 FDA applications had converted from accelerated to standard approval, and 1/13 EMA applications had converted from conditional or exceptional marketing authorization to standard approval. In four of the six products converted to full approval, this occurred at the same time or within less than a year of the second agency's approval.
Further, among the 21 products with different indications at time of initial marketing authorization, 10 had their indication changed post approval (usually in response to additional data submitted) and before our cohort cutoff date, which resulted in alignment of the indication. In 6 out of these 10 products, this alignment occurred at the same time or within less than a year of the second agency's approval.
Discussion
This is the first effort by the EMA and the FDA to compare the agencies’ decisions related to marketing applications and the reasons for any differences. A previous FDA evaluation of regulatory outcomes between January 2006 and October 2008 found that the EMA and the FDA had similar rates of approval (67% and 64%, respectively) and 64% of applications were approved by both agencies.10 However, another study of new applications between 1995 and 2007 suggested that despite the similar rates of overall approval, many of the applications approved by the FDA were not approved by the EMA, and vice versa.11 In our cohort of agency decisions from 2014 to 2016, we found high concordance (91%) in the agencies’ initial decisions on marketing approval. Concordance increased to 98% following review of resubmitted or reexamined applications, even though the agencies do not discuss most applications nor specifically strive to reach the same decisions.
In this cohort EMA had a higher rate of first‐cycle approval than FDA. The FDA's initial nonapproval decisions (13% FDA vs. 3% EMA) reflected a variety of factors, led by differing agency conclusions about efficacy, which is not surprising as some difference in judgement is expected from independent assessments. The effect of differences in conclusions about efficacy were more visible in types of approval (7 products) and approved indications (12 products).
Differences in clinical data submitted in support of an application was the second most common root of divergent FDA and EMA outcomes. Although our study did not focus on comparisons of time to submission of or agency approval of an application, we noted that some instances of dissimilarities in clinical data related to differential timing of submissions and availability of data. For example, for applications submitted to FDA first, it was not uncommon for data in the EMA submissions to be more mature, including trials that were ongoing at the time of submission to FDA, often because they were now completed. This also explained many cases of agency differences in type of marketing approval and approved indication.
We observed remarkable similarity in the basic scientific and data interpretation issues raised by the FDA and the EMA during reviews of the same applications. Specifically, most of the FDA's second cycle approvals (i.e., approvals after resubmission of the applications) were based on submission by the sponsor of the same additional data that EMA had received during its initial review either from the start or following request after clock‐stops. We did not compare agency times from first submission to approval of the marketing applications to assess the impact of these differences in procedures.
The only therapeutic areas that stood out in terms of outcome divergence overall were oncology and hematology. In these areas, submissions to the EMA often were later than to the FDA and, as noted above, often included additional clinical trials or more mature data from the same clinical trial than were submitted to the FDA. In those instances, the EMA was more likely than the FDA to grant standard approval (whereas the FDA issued accelerated approval) or a broader indication.
In addition to constituting the majority of products in our cohort, the preponderance of outcome differences in oncology/hematology may have been influenced by factors relevant to its time frame, including increasing use of expedited development and access tools, marked by the FDA Safety and Innovation Act which, among other things, established the FDA's Breakthrough Therapy designation in 2012, several years before establishment of the EMA's Priority Medicines (PRIME) scheme in 2016.12, 13
In assessing differences in indication, we observed themes of variation in EMA and FDA approaches. For example, among products for which supporting trials showed efficacy in patients who had failed first‐line therapy, the FDA's approved indications were typically limited to the population studied (treatment failures or relapses) while the EMA's also included patients for whom first‐line therapy was not appropriate. Other variations included descriptions of diagnostic criteria or pharmacogenomic characteristics (e.g., tumor subtype or hepatitis C virus genotype) to describe the recommended patient population, enough to make a notable difference in the target population. Since indications may impact clinical practice and medicines reimbursement, both of which have regional variation, there is room for further examination and discussion of the importance of some of the differences we observed.
