

Abstract
Sepsis is an infection-induced systemic inflammatory syndrome. The immune response in sepsis is characterized by the activation of both proinflammatory and anti-inflammatory pathways. When sepsis occurs, the expression and activity of many inflammatory cytokines are markedly affected. Xenobiotic receptors are chemical-sensing transcription factors that play essential roles in the transcriptional regulation of drug-metabolizing enzymes (DMEs). Xenobiotic receptors mediate the functional crosstalk between sepsis and drug metabolism because the inflammatory cytokines released during sepsis can affect the expression and activity of xenobiotic receptors and thus impact the expression and activity of DMEs. Xenobiotic receptors in turn may affect the clinical outcomes of sepsis. This review focuses on the sepsis-induced inflammatory response and xenobiotic receptors such as pregnane X receptor (PXR), aryl hydrocarbon receptor (AHR), glucocorticoid receptor (GR), and constitutive androstane receptor (CAR), DMEs such as CYP1A, CYP2B6, CYP2C9, and CYP3A4, and drug transporters such as p-glycoprotein (P-gp), and multidrug resistance-associated protein (MRPs) that are affected by sepsis. Understanding the xenobiotic receptor-mediated effect of sepsis on drug metabolism will help to improve the safe use of drugs in sepsis patients and the development of new xenobiotic receptor-based therapeutic strategies for sepsis.
Key words: Sepsis, Inflammatory cytokines, Xenobiotic receptors, Drug metabolism, Drug-metabolizing enzymes, Drug transporters
Abbreviations: AHR, aryl hydrocarbon receptor; AP-1, adaptor protein 1; ARNT, AHR nuclear translocator; CYPs, cytochrome P450s; COX-2, cyclooxygenase 2; CLP, cecum ligation and puncture; DMEs, drug-metabolizing enzymes; DREs, dioxin response elements; GC, glucocorticoid; GR, glucocorticoid receptor; GREs, glucocorticoid receptor response elements; Gsts, phase II glutathione S-transferase; HSP90, heat shock protein 90; IL-1β, interleukin-1β; IRF3, interferon regulatory factor 3; IRF7, interferon regulatory factor 7; IBD, inflammatory bowel disease; LPS, lipopolysaccharide; Mrp, phase III multidrug-resistant protein; NR, nuclear receptor; NF-κB, nuclear factor-kappa B; NOS, nitric oxide synthase; Oatp2, organic anion transport polypeptide 2; PXR, pregnane X receptor; PRRs, pattern recognition receptors; PKC, protein kinase C; P-gp, p-glycoprotein; PCN, pregnenolone-16α-carbonitrile; PAS, Per/ARNT/Sim; PLA2, phospholipase A2; TNF-α, tumor necrosis factor; SRC1, steroid receptor coactivator 1; STAT3, signal transducers and activators of transcription 3; Sult, sulfonyl transferase; Ugts, UDP-glucuronic transferase
Graphical abstract
The functional crosstalk between sepsis and drug metabolism is mediated by xenobiotic receptors. The inflammatory cytokines released during sepsis can affect the expression and activity of xenobiotic receptors and thus impact the expression and activity of DMEs.
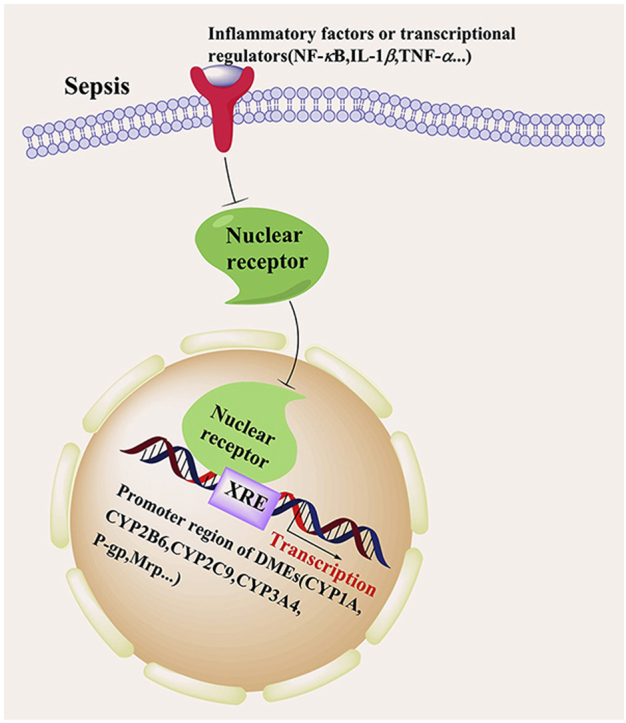
1. Introduction
Sepsis remains a major challenge in the clinic because of its high rates of morbidity and mortality. Despite advances in intensive care and supportive technologies, the mortality rate of sepsis still ranges from 15% to 80%1. There are 31.5 million cases of sepsis worldwide, resulting in approximately 5.3 million deaths per year1. Sepsis is the 10th leading cause of death in the United States2.
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection3. The pathogenesis of sepsis involves the release of endotoxins and exotoxins from pathogens. Endotoxins, such as lipopolysaccharide (LPS) and lipoteichoic acid, are structural components of the bacterial cell wall4. Exotoxins are actively secreted toxins mainly produced by gram-positive bacteria5. Endotoxins and exotoxins activate innate immune responses through pattern recognition receptors (PRRs) and transcription factors, such as nuclear factor-kappa B (NF-κB), and subsequently induce the production of inflammatory cytokines, such as tumor necrosis factor (TNF-α), interleukin-1β (IL-1β), interferon regulatory factor 3 (IRF3), interferon regulatory factor 7 (IRF7), and adaptor protein 1 (AP-1)6. The release of inflammatory cytokines induces the production of new cytokines, which in turn causes cell and organ damage. There are several commonly used mouse models of sepsis. The lipopolysaccharide (LPS) model simulates sepsis-induced inflammatory response. This toxemia model induced by i.p. injection of LPS can cause a sharp increase in cytokine production in animals at an early stage, and the inflammatory response disappears very quickly7. Cecal ligation and puncture (CLP) is an independent model of sepsis that induced by bacterial infection. In the CLP model, because of the pathophysiological process of cecal ligation and perforation, the dysregulation of inflammatory factor expression and the immune response is the closest to the pathophysiology of clinical sepsis. As such, CLP is currently the most commonly used animal model of sepsis8.
