

Abstract
Background
Diffuse panbronchiolitis (DPB) is a sinopulmonary disease mainly affecting Asian populations. Primary ciliary dyskinesia (PCD) is a genetically heterogeneous disorder impairing ciliary structure and function. These two disorders are not easily distinguished by clinical signs and symptoms.
Methods
In 105 Japanese patients with recurrent sinopulmonary infection, initially diagnosed with DPB, and 37 patients with recurrent airway infection diagnosed in adulthood, the deletion allele of DRC1 or CCDC164, recently recognized as a pathogenic PCD gene variant, was searched using a multiplexed PCR‐based method, and the deletion breakpoints and other variants around the gene were determined by Sanger sequencing and targeted resequencing.
Results
A large homozygous deletion in DRC1 was identified in three of the 142 patients. Furthermore, heterozygous carriers of the deletion with the same breakpoint were found with the allele frequency of 0.002 in the healthy Japanese population, indicating that this loss‐of‐function variant may be acting as a common mutation causing PCD in Japanese.
Conclusion
PCD caused by the DRC1 defect is not readily identified by either high‐speed video‐microscopy or ciliary ultrastructure analysis, posing significant difficulties in reaching a correct diagnosis without the aid of genetic tests. Careful investigation of the causes of sinopulmonary diseases is warranted in Asian populations.
Keywords: diffuse panbronchiolitis, founder mutation, loss‐of‐function variant, nexin‐dynein regulatory complex, primary ciliary dyskinesia, sinopulmonary disease
A large homozygous deletion in DRC1 was identified in three patients with recurrent sinopulmonary infection. Furthermore, heterozygous carriers of the DRC1 deletion with the same break point were found with the allele frequency of 0.002 in the healthy Japanese population, indicating that this loss‐of‐function variant may be acting as a common mutation causing PCD in Japanese.

1. INTRODUCTION
Diffuse panbronchiolitis (DPB) is a clinical entity characterized by sinopulmonary infection with a large amount of sputum production, and small inflammatory nodular lesions around the respiratory bronchioles in the lung (Homma et al., 1983). In a survey, the prevalence of DPB was estimated to be 13.8 and 6.6 in 100,000 in the 1980s and 1990s, respectively (Kono et al., 2012). DPB, a multifactorial disease, is associated with human leukocyte antigen (HLA) class‐I alleles. It affects mainly Asian populations, including Japan, Korea, and China (Ding et al., 2007; Keicho & Hijikata, 2011; Park et al., 1999), and is distinct from cystic fibrosis (CF) in encountered European descents.
Primary ciliary dyskinesia (PCD) is a genetically heterogeneous disorder of the ciliary structure and function (Afzelius, 1976; Lucas et al., 2017; Shapiro et al., 2016), and follows both autosomal recessive and X‐linked inheritance (Olcese et al., 2017). It is estimated to affect one in 15,000–30,000 individuals in the world (Bush et al., 2007). Approximately 40 genes that cause PCD have been identified so far. Its classical form with a triad of situs inversus, chronic sinusitis, and bronchiectasis, is known as Kartagener syndrome. Sinopulmonary diseases including PCD, DPB and CF, are characterized by leading symptoms such as productive cough, chronic rhinosinusitis, and recurrent infections of the upper and lower respiratory tract; these diseases are not easily distinguished clinically from each other in case of situs solitus, the normal position of organs (Amitani, Tomioka, Kurasawa, Ishida, & Kuze, 1990; Knowles, Zariwala, & Leigh, 2016).
Recently, we experienced an adult patient, who was followed based on initial diagnosis, as DPB. However, eventually, a homozygous loss‐of‐function mutation within DRC1 (CCDC164, MIM 615288), one of the PCD causative genes, was identified in the patient, who was re‐diagnosed with PCD (Morimoto et al., 2019). The first case of PCD caused by a DRC1 mutation was reported only six years ago (Wirschell et al., 2013). These findings prompted us to search for the same variants in DNA samples obtained from patients previously diagnosed as having DPB in Japan.
2. MATERIALS AND METHODS
The ethical committee approved the study protocol (RIT/IRB 29–28) at the Research Institute of Tuberculosis of the Japanese Anti‐Tuberculosis Association, and written informed consent was obtained from each individual for the purpose of disease gene research. Demographic and clinical information of the patients has been extracted from the metadata, retrospectively (Hijikata et al., 2011).
