

Abstract
Transient receptor potential vanilloid subtype 1 (TRPV1) is a nonselective cationic channel activated by painful stimuli such as capsaicin and noxious heat, and enriched in sensory neurons of the pain pathway. During inflammation, chemical mediators activate protein kinases (such as PKC) that phosphorylate TRPV1 and thereby enhance its function, with consequent increases in nociceptor sensitization. However, the causal relationships between TRPV1 phosphorylation and pathological pain remain unexplored. To directly investigate the roles of one specific TRPV1 phosphorylation event in vivo, we genetically altered a major PKC phosphorylation site, mouse TRPV1 S801, to alanine. The TRPV1 expression pattern in sensory neurons of S801A knock-in (KI) mice was comparable to that in WT controls. However, sensitization of capsaicin-mediated currents after the activation of PKC was substantially impaired in sensory neurons from KI mice. Thermal hyperalgesia induced by PMA or burn injury in KI was identical to WT. Inflammatory thermal hyperalgesia was only marginally attenuated in KI mice. In contrast, PMA-evoked nocifensive responses and sensitization of capsaicin responses were significantly attenuated in the hindpaws of KI mice. Ongoing pain from inflamed masseter muscle was also reduced in KI mice, and was further inhibited by the TRPV1 antagonist AMG9810. These results suggest that PKC-mediated phosphorylation of TRPV1 S801 contributes to inflammation-mediated sensitization of TRPV1 to ligand, but not heat, in vivo. Further, this suggests that interference with TRPV1 S801 phosphorylation might represent one potential way to attenuate inflammatory pain, yet spare basal sensitivity and produce fewer side effects than more general TRPV1 inhibition.
SIGNIFICANCE STATEMENT Transient receptor potential vanilloid subtype 1 (TRPV1) has been considered a potential target for pain intervention. Global inhibitors of TRPV1 function, however, produce side effects which could compromise their clinical utility. By precisely removing a unique PKC phosphorylation site (TRPV1 S801) in mice through CRISPR/Cas9 editing, we provide in vivo evidence for a highly specific inhibition that leaves basal TRPV1 function intact, yet alleviates some forms of hyperalgesia. These findings support inhibition of TRPV1 S801 phosphorylation as a potential intervention for pain management.
Keywords: inflammation, mouse genetics, pain, phosphorylation, protein kinase C, TRPV1
Introduction
Transient receptor potential vanilloid subtype 1 (TRPV1) is a nonselective cation channel enriched in nociceptive neurons that can be activated by polymodal stimuli such as capsaicin and noxious heat. In vivo, TRPV1 has been implicated in acute capsaicin and heat nociception and in thermal and mechanical hyperalgesia (Chung et al., 2011a). Our recent studies suggest that TRPV1 also mediates spontaneous pain from inflamed masseter muscle (Wang et al., 2017). These results rationalize targeting of TRPV1 for the treatment of hyperalgesia. However, side-effects such as hyperthermia and loss of protective thermal nociception have called into question the clinical promise of agents that inhibit all TRPV1 functions (Szolcsányi and Pintér, 2013). A better strategy might therefore be to selectively target TRPV1 involvement in pathological functions, without interfering with its physiological roles.
TRPV1 function in nociceptive primary afferents is dynamically regulated during inflammation or tissue injury. Various inflammatory mediators have been shown to activate multiple protein kinases that, in turn, phosphorylate TRPV1 (Huang et al., 2006; Levine and Alessandri-Haber, 2007). TRPV1 phosphorylation enhances the channel's responsiveness, such that mild agonists that minimally activate TRPV1 in its unphosphorylated state can do so robustly. Such functional regulation has been reported for multiple inflammatory mediators and it has therefore been suggested that TRPV1 phosphorylation is an integrative hub for nociceptor sensitization during inflammation (Levine and Alessandri-Haber, 2007).
Because TRPV1 phosphorylation is thought to be an important contributor to the pathological functions of TRPV1, the residues of TRPV1 subject to phosphorylation might be targeted to treat hyperalgesia. It is therefore critical to determine the contributions of different TRPV1 phosphorylation residues to hyperalgesia. The kinases that phosphorylate TRPV1 include PKC, PKA, CaMKII, Src, and cdk5 (Bhave et al., 2002, 2003; Numazaki et al., 2002; Mohapatra and Nau, 2003; Jung et al., 2004; Pareek et al., 2007). Among these, PKC mediates TRPV1 sensitization after the activation of receptors for key inflammatory mediators (Huang et al., 2006; Levine and Alessandri-Haber, 2007). PKC-induced TRPV1 phosphorylation enhances responses to capsaicin, acid, and heat (Vellani et al., 2001). These PKC effects are mediated mainly by phosphorylation of three residues (S502, T704, and S800) in rat TRPV1 (Numazaki et al., 2002; Bhave et al., 2003). Phosphorylation of different TRPV1 residues apparently results in different extents of sensitization in response to different stimulus modalities. For example, two serine residues, S502 and S800, are involved in sensitization of capsaicin-evoked responses induced by phorbol myristate acetate (PMA), an agonist of PKC (Numazaki et al., 2002; Bhave et al., 2003). In contrast, T704 phosphorylation mediates direct activation of TRPV1 by PMA and determines its basal heat sensitivity, but apparently does not contribute to its hypersensitivity to capsaicin (Bhave et al., 2003; Li et al., 2014). Recently, we extended the modality-specific analysis of TRPV1 sensitization by PKC (Wang et al., 2015). We found that TRPV1 S800 and T704, but not S502, mediate PMA-induced hypersensitivity to heat, with a lesser contribution from S800 than T704 (Wang et al., 2015). In contrast, PMA-induced hypersensitivity to acid was attenuated only in S800A, but not by mutation of S502 or T704. These results further suggest that mechanisms of PKC-induced TRPV1 hypersensitivity are modality-specific and that S800 is a polymodal sensitization site integrating multiple inflammatory signals in nociceptors.
The rat TRPV1 S800 site equivalent is conserved in TRPV1 orthologs across different species (Fig. 1A), including human, in all common variants (Wang et al., 2016). However, despite its clear implication in TRPV1 sensitization, the specific contribution of phosphorylation at S800 or any other single TRPV1 residue to pain or hyperalgesia in vivo is not known. In this study, we therefore generated a knock-in (KI) mouse line in which the corresponding residue, TRPV1 S801, was mutated into alanine by genome editing to prevent its PKC-mediated phosphorylation. Using this novel genetic model, we identified multiple contributions of TRPV1 S801 phosphorylation to nociception and inflammatory pain hypersensitivity in vivo.
Figure 1.

Generation of TRPV1 S801A KI mice using the CRISPR/Cas9 method. A, Sequence alignment of carboxy terminal domains of TRPV1 from different species. The orthologous mouse S801 residue is shown in red. B, Sequences of region of interest in trpv1 gene (top), the repair template (middle), and the resulting targeted trpv1 gene (bottom). Note that the PAM site was not mutated as intended. C, Enzymatic digestion of PCR products amplified from genomic DNA of a heterozygous founder (KI/WT) or C57BL/6 using XhoI (Xho) or MluI (Mlu) or undigested (un). Upper size marker, 300 bp; lower size marker, 200 bp. D, Sanger sequencing of PCR products amplified from genomic DNA of a heterozygous founder. E, Example of genotyping assay in WT, heterozygote KI, and homozygote KI using primers indicated in A, WT-specific primers, Comm-for and WT-rev; KI-specific primers, Comm-for and KI-rev. Upper size marker, 300 bp; lower size marker, 200 bp.
Materials and Methods
Procedures
All procedures were conducted in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were performed under protocols approved by The University of Maryland or Johns Hopkins University Animal Care and Use Committees.