In addition to their involvement in the inflammatory response, the inflammatory cytokines released in sepsis can also influence the expression and activity of xenobiotic receptors and further affect drug-metabolizing enzymes (DMEs) and transporters. In sepsis, the expression of hepatic and intestinal cytochrome p450 enzymes and drug transporters, such as CYP1A, CYP2B6, CYP2C9, CYP3A4, P-gp and MRPs, is downregulated, as is the uptake activity of trans carriers, such as organic anion transport polypeptide 3 (OATP3). Therefore, alterations in drug metabolism have been found in sepsis patients in the clinic. Carillo and colleagues9 reported that children with sepsis had a two-fold reduction in antipyrine clearance and that those with multiple organ failure had a four-fold decrease in antipyrine clearance. These results were consistent with the report that the clearance of antipyrine is inversely correlated with circulating level of IL-6 and multiorgan failure. Clinical studies have shown that pro-inflammatory cytokines (especially IL-6) and cytokine antagonists (especially anti-IL-6 monoclonal antibody, tocilizumab) can affect the pharmacokinetics of co-administered small molecule drug10. Treatment of rheumatoid arthritis patients with tocilizumab resulted in a 43% reduction in plasma exposure to the CYP3A4 substrate simvastatin. This reduction indicates reversal of IL-6 mediated CYP3A4 suppression in rheumatoid arthritis patients with tocilizumab exposure11.
Xenobiotic receptors are chemical-sensing transcriptional factors that play essential roles in the transcriptional regulation of DMEs. Many of the xenobiotic receptors, such as PXR, CAR, and GR12, 13, 14, belong to the nuclear receptor (NR) superfamily. NRs are proteins sensitive to steroids, thyroid hormones, or other molecules that are widespread in the organs of species from various genera. NRs are divided into orphan NRs, steroid hormone receptors and nonsteroid hormone receptors. NRs regulate the growth, balance, and metabolism of an organism by influencing gene expression15. The AHR is another xenobiotic receptor. Although the AHR does not belong to the NR superfamily, the mode of action by which the AHR senses chemicals and regulates DMEs is similar to that of the xenobiotic NRs. The primary function of xenobiotic receptors is to regulate the expression of DMEs and drug transporters, and in so doing, to affect the metabolism and disposition of drugs and other xenobiotics. DMEs, among which phase I cytochrome P450s (CYPs) are the most important, are involved in the oxidation, reduction, and hydrolysis of many clinical drugs16.
This review details which inflammatory cytokines are released and how they may interact with xenobiotic receptors and DMEs during sepsis.
2. Xenobiotic receptors mediate the effect of sepsis on drug metabolism
2.1. The role of PXR in mediating the effect of sepsis on drug metabolism
2.1.1. Effects of sepsis on the expression of PXR and its target DMEs
Infection and inflammation are known to inhibit drug metabolism and, in some cases, increase drug toxicity17. Recent evidence suggests that PXR plays an important role in mediating the suppressive effect of sepsis on drug metabolism18,19.
PXR is as a decisive xenobiotic receptor involved in the regulation of DMEs and transporter genes. PXR has been confirmed to be the core component involved in the transcriptional regulation of DME genes in a range of diverse species. The activation of PXR induces the expression of many DMEs and transporters, including phase I enzymes, phase II enzymes and phase III drug transporters. Examples of PXR target genes are phase I Cyp3a, Cyp2b6, Cyp2c9 and Cyp2c19; phase II glutathione S-transferase (Gsts), UDP-glucuronic transferase (Ugts), sulfonyl transferase (Sult); and phase III multidrug resistant protein (Mrp), organic anion transport polypeptide 2 (Oatp2) and p-glycoprotein (P-gp)20,21.
It has been suggested that sepsis inhibits drug metabolism by inhibiting the expression and activity of PXR. In the LPS model of sepsis in C57BL/6 mice, the mRNA and protein levels of PXR are significantly decreased, leading to the downregulation of the expression of the PXR target gene Cyp3a1122. It has been suggested that the sepsis-induced release of proinflammatory cytokines is responsible for the suppression of the expression of PXR and DMEs23. The suppression of PXR and DMEs is believed to be due to the sepsis-induced release of proinflammatory cytokines23. In support of this hypothesis, it has been reported that the treatment of wild-type mice with the proinflammatory cytokine IL-6 decreases the mRNA and protein expression of PXR. The hepatic mRNA expression levels of the PXR target genes Cyp3a, Mrp, and P-gp are also reduced in IL-6-treated mice. The effect of IL-6 on the expression of PXR target DME genes is abolished in PXR-knockout mice, suggesting that the inhibition of drug metabolism by IL-6 is PXR dependent (Fig. 1)24.
Figure 1.

Effects of sepsis on the expression of PXR and its target DMEs. The expression of IL-1β, IL-6 and TNF-α in LPS-induced inflammatory macrophages is induced through the NF-κB and PKC-NF-κB pathways in sepsis. The increase in IL-1β, IL-6, and TNF-α expression inhibits the activation of PXR in hepatocytes, leading to the downregulation of the activity of DMEs and drug transporters such as Cyp3a4, P-gp and Mrp.