2.1. DNA and clinical information about patients
DNA samples were available from 105 of 108 unrelated Japanese patients previously reported by Hijikata et al. (Hijikata et al., 2011). All fulfilled the following diagnostic criteria for DPB proposed in 1995 by a working group of the Ministry of Health and Welfare of Japan: persistent cough, sputum, and exertional dyspnea; history of, or current chronic sinusitis; bilateral diffuse small nodular shadows on a plain chest X‐ray film or centrilobular micronodules on chest computed tomography (CT) images; coarse crackles; FEV1.0/FVC less than 70% and PaO2 less than 80 Torr; and titer of cold hemagglutinin equal to or higher than 64 ×. Definite cases should fulfill the first three criteria and at least two other remaining criteria, excluding other chronic respiratory diseases (Keicho & Kudoh, 2002). Our screening was extended to 37 patients with recurrent infections of the upper and lower airways, recruited in the same period (mainly from 1997 to 1999), but not meeting the DPB criteria. The DNA panel of the healthy anonymous Japanese population, consisting of 499 samples was obtained from the Health Science Research Resources Bank (Japan Health Sciences Foundation).
2.2. PCR to detect a large deletion spanning exons 1–4 in DRC1
Genomic DNA was tested for the presence or absence of the DRC1 deletion allele using the multiplexed PCR‐based method as described recently (Morimoto et al., 2019). Briefly, the deletion allele was detected in a 515‐bp fragment using primers located outside the deletion, and the wild type allele was amplified in 600‐bp, 376‐bp, and 849‐bp fragments with primer pairs from flanking regions of exons 2, 3, and 4, respectively. PCR products were electrophoresed in 2% agarose gel and stained with GelRed (Biotium).
2.3. Sanger sequencing to confirm nucleotide positions of deletion junctions
The genomic region encompassing the deletion in DRC1 was amplified with primers 5′‐CAGTGGACTAAGGTAGGTGCTTGG‐3′ and 5′‐ACCAAATGGCTTTGACAAGGGTCA‐3′, with Tks Gflex DNA polymerase (TaKaRa) as recommended by the manufacturer, and the PCR conditions were 94ºC for 1 min followed by 35 cycles of 98ºC for 10 s, 60ºC for 15 s, and 68ºC for 1 min 30 s. The PCR product of 1,215 bp was purified with QIAquick PCR purification kit (QIAGEN) and sequenced bidirectionally with primers 5′‐GGCAGTCAGTCCTACATATCAAGG‐3′ or 5′‐TAAGAAGGCAATATACGGAATAGG‐3′, using BigDye terminator v3.1 cycle sequencing kit and SeqStudio Genetic Analyzer (Thermo Fisher Scientific).
2.4. Targeted resequencing for all exons of DRC1 and SELENOI genes
Seventeen exons of DRC1 and adjacent 10 exons of SELENOI were amplified by PCR using 15 primer sets (Table S1). In each analyzed patient or control, the PCR products were mixed, purified with QIAquick PCR purification kit and subjected to library preparation using QIAseq FX DNA Library Kit (QIAGEN). The appropriate library size was selected with AMPure XP beads (Beckman Coulter), and 2 × 150 bp paired‐end sequencing run was performed using MiSeq Reagent Kit Nano v2 (300 Cycle) and MiSeq Next‐Generation Sequencing System (Illumina). The sequence reads were trimmed, aligned to the human genome (hg38), and variants were called and listed using CLC Genomics Workbench version 11.0 (QIAGEN). Genotype data of Japanese in Tokyo (JPT) and Northern and Western European Ancestry (CEU) populations were downloaded from the 1,000 genomes project (http://www.1000genomes.org). Haplotypes consisting of the variants in exon regions of DRC1 and SELENOI were estimated using Haploview 4.2 (Barrett, Fry, Maller, & Daly, 2005).
3. RESULTS
3.1. Characteristics of the patients meeting the clinical diagnostic criteria for DPB
Demographic data are shown in Table 1. All of the 105 patients, reported previously (Hijikata et al., 2011), fulfilled the clinical diagnostic criteria for DPB described in the Materials and Methods. Raw data of clinical and demographic information about all the patients are shown in Table S2.
Table 1.
Characteristics of the patients meeting the clinical diagnostic criteria for DPB
| Patients | (N = 105) |
|---|---|
| Male/Female (n) | 58/46a |
| Age at diagnosis (year) | 52 (41–62)b |
| Duration from diagnosis to recruitment (year) | 6 (2–11)b |
| Persistent cough, sputum, and exertional dyspnea (n) | 105 |
| History of or current chronic sinusitis (n) | 105 |
| Bilateral diffuse small nodular shadows (n) | 105 (only on CT images = 10) |
| FEV1.0/FVC (%) | 60.0 (46.7–66.7)b |
| PaO2 (Torr) | 68.0 (60.2–74.0)b |
| Cold hemagglutinin (titer) | 512 × (128 × −1,024 ×)b |
| HLA‐B*5401 (n) | 39 (monoallelic = 35, biallelic = 4) |
| HLA‐B*5504 (n) | 4 (monoallelic = 4) |
one missing.