Generation of TRPV1 S801A KI mice
To generate TRPV1 S801A KI mice, the TRPV1 locus was edited using the CRISPR/Cas9 technique (Cong et al., 2013). An sgRNA recognition sequence (GGGACGCAAGCACTCGAGAT; highlighted in yellow at top sequence of Fig. 1A) adjacent to a protospacer adjacent motif (PAM) sequence (AGG) was selected to target the vicinity of the TRPV1 S801 codon. Complementary oligonucleotides encoding the recognition sequence (5′-CAC CGG GAC GCA AGC ACT CGA GAT-3′ and 5′-AAA CAT CTC GAG TGC TTG CGT CCC-3′) were annealed together and subcloned into pX330 (Cong et al., 2013) using BbsI sites. A DNA sequence encoding the full-length sgRNA was subsequently PCR amplified from this construct using the following two primers: 5′-TTA ATA CGA CTC ACT ATA GGG GGG ACG CAA GCA CTC GAG AT-3′ and 5′-AAAAGCACCGACTCGGTGCC-3′, in which the underlined portion of the first primer encodes an attached T7 promoter. The full-length sgRNA was then generated by in vitro reverse transcription using the T7 Quick High Yield RNA synthesis kit (New England Biolabs) and the MEGAclear clean-up kit, followed by precipitation with ammonium acetate and resuspension in nuclease free water. A 150 nucleotide custom synthesized single stranded DNA repair template (Dharmacon) (5′-TGC CTT TCA GTT TCA GGG AGA AAC TGG AAG AAC TTT GCC CTG GTT CCC CTT CTG AGG GAC GCA GCC ACG CGT GAT AGA CAT AGC ACC CAG CCG GAA GAA GTT CAG CTG AAG CAC TAT ACG GGA TCC CTT AAG CCA GAG GAT GCT GAG GTC-3′; Fig. 1A) was designed to span a region including part of TRPV1 exon 15 and a portion of the preceding intron. This template was identical to the corresponding TRPV1 locus, with the exception of a mutation of the codon encoding TRPV1 serine 801 to alanine, silent mutations changing an XhoI site to an MluI site, and disruption of the PAM sequence from AGG to AGA. The template was resuspended in nuclease free water to 1 μg/ml before use. Polyadenylated Cas9 mRNA was produced by in vitro transcription from NotI-linearized plasmid pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene plasmid # 42230) (Cong et al., 2013) using the mMESSAGE mMACHINE T7 ULTRA IVT kit (ThermoFisher, Cat# AM1345), precipitated using lithium chloride, and resuspended in nuclease-free water. The sgRNA, Cas9 mRNA and the repair template were diluted in microinjection buffer (Brinster et al., 1985). Two hundred and seventy single-cell embryos from C57BL/6 mice were microinjected into one pronucleus. Injected zygotes were implanted into 10 pseudopregnant ICR female mice to produce founder offspring. Initially, genomic DNA from the founders was amplified with two primers (5′-AGAACTTTGCCCTGGTTCCC-3′ and 5′-TCAACCCGGCTCTATTGCTC-3′) to yield a 247 bp fragment that spanned the intended mutagenesis site. This product was subsequently digested with either XhoI (to detect the WT allele) or MluI (to detect the mutant allele). Subsequent analysis of the mutated TRPV1 genomic sequence was performed in founders and in the offspring resulting from founder mating with WT C57BL6 mice by sequencing a 560 bp genomic PCR product that extended beyond either end of the homology repair template (using primers 5′-TCCGTGACCCATGGATCTCT-3′ and 5′-GCAGAGTACAGCCAGCCAACA-3′). To further confirm proper targeted recombination, we performed reverse transcription of mRNA harvested from dorsal root ganglia (DRG) of homozygous KI/KI mice from each of two founder-derived lines, followed by PCR amplification of the mutation-containing region using two primers (5′-ACACCAACGTGGGCATCATC-3′ and 5′-TGGTTAGATTCACAGCTCGCTTC-3′) annealing to exons 14 and 15, respectively (to exclude amplification of genomic DNA). The PCR products were sequenced to confirm the sole presence of the KI allele. Routine allele-specific genotyping was later performed on the colonies using a common forward primer (Comm-for, 5′-TCCGTGACCCATGGATCTCT-3′) that anneals upstream of the repair template with either a WT-specific reverse primer (WT-rev, 5′-ATGCCTATCTCGAGTGCT-3′) or a mutant-specific reverse primer (KI-rev, 5′-ATGCCTATCACGCGTGGC 3′) (Fig. 1B). We obtained two founder lines. To minimize the impact of potential unintended mutations from the CRISPR/Cas9 gene editing, both founders were backcrossed with C57BL/6 mice. For the CFA inflammatory hindpaw model, we used mice backcrossed a minimum of two generations. For all other experiments, we used mice backcrossed a minimum of four generations. Both S801A mouse lines were used in all experiments, and the data were pooled as we did not find a difference between them.
Dissociation of mouse sensory neurons
Mice (4–9 weeks old) were anesthetized using a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg). DRG were dissected out and collected in cold Puck's saline (171 mm NaCl, 6.7 mm KCl, 1.4 mm Na2HPO4, 0.5 mm KH2PO4, 6.0 mm glucose, pH 7.3). The ganglia were incubated in 5 ml of DMEM/F12 medium containing collagenase type IV (1 mg/ml, Millipore Sigma) at 37°C for 30 min. The ganglia were incubated for an additional 15 min after the addition of trypsin (0.25%) and EDTA (0.025%). The tissues were triturated with flame-polished Pasteur pipettes. The neurons were plated onto glass coverslips (8 mm) coated with poly-ornithine and laminin. Dissociated neurons were maintained with DMEM/F12 containing 10% FBS, 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator. Electrophysiological recordings were performed after 1–3 d.
Whole-cell voltage-clamp techniques
Whole-cell voltage-clamp techniques were performed as described previously (Chung and Wang, 2011; Wang et al., 2012). The recording pipettes (2–3 MΩ) were pulled from borosilicate glass using a P-97 (Sutter Instrument). In analyses of HEK293 cells, the pipettes were filled with internal solution (150 mm NaCl, 1 mm MgCl2, 10 mm HEPES, 5 mm EGTA, pH 7.4). Unless otherwise indicated, the recording bath contained an external solution (140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, 10 mm glucose, pH 7.4). In analyses of sensory neurons, the pipette was filled with a solution containing 140 mm KCl, 5 mm NaCl, 1 CaCl2, 1 MgCl2, 2.5 Mg-ATP, 10 mm EGTA, and 10 mm HEPES, pH 7.3. Osmolarity of each solution was measured by a vapor pressure osmometer (Wescor), and was adjusted with mannitol to 290 to 310 mOsm as necessary. Unless otherwise indicated, all recordings were performed at room temperature.
Immunohistochemistry
Immunohistochemical staining was performed as described previously (Chung et al., 2012). Mice were transcardially perfused with 3.7% paraformaldehyde. TG and DRG were dissected, cryoprotected, and sectioned at 12 μm intervals. The lumbar spinal cord was sectioned at 30 μm intervals. Conventional immunohistochemical procedures were performed with rabbit anti-TRPV1 (1:1000) (Tominaga et al., 1998), guinea pig anti-CGRP (1:1000; Peninsula Labs), and mouse anti-NF200 (1:1000; Sigma-Aldrich) followed by appropriate secondary antibodies (Invitrogen). To label nonpeptidergic nociceptive afferents, sections were exposed to IB4-biotin (10 μg/ml, Invitrogen), followed by streptavidin-conjugated fluorophore (Invitrogen). Images were acquired by optical sectioning fluorescence microscopy (Axiovert; Carl Zeiss Microimaging).
Western blot assay
Mice were killed and whole DRG were excised into 1.5 ml of cold Ca2+ and Mg2+-free Hank's balanced salt solution. The buffer was removed and replaced with 0.2 ml of radioimmunoprecipitation assay buffer (Thermo Scientific, 89900) containing protease inhibitor mixture (Cell Signaling Technology, 5872). The DRG were mixed in the buffer and sonicated (25 amplitude setting, 10 pulses, Qsonica, Q55). Samples were centrifuged at 12,000 × g for 10′ in a tabletop centrifuge precooled to 4°C to remove debris. The supernatant was collected into 1.5 ml tubes. Sample lysates were loaded onto 4–12% Bis-Tris NuPAGE gels (Invitrogen) at 30 μg/well and blotted onto PVDF membranes. The blot was blocked and then probed with antibodies against TRPV1 [Proteintech, 22686-1-AP, rabbit, 1:700; custom (Tominaga et al., 1998), rabbit, 1:800] and GAPDH (EMD, CB1001, mouse, 1:10000), and incubated at 4°C overnight. The blot was washed and fluorescently labeled with goat-anti-mouse (1:20,000) (Li-Cor, 926–68020) and goat-anti-rabbit (1:1000) (Li-Cor, 926–32211). After a wash, the wet blot was scanned using an Odyssey imager and Image Studio software version 5.2. The images were quantified using ImageJ.
Behavioral pain measurements
Adult (>8 weeks old) mice were randomly allocated into different experimental groups. Both male and female mice were used. The experimenter was blinded to the experimental groups.
Acute hindpaw nocifensive behaviors.
Twenty microliters of PMA (3 ng/μl) in PBS or PBS alone (vehicle) was injected intraplantarly to a hindpaw. An anesthetic was not used for injections. The mice were then immediately put into plastic boxes (10 × 10 × 14 cm) on a lab bench with Whatman paper (Millipore-Sigma, 3030917) under the box and observed for 30 min, with video recordings to evaluate nociceptive behavior and quantify time spent for licking and biting of the injected paw. The mice were then injected with 1 μg of capsaicin in a buffer containing 135 mm NaCl, 3 mm KCl, 1 mm Na2HPO4, 1 mm MgSO4, 1.2 mm CaCl2, pH 7.4, 10 μl, into the same hindpaw, and placed back into the box for an additional 15 min of recording. Quantifications were done by an observer blinded to the genotypes of the mice.