At the mechanistic level, the sepsis-induced activation of NF-κB and PKC may be responsible for the inhibition of PXR. NF-κB, a major nuclear transcription factor that regulates the expression of inflammatory genes, is composed of p65 (RelA) and P50 heterodimers. Under nonactivation conditions, NF-κB, together with IκB (a protein that inhibits NF-κB), can be polymerized into tripolymeric P65-P50-IκB and reside in the cytoplasm. The activation of NF-κB by LPS and TNF-α causes the formation of the p65/p50 dimer, which binds to DNA. Previous studies have shown that NF-κB p65 interferes with the binding of the PXR-RXRα complex to DNA, thus inhibiting the activity of PXR. The mechanism of this process involves the direct interaction of p65 with the DNA-binding domain of RXRα, leading to steric hindrance that blocks its binding to the PXR protein. Subsequently, the activation of the PXR–RXRα heterodimer is inhibited, the transcriptional activity of PXR is decreased, and DME expression is downregulated25.
The activation of the PKC signaling pathway can inhibit PXR activity through changes in the PXR–NR cofactor complex, which may be directly changed by the phosphorylation of NR corepressor protein (NCoR), and steroid receptor coactivator 1 protein (SRC1)26. LPS and inflammatory cytokines directly participate in PKC activation; furthermore, activated PKC facilitates the activation and phosphorylation of the kinase Iκκ, leading to the activation of NF-κB. Activated NF-κB inhibits PXR activity as described above. Additionally, activated PKC can also inhibit PXR activity by disrupting the interaction between PXR and the NR coactivator steroid receptor coactivator 1 (SRC1), enhancing the interaction between PXR and the NR corepressor NCoR27,28. As summarized in Fig. 1, both NF-κB and PKC may mediate the effect of sepsis on the expression and activity of PXR.
2.1.2. Reciprocal effects of PXR on inflammation and sepsis
Interestingly, the effect of sepsis on PXR is bidirectional, meaning that PXR can also affect inflammation and the outcome of sepsis. Activated PXR exerts anti-inflammatory effects by inhibiting NF-κB and thus reduce inflammation caused by the inflammatory cytokines IL-6 and IL-129. As a key regulator of inflammation, activated NF-κB is frequently detected in various inflammatory diseases and tumors. The gene expression levels of proinflammatory cytokines, including Il-6, Il-2, Tnf-α and Cox-2, in hepatocytes of Pxr-knockout mice are significantly higher than those of wild-type (WT) mice, which can be explained by the failure of PXR to inhibiting NF-κB in vivo. The induction of IκBα and TNF-α by TNF-α is significantly inhibited in primary hepatocytes isolated from WT mice after 24 h of treatment with the mouse PXR activator pregnenolone-16α-carbonitrile (PCN)30. However, in hepatocytes isolated from Pxr-knockout mice, the abovementioned inhibition is lost, indicating that the inhibitory effect of PCN on the expression of NF-κB target genes is mediated by PXR. Together, these results suggest that the activation of PXR can inhibit NF-κB signal transduction NF-κB and target gene expression in vivo30.
Because of the anti-inflammatory activity of PXR, it has been suggested that pharmacological activation of PXR may be useful to treat sepsis. PXR is considered a potential therapeutic target in inflammatory diseases such as sepsis and inflammatory bowel disease (IBD)31. PXR plays an important role in promoting immune and inflammatory responses. In addition, activated PXR can inhibit the activity of NF-κB by negative feedback, thereby inhibiting the expression and secretion of IL-6, blocking the adhesion and migration of inflammatory cells32. Therefore, the balance between inflammation and immunity in the pathogenesis of sepsis can be restored by the activation of PXR (Fig. 1).
2.2. The role of AHR in mediating the effect of sepsis on drug metabolism
2.2.1. Effect of AHR on sepsis
The AHR is a period circadian protein, Per/aryl hydrocarbon receptor nuclear translocator protein, ARNT/single-minded protein, Sim (PAS) domain transcriptional factor and typical xenobiotic receptor. Although the AHR does not belong to the NR superfamily, the mode of action by which the AHR senses chemicals and regulates DMEs is similar to that of the xenobiotic NRs. In addition to its xenobiotic functions, the AHR also plays an important role in maintaining physiological homeostasis and participates in cell proliferation and differentiation, the immune response, gene expression regulation, hormone metabolism, inflammation, immune self-recognition and reactions to external stimuli. The AHR is also a key regulator of inflammation and immunity in severe pneumonia and other types of sepsis33, 34, 35, 36, 37. The expression of the AHR, which is indispensable for the LPS-induced signaling cascade of the inflammatory response and sepsis tolerance, is closely linked to the release of inflammatory factors38, 39, 40. The activation or upregulation AHR expression can regulate the expression of the anti-inflammatory cytokine IL-10 and downregulate the expression of the proinflammatory cytokines IL-6 and IL-8, thereby inhibiting the inflammatory response, maintaining immune homeostasis, and altering the occurrence and prognosis of sepsis (Fig. 2).
Figure 2.

Effects of sepsis on AHR and inflammatory factors. During sepsis, the activation of NF-κB increases the expression of inflammatory factors IL-1β and IL-10 in LPS-stimulated inflammatory macrophages, and the expression of AHR is upregulated through the NF-κB pathway. Concurrently, the activation of AHR can enhance the phosphorylation of SRC; then, p-SRC upregulates STAT3 phosphorylation, and p-STAT3 further positively regulates the production of IL-10 by macrophages.
IL-10, which plays an important negative regulatory role in the early progression of sepsis, can inhibit the inflammatory response induced by various immune cells and limit tissue damage and immunopathological changes during infection41. IL-10 is regulated by multiple transcription factors and signaling pathways. An increasing number of studies have shown that the AHR is the key regulator of IL-10 expression. Src tyrosine kinase (SRC), one of nine nonreceptor tyrosine kinases, is a known intracellular target protein that mediates tyrosine phosphorylation. SRC can induce the production of IL-10 and downregulate the expression of the proinflammatory factors IL-1β, IL-6, IL-18, and TNF-α42. Signal transducers and activators of transcription 3 (STAT3) are known transcriptional regulators of LPS-induced inflammatory factor expression. STAT3 regulates IL-10 expression by binding to intron 4, and STAT3 is also a downstream regulatory factor of IL-10. Moreover, IL-10 can activate STAT3 in an autocrine regulatory manner43.
