

Abstract
Background
X‐linked hydrocephalus (XLH), characterized by mental retardation and bilateral adducted thumbs, often come out to be a genetic disorder of L1CAM. It codes the protein L1 cell adhesion molecule (L1CAM), playing a crucial role in the development of the nervous system. The objective of the study was to report a new disease‐causing mutation site of L1CAM, and gain further insight into the pathophysiology of hydrocephalus.
Methods
We collect the samples of a couple and their second hydrocephalic fetus. Then, the whole‐exome sequencing and in‐depth mutation analysis were performed.
Results
The variant c.2491delG (p.V831fs), located in the exon 19 of L1CAM (chrX:153131214), could damage the L1CAM function by producing a frameshift in the translation of fibronectin type‐III of L1CAM.
Conclusion
We identified a novel disease‐causing mutation in L1CAM for the first time, which further confirmed L1CAM as a gene underlying XLH cases.
Keywords: frameshift mutation, L1CAM, X‐linked hydrocephalus
We identified a novel disease‐causing mutation in L1CAM by first time, which further confirmed L1CAM as a gene underlying XLH cases.
1. INTRODUCTION
Hydrocephalus, the abnormal accumulation of intracranial cerebrospinal fluid (CSF), is a common malformation of fetuses. Accompanied by other structural brain lesions, it affects approximately one in every 1,000 children born (Tully & Dobyns, 2014; Warf, 2005). The pathogenesis of this process remains to be fully elucidated; nonetheless, a few points are established.
A large part of hydrocephalic patients show the existence of chromosome abnormalities. Researchers has proved that there were mutations in L1CAM (OMIM 308840)(Marin et al., 2015; Patzke, Acuna, Giam, Wernig, & Sudhof, 2016), which code the protein L1 cell adhesion molecule (L1CAM), a neuronal cell adhesion molecule belonging to the immunoglobulin superfamily (IgSF) and playing a key role in the development of the nervous system (Chang, Rathjen, & Raper, 1987; Rathjen & Schachner, 1984).
Mutations in L1CAM can result in different X‐linked neurological syndromes, known as L1 syndrome (Lyonnet et al., 1992; Schrander‐Stumpel, Legius, Fryns, & Cassiman, 1990).
The diagnosis of hydrocephalus is mainly based on the result of ultrasound detection, which is neither precise nor timely. Thus, the use of genetic sequencing is increasingly popular and important in recent years. The purpose of the study was to report a new disease‐causing mutation site of L1CAM, making a small step forward in the pathogenesis of hydrocephalus.
2. MATERIALS AND METHODS
2.1. Ethical compliance
The research was approved by the Institutional Committee for the Protection of Human Subjects (Institutional Review Board of Sichuan Provincial Hospital for Women and Children), and all patients signed the informed consent.
2.2. Sample collection
The blood samples of the parents and the tissue of their fetus were collected and kept at −80°C.
2.3. Mutation analysis
Genomic DNA was extracted from tissue and blood samples according to standard protocols. Applied Biosystems 3730xl DNA Analyzer was used to sequence the result of PCR amplification. Then, we found out the sites that need to be sequenced on the peak of Sanger sequencing, specific primers were designed according to the site information on USCS via Prime Primer 5, and to confirm whether they have variation. The library was further constructed by using Roche SeqCap EZ MedExome Enrichment kit and sequenced on an Illumina HiSeq X machine. Raw reads were mapped to the human reference genome GRCh37/hg19 using BWA (v0.7.12‐r1039) (Li & Durbin, 2009), and the SAM files were transformed to BAM files and sorted by using SAMtools (v0.1.18). Then Picard v1.134 (http://broadinstitute.github.io/picard/) was used to mark duplicate reads. Variants were called by GenomeAnalysisTK (GATK v3.7) (McKenna et al., 2010) and annotated by ANNOVAR (2016Jul16 version). The Exome Aggregation Consortium (ExAC Version 0.3.1), Genomes 1,000 Project, ESP6500, and other public database were used to filter the variants. The candidate pathogenic mutations were verified by Sanger sequencing.