Eye-wiping test.
To test chemical sensitivity of ophthalmic nociceptors, an eye wiping test was performed. Mice were placed in plastic containers (9 × 9×13 cm high) with two mirror back walls, affording the camera a four-sided view. A digital video camera (Sony HDR-CX230/B High Definition Handycam Camcorder) was placed at a fixed distance from the cubicle to record their behavior. Free behaviors of mice were videotaped 5 min before and 5 min after the application of 20 μl of capsaicin solution (0.03% in H2O with 3.3% PEG300). The number of eye wipes with the forepaw was counted for 5 min.
Complete Freund's adjuvant (CFA)-mediated hindpaw and masseter muscle inflammation.
Mice were briefly anesthetized using 3% isoflurane and CFA (20 μl, 1:1 in PBS) was injected unilaterally into masseter muscle or hindpaw as indicated in each experiment. CFA invariably produced swelling of the masseter muscle and hindpaw.
Carrageenan-induced hindpaw inflammation.
Mice were anesthetized with isoflurane (3%) and one hindpaw was cleaned with betadine and alcohol, followed by unilateral carrageenan injection (20 μl, 1% in PBS, using a 30 gauge needle). The injection site was near the proximal footpad. The carrageenan was prepared by heating PBS to 50°C, then slowly adding the carrageenan and stirring for 1 h.
Hargreaves' radiant paw-heating assay.
Mice were acclimated to the testing environment for at least 30 min each day for 2 d by placing them on the glass platform (30°C) of a Hargreaves device (PAW Thermal Stimulator, UC San Diego or Plantar Analgesia Meter, IITC Life Sciences) under an acrylic box. For testing, the mice were placed on the glass platform under an acrylic box for 10 min until they settled. Baseline latency of the radiant heat source was adjusted to a range of 10–12 s with a cutoff time of 20.5 s to prevent tissue damage. Paw withdrawal latency in response to the heat stimulus was measured in both hindpaws 3 times each, with a 10 min interstimulus interval, and the average of the three latencies was used for analysis.
Measuring noxious heat threshold.
Hindpaw noxious heat threshold was assessed as described previously (Bölcskei et al., 2005) using a temperature-ramp hot plate device (IITC Life Science). The temperature of the plate was increased from 30°C to 50°C linearly (6°C/min). The mice were habituated on the metal plate (20 × 15 cm) maintained at 30°C under an acrylic observation chamber (20 × 15 × 20 cm) for 30 min on each of 2 d. The test lasted until the animal showed nocifensive behaviors (licking, shaking, or lifting) involving either hindpaw, then the mouse was immediately removed from the plate. The heat threshold measurement was repeated after 30 min and the mean of the two thresholds was considered the noxious heat threshold of the mouse.
Von Frey measurement in hindpaw skin.
Mice were placed under acrylic boxes on an elevated wire mesh platform and habituated in a behavioral room for at least 30 min per day for 3 d before testing. A series of von Frey filaments were applied perpendicularly to the hindpaw plantar surface. The filaments had bending forces ranging from 0.008 to 4 g. Lifting of the hindpaw or flinching immediately upon removal of the filament was defined as a response. Each filament was applied 5 times at intervals of a few seconds. The response frequencies [(number of responses/number of stimuli) × 100%] to a range of filament forces were determined and stimulus-response frequency curves were plotted. The plots were fitted with a logistic function from which EF50, the mechanical force that produced a 50% response frequency, was obtained.
Paw-pinching assay.
For the pinch assay (Huang et al., 2019), each mouse was confined in a Plexiglas chamber (7.5 cm long × 7.5 cm wide × 10 cm high) which was placed onto a glass surface, allowing video recording from the bottom. An alligator clip (Generic Micro Steel toothless alligator test clips 5AMP) was applied to the ventral skin surface between the footpad and the heel. The animal was put back into the chamber and video recorded for 60 s to determine duration of nocifensive behavior toward the hindpaw.
Tail immersion assay.
Mice were wrapped in a towel while leaving the tail exposed for most of its length. While the mouse was being held, the last 2 cm of tail was dipped into a water bath maintained at 50°C. The temperature was confirmed throughout the experiment with a thermometer (BAT-12; Physitemp Instruments). The time for the mouse to withdraw or flick its tail was measured, and the average withdrawal latency was determined for each group.
Hot plate assay.
Mice were placed on a hot plate apparatus (series 8, PE34) (IITC Life Science) with the plate set to a temperature of 53°C. The latency was measured for the mouse to show signs of nocifensive behavior, such as licking of hindpaws or jumping from the heated plate. A cutoff of 20 s was used to prevent serious burn injury.
PMA-induced thermal hyperalgesia of hindpaw.
To test PMA-mediated thermal hyperalgesia, 2 ng of PMA (20 μl of 0.1 μg/ml in PBS) was injected intraplantarly into one hindpaw. A Hargreaves' test was performed before and 3–4 h after the injection.
Mild hindpaw thermal injury.
A mild thermal injury model was performed as described previously (Bölcskei et al., 2005). After measurement of basal heat sensitivity, the mice were anesthetized using isoflurane. The left hindpaw was immersed into a water bath at 51°C for 15 s to a level above the ankle. The mice were returned to their home cage and allowed to recover from anesthesia. To evaluate changes in thermal sensitivity induced by mild heat injury, we evaluated thermal threshold again 30 and 60 min after heat injury.
Mouse grimace scale (MGS) measurements.
Previously we showed that masseter injection of CFA increases MGS scores. This was substantially attenuated by pharmacological or genetic inhibition of TRPV1 or transient receptor potential ankyrin subtype 1 (TRPA1), ablation or chemogenetic inhibition of TRPV1+ primary afferents, or ablation of NK1+ second order neurons (Asgar et al., 2015; Wang et al., 2017, 2018). These studies suggest that masseter inflammation-induced changes in MGS are mediated by nociceptive inputs from masseter muscle, and supports MGS as a method for assessing spontaneous pain during masseter inflammation. The MGS was used as previously described (Wang et al., 2017, 2018). The mice were videotaped for 30 min for each experimental time point. To capture facial images of mice in an unbiased manner, image extraction was performed by blinded experimenters. Images containing a clear view of the entire face were manually captured every 3 min during the video recording (10 images per 30 min session). The scores of the five action units in each photograph were averaged, and a mean MGS score was obtained from the 10 images, which was presumed to reflect the level of spontaneous pain. The mean MGS score of each mouse before CFA treatment was used as the baseline value.
Bite force assay.
To assess bite-evoked pain associated with craniofacial muscle inflammation in mice, we performed a bite force assay as previously described (Wang et al., 2017, 2018; Guo et al., 2019). Mice were acclimated to the testing environment and handling for 2 d before behavioral testing. Mice were placed in a modified 60 ml plastic syringe with a wide opening at one end to accommodate the head of the mouse. To prevent the mouse from escaping, the syringe plunger was inserted into the syringe to loosely restrain the mouse inside the syringe. To minimize stress, the mouse was released immediately from the syringe if it vigorously moved or tried to hide inside the syringe. The syringe containing the mouse was held manually and moved slowly at 0.5–1 cm/s toward bite plates so that the mouse could bite the plates. Spike 2 software was used to measure the voltage changes from transducer displacement. SigmaPlot 8.0 was used to convert the voltage change into force based on calibration using standard weights. Bite force was recorded for 120 s per session and the top five force measurements were averaged.
Experimental design and statistical analysis
The method of statistical analysis used in each dataset is indicated in the figure legend. Data from two groups were compared using Student's t test. Data from three or more groups were compared using one-way ANOVA followed by Bonferroni's post hoc test. The effects of pharmacological or genetic manipulations in different time points were analyzed with two-way ANOVA with repeated measures. All multiple-group comparisons were performed by Bonferroni's post hoc test. Data are presented as means ± SEM. The criterion for statistical significance was p < 0.05. All statistical analyses were performed using GraphPad Prism 6.0.