We recently reported that both the phosphorylation of SRC and STAT3 and the expression of IL-10 are decreased in inflammatory macrophages derived from Ahr-knockout mice. These results suggest that the AHR positively regulates the expression of IL-10 through a nongenomic pathway, and this regulation depends on the tyrosine phosphorylation-mediated activation of SRC and STAT344. We have shown that the positive regulation of IL-10 by AHR is reduced by specific SRC inhibitors and that the phosphorylation of STAT3 is downregulated by Src inhibitors, suggesting that SRC can induce IL-10 production upstream of STAT344. These results indicated that AHR activation enhances the phosphorylation of SRC; then, p-SRC upregulates STAT3 phosphorylation, and p-STAT3 further positively regulates the production of IL-10 by macrophages. Thus, the inhibition of the inflammatory reaction by AHR activation can improve tolerance to LPS, regulate immune functions, and reduce sepsis-induced mortality.
Previous studies also found that the AHR is involved in LPS-induced inflammatory gene expression. Ahr-knockout mice are hypersensitive to LPS-induced sepsis, mainly as a result of macrophage dysfunction. LPS-treated Ahr-knockout mice show markedly higher plasma levels of IL-1β, IL-18, IL-6, and TNF-α than WT control mice45. Activated AHR also plays a central role in limiting endotoxin-induced inflammation46. The activation of the AHR by endogenous and exogenous ligands upregulates the expression of anti-inflammatory factors such as IL-10 and downregulates the expression of proinflammatory factors such as IL-1β, IL-6, IL-18 and TNF-α. These anti-inflammatory and proinflammatory factors can regulate cytokine production by immune cells and thus affect the inflammatory response and homeostasis of the immune system. Therefore, we believe that the protective effects of AHR via the regulation of the SRC-STAT3-IL-10 signaling pathway may be a potential therapeutic target for the early intervention and treatment of severe pneumonia and sepsis.
2.2.2. Effects of sepsis on the expression of AHR and its target DMEs
Cyp1a and Cyp1b are primary AHR target genes. The hepatic expression of Ahr and Cyp1a2, the predominant Cyp1a isoform in the rat liver, which plays an important role in the sepsis-induced inflammatory response and liver injury, is downregulated in cecum ligation and puncture (CLP)-induced sepsis47, 48, 49, 50, 51. Transcription of the Cyp1a2 gene is mediated by the AHR, together with its heterodimerization partner AHR nuclear translocator (ARNT) and the chaperone heat shock protein 90 (HSP90)52, 53,52, 53. Upon ligand stimulation, the AHR–HSP90 complex enters the nucleus and subsequently dissociates, enabling the phosphorylation of the AHR by tyrosine kinase. The activated AHR then forms a heterodimeric complex with ARNT54. Within the nucleus, the AHR–ARNT complex recognizes and binds to specific regulatory sequences known as dioxin response elements (DREs) in the promoter region and initiates the transcription of the Cyp1a2 gene (Fig. 3)55, 56, 57, 58.
Figure 3.

Effects of sepsis on the expression of the AHR and the GR and their target DMEs. In sepsis, the activation of GR and AHR in hepatocytes is inhibited due to the production of inflammatory factors TNF-1 and IL-1β. The inhibition of GR expression leads to the downregulation of CYP2C9 gene expression. The inhibition of GR can also reduce PXR expression, thereby inhibiting the expression of CYP2B6 and CYP3A4. The suppression of AHR has an effect on the phosphorylation of AHR, leading to the downregulation of the expression of Cyp1a2.
The downregulation of hepatic AHR and Cyp1a2 expression in septic animals does not appear to be due to elevated endotoxin levels because the treatment of hepatocytes with TNF-α and IL-1β is sufficient to downregulate the protein expression of Cyp1a2 and the AHR59. These results suggest that endotoxin itself may not be required to decrease the expression of the Ahr and Cyp1a2 genes after CLP but rather that the decreased expression of Ahr and Cyp1a2 is caused by the proinflammatory factors TNF-α and IL-1β60, 61, 62. Interestingly, the effect of sepsis on the expression of AHR and Cyp1a2 is model specific. The suppression of AHR and Cyp1a2 expression has been found only in the CLP model of sepsis. In a previous study, we found that LPS-induced sepsis upregulates rather than downregulates the expression of the AHR and Cyp1a2. The induction of AHR expression in LPS-stimulated macrophages is attenuated in cells cotreated with an NF-κB inhibitor (PDTC), suggesting that AHR induction is NF-κB dependent.
In the CLP model of sepsis, the decreased expression of the Ahr gene precedes the downregulation of Cyp1a2 expression, suggesting that the decreased expression of Ahr may be responsible for the downregulation of Cyp1a2 expression63. As AHR induces the expression of specific genes by binding to their promoters, the translocation of AHR to the nucleus in sepsis suggests that alterations in the DNA binding of Ahr may have contributed to downregulated Cyp1a2 expression in sepsis59. Therefore, during CLP-induced sepsis, the release of proinflammatory factors, such as TNF-α and IL-1β, decreases the expression of AHR. The decreased expression of AHR in turn downregulates the expression of Cyp1a2. In the LPS model of sepsis, LPS may increase the expression of AHR through the activation of NF-κB, which increases the expression and activity of Cyp1a2. Cyp1a2 has both endobiotic and xenobiotic substrates. Future studies are necessary to define the pathophysiological consequence of the sepsis-induced regulation of Ahr and Cyp1a2.