3. RESULTS
A 25‐year‐old woman was referred to our department for having one spontaneous abortion and two voluntary terminations of pregnancy due to fetal hydrocephalus. Blood samples of this couple and tissue of the last hydrocephalic fetus were collected. The familial pedigree was consistent with X‐linked recessive inheritance (Figure 1).
Figure 1.
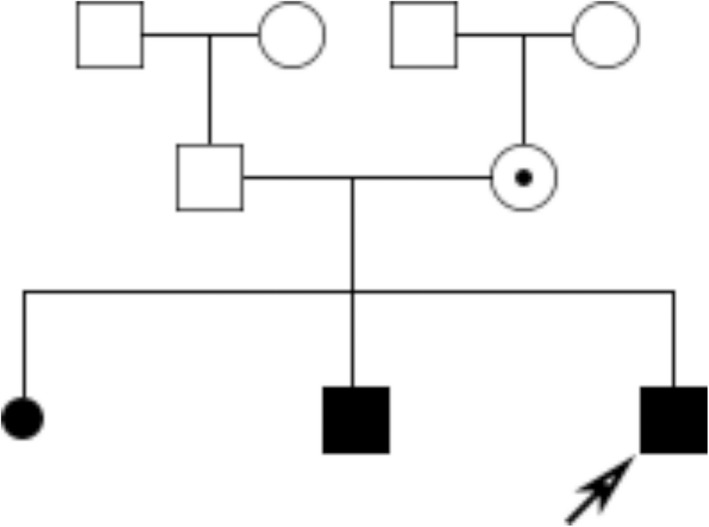
Pedigree of the family
During the first pregnancy, a natural abortion happened around 11 weeks of gestation. As for the second pregnancy, a fetal ultrasound scan at 24+ weeks of gestation proved the presence of hydrocephalus, and the woman required an interruption of pregnancy.
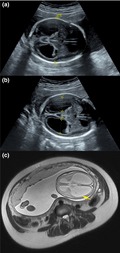
The third pregnancy was similar to the second one. The ultrasound scan evaluation at 25 + 4 gestation weeks revealed the bilateral ventriculomegaly with dilatation of the third ventricle and polyhydramnios. The magnetic resonance imaging (MRI) further proved the presence of callosal agenesis and lissencephaly (Figure 2).
Figure 2.
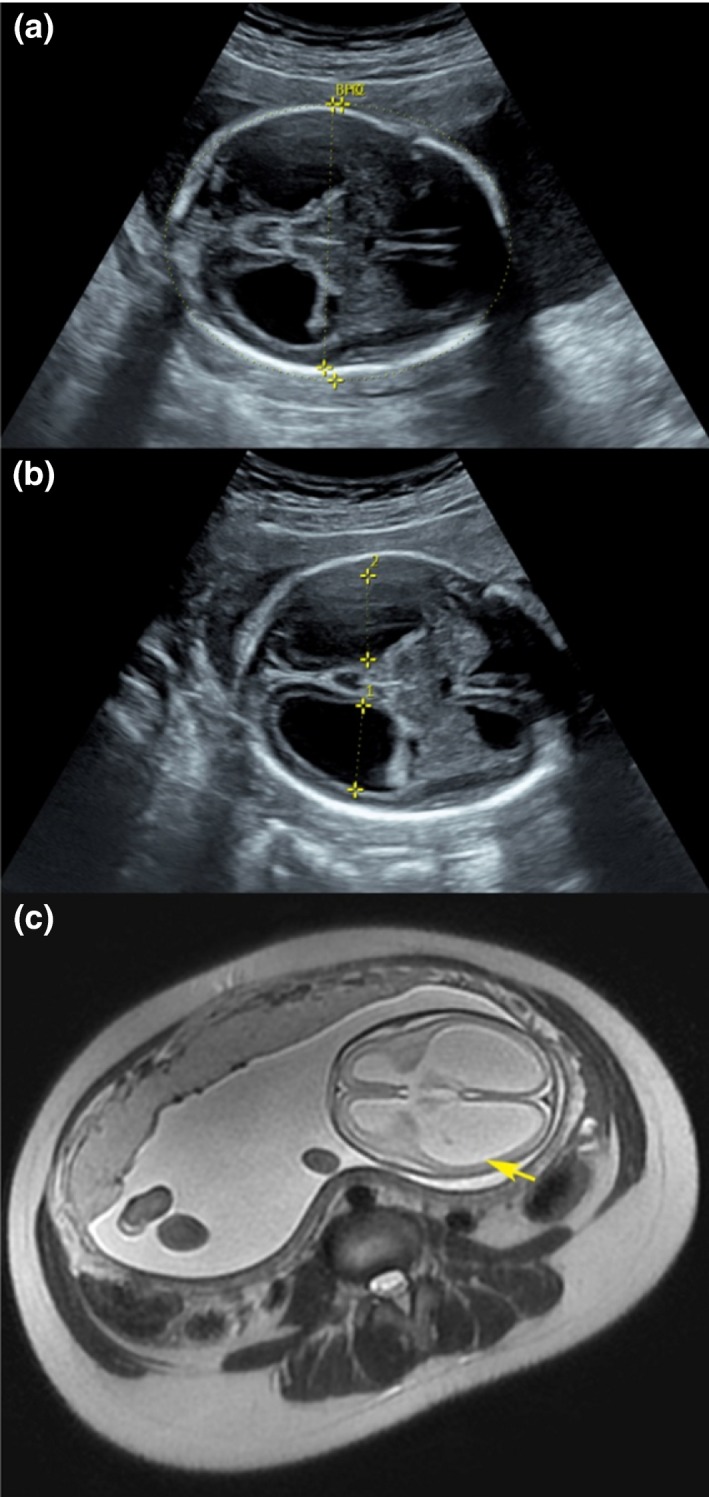
The ultrasound scan and magnetic resonance imaging (MRI) scan. (a) The ultrasound scan, BPD = 6.54 cm, HC = 24.36 cm; (b) The ultrasound scan, 1D = 2.09 cm, 2D = 2.08 cm; (c) MRI scan
After collecting the samples of the couple and the third fetus, we used the whole‐exome sequencing and in‐depth mutation analysis to find the potential causes. The variant was confirmed in DNA extracted from the fetus and mother (Figure 3). Potential variants were called by Genome Analysis TK, public databases were used to filter the variants, and the effects of these variants were annotated by ANNOVAR programs.