Results
TRPV1 S801A KI mice were generated using CRISPR/Cas9
To assess the role of TRPV1 phosphorylation at a single residue in hyperalgesia, we focused our attention on S801 in mouse TRPV1. We selected this residue because our previous study suggested it as a polymodal sensitization site (Wang et al., 2015), and alanine mutation at this site does not produce basal functional changes in TRPV1 (Bhave et al., 2003; Wang et al., 2015). To generate a mouse line lacking phosphorylation at TRPV1 S801, we edited the TRPV1 locus using the CRISPR/Cas9 technique following the strategy shown in Figure 1B. A guide RNA sequence was selected to direct Cas9-mediated cleavage ∼6 nt downstream of the TRPV1 S801 codon to be targeted. A homology repair template used to direct repair of the resulting double strand DNA break was designed to introduce several changes in the TRPV1 gene: mutation of the S801 codon to one encoding alanine; the silent replacement of an XhoI site with an MluI site; and the disruption of the PAM sequence. Candidate founder mice and their offspring were genotyped using three complementary methods. As an initial step to detect successful KI of the repair template, we performed enzymatic digestion of PCR products amplified from the targeted locus with XhoI or MluI, to identify mice in which the MluI site had been successfully inserted. Whereas the PCR product from WT mice could be digested only with XhoI, we identified several founder mice that also exhibited partial MluI sensitivity within the corresponding PCR product, suggestive of the presence of one copy of the desired mutant allele (Fig. 1C). Two of these founders were subsequently mated against C57BL/6 mice to generate two parallel lines of candidate KI mice. We amplified PCR products encompassing a genomic DNA segment extending beyond that corresponding to the homology repair template from both the founders and their offspring. Sequencing these products confirmed the introduction of most of the intended nucleotide substitutions into the TRPV1 locus in both lines (Fig. 1D). Namely, the S801 codon was successfully changed to an alanine codon, and the XhoI site was silently replaced with an MluI site, without alteration of the remaining nearby translational code. Interestingly, the intended mutation of the PAM sequence was not achieved in either line, suggesting that in both founders the homologous recombination had resolved itself between the successfully introduced mutations and the PAM site. Heterozygous KI mice within each of the two mutant lines were independently interbred to generate homozygous KI mice from both lines. Inheritance of two copies of the KI allele was confirmed by sequencing of genomic DNA-derived PCR products and separately through the use of allele-specific PCR primers designed to yield a product only in the presence of the WT or KI allele, respectively (Fig. 1B,E). Finally, to confirm that the KI allele was expressed, and that no residual WT TRPV1 mRNA was produced in homozygous KI animals, we performed RT-PCR of mRNA collected from DRG of KI homozygous mice followed by sequencing of the PCR products. In this assay, we detected only the sequence of the KI allele, without evidence of contaminating WT sequence (data not shown). Together, these results confirmed our successful introduction of the S801A mutation into the TRPV1 gene. Both heterozygous and homozygous KI mice were healthy and fertile, without overt differences in appearance or behavior from their WT littermates.
TRPV1 expression is normal in TRPV1 S801A KI mice
We next investigated whether the expression pattern of the TRPV1 protein is changed in trigeminal ganglia (TG) of TRPV1 S801A KI mice. Immunohistochemical labeling of TRPV1 in TG showed that neurochemical properties of TRPV1-expressing neurons of KI mice were not different from those of WT (Fig. 2A–D). In both KI and WT, the majority of TRPV1+ neurons (∼65%) were colabeled with CGRP but only ∼15% and 12% of neurons were colabeled with isolectin B4 (IB4) and neurofilament heavy chain (NF200), respectively. The size of neurons expressing TRPV1 was also comparable between KI and WT (Fig. 1E). These findings were also similar in DRG (data not shown). TRPV1+ central terminals also appeared similar between WT and KI mice, in both cases projecting into the superficial layer of the spinal cord dorsal horn, and partially overlapping with CGRP and IB4+ terminals (Fig. 1F). The total amount of TRPV1 protein in whole dorsal root ganglion lysates also showed no difference by immunoblot between KI and WT (Fig. 1G,H). These results suggest that TRPV1 expression was not altered in TRPV1 S801A KI mice.
Figure 2.

TRPV1 S801A KI mice show no altered expression of TRPV1. A–C, Representative images of double labeling for TRPV1 and CGRP (A), IB4 (B), or NF200 (C) in TG section obtained from WT or TRPV1 S801A KI mice. Arrowheads highlight representative neurons exhibiting colocalization of TRPV1 and the respective markers. Scale bar, 50 μm. D, Quantification of the proportion of neurons showing colocalization of TRPV1 and neurochemical markers in TG, n = 3–4 mice in each group. E, Size distribution of TRPV1+ neurons in TG from WT and TRPV1 S801A KI mice. Number of mice is in parentheses. F, Triple labeling of TRPV1, IB4 and CGRP in the spinal cord of WT and TRPV1 S801A KI mice. Scale bar, 100 μm. G, Western blot analysis of TRPV1 using protein extract of whole DRG from WT or TRPV1 S801A KI mice. Representative gel image of Western blot using two antibodies against TRPV1 [upper panels in left, Proteintech; right, custom (Tominaga et al., 1998)] or an antibody against GAPDH (bottom) in WT, KI and TRPV1 KO DRG, Arrow, TRPV1 band. At right, only the upper band of the doublet is consistently absent from TRPV1 KO lysates. H, Relative quantification of TRPV1 using TRPV1 antibody against TRPV1 from Proteintech, n = 7 for WT and n = 6 for KI.
TRPV1 S801A KI mice showed impaired sensitization of capsaicin-evoked currents
To compare the properties of capsaicin-evoked currents in sensory neurons between WT and TRPV1 S801A mice, we performed whole-cell voltage-clamp recordings on dissociated DRG neurons. Two different doses of capsaicin (0.4 and 10 μm) produced current densities that were similar between genotypes when comparing KI to WT (Fig. 3A). Times taken for 10–90% rise of currents evoked by capsaicin (10 μm) were also not different between WT and KI (Fig. 3B). The proportions of dissociated DRG neurons responsive to capsaicin (1 μm) in Ca2+ imaging experiments were also similar (WT, 49.3%; KI, 46.2%). These data are consistent with the identical capsaicin sensitivities of heterologously expressed rat TRPV1 WT and S800A (Numazaki et al., 2002; Bhave et al., 2003). The application of PMA sensitizes TRPV1. This effect is mediated through the activation of PKC, since a PKC inhibitor blocks the effects of PMA and an inactive PMA analog does not produce sensitization (Bhave et al., 2003). To evaluate whether PKC-mediated sensitization of TRPV1 is altered in TRPV1 S801A KI neurons, we determined the extent of PMA-induced increases of peak-current densities evoked by a low-dose of capsaicin (Fig. 3C–E). Brief application of capsaicin (0.4 μm) evoked small current responses in WT (Fig. 3C), homozygous TRPV1 S801A KI (Fig. 3D) and heterozygous (Het, data not shown) neurons. PMA (0.3 μm) was subsequently applied for 2 min, followed by a second application of 0.4 μm capsaicin. A saturating dose of 10 μm capsaicin was applied at the end of the recording period, to permit normalization of the currents evoked by the lower capsaicin dose. Under these conditions, the normalized response (peak-current densities) to the second 0.4 μm capsaicin application was significantly greater than that to the first application in all experimental groups. However, the extent of sensitization by PMA varied between genotypes (Fig. 3E; time effect, F(1,20) = 106.68, p < 0.0001; genotype effect, F(2,20) = 11.74, p = 0.0004; interaction, F(2,20) = 11.62, p = 0.0004; two-way repeated-measures ANOVA). Specifically, post hoc analysis showed the extent of PMA-induced sensitization was significantly smaller in Het and KI neurons than in WT (Fig. 3E), supporting a role for TRPV1 S801 phosphorylation in this process. The small but significant PMA-induced sensitization in KI neurons might reflect overlapping contributions from one or more additional phosphorylated serine residues, likely including S502 (Numazaki et al., 2002; Bhave et al., 2003).
Figure 3.

TRPV1 S801A KI mice show attenuated sensitization of capsaicin-evoked current response by PMA. A, Densities of currents evoked by 0.4 or 10 μm capsaicin in dissociated DRG neurons from WT and KI mice. The numbers within parentheses represent the number of neurons. NS, not significant (left, p = 0.61; right, p = 0.63 in Student's t test). B, Rising time (10–90%) of currents evoked by 10 μm capsaicin in dissociated DRG neurons from WT and KI mice. NS, not significant (p = 0.73 in Student's t test). C, D, Representative current traces evoked by capsaicin in dissociated DRG neurons from WT (C) or KI (D) mice. The currents were recorded by whole-cell voltage clamp at −60 mV and 0.4 or 10 μm capsaicin and 0.3 μm PMA was applied as indicated. Current amplitudes were normalized to the current amplitude evoked by 10 μm capsaicin in each neuron. E, Extent of PMA-mediated sensitization of responses evoked by first and second application of 0.4 μm capsaicin that were normalized to the response by 10 μm capsaicin in each neuron. ***p < 0.001; ****p < 0.0001 in Bonferroni's post hoc test after two-way repeated-measures ANOVA, #p < 0.05; ###p < 0.001; ####p < 0.0001 in Bonferroni's post hoc test between first and second currents. Numbers within parentheses represent the numbers of cells analyzed.