2.3. The role of GR in mediating the effect of sepsis on drug metabolism
2.3.1. Effect of sepsis on GR expression
Glucocorticoid (GC) therapy has been widely used in the clinic to treat sepsis. The pharmacological activity of GCs is mediated by the glucocorticoid receptor (GR). The GR is a nuclear hormone receptor with a wide range of roles in both health and disease. The GR is a ligand-binding transcription factor, but in the absence of ligand, the GR resides in the cytoplasm bound to HSP90 and other stabilizing cofactors64. When GCs and other GR ligands cross the cell membrane, they bind to free GR in the cytoplasm. The ligand-activated GR then rapidly translocates to the nucleus, where the transcription of target genes is initiated. Two molecules of GR bind to glucocorticoid receptor response elements (GREs) in the promoter regions of steroid-sensitive genes as a homodimer, leading to the transcription of genes encoding anti-inflammatory mediators (e.g., IL-10) and the inhibition of NF-κB65. GCs can also suppress the synthesis of the phospholipase A2 (PLA2), cyclooxygenase 2 (COX-2), and nitric oxide synthase (NOS) genes, decreasing the production of prostanoids, platelet-activating factor, and nitric oxide, which are key molecules in the inflammatory pathway. Therefore, GCs attenuate the inflammatory response and nearly preserve or restore the histology of both lung and systemic organs, thereby promoting sepsis recovery (Fig. 4)66, 67, 68, 69.
Figure 4.

Effects of sepsis on GR and inflammatory factors. During sepsis, the expression of GR is downregulated in LPS-stimulated inflammatory macrophages. Decreased GR expression results in the activation of NF-κB, and the sustained activation of NF-κB, leading to the upregulation of iNOS, TNF-α and IL-6 expression.
Some studies have shown that in the LPS-induced sepsis model, GR expression is markedly reduced in the lung, liver, and kidney. Decreased GR expression results in the activation of NF-κB, and the sustained activation of NF-κB eventually upregulates the expression of iNOS, TNF-α, and IL-6. Decreased GR expression may reduce the therapeutic effect of GCs in sepsis, further decreasing the sensitivity to GCs70,71.
Three different 3′-splice variants of the GR have been reported: GR-α, the most abundant, which binds to ligands and is functionally active; GR-P, which is thought to enhance the function of GR-α72; and GR-β, a dominant-negative inhibitor of GR-α73,74. A study found that the mRNA levels of the GR-α and GR-p splice variants in neutrophils from children with sepsis were reduced on day 0 compared to their levels after recovery75. GR-α and GR-P mRNA levels gradually increased on days 3 and 7 and normalized after recovery. GR-β mRNA levels did not change significantly during sepsis. These results suggest that GC treatment that takes into account the timing and tissue-specific regulation of GR splice variants would benefit patients with sepsis.
2.3.2. Effects of sepsis on the expression of GR and its target DMEs
Studies have confirmed the existence of a GR-PXR-P450 cascade and illustrated the central contribution of the GR and this cascade to the maintenance of a significant level of xenobiotic metabolism in the liver76. First, physiological concentrations of GCs induce CYP2C9 expression by activating the GR. The activated GR also increases the expression of PXR and its heterodimerization partner RXR, leading to increased basal transcriptional activation of the PXR target gene CYP3A4. Second, PXR can be activated by xenobiotics or high concentrations of GCs, which increases the expression of CYP2B6 and CYP3A477. Therefore, the activation of the GR results in the enhanced expression of CYP2B6, CYP3A4, and CYP2C9. GCs increase the mRNA and protein levels of PXR and RXR, potentiating the xenobiotic receptor-mediated induction of CYP2B6, CYP2C9 and CYP3A4 expression78,79. Since the expression of the GR is suppressed by sepsis, the inhibition of the GR by sepsis may downregulate the expression of CYP2B6, CYP2C9 and CYP3A4 genes, thereby inhibiting drug metabolism (Fig. 4).
3. Conclusions and perspectives
Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection. Drug metabolism is known to be compromised during sepsis, but the underlying mechanism needs to be clarified. In this article, we reviewed the effect of the sepsis-induced release of inflammatory factors, such as IL-1β, IL-6 and TNF-α, on the expression and activity of xenobiotic receptors, such as PXR, AHR, and GR, as well as their target DMEs. Accumulating evidence suggests that xenobiotic receptors are important mediators of the inhibitory effect of sepsis on drug metabolism. The effect of sepsis on DMEs has potential implications in drug metabolism and disposition, therapeutic outcomes, drug toxicity, and drug–drug interactions. It would be interesting to know whether the pharmacological activation of PXR can be used to mitigate the sepsis-induced inhibition of drug metabolism. However, the expression and activity of xenobiotic receptors can in return affect inflammation and the clinical outcome of sepsis. It would be equally interesting to know whether xenobiotic receptors, such as PXR and AHR, can be used as therapeutic targets for sepsis. Among the limitations of the studies herein, many of the observations have been made in animal models of sepsis. Thus, many of the mechanistic insights need to be validated in human studies.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (8140130969and 8176130232), and Hainan Provincial Science and Technology Major Project (ZDKJ201804, China).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Chuanzhu Lv, Email: lvchuanzhu677@126.com.
Ling Huang, Email: puer6@163.com.
Author contributions
Ling Huang contributed significantly to manuscript preparation and wrote the manuscript. Chuanzhu Lv contributed to the conception of the manuscript and revised the manuscript. All authors edited the article.
Conflicts of interest
The authors declare there is no conflicts of interest regarding the publication of this paper.