Figure 3.
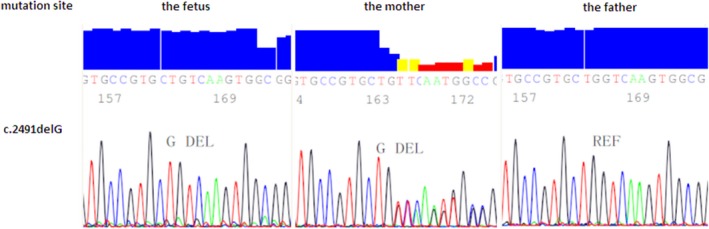
Sequence analysis results (GenBank ID number M77640.1)
The proband was found to be hemizygous for L1CAM with a mutation NM_000425.5:c.2491del:p.(Val831Serfs*20) and the mother was found to be heterozygote. Computational analysis predicted that this was a frameshift mutation, located in the exon 19 of L1CAM (chrX:153131214), coding the fibronectin type‐III of L1CAM. The mutation will lead to the translation errors of amino acid and an early translation termination, belonging to the loss‐of‐function mutation. Moreover, the ClinGen haploinsufficiency score and pLI in ExAC of L1CAM was 3 and 1, respectively, (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/clingen_gene.cgi?sym=L1CAM&subject=, and http://exac.broadinstitute.org/gene/ENSG00000198910), suggesting a strong relationship between the loss‐of‐function mutation and disease. In addition, by now, this mutation had not been reported in gnomAD or 1,000 Genomes Project yet. Besides, according to OMIM, diseases related to L1CAM are X‐linked recessive, and the phenotype was consistent with that of the proband. The genetic pattern was consistently with “the L1 syndrome.” According to the guide of ACMG (American society of medical genetics and genomics), with the evidence of a PVS1, a PM2, and a PP4, it was an X‐linked and pathogenic mutation (Figure 3).