Our findings should be considered within the limitations of the study. The cohort covered a short time period and results reflect prevalent therapeutic areas such as oncology (25% of cohort). This field has undergone an explosion of new research and factors that impact marketing authorization decisions, such as identification of new treatment subpopulations and consideration of acceptable endpoints in clinical trials in addition to the already mentioned emphasis on use of expedited development pathways. Our cohort also did not include blood products, vaccines, or regenerative medicines, a decision made simply because most previous comparisons have also excluded these, but it is an area that could be interesting for future exploration.
Some might consider it limiting that determination of reasons for agency discordance was based solely on information in EMA and FDA written reviews. We did not include other documents as primary sources of information, such as FDA or EMA Scientific Advisory Committee meeting minutes, nor did we systematically interview agency staff. Nevertheless, EMA European Public Assessment Reports and FDA reviews are official records documenting the agencies’ rationale for regulatory decisions, and we found them complete and informative. Reading them revealed striking similarities in how the agencies approached the same information, including assumptions, questions, and contextual considerations. We did not evaluate whether the two agencies’ experts convened to discuss development programs or applications under review. This would have been most informative if we compared concordant outcomes, a more complex exploration. We do know that such discussions during 2014–2016 were not the norm, other than perhaps for some oncology application reviews.
We did not assess divergence between the EMA and the FDA with respect to labeled contraindications or other limitations of use, risk minimization requirements, and requirements for postapproval safety and efficacy studies. We concluded that such analyses would be minimally informative on such a broad scale, as the two agencies have distinctly different labeling practices and regulatory authorities in these areas. For example, whereas the EMA can require applicants to conduct postauthorization studies to further evaluate efficacy of any drug, the FDA can only require confirmatory trials for drugs under accelerated approval.
Lastly, we did not systematically compare order, timing, duration of application reviews, or time to approval across the two agencies. Although in some cases differential timing of submissions clearly influenced a small number of regulatory outcomes, in those cases it mostly reflected data availability or maturity. We believe evaluation of the rationale for differences in regulatory decision making is more informative for understanding regulatory science and the impact and direction of our collaborative regulatory efforts.
Methods
We identified all marketing applications for new active substances, NCEs, and original therapeutic biologic products submitted by the same applicant with a regulatory outcome at both agencies in the period 2014 to 2016 and those with an outcome at one agency during the study period and at the other agency outside of the study period. A regulatory outcome was defined as approval, nonapproval, or applicant withdrawal of the marketing authorization application.
We did not include applications for nontherapeutic biologics (i.e., vaccines and blood products); advanced therapy medicinal products, such as cell and gene therapies; or those for which one agency's regulatory outcome was still pending at the time of our data lock for cohort selection (January 30, 2018). Information was extracted about each marketing application and its regulatory outcome from FDA and EMA internal databases and public websites. The agencies’ internal reviews and correspondences allowed for more detailed assessment of the scientific and regulatory reasons underlying differences in regulatory outcomes than were in the publicly available reviews.
General drug characteristics were summarized, namely, the therapeutic area and whether the drug was an NCE or biologic. Because of differences in EMA and FDA regulations, we did not compare application characteristics related to orphan designation, expedited development pathways (FDA breakthrough or EMA priority medicines designation), or expedited review pathways (FDA priority review and EMA accelerated assessment).13, 14
Concordant outcomes were defined as those for which both the FDA and the EMA had the same regulatory result, namely: both agencies approved the application; both agencies did not approve the application, the application was withdrawn (by the applicant) from both agencies prior to a decision, or the application was not approved by one agency and withdrawn at the other agency. Withdrawal was considered concordant with nonapproval when an applicant withdrew its marketing application in anticipation of a nonapproval decision by that agency. Discordant outcomes were defined as those for which the FDA and the EMA had different regulatory conclusions: one agency approved the application while the other did not approve it, or one agency approved the application whereas it was withdrawn from the other agency.