References
- 1.Cheng B.L., Hoeft A.H., Book M., Shu Q., Pastores S.M. Sepsis: pathogenesis, biomarkers, and treatment. BioMed Res Int. 2015;2015 doi: 10.1155/2015/846935. 846935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaieski D.F., Edwards J.M., Kallan M.J., Carr B.G. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41:1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 3.Coopersmith C.M., Deutschman C.S. The new sepsis definitions: implications for the basic and translational research communities. Shock. 2017;47:264–268. doi: 10.1097/SHK.0000000000000763. [DOI] [PubMed] [Google Scholar]
- 4.Rhodes A., Evans L.E., Alhazzani W., Levy M.M., Antonelli M., Ferrer R. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43:304–377. doi: 10.1007/s00134-017-4683-6. [DOI] [PubMed] [Google Scholar]
- 5.Yadav H., CartinCeba R. Balance between hyperinflammation and immunosuppression in sepsis. Semin Respir Crit Care Med. 2016;37:42–50. doi: 10.1055/s-0035-1570356. [DOI] [PubMed] [Google Scholar]
- 6.Chousterman B.G., Swirski F.K., Weber G.F. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39:517–528. doi: 10.1007/s00281-017-0639-8. [DOI] [PubMed] [Google Scholar]
- 7.Li W., Zhu S., Zhang Y., Li J.H., Sama A.E., Wang P. Use of animal model of sepsis to evaluate novel herbal therapies. J Vis Exp. 2012;62:3926. doi: 10.3791/3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doi K. How to replicate the complexity of human sepsis: development of a new animal model of sepsis. Crit Care Med. 2012;40:2722–2723. doi: 10.1097/CCM.0b013e31825bc83f. [DOI] [PubMed] [Google Scholar]
- 9.Carcillo J.A., Doughty L., Kofos D., Frye R.F., Kaplan S.S., Sasser H. Cytochrome P450 mediated-drug metabolism is reduced in children with sepsis-induced multiple organ failure. Intensive Care Med. 2003;29:980–984. doi: 10.1007/s00134-003-1758-3. [DOI] [PubMed] [Google Scholar]
- 10.Schmitt C., Kuhn B., Zhang X., Kivitz A., Grange S. Disease drug–drug-interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2012;89:735–740. doi: 10.1038/clpt.2011.35. [DOI] [PubMed] [Google Scholar]
- 11.Schmitt C., Kuhn B., Zhang X., Kivitz A., Grange S. Tocilizumab has no clinically relevant effect on methotrexate pharmacokinetics in patients with rheumatoid arthritis. Int J Clin Pharm. 2012;50:218–223. doi: 10.5414/cp201613. [DOI] [PubMed] [Google Scholar]
- 12.He J.H., Wen X. Chapter 3. Nuclear xenobiotic receptors. Prog Nucleic Acid Res. 2009;87:87–116. [Google Scholar]
- 13.Neher M.D., Weckbach S., Huber-Lang M.S., Stahel P.F. New insights into the role of peroxisome proliferator-activated receptors in regulating the inflammatory response after tissue injury. PPAR Res. 2012;5 doi: 10.1155/2012/728461. 728461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Irigoyen O., Moschetta A. A novel protective role for FXR against inflammasome activation and endotoxemia. Cell Metabol. 2017;25:763–764. doi: 10.1016/j.cmet.2017.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Kliewer S.A., Umesono K., Noonan D.J., Heyman R.A., Evans R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zanger U.M., Schwab M. Cytochrome P450 enzymes in drug metabolism. Pharmacol Ther. 2013;138:103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Oladimeji P., Chen T. PXR: more than just a master xenobiotic receptor. Mol Pharmacol. 2017;93:119–127. doi: 10.1124/mol.117.110155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgan E.T., Goralski K.B., Piquette-Miller M., Renton K.W., Robertson G.R., Chaluvadi M.R. Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab Dispos. 2008;36:205–216. doi: 10.1124/dmd.107.018747. [DOI] [PubMed] [Google Scholar]
- 19.Kacevska M., Robertson G.R., Clarke S.J., Liddle C. Inflammation and CYP3A4-mediated drug metabolism in advanced cancer: impact and implications for chemotherapeutic drug dosing. Expert Opin Drug Metabol Toxicol. 2008;4:137–149. doi: 10.1517/17425255.4.2.137. [DOI] [PubMed] [Google Scholar]
- 20.Chai X., Zeng S., Xie W. Nuclear receptors PXR and CAR: implications for drug metabolism regulation, pharmacogenomics and beyond. Expert Opin Drug Met. 2013;9:253–266. doi: 10.1517/17425255.2013.754010. [DOI] [PubMed] [Google Scholar]
- 21.Falkner K.C., Pinaire J.A., Xiao G.H., Geoghegan T.E., Prough R.A. Regulation of the rat glutathione S-transferase A2 gene by glucocorticoids:involvement of both the glucocorticoi and pregnane X receptors. Mol Pharmacol. 2001;60:611–619. [PubMed] [Google Scholar]
- 22.Sun H.Y., Yan Y.J., Li Y.H., Lv L. Reversing effects of ginsenosides on LPS-induced hepatic CYP3A11/3A4 dysfunction through the pregnane X receptor. J Ethnopharmacol. 2019;229:246–255. doi: 10.1016/j.jep.2018.09.041. [DOI] [PubMed] [Google Scholar]
- 23.Teng S., Piquette-Miller M. Involvement of the pregnane X receptor in regulation of transporters during inflammation. Clin Pharmacol Ther. 2004;75:51. [Google Scholar]
- 24.Teng S., Piquette-Miller M. The involvement of the pregnane X receptor in hepatic gene regulation during inflammation in mice. J Pharmacol Exp Ther. 2005;312:841–848. doi: 10.1124/jpet.104.076141. [DOI] [PubMed] [Google Scholar]
- 25.Gu X., Ke S., Liu D., Sheng T., Thomas P.E., Rabson A.B. Role of NF-kappaB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J Biol Chem. 2006;281:17882–17889. doi: 10.1074/jbc.M601302200. [DOI] [PubMed] [Google Scholar]
- 26.Ding X., Staudinger J.L. Repression of PXR-mediated induction of hepatic CYP3A gene expression by protein kinase C. Biochem Pharmacol Mar. 2005;69:867–873. doi: 10.1016/j.bcp.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 27.Hyrsova L., Smutny T., Carazo A., Moravcik S., Mandikova J., Trejtnar F. The pregnane X receptor down-regulates organic cation transporter1 (SLC22A1) in human hepatocytes by competing for (“squelching”) SRC-1coactivator. Br J Pharmacol. 2016;173:1703–1715. doi: 10.1111/bph.13472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bankey P., Carlson A., Ortiz M., Singh R., Cerra F. Tumor necrosis factor production by Kupffer cells requires protein kinase C activation. J Surg Res. Sep 1990;49:256–261. doi: 10.1016/0022-4804(90)90130-t. [DOI] [PubMed] [Google Scholar]