Median and 25 to 75 percentiles in parenthesis.
3.2. Detection of loss‐of‐function mutations of the DRC1 gene in patients
Using a multiplexed PCR‐based method, the presence or absence of a large homozygous deletion spanning exons 1–4 of DRC1 was detected. Of the 105 patients with DPB, two were homozygous for the deletion allele (Figure 1: DPB002 and DPB023), and two others had heterozygous deletion alleles (Figure 1: DPB058 and DPB132). For these four patients, no other pathogenic or likely pathogenic variants were found in any exons and exon‐intron boundaries of DRC1 by targeted resequencing and Sanger sequencing (Table S3).
Figure 1.
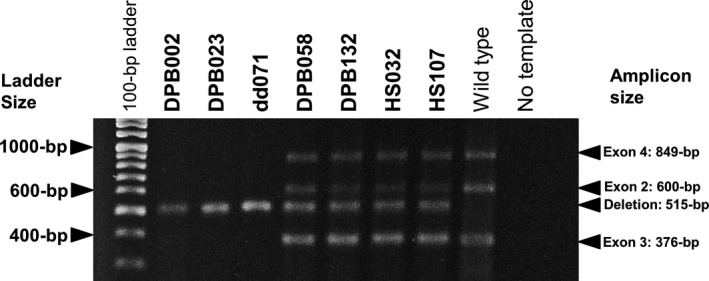
Detection of PCR products from the multiplexed PCR‐based method by agarose gel electrophoresis. Only deletion alleles were amplified from genomic DNA samples of two DPB patients (DPB002 and DPB023) and of a patient with recurrent airway infections (dd071). Both wild type and deletion alleles were amplified from those of two other DPB patients (DPB058 and DPB132) and two healthy individuals (HS032 and HS107). One reference sample showing homozygous insertion (wild type) and PCR products with no genomic DNA (No template) were also shown as positive and negative controls, respectively
3.3. Clinical signs and symptoms of the patients carrying the large deletion in the DRC1 gene
As shown in Table 2, all of the patients had typical signs and symptoms as sinopulmonary disease, and the former two patients' age at diagnosis was 43 and 29 years old. The two patients did not have the HLA‐B*5401 or HLA‐B*5504 alleles, known as DPB susceptibility alleles (Keicho et al., 1998). These HLA‐B alleles are known to be disease markers for DPB, and a major disease susceptibility gene is suspected to be located near the HLA‐B locus on human chromosome 6 (Hijikata et al., 2011; Keicho et al., 2000). In addition, a cold hemagglutinin test that often shows high titers in DPB (Sugiyama, Kudoh, Maeda, Suzaki, & Takaku, 1990) was low or borderline in these patients. Consanguinity was not described. Situs inversus was not recorded. Although a long‐term macrolide treatment was effective in most of the 105 patients, according to the description by doctors‐in‐charge, the treatment effect such as an improvement in signs and symptoms, including sputum production, was not substantial in the second homozygous patient (DPB023), showing diffuse centrilobular micronodules with focal bronchiectasis.
Table 2.
Clinical signs and symptoms of the patients homozygous (DPB002 and DPB023) and heterozygous (DPB058 and DPB132) for the large deletion in the DRC1 gene
| DPB002 | DPB023 | DPB058 | DPB132 | |
|---|---|---|---|---|
| Male/Female | Female | Female | Female | Male |
| Age at diagnosis (year) | 43 | 29 | 43 | 62 |
| Duration from diagnosis to recruitment (year) | 0 | 3 | 26 | 1 |
| Persistent cough, sputum, and exertional dyspnea | Yes | Yes | Yes | Yes |
| History of or current chronic sinusitis | Yes | Yes | Yes | Yes |
| Bilateral diffuse small nodular shadows | Yes | Yes | Yes | Yes |
| FEV1.0/FVC (%) (normal range ≥70%) | 43.0 | 69.1 | 53.0 | 70.9 |
| PaO2 (Torr) (normal range ≥80 Torr) | 71 | NA | 64 | 82 |
| Cold hemagglutinin (titer) (normal range <64 ×) | 4 × | 64 × | NA | 128 × |
| HLA‐B alleles | HLA‐B*4801/HLA‐B*5101 | HLA‐B*4801/HLA‐B*5201 | HLA‐B*0702/HLA‐B*5401 | HLA‐B*0701/HLA‐B*5401 |
| Large DRC1 deletion | homozygous | homozygous | heterozygous | heterozygous |
Abbreviations: NA, not available.
Of the 37 patients with recurrent infections of the upper and lower airways recruited in the same period but not meeting the DPB criteria, another 41‐year‐old male patient also had the homozygous deletion in DRC1 (Figure 1: dd071); however, detailed information about this patient was not obtained.