4. DISCUSSION
Hydrocephalus, including X‐linked hydrocephalus (XLH), often comes out to be a genetic disorder, characterized by mental retardation and bilateral adducted thumbs (Okamoto et al., 2004). The features of XLH include the enlargement of both third and lateral ventricles, agenesia of corpus callosum, atrophy of corticospinal descending pathways in the pons and medulla, and spasticity of upper and mostly lower limbs (Itoh & Fushiki, 2015; Tonosaki et al., 2014). Despite of its unclear pathogenesis, most cases reported the strong link between mutations of L1CAM and XLH.
L1CAM consists of six immunoglobulin, five fibronectin III‐like domains, a single pass transmembrane domain, and a short cytoplasmic domain (Moos et al., 1988). It produces a variety of molecular and cellular effects, crucial to brain development (Chang et al., 1987; Rathjen & Schachner, 1984). The dysfunction of L1CAM can lead to “the L1 syndrome,” which is X‐linked, including hydrocephalus with stenosis of the Sylvius aqueduct (HSAS; phenotype MIM number 307000), MASA (mental retardation, aphasia, spastic paraplegia, adducted thumbs) syndrome (phenotype MIM number 303350), complicated hereditary spastic paraplegia type 1 (SPG1, phenotype MIM number 303350), and agenesis of the corpus callosum (phenotype MIM number 308840) (Basel‐Vanagaite et al., 2006).
According to Vos et al.’s report (Vos et al., 2010), 85% of hydrocephalus fetus were facing L1CAM mutation when they had three or more L1 syndrome‐related morphological alterations, and more than one affected relative. Ferese et al. (2016) reported that a splicing mutation (NM_000425.4:c.1267 + 5delG) in L1CAM, which produced the skipping of exon 10, could result in hydrocephalus. In Liebau's study (Liebau, Gal, Superti‐Furga, Omran, & Pohl, 2007), a mutation at the beginning of intron 18 of L1CAM was related to the agenesis of corpus callosum, adducted thumbs, hydrocephalus, and mental retardation. Hübner et al. (2004) found out that a mutation of L1CAM in two unrelated families resulted in a frame shift due to insertion of the first 10 bp of intron 5 in the mature mRNA of L1CAM, leading to a largely truncated protein. In our study, we found a NM_000425.5:c.2491del:p.(Val831Serfs*20) variant, located in the exon 19 of L1CAM (chrX:153131214), that could damage the L1CAM function by producing a frameshift in the translation of fibronectin type‐III of L1CAM, resulting in the bilateral ventriculomegaly with dilatation of the third ventricle, polyhydramnios, callosal agenesis, and lissencephaly.
In summary, we identified a novel XLH‐causing mutation NM_000425.5:c.2491del:p.(Val831Serfs*20) in L1CAM for the first time. The L1CAM mutations are manifold, and most of them are unique for each family (Vos et al., 2010). The more disease‐causing mutations we found, the more accurate predictions we are able to make.
CONFLICT OF INTEREST
The authors declared that they have no conflicts of interest to this work. We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
AUTHORS' CONTRIBUTIONS
Dr. Xueyan Wang and Dr. Weiqi Kong had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
ACKNOWLEDGMENT
This work was supported by the Fundamental Research Funds of the Science and Technology Department (No. 18YYJC0624).