- 29.Sun M., Cui W., Woody S.K., Staudinger J.L. Pregnane X receptor modulates the inflammatory response in primary cultures of hepatocytes. Drug Metab Dispos. 2014;43:335–343. doi: 10.1124/dmd.114.062307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou C., Tabb M.M., Nelson E.L., Grün F., Verma S., Sadatrafiei A. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Investig. 2006;116:2280–2289. doi: 10.1172/JCI26283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng J., Shah Y.M., Gonzalez F.J. Pregnane X receptor as a target for treatment of inflammatory bowel disorders. Trends Pharmacol Sci. 2012;33:323–330. doi: 10.1016/j.tips.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J., Xie W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol Sci. 2012;33:552–558. doi: 10.1016/j.tips.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheeler M.A., Rothhammer V., Quintana F.J. Control of immune-mediated pathology via the aryl hydrocarbon receptor. J Biol Chem. 2017;292:12383–12389. doi: 10.1074/jbc.R116.767723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shinde R., Hezaveh K., Halaby M.J., Kloetgen A., Chakravarthy A., Medina T.D.A. Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat Immunol. 2018;19:571–582. doi: 10.1038/s41590-018-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nebert D.W. Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog Lipid Res. 2017;67:38–57. doi: 10.1016/j.plipres.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esser C., Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev. 2015;67:259–279. doi: 10.1124/pr.114.009001. [DOI] [PubMed] [Google Scholar]
- 37.Pot C., Quintana F.J., Kuchroo V.K. Fine tuning of the immune response by the aryl hydrocarbon receptor. Semin Immunopathol. 2013;35:613. [Google Scholar]
- 38.Weber G.F., Chousterman B.G., He S., Fenn A.M., Nairz M., Anzai A. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science. 2015;347:1260–1265. doi: 10.1126/science.aaa4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffmann J., Machado D., Terrier O., Pouzol S., Messaoudi M., Basualdo W. Viral and bacterial co-infection in severe pneumonia triggers innate immune responses and specifically enhances IP-10: a translational study. Sci Rep. 2016;6:38532. doi: 10.1038/srep38532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steel H.C., Cockeran R., Anderson R., Feldman C. Overview of community-acquired pneumonia and the role of inflammatory mechanisms in the immunopathogenesis of severe pneumococcal disease. Mediat Inflamm. 2013:1–18. doi: 10.1155/2013/490346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabrysova L., Howes A., Saraiva M., O'Garra A. The regulation of IL-10 expression. Curr Top Microbiol Immunol. 2014;380:157–190. doi: 10.1007/978-3-662-43492-5_8. [DOI] [PubMed] [Google Scholar]
- 42.Smolinska M.J., Horwood N.J., Page T.H., Smallie T., Foxwell B.M.J. Chemical inhibition of Src family kinases affects major LPS-activated pathways in primary human macrophages. Mol Immunol. 2008;45:990–1000. doi: 10.1016/j.molimm.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 43.Niemand C., Nimmesgern A., Haan S., Fischer P., Schaper F., Rossaint R. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J Immunol. 2003;170:3263–3272. doi: 10.4049/jimmunol.170.6.3263. [DOI] [PubMed] [Google Scholar]
- 44.Zhu J., Luo L., Tian L., Yin S., Ma X., Cheng S. Aryl hydrocarbon receptor promotes IL-10 expression in inflammatory macrophages through Src-STAT3 signaling pathway. Front Immunol. 2018;19:2033. doi: 10.3389/fimmu.2018.02033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sekine H., Mimura J., Oshima M., Okawa H., Kanno J., Igarashi K. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Cell Mol Biol. 2009;29:6391–6400. doi: 10.1128/MCB.00337-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bessede A., Gargaro M., Pallotta M.T., Matino D., Servillo G., Brunacci C. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. 2014;511:184–190. doi: 10.1038/nature13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spatzenegger M., Horsmans Y., Verbeeck R.K. Differential activities of CYP1A isozymes in hepatic and intestinal microsomes of control and 3-methylcholanthrene-induced rats. Pharmacol Toxicol. 2000;86:71–77. doi: 10.1034/j.1600-0773.2000.d01-14.x. [DOI] [PubMed] [Google Scholar]
- 48.Crawford J.H., Yang S., Zhou M., Simms H.H., Wang P. Down-regulation of hepatic CYP1A2 plays an important role in inflammatory responses in sepsis. Crit Care Med. 2004;32:502–508. doi: 10.1097/01.CCM.0000109453.57709.E2. [DOI] [PubMed] [Google Scholar]
- 49.Hankakoski P., Negishi M. Regulation of cytochrome P450 (CYP) genes by nuclear receptors. Biochem J. 2000;347:321–337. doi: 10.1042/0264-6021:3470321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waxman D.J. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR and PPAR. Arch Biochem Biophys. 1999;369:11–23. doi: 10.1006/abbi.1999.1351. [DOI] [PubMed] [Google Scholar]
- 51.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 52.Honkakoski P., Negishi M. Regulation of cytochrome P450 (CYP) genes by nuclear receptors. Biochem J. 2000;341:321–337. doi: 10.1042/0264-6021:3470321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waxman D.J. P450 gene induction by structurally diverse xenochemicals: central role of nuclear CAR, PXR, and PPAR. Arch Biochem Biophys. 1999;369:11–23. doi: 10.1006/abbi.1999.1351. [DOI] [PubMed] [Google Scholar]