PMA not only sensitizes heterologously expressed TRPV1 but also reverses desensitization produced by a high concentration of capsaicin (Mandadi et al., 2004, 2006). This reversal phenomenon is abolished in the rat TRPV1 S502A/S800A double mutant (Mandadi et al., 2006). However, it is unknown whether this effect of PMA occurs in sensory neurons, or the extent to which it is attributable to phosphorylation of S800. We therefore investigated whether PKC-mediated reversal of capsaicin-induced desensitization is affected in DRG neurons dissociated from KI mice. Initial stimulation with 10 μm capsaicin evoked a robust and reversible activation of TRPV1 in both WT and KI neurons (Fig. 4A). A second application of 10 μm capsaicin reproducibly evoked a second current response, but the amplitude of this response was only ∼60% that of the initial current, indicative of TRPV1 desensitization. The extent of capsaicin-induced desensitization was not significantly different between genotypes (Fig. 4A,C), suggesting that capsaicin-induced desensitization does not require TRPV1 S801. In WT neurons, PMA application before the second application of capsaicin prevented desensitization and resulted in a tendency of the second response to be even larger than the first response (Fig. 4B), which was significantly different from the condition without PMA treatment (Fig. 4C). However, in Het neurons, the second 10 μm capsaicin response (after PMA) was almost equal to the first response. In KI neurons, the second response was smaller than the first response, showing desensitization even after PMA treatment. As a result, the ratios of first/second responses were not significantly different between PMA and Veh groups in either the Het or KI neurons (Fig. 4C). These results suggest that PMA-mediated reversal of capsaicin-induced desensitization is impaired in TRPV1 S801A KI mice.
Figure 4.

TRPV1 S801A KI mice show attenuated PMA mediated recovery from desensitization to capsaicin. A, Representative current responses evoked by consecutive application of 10 μm capsaicin in dissociated DRG neurons from WT (left) or KI (right) mice. The currents were recorded by whole-cell voltage clamp at −60 mV. Scale bars, 50 pA/pF and 30 s. B, Representative current responses by consecutive application of 10 μm capsaicin with 0.3 μm PMA before the second capsaicin application in dissociated DRG neurons from WT (left) or KI (right) mice. Scale bars, 50 pA/pF and 1 min. C, The extent of PMA-mediated recovery of desensitization. *p < 0.05 in Bonferroni's post hoc test after two-way ANOVA, Numbers within parentheses represent the numbers of cells analyzed.
TRPV1 S801A mutation only marginally affected inflammatory thermal hyperalgesia
Next we examined the somatosensory phenotypes of TRPV1 S801A KI mice. Paw withdrawal latency from a radiant heat source (Hargreaves' assay; Fig. 5A) or from a hot plate (53°C; Fig. 5B) was comparable between WT and KI mice. Latency to tail flick after immersion into hot water (50°C; Fig. 5C) was also not different. Paw licking duration after pinching of the plantar surface of hindpaw using an alligator clip was also comparable between WT and KI mice (Fig. 5D). Mechanical threshold of hindpaw determined by Von Frey assay (Fig. 5A) and nociceptive threshold on a variable-temperature hot plate (Fig. 5F) were also not significantly different between WT and KI mice. Together, these findings argue against a role for TRPV1 S801 phosphorylation in basal mechanical or heat pain sensitivity. To assay for inflammation-induced hyperalgesia, the hindpaw was injected with carrageenan, which induced mechanical and thermal hyperalgesia both in WT and KI mice (Fig. 5E,F). Yet, the extent of change was not different between genotypes. To examine PKC-related thermal hyperalgesia, we injected PMA (2 ng) into the hindpaw. To our surprise, PMA decreased paw withdrawal latency in the Hargreaves assay in both WT and KI mice, without a significant difference between genotypes (Fig. 5G). To test inflammation-mediated thermal hyperalgesia, we injected CFA into the hindpaw. Paw withdrawal latency was decreased at 1 d after intraplantar CFA in the ipsilateral hindpaws of both WT and KI mice. The extent of decrease was slightly less in KI mice compared with WT (interaction, F(2,74) = 3.16, p = 0.0483, two-way RM ANOVA), but there was no significant difference between groups in post hoc analysis (Fig. 5H). To further test whether pathological heat hyperalgesia is affected in KI mice, we used a mild thermal injury model, in which heat hyperalgesia is partially mediated by TRPV1 (Bölcskei et al., 2005). Mild thermal injury of the hindpaw resulted in decreased withdrawal latency (Fig. 5I) and heat pain threshold (Fig. 5J) in both WT and KI mice after 30 min, without significant differences between genotypes. These results suggest that ablation of S801 phosphorylation on TRPV1 marginally affects inflammatory heat hyperalgesia but is not sufficient to robustly attenuate it.
Figure 5.

The effects of TRPV1 S801A mutation on normal heat and mechanical sensitivity and hyperalgesia in hindpaw. A–D, Basal nocifensive behaviors of WT and KI mice in Hargreaves assay (A), hot plate assay (55°C; B), tail immersion assay (50°C; C), and paw-pinching assay using an alligator clip (D). Note that lamp intensity was higher in A than in G–I. Number of mice is in parentheses. p = 0.29 (A), 0.65 (B), 0.52 (C), and 0.63 (D) in unpaired Student's t test. E, F, Mechanical (E) or thermal (F) sensitivity before and 4 h after the intraplantar injection of Carrageenan (Cg). **p < 0.01; NS, not significant (p = 0.99 in E and F) in Bonferroni's post hoc analysis after two-way repeated-measures ANOVA. G, Thermal hyperalgesia evaluated using the Hargreaves' test after intraplantar injection of PMA (2 ng). Number of mice is in parentheses. **p < 0.01; NS, not significant (p = 0.71) in Bonferroni's post hoc analysis after two-way repeated-measures ANOVA. H, Thermal hyperalgesia evaluated using the Hargreaves' test after intraplantar injection of CFA (20 μl, 1:1 in PBS). *p < 0.05 in two-way repeated-measures ANOVA, NS, not significant (p = 0.17 in 1 d and 0.94 in 2 d); ****p < 0.0001 in Bonferroni's post hoc analysis. I, J, Hindpaw withdrawal latency in the Hargreaves' test (I) and nociceptive heat threshold on the variable hot plate (J) in a mild thermal injury model. **p < 0.01; ****p < 0.0001; NS, not significant (p = 0.99 in I and 0.96 in J) in Bonferroni's post hoc analysis after two-way repeated-measures ANOVA.
TRPV1 S801A mutation attenuates PMA-induced nociception and sensitization of capsaicin responses
We next assessed the impact of TRPV1 S801 mutation on acute responses to noxious chemical stimulation. The eye wiping behavioral response to capsaicin was not different between WT and KI mice (Fig. 6A). Intraplantar injection of capsaicin (1 μg) similarly evoked nocifensive behaviors that were not statistically different between genotypes (Fig. 6B). These results suggest that the basal sensitivity of KI mice to TRPV1 ligands is comparable to that of WT. PMA injection is known to evoke a nocifensive behavioral response in mice that depends on TRPV1 (Bölcskei et al., 2005; Ferreira et al., 2005). This response has been proposed to involve protein kinases, since it could be attenuated by a PKC inhibitor (Ferreira et al., 2005) or segregation of TRPV1 from the adaptor protein AKAP, which mediates TRPV1-protein kinase interactions (Zhang et al., 2008). When we administered PMA (60 ng) into the hindpaw, WT mice showed robust nocifensive behavior (Fig. 6C). Such behavior was completely absent in TRPV1 KO mice (data not shown), a finding consistent with a previous report (Bölcskei et al., 2005). In KI mice, PMA produced only a modest response that was significantly smaller than that in WT (Fig. 6C). Furthermore, when capsaicin was injected into a hindpaw that had been preinjected with PMA, nocifensive behaviors were significantly less in KI mice, compared with WT mice (Fig. 6D). Importantly, when the paw was pretreated with vehicle, capsaicin-induced nocifensive behaviors were not significantly different between WT and KI mice (Fig. 6D). There was a tendency toward greater capsaicin-induced nocifensive behaviors after PMA injection than that after vehicle injection, but this trend did not reach statistical significance. This is in contrast to the clear PMA-induced sensitization of capsaicin-evoked currents in Figure 3. Although the source of this discrepancy is not clear, it might be due to the complicated action of PMA in vivo. PMA injection evokes broad inflammatory signaling, which may lead to the eventual activation of TRPV1 and, thereby, induce long-lasting nociception (Bölcskei et al., 2005; Ferreira et al., 2005). However, subsequent TRPV1 desensitization might suppress nocifensive behaviors evoked by later capsaicin injection. It is thus possible that, under our experimental conditions, PMA-induced reversal of the desensitization was not sufficient to overcome the effects of desensitization produced by preceding PMA. Nonetheless, these results suggest that TRPV1 KI and WT mice show identical capsaicin responses under naive conditions, but that loss of TRPV1 S801 phosphorylation significantly attenuates both acute PMA-mediated nociception and the PKC-mediated sensitization of capsaicin evoked nociceptive responses in vivo.
Figure 6.