Several applications were either resubmitted to the FDA after an initial nonapproval decision,15 or reexamined after an initial negative opinion by the EMA's CHMP prior to decision by the European Commission.16 We therefore explored the impact of resubmission or reexamination on the extent of EMA and FDA concordance by assessing both initial and final regulatory outcomes. For applications that were not ever resubmitted to the FDA or reexamined by the CHMP, the initial outcome was also considered to be the final outcome.
For marketing applications that were approved by both agencies (final concordant approval outcomes), we also compared the type of marketing approval granted. For the FDA, the type of marketing approval refers to standard approval or accelerated approval17; for the EMA, the type of approval refers to standard marketing authorization, conditional marketing authorization, or marketing authorization granted under exceptional circumstances.18, 19 We considered the FDA's accelerated approval and the EMA's conditional marketing authorization to be similar—each is an approval mechanism for drugs intended for a serious or life‐threatening disease or condition with an unmet medical need, and for which postauthorization studies are expected to provide additional data to support standard approval.
We also assessed for differences in approved indications across agencies, defining indication as the medical disease or condition and population for which use of the drug is approved. Information about the approved indication was obtained from the European Summary of Product Characteristics and the FDA‐approved Prescribing Information. Indications were considered similar when they were for the same general intended use and there were only differences in wording or organization of the information. For products with multiple approved uses, we compared each distinct indication.
A “notable difference” in indication was defined as a difference in disease or condition stage, severity, or sub‐group; target population (age group, pharmacogenomic subgroup, previous treatment failure, other patient characteristics); and/or other conditions of use. Other conditions of use comprised type of therapy (symptomatic, disease‐modifying); use with other products (monotherapy, combination therapy, adjuvant therapy); and place in or line of therapy.
Judgment on “notable” difference in indication was made using a multistep process that began by two researchers independently comparing the verbatim indication statements, and other sections of the labels as needed, to preliminarily identify those products with notable differences. Indications for which the two researchers were uncertain or had divergent interpretations were then independently evaluated by the other researchers using the same criteria for notable differences. Discrepancies were resolved through consensus.
Finally, for applications with discordant outcomes and concordant approvals with differences in the indication or type of marketing approval, we undertook detailed assessments to evaluate the reasons for each using a similar multistep approach to the one described above. Reasons for discordance were grouped into six categories:
Clinical data differences were defined as differences in the main (pivotal) trials considered in support of efficacy—these could be different trials submitted or assessed or, if the same main trial was evaluated, differences in the trial data evaluated. When the same clinical trial and major analyses were submitted to both agencies, we did not assess further details about which specific additional analyses were conducted or requested by one agency and not the other.
Efficacy conclusion differences were defined as differences in FDA and EMA conclusions about the strength or clinical relevance of evidence to support efficacy, when the same clinical trial data were submitted to both agencies
Safety conclusion differences were defined as differences in conclusions about the strength of evidence to support safety or acceptability of the drug's toxicity when the same clinical trial data were submitted to both agencies.
Regulatory authority differences referred specifically to the EMA's approval under Exceptional Circumstances, an option with no parallel at the FDA.
Quality differences included divergent conclusions about the quality of the drug substance or product, acceptability of the manufacturing process, or applicant compliance with good manufacturing practices.
Data integrity differences included divergent conclusions about some aspect of data integrity in the application, such as nonclinical or clinical trial data or applicant compliance with good clinical practices.
Regional differences in medical practice or standard of care were considered factors that could influence agency conclusions about safety or efficacy but not as a separate category.
We began by identifying all factors that contributed to the differences between EMA and FDA outcomes and, whenever possible, drilled down to the primary driver for the divergence. If the agencies’ decisions on the applications were based on review of differing clinical data, then this was the primary reason for the different regulatory outcomes. This is because our study objective was to determine whether the agencies varied in their conclusions about an application, and such an assessment can only be made when the conclusions are based on the same information.