- 54.Tomita S., Signal C.J., Yim S.H., Gonzales F.J. Conditional disruption of the aryl hydrocarbon receptor nuclear translocator (Arnt) gene leads to loss of target gene induction by the aryl hydrocarbon receptor and hypoxia-inducible factor l alpha. Mol Endocrinol. 2000;14:1674–1681. doi: 10.1210/mend.14.10.0533. [DOI] [PubMed] [Google Scholar]
- 55.Reyes H., Reisz-Porszasz S., Hankinson O. Identification of the AH receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the AH receptor. Science. 1992;256:1193–1195. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 56.Poellinger L. Ligand-dependent recruitment of the Arnt coregulator determines DNA recognition by the dioxin receptor. Mol Cell Biol. 1993;13:2504–2514. doi: 10.1128/mcb.13.4.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coumailleau P., Poellinger L., Gustafsson J.A., Whitelaw M.L. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. J Biol Chem. 1995;270:5291–5300. doi: 10.1074/jbc.270.42.25291. [DOI] [PubMed] [Google Scholar]
- 58.Whitelaw M.L., McGuire J., Picard D., Gustafsson J.A., Poellinger L. Heat shock protein hsp90 regulates dioxin receptor function in vivo. Proc Natl Acad Sci USA. 1995;92:4437–4441. doi: 10.1073/pnas.92.10.4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou M., Maitra S.R., Wang P. The potential role of transcription factor aryl hydrocarbon receptor in downregulation of hepatic cytochrome P-450 during sepsis. J Mol Med. 2008;21:423–428. [PMC free article] [PubMed] [Google Scholar]
- 60.Morgan E.T. Regulation of cytochromes P450 during inflammation and infection. Drug Metab Rev. 1997;29:60. doi: 10.3109/03602539709002246. [DOI] [PubMed] [Google Scholar]
- 61.Barker C.W., Fagan J.B., Pasco D.S. Interleukin-1 beta suppresses the induction of P4501A1 and P4501A2 mRNAs in isolated hepatocytes. J Biol Chem. 1992;267:8050–8055. [PubMed] [Google Scholar]
- 62.Nicholson T.E., Renton K.W. Role of cytokines in the lipopolysaccharide-evoked depression of cytochrome P450 in the brain and liver. Biochem Pharmacol. 2001;62:1709–1717. doi: 10.1016/s0006-2952(01)00859-0. [DOI] [PubMed] [Google Scholar]
- 63.Crawford J.H., Yang S., Zhou M., Simms H., Wang P. Down-regulation of hepatic CYP1A2 plays an important role in inflammatory responses in sepsis. Crit Care Med. 2004;32:502–508. doi: 10.1097/01.CCM.0000109453.57709.E2. [DOI] [PubMed] [Google Scholar]
- 64.Groeneweg F.L., Karst H., Kloet E.R.D. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol. 2012;350:299–309. doi: 10.1016/j.mce.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 65.Vardas K., Ilia S., Sertedaki A., Charmandari E., Briassouli E., Goukos D. Increased glucocorticoid receptor expression in sepsis is related to heat shock proteins, cytokines, and cortisol and is associated with increased mortality. Intens Care Med Exp. 2017;5:10–27. doi: 10.1186/s40635-017-0123-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chrousos G.P. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- 67.Barnes P.J., Greening A.P., Crompton G.K. Glucocorticoid resistance in asthma. Am J Respir Crit Care Med. 1995;152:125–142. doi: 10.1164/ajrccm/152.6_Pt_2.S125. [DOI] [PubMed] [Google Scholar]
- 68.Pruzanski W., Vadas P. Phospholipase A2—a mediator between proximal and distal effectors of inflammation. Immunol Today. 1991;12:143–146. doi: 10.1016/S0167-5699(05)80042-8. [DOI] [PubMed] [Google Scholar]
- 69.Albina J.E., Reichner J.S. Nitric oxide in inflammation and immunity. New Hori. 1995;3:46–64. [PubMed] [Google Scholar]
- 70.Da J., Chen L., Hedenstierna G. Nitric oxide up-regulates the glucocorticoid receptor and blunts the inflammatory reaction in porcine endotoxin sepsis. Crit Care Med. 2007;35:26–32. doi: 10.1097/01.CCM.0000250319.91575.BB. [DOI] [PubMed] [Google Scholar]
- 71.Goodwin J.E., Feng Y., Velazquez H., Sessa W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci U S A. 2013;110:306–311. doi: 10.1073/pnas.1210200110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Lange P., Segeren C.M., Koper J.W., Wiemer E., Sonneveld P., Brinkmann A.O. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor mediated effects in transfected cells. Cancer Res. 2001;61:3937–3941. [PubMed] [Google Scholar]
- 73.Zhou J., Cidlowski J.A. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 2005;70:407–417. doi: 10.1016/j.steroids.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 74.Bamberger C.M., Bamberger A.M., de Castro M., Chrousos G.P. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Investig. 1995;95:2435–2441. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van den Akker E.L., Koper J.W., Joosten K., de Jong F.H., Hazelzet J.A., Lamberts S.W. Glucocorticoid receptor mRNA levels are selectively decreased in neutrophils of children with sepsis. Intensive Care Med. 2009;35:1247–1254. doi: 10.1007/s00134-009-1468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dvorak Z. Colchicine down-regulates cytochrome P450 2B6, 2C8, 2C9, and 3A4 in human hepatocytes by affecting their glucocorticoid receptor-mediated regulation. Mol Pharmacol. 2003;64:160–169. doi: 10.1124/mol.64.1.160. [DOI] [PubMed] [Google Scholar]
- 77.Pascussi J.M., Gerbalchaloin S., Drocourt L., Maurel P., Vilarem M.J. The expression of CYP2B6, CYP2C9 and CYP3A4 genes: a tangle of networks of nuclear and steroid receptors. BBA Gen Subjects. 2003;1619:243–253. doi: 10.1016/s0304-4165(02)00483-x. [DOI] [PubMed] [Google Scholar]
- 78.Pascussi J.M., Gerbalchaloin S., Fabre J.M., Maurel P., Vilarem M.J. Dexamethasone enhances constitutive androstane receptor expression in human hepatocytes: consequences on cytochrome P450 gene regulation. Mol Pharmacol. 2000;58:1441–1450. doi: 10.1124/mol.58.6.1441. [DOI] [PubMed] [Google Scholar]
- 79.Pascussi J.M., Drocourt L., Fabre J.M., Maurel P., Vilarem M.J. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol Pharmacol. 2000;58:361–372. doi: 10.1124/mol.58.2.361. [DOI] [PubMed] [Google Scholar]