3.4. Allele frequency distribution of the large DRC1 deletion in healthy Japanese, and confirmation of the deletion junctions
Carriers of the identical genetic deletion were found in two of 499 healthy Japanese (Figure 1: HS032 and HS107). An estimated allele frequency in the Japanese population of the large deletion in DRC1 was, thus, 0.002.
To identify the 27,748 bp deletion, the PCR products encompassing the deletion were directly sequenced by the Sanger sequencing method, with breakpoints in SELENOI‐DRC1 intergenic region and intron 4 of DRC1, identical to the deletion found in our initially reported cases (Morimoto et al., 2019), and shared among all deletion alleles, including those of two healthy carriers (Figure S1). An overall profile of targeted resequencing of the above seven cases is illustrated in Figure 2a, and observed variations are summarized in Table S4. Three patients with the homozygous deletions (six alleles in total) carried the same homozygous genotypes for all the variations listed in Table S4; therefore, only one haplotype could be estimated from them. The haplotype estimation using JPT data of the listed positions in Table S4 revealed that the deletion was carried by a major haplotype of the JPT population, as shown in Figure 2b. The same haplotype was also expected in the CEU population. However, the estimated frequency (9.1%) in this population was much lower than that observed in the JPT (60.1%).
Figure 2.

Recurring large deletions in DRC1 (a) A targeted resequencing profile around SELENOI and DRC1. The first three lanes (DPB002, DPB023, and dd071) indicate large homozygous deletions spanning exons 1–4 of DRC1. The rest (DPB058, DPB132, HS032, and HS107) had heterozygous deletions, and PCR products were thus mapped to all exons because of the presence of the insertion allele. Deletion breakpoints are illustrated as dotted lines within the SELENOI‐DRC1 intergenic region and within intron 4 of DRC1. The deletion size between the dotted lines was 27,848 bp, according to hg38. (b) The D', r2 values and haplotype estimation using JPT (left) and CEU (right) data of the positions listed in Table S4. The deletion in DRC1 was carried by the major haplotype (in the red box) of JPT. The same haplotype was also found in the CEU population but with a reduced frequency than in the JPT
4. DISCUSSION
We identified a large homozygous deletion in DRC1 in two (1.9%) of 105 Japanese adult patients clinically diagnosed as having DPB in the past and a heterozygous deletion in two other patients, in which other loss‐of‐function mutations have not yet been found within our search. During the same period, another Japanese patient with recurrent airway infection was also found to have the homozygous deletion in DRC1. Surprisingly, carriers of the large deletion with the same breakpoint were found with the allele frequency of 0.002, which was observed in a major haplotype in the Japanese population. However, the same deletions were not found in more than 20,000 non‐Asians (Morimoto et al., 2019), indicating that this loss‐of‐function variant may be a major cause of PCD in Japan, presumably acting as a founder mutation. Clinical features of these patients are hardly distinguishable from those of classical DPB, posing significant difficulties in reaching a correct diagnosis without the aid of genetic tests (Morimoto et al., 2019; Wirschell et al., 2013).
It is known that episodes of neonatal respiratory distress at full‐term birth are common in PCD (Knowles et al., 2016; Mullowney et al., 2014), but are less frequent in Japanese PCD patients as a whole. In addition, congenital heart disease and otitis media are also not commonly observed in Japan (Amitani et al., 1990; Inaba et al., 2019). This may indicate two possibilities: first, pediatricians are mostly unaware of PCD until adulthood; or second, alternatively, signs and symptoms of PCD in Japanese are relatively mild and somewhat controllable by early treatment with antibiotics in infancy or childhood. Mild clinical signs and symptoms depending on a particular genotype have recently been reported in PCD caused by RSPH1 mutations (Knowles et al., 2014).
In the present study, using a multiplexed PCR‐based method, we found a large homozygous deletion spanning exons 1–4 of DRC1 in two of 105 patients with definite DPB according to the current clinical criteria, and another patient with recurrent airway infections not fulfilling the criteria. In the subsequent analysis of the healthy Japanese population, 0.2% of alleles had a large deletion with identical breakpoints, expecting 0.0004% as homozygotes by the Hardy‐Weinberg principle. If prevalence of Japanese PCD is estimated at 1:20,000 ( = 0.005%) (Takeuchi et al., 2018), PCD caused by this deletion in DRC1 accounts for 8% ( = 0.0004/0.005), indicating that this DRC1 mutation may be the most common cause of PCD in Japan, since, so far, no mutation hotspots of known PCD genes have been identified in Japan (Takeuchi et al., 2018). In the two patients heterozygous for the same deletion, no other pathogenic or likely pathogenic variants were found in any exons and exon‐intron boundaries of DRC1. Further investigation would be necessary to determine whether pathogenic variants for compound heterozygosity are hidden in unscreened regions of the gene.