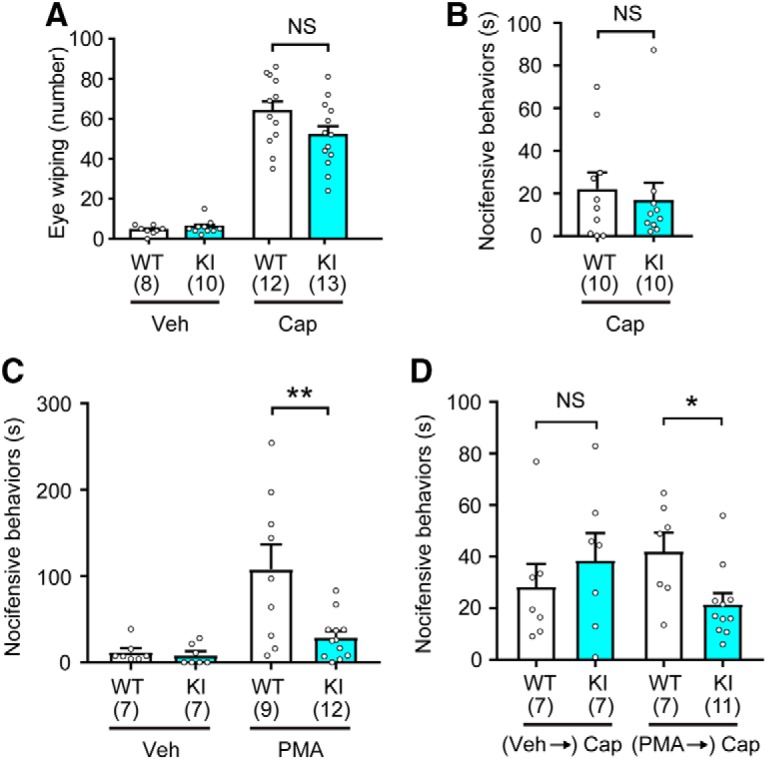
TRPV1 S801A mice show attenuation of acute PMA-induced nocifensive behavior and PMA-induced sensitization of capsaicin-mediated nocifensive behavior. A, Number of eye-wiping responses during the 5 min after ocular application of vehicle (20 μl of saline or PBS) or capsaicin (0.03%; 20 μl). Number of mice is in parentheses; NS, not significant (p = 0.09) in Student's t test. B, Duration of hindpaw nocifensive behaviors (licking and shaking) over 15 min after intraplantar injection of 1 μg of capsaicin; NS, not significant (p = 0.65) in Student's t test. C, Duration of hindpaw nocifensive behaviors (licking and shaking) over 30 min after intraplantar injection of vehicle (PBS) or PMA (60 ng). **p < 0.01 in Student's t test. D, Duration of hindpaw nocifensive behaviors (licking and shaking) over 5 min after intraplantar injection of capsaicin (1 μg). The hindpaws were pretreated with either vehicle or PMA (60 ng) 30 min before capsaicin injection. *p < 0.05; NS, not significant (p = 0.47) in Student's t test.
TRPV1 S801A attenuates spontaneous pain from inflamed masseter muscle
To examine the role of TRPV1 S801 phosphorylation in spontaneous pain, we used the MGS in a masseter inflammation model (Fig. 7A). Unilateral injection of CFA into the masseter muscle of WT mice significantly increased MGS scores on postinjection days 1 and 3 compared with vehicle (Fig. 7A), consistent with our previous results (Wang et al., 2017). The injection of vehicle into the masseter muscle of KI mice slightly increased MGS scores to a similar extent as in WT mice. Further, whereas the injection of CFA into the masseter muscle of KI mice produced significantly greater MGS scores than injection of the vehicle, the extent of increase by CFA was significantly less in KI mice than that in WT mice (interaction, F(9,36) = 24.4, p < 0.0001; genotype effect, F(3,12) = 46.1, p < 0.0001; time effect, F(3,36) = 235.3, p < 0.0001; two-way RM ANOVA), suggesting a partial role for TRPV1 S801 in this model of inflammation-induced spontaneous pain.
Figure 7.

TRPV1 S801A mice show attenuated spontaneous ongoing pain after masseter inflammation. A, MGS score before and after masseter injection of CFA or vehicle (Veh) in WT or TRPV1 S801A KI mice. ****p < 0.0001 in post hoc analysis after two-way repeated-measures ANOVA, Number of mice is in parentheses. B, Effects of masseter injection of AMG9810 (200 nmol) on MGS scores 1 d after masseter CFA injection in WT and KI mice. The effects of pharmacological manipulation were analyzed with two-way repeated-measures ANOVA, ****p < 0.0001 with Bonferroni's post hoc test. C, Changes in bite force before and after masseter injection of CFA in WT or TRPV1 S801A KI mice. Data were obtained from the same mice as in B.
To further estimate the proportion of TRPV1-mediated pain that could be attributed to S801 phosphorylation in this model of spontaneous pain, we tested the effects of AMG9810, a small molecule antagonist of TRPV1 function, in both WT and KI mice (Fig. 7B). One day after masseter injection of CFA, MGS scores were evaluated before and 1 h after the injection of AMG9810 into the masseter muscle. The effects of genotype and AMG9810 on MGS scores were significantly different between KI and WT mice (interaction, F(1,18) = 5.8, p < 0.027; genotype effect, F(1,18) = 43.88, p < 0.0001; time effect, F(1,18) = 76.66, p < 0.0001; two-way RM ANOVA). CFA once again produced robust increases in MGS scores after 1 d in both genotypes, albeit to a lesser extent in KI mice. Masseter injection of AMG9810 significantly attenuated MGS in both KI and WT mice, but the extent of inhibition was greater in WT than KI, which resulted in a similar level of residual MGS score in WT and KI mice. These results are not likely confounded by differential CFA evoked changes in TRPV1 expression between genotypes, since when we examined TRPV1 mRNA in TG 1 d after CFA injection, ipsilateral TRPV1 mRNA was not significantly different between KI and WT ganglia (TRPV1/GAPDH; 1.48 ± 0.01 in WT; 1.51 ± 0.01 in KI; n = 5 in WT and KI; t(8) = 2.03, p = 0.77, unpaired t test). Our previous study showed that reduction of bite force during masseter inflammation was modestly attenuated in TRPV1 KO mice (Wang et al., 2017). We therefore investigated whether CFA-induced changes in bite-evoked pain might be affected by the lack of phosphorylation at S801 (Fig. 7C). During masseter inflammation, the reduction of bite force was not significantly different between genotypes (WT, 30.6 ± 5.3%; KI, 34.5 ± 9.1%; n = 10 in WT and KI; t(18) = 0.37, p = 0.71, unpaired t test), arguing that this phenomenon does not depend on TRPV1 S801 phosphorylation.
Discussion
In this study, we examined the in vivo importance of phosphorylation of TRPV1 S801 by generating a KI mouse line with that residue mutated to alanine. Overall TRPV1 expression levels and the TRPV1 expression pattern in sensory ganglia were not affected in the KI mice. However, electrophysiological recordings in sensory neurons derived from these mice clearly showed attenuation, but not elimination, of PKC-mediated sensitization of capsaicin responses and impaired reversal of capsaicin-induced desensitization by PMA. These findings suggest that sensory neurons of KI mice with the S801A mutation are functionally impaired in PKC-mediated sensitization. However, we cannot exclude the possibility that the membrane expression of TRPV1 is altered in KI, which needs to be clarified in the future.
Our macroscopic recording of neurons from HET mice showed an intermediate phenotype between WT and KI, supporting biallelic trpv1 expression. However, our experiments do not allow us to presume any subunit association events underlying such a phenotype. For example, HET neurons might contain only two homomeric populations of TRPV1, with the S801A mutation or the WT. Alternatively, the assembly of heteromeric tetramers of S801A and WT in varying stoichiometries could exist in HET neurons. The latter possibility could be potentially important if there is a cooperativity between phosphorylation sites, either between S801 sites on different subunits or between S801 and other sites on the same or different subunits. Although we have no evidence for cooperativity of TRPV1 phosphorylation, the possibilities raised by the in vitro HET phenotype are intriguing.
Basal withdrawal responses to natural stimuli (heat and mechanical force) were indistinguishable between WT and KI mice, as was the nocifensive response to capsaicin, suggesting that alanine mutation of S801 has little or no effect on normal TRPV1 channel function. This also suggests that basal phosphorylation of TRPV1 S801 does not greatly influence behavioral responsiveness to these stimuli under physiological conditions. KI mice showed only a slight reduction of CFA-mediated thermal hyperalgesia in the hindpaw and no alteration in thermal hyperalgesia in a mild thermal injury model.