Kong W, Wang X, Zhao J, Kang M, Xi N, Li S. A new frameshift mutation in L1CAM producing X-linked hydrocephalus. Mol Genet Genomic Med. 2020;8:e1031 10.1002/mgg3.1031
REFERENCES
- Basel‐Vanagaite, L. , Straussberg, R. , Friez, M. J. , Inbar, D. , Korenreich, L. , Shohat, M. , & Schwartz, C. E. (2006). Expanding the phenotypic spectrum of L1CAM‐associated disease. Clinical Genetics, 69(5), 414–419. [DOI] [PubMed] [Google Scholar]
- Chang, S. , Rathjen, F. G. , & Raper, J. A. (1987). Extension of neurites on axons is impaired by antibodies against specific neural cell surface glycoproteins. Journal of Cell Biology, 104(2), 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferese, R. , Zampatti, S. , Griguoli, A. M. , Fornai, F. , Giardina, E. , Barrano, G. , … Gambardella, S. (2016). A new splicing mutation in the L1CAM gene responsible for x‐linked hydrocephalus (HSAS). Journal of Molecular Neuroscience: MN, 59(3), 376–381. [DOI] [PubMed] [Google Scholar]
- Hübner, C. A. , Utermann, B. , Tinschert, S. , Krüger, G. , Ressler, B. , Steglich, C. , … Gal, A. (2004). Intronic mutations in the L1CAM gene may cause X‐linked hydrocephalus by aberrant splicing. Human Mutation, 23(5), 526. [DOI] [PubMed] [Google Scholar]
- Itoh, K. , & Fushiki, S. (2015). The role of L1cam in murine corticogenesis, and the pathogenesis of hydrocephalus. Pathology International, 65(2), 58–66. [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics (Oxford, England), 25(14), 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebau, M. C. , Gal, A. , Superti‐Furga, A. , Omran, H. , & Pohl, M. (2007). L1CAM mutation in a boy with hydrocephalus and duplex kidneys. Pediatric Nephrology (Berlin, Germany), 22(7), 1058–1061. [DOI] [PubMed] [Google Scholar]
- Lyonnet, S. , Pelet, A. , Royer, G. , Delrieu, O. , Serville, F. , le Marec, B. , … Dubay, C. (1992). The gene for X‐linked hydrocephalus maps to Xq28, distal to DXS52. Genomics, 14(2), 508–510. [DOI] [PubMed] [Google Scholar]
- Marin, R. , Ley‐Martos, M. , Gutierrez, G. , Rodriguez‐Sanchez, F. , Arroyo, D. , & Mora‐Lopez, F. (2015). Three cases with L1 syndrome and two novel mutations in the L1CAM gene. European Journal of Pediatrics, 174(11), 1541–1544. [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moos, M. , Tacke, R. , Scherer, H. , Teplow, D. , Fruh, K. , & Schachner, M. (1988). Neural adhesion molecule L1 as a member of the immunoglobulin superfamily with binding domains similar to fibronectin. Nature, 334(6184), 701–703. [DOI] [PubMed] [Google Scholar]
- Okamoto, N. , Del Maestro, R. , Valero, R. , Monros, E. , Poo, P. , Kanemura, Y. , & Yamasaki, M. (2004). Hydrocephalus and Hirschsprung's disease with a mutation of L1CAM. Journal of Human Genetics, 49(6), 334–337. [DOI] [PubMed] [Google Scholar]
- Patzke, C. , Acuna, C. , Giam, L. R. , Wernig, M. , & Sudhof, T. C. (2016). Conditional deletion of L1CAM in human neurons impairs both axonal and dendritic arborization and action potential generation. The Journal of Experimental Medicine, 213(4), 499–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathjen, F. G. , & Schachner, M. (1984). Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. The EMBO Journal, 3(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrander‐Stumpel, C. , Legius, E. , Fryns, J. P. , & Cassiman, J. J. (1990). MASA syndrome: New clinical features and linkage analysis using DNA probes. Journal of Medical Genetics, 27(11), 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonosaki, M. , Itoh, K. , Umekage, M. , Kishimoto, T. , Yaoi, T. , Lemmon, V. P. , & Fushiki, S. (2014). L1cam is crucial for cell locomotion and terminal translocation of the Soma in radial migration during murine corticogenesis. PLoS ONE, 9(1), e86186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully, H. M. , & Dobyns, W. B. (2014). Infantile hydrocephalus: A review of epidemiology, classification and causes. European Journal of Medical Genetics, 57(8), 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos, Y. J. , de Walle, H. E. , Bos, K. K. , Stegeman, J. A. , Ten Berge, A. M. , Bruining, M. , … Hofstra, R. M. (2010). Genotype‐phenotype correlations in L1 syndrome: A guide for genetic counselling and mutation analysis. Journal of Medical Genetics, 47(3), 169–175. [DOI] [PubMed] [Google Scholar]
- Warf, B. C. (2005). Hydrocephalus in Uganda: The predominance of infectious origin and primary management with endoscopic third ventriculostomy. Journal of Neurosurgery, 102(1 Suppl), 1–15. [DOI] [PubMed] [Google Scholar]