So far, ciliary defects in PCD can be classified into five main ultrastructural and other phenotypes as follows: the absence of outer dynein arms (ODAs), due to mutations in genes encoding ODA subunits; the absence of both ODAs and inner dynein arms (IDAs), caused by mutations related to dynein arm assembly; the absence of IDAs with microtubule disorganization, related to CCDC39 and CCDC40, essential to the docking of IDAs; an intermittent absence of the central pair and radial spoke defects in mutations of the radial spoke proteins; and an apparent absence of nexin link or even no detectable abnormalities on electron microscopy (EM) analysis but related to mutations of a subunit of nexin‐dynein regulatory complexes (N‐DRCs), involved in the connection between the IDA, radial spokes, and outer doublets (Shapiro & Leigh, 2017). PCD causative mutations in this fifth category have recently been reported in three human genes, DRC1, CCDC65 (DRC2), and GAS8 (DRC4) of the N‐DRC subunits (Austin‐Tse et al., 2013; Horani et al., 2013; Jeanson et al., 2016; Morimoto et al., 2019; Olbrich et al., 2015; Wirschell et al., 2013).
Although homozygous nonsense mutations in the human DRC1 gene were clearly demonstrated in 2013 with immunofluorescence staining results of N‐DRC subunits in four patients as one type of PCD (Wirschell et al., 2013), the subsequent case report has not yet been published. Strikingly, all of the 13 patients with N‐DRC subunit defects (DRC1, CCDC65, and GAS8) reported previously had situs solitus, presumably because subtle beating pattern abnormalities caused by these defects would be efficient enough to propel the extra‐embryonic fluid toward the left side of the embryo (Jeanson et al., 2016; Shapiro & Leigh, 2017). All of our cases without situs inversus may support the above possibility, though more case studies have been awaited (Morimoto et al., 2019). Furthermore, as compared with other forms of the disease, including those with CCDC39 and CCDC40 mutations (Davis et al., 2015), DRC1 mutations may have a mild clinical phenotype, at least in Japanese, with a delayed onset of symptoms and long survival presented in our report, considering a seemingly adult onset of the disease. It is not known yet whether the frequency of infertility caused by such subtle beating pattern defect is lower than that of other structural defects with ciliary immotility.
This type of PCD is not readily identified by either high‐speed video‐microscopy or EM analysis (Olbrich et al., 2015; Shapiro & Leigh, 2017) but rather associated with a secondary IDA defect due to inflammatory changes or artifact (O'Callaghan, Rutman, Williams, & Hirst, 2011). Nasal nitric oxide (NO) levels have been reported to be low for all N‐DRC defects, including DRC1 mutations (Morimoto et al., 2019; Wirschell et al., 2013). This finding is worthwhile because nasal NO is a useful noninvasive test in suspecting PCD (Leigh et al., 2013), even though the patients do not reveal any specific findings clinically. DPB also shows relatively low nasal NO levels in one report (Nakano et al., 2000), but a future comparison study may determine whether an appropriate cutoff value distinguishes most PCD types from classical DPB, after excluding extremely rare CF in Asia (Yamashiro et al., 1997).
Based on the references (Amitani et al., 1990; Austin‐Tse et al., 2013; Homma et al., 1983; Horani et al., 2013; Jeanson et al., 2016; Keicho & Kudoh, 2002; Olbrich et al., 2015; Wirschell et al., 2013), we summarized clinical similarities and possible differences between classical DPB and PCD with N‐DRC mutations in Table S5. Clinical signs and symptoms are similar to each other, but classical DPB has been regarded as an adult disease; lower respiratory symptoms are commonly observed in people in their 40s or 50s, even though chronic rhinosinusitis is experienced often from childhood, and abnormalities at birth have not been reported yet.
Pulmonary lesions on CT images are not easily distinguished between the two diseases when bilateral centrilobular nodules are observed. It is not known whether bronchiectasis predominantly affecting the middle lobe or lingula is more common in PCD than DPB (Kennedy et al., 2007). Pathologically, Homma previously reported that main pulmonary lesions shown as diffuse centrilobular nodules on chest CT (Amitani et al., 1990) are membranous bronchiolitis in PCD with Kartagener triad, distinct from respiratory bronchiolitis in classical DPB (Homma et al., 1999); however, no pathological reports on PCD caused by mutations of an N‐DRC subunit have been published yet. In classical DPB, cilia function may be secondarily affected by chronic airway inflammation (Amitani et al., 1990), but the motion of sperm is not affected, and consequently, infertility is usually not observed.