Considering the contribution of TRPV1 and the proposed role of PKC in thermal hyperalgesia and in phosphorylation of TRPV1 S801 (Huang et al., 2006; Mandadi et al., 2006), such a minor contribution of TRPV1 S801 to thermal hyperalgesia was rather surprising. However, S801 is only one of several PKC phosphorylation sites in TRPV1. Indeed, PMA-induced sensitization of TRPV1 responsiveness to heat is only partially impaired by alanine mutation of rat TRPV1 S800 in vitro (Wang et al., 2015). In contrast, PMA-induced sensitization of TRPV1 to heat was profoundly impaired when another PKC-mediated phosphorylation site, T705 (or T704 in rat), was mutated (Wang et al., 2015). In vitro findings have suggested that mutation of rat TRPV1 S800 to alanine alone suppressed the phosphorylation of a c-terminal fragment of TRPV1 to a similar level as quadruple mutations of rat TRPV1 S800/T704/S774/S820 (Bhave et al., 2003), raising the possibility that the rat TRPV1 S800A mutation also affects phosphorylation at T704. Although we could not directly test this possibility, any such influence of S800 on T704 phosphorylation might be weaker in intact channels, limiting the impact of individual S800 or S801 mutations on heat hypersensitivity in vitro or heat hyperalgesia in vivo.
Although direct in vivo evidence is lacking, previous studies support the idea that phosphorylation of mouse TRPV1 T705 plays a dominant role in inflammatory thermal hyperalgesia (Li et al., 2014; Wang et al., 2015). Ablation of phosphorylation at T705 alone or in combination with that at S801 might therefore be predicted to substantially impair thermal hyperalgesia. This approach, however, would be confounded by the fact that the T705A mutation alters basal TRPV1 heat and acid sensitivity (Li et al., 2014; Wang et al., 2015). Unlike TRPV1 S801 and T705, mouse TRPV1 S503 can be phosphorylated by both PKC and PKA (Bhave et al., 2002, 2003). However, alanine mutation of this residue does not impair PMA-induced sensitization to heat (Wang et al., 2015). Interestingly, alanine or aspartate mutation of rat TRPV1 S502 impairs forskolin-induced sensitization of TRPV1 to heat (Rathee et al., 2002). Thus during CFA-induced inflammation, PKA-mediated phosphorylation of TRPV1 S502, along with other PKA-dependent residues, possibly sensitizes TRPV1 to heat and limits loss of function in TRPV1 S801A KI mice. Moreover, PKC and PKA are not the only kinases capable of phosphorylating TRPV1. In fact, inflammation can potentially promote phosphorylation of at least eight residues in TRPV1 through a collection of protein kinases that also include CaMKII, cdk5, and src (Bhave et al., 2002; Mohapatra and Nau, 2003; Jung et al., 2004; Zhang et al., 2005; Pareek et al., 2007). Our experiments do not exclude roles for TRPV1 residues phosphorylated by these protein kinases during CFA-induced thermal hyperalgesia. Additional TRPV1 S801-independent contributors to heat sensation and thermal hyperalgesia likely include inflammation-induced increases in TRPV1 expression (Ji et al., 2002; Chung et al., 2016), receptor tyrosine kinase mediated TRPV1 membrane trafficking (Zhang et al., 2005), and non-TRPV1 heat gated ion channels (Vandewauw et al., 2018). All these mechanisms of thermal sensation and hyperalgesia could contribute to the lack of a dominant thermal phenotype in the KI mouse.
Although PMA is known to sensitize but not directly activate TRPV1 (Bhave et al., 2003), it produces nocifensive behavioral responses in vivo that are entirely or partially dependent on TRPV1 (Bölcskei et al., 2005; Ferreira et al., 2005). Such PMA-induced nocifensive behaviors are dependent on PKC, mast cell degranulation, multiple cytokines, and growth factors at the site of injection (Ferreira et al., 2005). Given these numerous contributors, it is striking that our data support a major role for TRPV1 S801 phosphorylation in acute PMA-induced hindpaw nocifensive behavior. In addition, when capsaicin-induced nocifensive behavior was compared after pretreatment with PMA, the KI mice showed clear attenuation of sensitized behavior. Since PMA itself causes TRPV1-dependent nocifensive behaviors, the responses to subsequent capsaicin application could reflect mixed consequences of sensitization and reversal of desensitization by the preceding PMA application. Interestingly, PMA-mediated thermal hyperalgesia was not affected in KI mice, again suggesting the involvement of S801-independent mechanisms (e.g., T705).
Ablation of TRPV1 S801 phosphorylation clearly attenuated ongoing pain caused by masseter inflammation without altering bite-evoked pain, which is consistent with differential contributions of TRPV1 to spontaneous vs bite-evoked pain (Wang et al., 2017). Combined pharmacological inhibition of total TRPV1 function with genetic ablation of S801 phosphorylation revealed phosphorylation at this residue accounts for approximately half of all TRPV1-mediated spontaneous pain during masseter inflammation. The magnitude of this effect is surprising given the multiplicity of TRPV1 sensitization mechanisms described above, and suggests a predominant contribution of phosphorylation at this single site in spontaneous pain in this model. How might the TRPV1 S801 mutation alter ongoing masseter pain after CFA injection? Inflammation upregulates TRPV1 expression in trigeminal ganglia (Chung et al., 2011b, 2016). However, the transcriptional level of TRPV1 in the TG of KI mice was not different from that of WT mice after CFA injection, arguing the KI phenotype is not likely due to differential TRPV1 upregulation. Tissue inflammation also produces putative endogenous agonists of TRPV1, such as hydroxyoctadecadienoic acid (HODE) (Patwardhan et al., 2010), and modulators such as glutamate (Chung et al., 2015). HODE depletion in inflamed masseter muscle has been reported to attenuate spontaneous pain, suggesting that constitutive activation of TRPV1 by endogenous agonists could lead to spontaneous pain (Wang et al., 2018). Ongoing pain might, therefore, be mediated by enhanced functionality of cell surface-expressed TRPV1 in this setting, coupled with increased availability of endogenous agonists. However, TRPV1 phosphorylation can also enhance its surface localization (Zhang et al., 2005; Camprubí-Robles et al., 2009). Our experiment did not dissociate these possible mechanisms of TRPV1 phosphorylation-dependent hyperalgesia.
The contribution of TRPV1 S801 phosphorylation to spontaneous pain that we observed after masseter inflammation may not be generalizable to other inflammatory pain models, since the overall contribution of TRPV1 to spontaneous pain after tissue injury or inflammation varies among models. For example, inhibition of TRPV1 did not affect guarding after skin incision (Wu et al., 2008) and was variably effective on conditioned place preference in knee joint arthritis (Okun et al., 2011). TRPV1 inhibition did attenuate mouth rubbing in oral mucositis and guarding in bone cancer (Ghilardi et al., 2005; Yamaguchi et al., 2016). The involvement of TRPV1 S801 phosphorylation in different pain models might thus be context dependent.
In conclusion, we used the CRISPR/Cas9 editing method to develop a KI mouse model with unmatched specificity for inhibition of TRPV1 phosphorylation, removing a single phosphorylation site by mutation S801A. The genetic ablation of S801 attenuates PKC-mediated sensitization of TRPV1, PMA-induced acute hindpaw nociception, and inflammation-mediated spontaneous pain from masseter muscle in vivo. These results suggest that small molecules selectively interfering with phosphorylation at TRPV1 S801 might provide an effective means of attenuating pain at sites of inflammation or injury without altering physiological pain sensation. Moreover, TRPV1 S801A mice should serve as a salient model for validating the specificity of such pharmacological modulators.
Footnotes
This work was supported by the National Institutes of Health (Grants DE023846 and R01 DE027731 to M.-K.C. and Grant F31 DE027270 to J.J.) and by the Neurosurgery Pain Research Institute at Johns Hopkins. We thank the Johns Hopkins Transgenic Mouse Core and Joel Pomerantz for help with the design and generation of the Crispr/CAS9 KI mice, Feng Zhang for kindly providing the Cas9 expression plasmid, Amelia Renehan and Izzy Kachik for help with video analysis, and Youping Zhang and Dennis Chang for help with behavioral and biochemical assays, respectively.
M.J.C. is an inventor on a patent related to TRPV1 that is licensed through UCSF and Merck and may be entitled to royalties on that patent. This potential conflict is being managed by the Johns Hopkins Office of Policy Coordination. The remaining authors declare no competing financial interests.