Funding
This work was funded by US Food and Drug Administration and the European Medicines Agency.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
M.K., Z.H., J.L., and S.L.K. wrote the manuscript; M.K., Z.H., R.V., C.B., J.L., and S.Y. designed the research; M.K., Z.H., and S.Y. performed the research; M.K., Z.H., R.V., C.B., J.L., and S.Y. analyzed the data.
Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the agencies or organizations with which the authors are affiliated.
Acknowledgments
We thank Dr. Youness Ameur of the European Medicines Agency for his contributions to the collection of data for the research reported in this article.
References
- 1. Tominaga, T. The ICH, the GHTF, and the future of harmonization initiatives. Ther. Innov. Sci. 47, 572–580 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Downing, N.S. , Zhang, A.D. & Ross, J.S. Regulatory review of new therapeutic agents — FDA versus EMA, 2011–2015. N. Engl. J. Med. 376, 1386–1387 (2017). [DOI] [PubMed] [Google Scholar]
- 3. Samuel, N. & Verma, S. Cross‐comparison of cancer drug approvals at three international regulatory agencies. Curr. Oncol. 23, e454–e460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alqahtani, S. , Seoane‐Vazquez, E. , Rodriguez‐Monguio, R. & Eguale, T. Priority review drugs approved by the FDA and the EMA: time for international regulatory harmonization of pharmaceuticals? Pharmacoepidemiol. Drug Saf. 24, 709–715 (2015). [DOI] [PubMed] [Google Scholar]
- 5. Sacks, L.V. , Shamsuddin, H.H. , Yasinskaya, Y.I. , Bouri, K. , Lanthier, M.L. & Sherman, R.E. Scientific and regulatory reasons for delay and denial of FDA approval of initial applications for new drugs, 2000‐2012. JAMA 311, 378–384 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Tafuri, G. , Trotta, F. , Leufkens, H.G. & Pani, L. Disclosure of grounds of European withdrawn and refused applications: a step forward on regulatory transparency. Br. J. Clin. Pharmacol. 75, 1149–1151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tafuri, G. et al How do the EMA and FDA decide which anticancer drugs make it to the market? A comparative qualitative study on decision makers’ views. Ann. Oncol. 25, 265–269 (2014). [DOI] [PubMed] [Google Scholar]
- 8. ICH . ICH – harmonization for better health. <http://www.ich.org/home.html> (2018). Accessed April 20, 2018.
- 9. European Medicines Agency – Cluster Activities . <http://www.ema.europa.eu/ema/index.jsp?curl=pages/partners_and_networks/general/general_content_000655.jsp%26mxml:id=WC0b01ac0580953d9>
- 10. Jenkins, JK . New Drug Review: 2008 Update. FDA/CMS Summit, Washington, DC (2008).
- 11. Huckle, P. Are today's regulatory submissions flawed? An industry viewpoint. Workshop Report: Predictable Outcomes – Why do Potential Winners Fail? (CMR International Institute for Regulatory Science, 2008).
- 12. European Medicines Agency. Enhanced early dialogue to facilitate accelerated assessment of PRIority Medicines (PRIME) EMA/CHMP/57760/2015, Rev. 1). <https://www.ema.europa.eu/documents/regulatory-procedural-guideline/enhanced-early-dialogue-facilitate-accelerated-assessment-priority-medicines-prime_en.pdf> (May 2018).
- 13. US Food and Drug Administration. FDA guidance for industry, expedited programs for serious conditions – drugs and biologics. <https://www.fda.gov/downloads/Drugs/Guidances/UCM358301.pdf> (September 2017).
- 14. Article 14(9) of Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency, as amended. March 2004.
- 15. US Code of Federal Regulations, Title 21, Section 314.110(b)(1) (21 CFR 314.110(b)(1)).
- 16. Article 9(2) of Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency, as amended. March 2004.
- 17. US Code of Federal Regulations, Title 21, Section 314.500 (21 CFR 314.500) .
- 18. Article 14(7) of Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency, as amended. March 2004.
- 19. Article 14(8) of Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency, as amended. March 2004.