In the treatment of DPB, long‐term macrolide therapy has strikingly improved the prognosis (Kudoh, Azuma, Yamamoto, Izumi, & Ando, 1998), whereas, in PCD, the effect seems to be variable (Knowles et al., 2016). In the present study, treatment effects were hardly observed in one of the two patients. Considering similarities and differences of the two diseases and impaired mucociliary clearance, pathogenesis including disease susceptibility genes of DPB should also be revisited. This is also important because a major disease susceptibility gene of DPB has been hypothesized in a mucin gene cluster between the HLA‐A and HLA‐B loci (Hijikata et al., 2011; Keicho & Hijikata, 2011; Keicho et al., 2000), but has not been specified yet.
Since this was a retrospective analysis, we used a panel of DNA samples and their corresponding data records, and could not access patients' detailed information or other test results, which limited our study. No parents' or sibling's DNA was, thus, obtained. Although a major JPT haplotype that carries the deletion in DRC1 was a minor haplotype in the CEU population, a disease‐specific haplotype exclusively in Asians was not determined. To characterize patients with PCD in Asia further, prospective studies on sinopulmonary diseases including nasal NO, genetic tests, and ideally, immunofluorescence staining of cilia subunits, should be conducted.
We identified the same homozygous deletions in DRC1 in Japanese patients initially diagnosed as having DPB and demonstrated that this mutation may be one of the common causes of PCD in Japan. Since this type of PCD is not readily identified by high‐speed video‐microscopy or EM analysis, nasal NO measurement with appropriate genetic testing would be useful in differential diagnosis. Investigation on sinopulmonary diseases including search for founder genetic mutations that cause ethnic variants of PCD may be worth considering in Asian populations.
CONFLICT OF INTEREST
There are no conflicts of interest to declare.
Supporting information
ACKNOWLEDGMENT
We thank Keiko Wakabayashi and Akiko Miyabayashi for their technical assistance in the study. The authors also thank Drs. Atsushi Inaba and Masanori Furuhata for their helpful discussions. This study was supported by a grant from the Ministry of Health, Labour and Welfare of Japan awarded to the Study Group on Diffuse Pulmonary Disorders, Scientific Research/Research on intractable diseases, and JSPS KAKENHI Grant Number JP18K08163.
Keicho N, Hijikata M, Morimoto K, et al. Primary ciliary dyskinesia caused by a large homozygous deletion including exons 1–4 of DRC1 in Japanese patients with recurrent sinopulmonary infection. Mol Genet Genomic Med. 2020;8:e1033 10.1002/mgg3.1033
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- Afzelius, B. A. (1976). A human syndrome caused by immotile cilia. Science, 193(4250), 317–319. 10.1126/science.1084576 [DOI] [PubMed] [Google Scholar]
- Amitani, R. , Tomioka, H. , Kurasawa, T. , Ishida, T. , & Kuze, F. (1990). Clinical and ultrastructural study on primary ciliary dyskinesia. Nihon Kyobu Shikkan Gakkai Zasshi, 28(2), 300–307. [PubMed] [Google Scholar]
- Austin‐Tse, C. , Halbritter, J. , Zariwala, M. A. , Gilberti, R. M. , Gee, H. Y. , Hellman, N. , … Hildebrandt, F. (2013). Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. American Journal of Human Genetics, 93(4), 672–686. 10.1016/j.ajhg.2013.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, J. C. , Fry, B. , Maller, J. , & Daly, M. J. (2005). Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics, 21(2), 263–265. 10.1093/bioinformatics/bth457 [DOI] [PubMed] [Google Scholar]
- Bush, A. , Chodhari, R. , Collins, N. , Copeland, F. , Hall, P. , Harcourt, J. , … Phillips, G. (2007). Primary ciliary dyskinesia: Current state of the art. Archives of Disease in Childhood, 92(12), 1136–1140. 10.1136/adc.2006.096958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, S. D. , Ferkol, T. W. , Rosenfeld, M. , Lee, H.‐S. , Dell, S. D. , Sagel, S. D. , … Leigh, M. W. (2015). Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. American Journal of Respiratory and Critical Care Medicine, 191(3), 316–324. 10.1164/rccm.201409-1672OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, K. , Liu, M. B. , Wu, J. L. , Ma, H. Q. , Fang, X. Y. , Miao, G. B. , & Zhang, L. (2007). Diffuse panbronchiolitis in China: Analysis of 45 cases. Chinese Medical Journal, 120(22), 2046–2048. 10.1097/00029330-200711020-00021 [DOI] [PubMed] [Google Scholar]