References
- Asgar J, Zhang Y, Saloman JL, Wang S, Chung MK, Ro JY (2015) The role of TRPA1 in muscle pain and mechanical hypersensitivity under inflammatory conditions in rats. Neuroscience 310:206–215. 10.1016/j.neuroscience.2015.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW 4th (2002) cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35:721–731. 10.1016/S0896-6273(02)00802-4 [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW 4th (2003) Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proc Natl Acad Sci U S A 100:12480–12485. 10.1073/pnas.2032100100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bölcskei K, Helyes Z, Szabó A, Sándor K, Elekes K, Németh J, Almási R, Pintér E, Petho G, Szolcsányi J (2005) Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain 117:368–376. 10.1016/j.pain.2005.06.024 [DOI] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD (1985) Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc Natl Acad Sci U S A 82:4438–4442. 10.1073/pnas.82.13.4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camprubí-Robles M, Planells-Cases R, Ferrer-Montiel A (2009) Differential contribution of SNARE-dependent exocytosis to inflammatory potentiation of TRPV1 in nociceptors. FASEB J 23:3722–3733. 10.1096/fj.09-134346 [DOI] [PubMed] [Google Scholar]
- Chung MK, Wang S (2011) Cold suppresses agonist-induced activation of TRPV1. J Dent Res 90:1098–1102. 10.1177/0022034511412074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Jung SJ, Oh SB (2011a) Role of TRP channels in pain sensation. Adv Exp Med Biol 704:615–636. 10.1007/978-94-007-0265-3_33 [DOI] [PubMed] [Google Scholar]
- Chung MK, Lee J, Duraes G, Ro JY (2011b) Lipopolysaccharide-induced pulpitis up-regulates TRPV1 in trigeminal ganglia. J Dent Res 90:1103–1107. 10.1177/0022034511413284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Jue SS, Dong X (2012) Projection of non-peptidergic afferents to mouse tooth pulp. J Dent Res 91:777–782. 10.1177/0022034512450298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Lee J, Joseph J, Saloman J, Ro JY (2015) Peripheral group I metabotropic glutamate receptor activation leads to muscle mechanical hyperalgesia through TRPV1 phosphorylation in the rat. J Pain 16:67–76. 10.1016/j.jpain.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Asgar J, Park J, Ro JY (2016) Transcriptome analysis of trigeminal ganglia following masseter muscle inflammation in rats. Mol Pain 12:1744806916668526. 10.1177/1744806916668526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira J, Trichês KM, Medeiros R, Calixto JB (2005) Mechanisms involved in the nociception produced by peripheral protein kinase c activation in mice. Pain 117:171–181. 10.1016/j.pain.2005.06.001 [DOI] [PubMed] [Google Scholar]
- Ghilardi JR, Röhrich H, Lindsay TH, Sevcik MA, Schwei MJ, Kubota K, Halvorson KG, Poblete J, Chaplan SR, Dubin AE, Carruthers NI, Swanson D, Kuskowski M, Flores CM, Julius D, Mantyh PW (2005) Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci 25:3126–3131. 10.1523/JNEUROSCI.3815-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Zou S, Mohammad Z, Wang S, Yang J, Li H, Dubner R, Wei F, Chung MK, Ro JY, Ren K (2019) Voluntary biting behavior as a functional measure of orofacial pain in mice. Physiol Behav 204:129–139. 10.1016/j.physbeh.2019.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang X, McNaughton PA (2006) Inflammatory pain: the cellular basis of heat hyperalgesia. Curr Neuropharmacol 4:197–206. 10.2174/157015906778019554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Lin SH, Malewicz NM, Zhang Y, Zhang Y, Goulding M, LaMotte RH, Ma Q (2019) Identifying the pathways required for coping behaviours associated with sustained pain. Nature 565:86–90. 10.1038/s41586-018-0793-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36:57–68. 10.1016/S0896-6273(02)00908-X [DOI] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U (2004) Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem 279:7048–7054. 10.1074/jbc.M311448200 [DOI] [PubMed] [Google Scholar]
- Levine JD, Alessandri-Haber N (2007) TRP channels: targets for the relief of pain. Biochim Biophys Acta 1772:989–1003. 10.1016/j.bbadis.2007.01.008 [DOI] [PubMed] [Google Scholar]
- Li L, Hasan R, Zhang X (2014) The basal thermal sensitivity of the TRPV1 ion channel is determined by PKCbetaII. J Neurosci 34:8246–8258. 10.1523/JNEUROSCI.0278-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandadi S, Numazaki M, Tominaga M, Bhat MB, Armati PJ, Roufogalis BD (2004) Activation of protein kinase C reverses capsaicin-induced calcium-dependent desensitization of TRPV1 ion channels. Cell Calcium 35:471–478. 10.1016/j.ceca.2003.11.003 [DOI] [PubMed] [Google Scholar]
- Mandadi S, Tominaga T, Numazaki M, Murayama N, Saito N, Armati PJ, Roufogalis BD, Tominaga M (2006) Increased sensitivity of desensitized TRPV1 by PMA occurs through PKCepsilon-mediated phosphorylation at S800. Pain 123:106–116. 10.1016/j.pain.2006.02.016 [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C (2003) Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J Biol Chem 278:50080–50090. 10.1074/jbc.M306619200 [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M (2002) Direct phosphorylation of capsaicin receptor VR1 by protein kinase cepsilon and identification of two target serine residues. J Biol Chem 277:13375–13378. 10.1074/jbc.C200104200 [DOI] [PubMed] [Google Scholar]
- Okun A, DeFelice M, Eyde N, Ren J, Mercado R, King T, Porreca F (2011) Transient inflammation-induced ongoing pain is driven by TRPV1 sensitive afferents. Mol Pain 7:4. 10.1186/1744-8069-7-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek TK, Keller J, Kesavapany S, Agarwal N, Kuner R, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB (2007) Cyclin-dependent kinase 5 modulates nociceptive signaling through direct phosphorylation of transient receptor potential vanilloid 1. Proc Natl Acad Sci U S A 104:660–665. 10.1073/pnas.0609916104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan AM, Akopian AN, Ruparel NB, Diogenes A, Weintraub ST, Uhlson C, Murphy RC, Hargreaves KM (2010) Heat generates oxidized linoleic acid metabolites that activate TRPV1 and produce pain in rodents. J Clin Invest 120:1617–1626. 10.1172/JCI41678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathee PK, Distler C, Obreja O, Neuhuber W, Wang GK, Wang SY, Nau C, Kress M (2002) PKA/AKAP/VR-1 module: a common link of gs-mediated signaling to thermal hyperalgesia. J Neurosci 22:4740–4745. 10.1523/JNEUROSCI.22-11-04740.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolcsányi J, Pintér E (2013) Transient receptor potential vanilloid 1 as a therapeutic target in analgesia. Expert Opin Ther Targets 17:641–657. 10.1517/14728222.2013.772580 [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21:531–543. 10.1016/S0896-6273(00)80564-4 [DOI] [PubMed] [Google Scholar]
- Vandewauw I, De Clercq K, Mulier M, Held K, Pinto S, Van Ranst N, Segal A, Voet T, Vennekens R, Zimmermann K, Vriens J, Voets T (2018) A TRP channel trio mediates acute noxious heat sensing. Nature 555:662–666. 10.1038/nature26137 [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA (2001) Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol 534:813–825. 10.1111/j.1469-7793.2001.00813.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lee J, Ro JY, Chung MK (2012) Warmth suppresses and desensitizes damage-sensing ion channel TRPA1. Mol Pain 8:22. 10.1186/1744-8069-8-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Joseph J, Ro JY, Chung MK (2015) Modality-specific mechanisms of protein kinase C-induced hypersensitivity of TRPV1: S800 is a polymodal sensitization site. Pain 156:931–941. 10.1097/j.pain.0000000000000134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Joseph J, Diatchenko L, Ro JY, Chung MK (2016) Agonist-dependence of functional properties for common nonsynonymous variants of human transient receptor potential vanilloid 1. Pain 157:1515–1524. 10.1097/j.pain.0000000000000556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lim J, Joseph J, Wang S, Wei F, Ro JY, Chung MK (2017) Spontaneous and bite-evoked muscle pain are mediated by a common nociceptive pathway with differential contribution by TRPV1. J Pain 18:1333–1345. 10.1016/j.jpain.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Brigoli B, Lim J, Karley A, Chung MK (2018) Roles of TRPV1 and TRPA1 in spontaneous pain from inflamed masseter muscle. Neuroscience 384:290–299. 10.1016/j.neuroscience.2018.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Gavva NR, Brennan TJ (2008) Effect of AMG0347, a transient receptor potential type V1 receptor antagonist, and morphine on pain behavior after plantar incision. Anesthesiology 108:1100–1108. 10.1097/ALN.0b013e31817302b3 [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Ono K, Hitomi S, Ito M, Nodai T, Goto T, Harano N, Watanabe S, Inoue H, Miyano K, Uezono Y, Matoba M, Inenaga K (2016) Distinct TRPV1- and TRPA1-based mechanisms underlying enhancement of oral ulcerative mucositis-induced pain by 5-fluorouracil. Pain 157:1004–1020. 10.1097/j.pain.0000000000000498 [DOI] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA (2005) NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J 24:4211–4223. 10.1038/sj.emboj.7600893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li L, McNaughton PA (2008) Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron 59:450–461. 10.1016/j.neuron.2008.05.015 [DOI] [PubMed] [Google Scholar]