- Hijikata, M. , Matsushita, I. , Tanaka, G. , Tsuchiya, T. , Ito, H. , Tokunaga, K. , … Keicho, N. (2011). Molecular cloning of two novel mucin‐like genes in the disease‐susceptibility locus for diffuse panbronchiolitis. Human Genetics, 129(2), 117–128. 10.1007/s00439-010-0906-4 [DOI] [PubMed] [Google Scholar]
- Homma, H. , Yamanaka, A. , Tanimoto, S. , Tamura, M. , Chijimatsu, Y. , Kira, S. , & Izumi, T. (1983). Diffuse panbronchiolitis. A disease of the transitional zone of the lung. Chest, 83(1), 63–69. 10.1378/chest.83.1.63 [DOI] [PubMed] [Google Scholar]
- Homma, S. , Kawabata, M. , Kishi, K. , Tsuboi, E. , Narui, K. , Nakatani, T. , … Nakata, K. (1999). Bronchiolitis in Kartagener's syndrome. European Respiratory Journal, 14(6), 1332–1339. 10.1183/09031936.99.14613329 [DOI] [PubMed] [Google Scholar]
- Horani, A. , Brody, S. L. , Ferkol, T. W. , Shoseyov, D. , Wasserman, M. G. , Ta‐shma, A. , … Kerem, E. (2013). CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS ONE, 8(8), e72299 10.1371/journal.pone.0072299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba, A. , Furuhata, M. , Morimoto, K. , Rahman, M. , Takahashi, O. , Hijikata, M. , … Keicho, N. (2019). Primary ciliary dyskinesia in Japan: Systematic review and meta‐analysis. BMC Pulmonary Medicine, 19(1), 135 10.1186/s12890-019-0897-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanson, L. , Thomas, L. , Copin, B. , Coste, A. , Sermet‐Gaudelus, I. , Dastot‐Le Moal, F. , … Legendre, M. (2016). Mutations in GAS8, a gene encoding a nexin‐dynein regulatory complex subunit, cause primary ciliary dyskinesia with axonemal disorganization. Human Mutation, 37(8), 776–785. 10.1002/humu.23005 [DOI] [PubMed] [Google Scholar]
- Keicho, N. , & Hijikata, M. (2011). Genetic predisposition to diffuse panbronchiolitis. Respirology, 16(4), 581–588. 10.1111/j.1440-1843.2011.01946.x [DOI] [PubMed] [Google Scholar]
- Keicho, N. , & Kudoh, S. (2002). Diffuse panbronchiolitis: Role of macrolides in therapy. American Journal of Respiratory Medicine, 1(2), 119–131. 10.1007/BF03256601 [DOI] [PubMed] [Google Scholar]
- Keicho, N. , Ohashi, J. , Tamiya, G. , Nakata, K. , Taguchi, Y. , Azuma, A. , … Kudoh, S. (2000). Fine localization of a major disease‐susceptibility locus for diffuse panbronchiolitis. American Journal of Human Genetics, 66(2), 501–507. 10.1086/302786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keicho, N. , Tokunaga, K. , Nakata, K. , Taguchi, Y. , Azuma, A. , Bannai, M. , … Kudoh, S. (1998). Contribution of HLA genes to genetic predisposition in diffuse panbronchiolitis. American Journal of Respiratory and Critical Care Medicine, 158(3), 846–850. 10.1164/ajrccm.158.3.9712125 [DOI] [PubMed] [Google Scholar]
- Kennedy, M. P. , Noone, P. G. , Leigh, M. W. , Zariwala, M. A. , Minnix, S. L. , Knowles, M. R. , & Molina, P. L. (2007). High‐resolution CT of patients with primary ciliary dyskinesia. American Journal of Roentgenology, 188(5), 1232–1238. 10.2214/ajr.06.0965 [DOI] [PubMed] [Google Scholar]
- Knowles, M. R. , Ostrowski, L. E. , Leigh, M. W. , Sears, P. R. , Davis, S. D. , Wolf, W. E. , … Zariwala, M. A. (2014). Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. American Journal of Respiratory and Critical Care Medicine, 189(6), 707–717. 10.1164/rccm.201311-2047OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, M. R. , Zariwala, M. , & Leigh, M. (2016). Primary ciliary dyskinesia. Clinics in Chest Medicine, 37(3), 449–461. 10.1016/j.ccm.2016.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono, C. , Yamaguchi, T. , Yamada, Y. , Uchiyama, H. , Kono, M. , Takeuchi, M. , … Tatsumi, K. (2012). Historical changes in epidemiology of diffuse panbronchiolitis. Sarcoidosis Vasc Diffuse Lung Dis, 29(1), 19–25. [PubMed] [Google Scholar]
- Kudoh, S. , Azuma, A. , Yamamoto, M. , Izumi, T. , & Ando, M. (1998). Improvement of survival in patients with diffuse panbronchiolitis treated with low‐dose erythromycin. American Journal of Respiratory and Critical Care Medicine, 157(6 Pt 1), 1829–1832. 10.1164/ajrccm.157.6.9710075 [DOI] [PubMed] [Google Scholar]
- Leigh, M. W. , Hazucha, M. J. , Chawla, K. K. , Baker, B. R. , Shapiro, A. J. , Brown, D. E. , … Knowles, M. R. (2013). Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia. Annals of the American Thoracic Society, 10(6), 574–581. 10.1513/AnnalsATS.201305-110OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, J. S. , Barbato, A. , Collins, S. A. , Goutaki, M. , Behan, L. , Caudri, D. , … Kuehni, C. E. (2017). European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. European Respiratory Journal, 49(1), 10.1183/13993003.01090-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto, K. , Hijikata, M. , Zariwala, M. A. , Nykamp, K. , Inaba, A. , Guo, T. C. , … Keicho, N. (2019). Recurring large deletion in DRC1 (CCDC164) identified as causing primary ciliary dyskinesia in two Asian patients. Molecular Genetics & Genomic Medicine, 7(8), e838 10.1002/mgg3.838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullowney, T. , Manson, D. , Kim, R. , Stephens, D. , Shah, V. , & Dell, S. (2014). Primary ciliary dyskinesia and neonatal respiratory distress. Pediatrics, 134(6), 1160–1166. 10.1542/peds.2014-0808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, H. , Ide, H. , Imada, M. , Osanai, S. , Takahashi, T. , Kikuchi, K. , & Iwamoto, J. (2000). Reduced nasal nitric oxide in diffuse panbronchiolitis. American Journal of Respiratory and Critical Care Medicine, 162(6), 2218–2220. 10.1164/ajrccm.162.6.2003051 [DOI] [PubMed] [Google Scholar]
- O'Callaghan, C. , Rutman, A. , Williams, G. M. , & Hirst, R. A. (2011). Inner dynein arm defects causing primary ciliary dyskinesia: Repeat testing required. European Respiratory Journal, 38(3), 603–607. 10.1183/09031936.00108410 [DOI] [PubMed] [Google Scholar]
- Olbrich, H. , Cremers, C. , Loges, N. T. , Werner, C. , Nielsen, K. G. , Marthin, J. K. , … Omran, H. (2015). Loss‐of‐function GAS8 mutations cause primary ciliary dyskinesia and disrupt the nexin‐dynein regulatory complex. American Journal of Human Genetics, 97(4), 546–554. 10.1016/j.ajhg.2015.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcese, C. , Patel, M. P. , Shoemark, A. , Kiviluoto, S. , Legendre, M. , Williams, H. J. , … Mitchison, H. M. (2017). X‐linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nature Communications, 8, 14279 10.1038/ncomms14279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, M. H. , Kim, Y. W. , Yoon, H. I. , Yoo, C. G. , Han, S. K. , Shim, Y. S. , & Kim, W. D. (1999). Association of HLA class I antigens with diffuse panbronchiolitis in Korean patients. American Journal of Respiratory and Critical Care Medicine, 159(2), 526–529. 10.1164/ajrccm.159.2.9805047 [DOI] [PubMed] [Google Scholar]
- Shapiro, A. J. , & Leigh, M. W. (2017). Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: Genetic defects with normal and non‐diagnostic ciliary ultrastructure. Ultrastructural Pathology, 41(6), 373–385. 10.1080/01913123.2017.1362088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, A. J. , Zariwala, M. A. , Ferkol, T. , Davis, S. D. , Sagel, S. D. , Dell, S. D. , … Leigh, M. W. (2016). Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatric Pulmonology, 51(2), 115–132. 10.1002/ppul.23304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama, Y. , Kudoh, S. , Maeda, H. , Suzaki, H. , & Takaku, F. (1990). Analysis of HLA antigens in patients with diffuse panbronchiolitis. American Review of Respiratory Disease, 141(6), 1459–1462. 10.1164/ajrccm/141.6.1459 [DOI] [PubMed] [Google Scholar]
- Takeuchi, K. , Kitano, M. , Kiyotoshi, H. , Ikegami, K. , Ogawa, S. , Ikejiri, M. , … Nakatani, K. (2018). A targeted next‐generation sequencing panel reveals novel mutations in Japanese patients with primary ciliary dyskinesia. Auris, Nasus, Larynx, 45(3), 585–591. 10.1016/j.anl.2017.09.007 [DOI] [PubMed] [Google Scholar]
- Wirschell, M. , Olbrich, H. , Werner, C. , Tritschler, D. , Bower, R. , Sale, W. S. , … Omran, H. (2013). The nexin‐dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nature Genetics, 45(3), 262–268. 10.1038/ng.2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro, Y. , Shimizu, T. , Oguchi, S. , Shioya, T. , Nagata, S. , & Ohtsuka, Y. (1997). The estimated incidence of cystic fibrosis in Japan. Journal of Pediatric Gastroenterology and Nutrition, 24(5), 544–547. 10.1097/00005176-199705000-00010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on request from the authors.