

Abstract
Background
Refractory congestive heart failure (CHF) and associated diuretic resistance are not well defined.
Objectives
To characterize renal function, electrolyte concentrations, indices of diuretic efficacy, and renin‐angiotensin‐aldosterone system (RAAS) activation in dogs with naturally occurring heart disease (HD) in American College of Veterinary Internal Medicine stages B1, B2, C, and D and to determine their usefulness in defining HD stages.
Animals
Group 1:149 dogs with HD stages B1, B2, C, and D. Group 2:22 dogs with HD stages C and D.
Methods
Group 1: Renal parameters, serum and urine electrolyte and diuretic concentrations, and urine aldosterone concentrations were measured. Medication dosages and measured variables were compared among stages. Correlation of furosemide dosages to serum concentrations was explored. Group 2: Angiotensin‐converting enzyme activity and RAAS components were measured and compared among CHF stages.
Results
Serum chloride concentration was the best differentiator of HD stage. Furosemide PO dosages (≤6 mg/kg/day) were weakly correlated with serum furosemide concentrations, whereas higher dosages were not significantly correlated. Angiotensin‐converting enzyme inhibitor dosage and RAAS inhibition were greater in stage D, compared to stage C dogs.
Conclusions and Clinical Importance
Hypochloremia is a useful marker for stage D HD in dogs. Poor furosemide dosage correlation to serum concentration may indicate variable and poor absorption, especially at higher dosages, advanced disease, or both. A small number of stage D dogs met proposed criteria for diuretic resistance. Greater RAAS inhibition in stage D versus stage C indicates effectiveness of RAAS‐suppressive treatments in this group of dogs with refractory CHF.
Keywords: ACVIM, congestive heart failure, diuretic, refractory
Abbreviations
- ACE
angiotensin‐converting enzyme
- ACVIM
American College of Veterinary Internal Medicine
- BUN
blood urea nitrogen
- CHF
congestive heart failure
- FE Na
fractional excretion of sodium
- HD
heart disease
- LC‐MS/MS
liquid chromatography‐mass spectrometry/mass‐spectroscopy
- RAAS
renin‐angiotensin‐aldosterone system
- SDMA
symmetric dimethylarginine
- sFur:uFur
serum furosemide concentration to urine furosemide concentration
- sTor:uTor
serum torsemide concentration to urine torsemide concentration
- UAldo:C
urine aldosterone to creatinine
- uK
urine potassium concentration
- uNa
urine sodium concentration
- uNa:uFur
urine sodium concentration to urine furosemide concentration
- uNa:uK
urine sodium concentration to urine potassium concentration
- uNa:uTor
urine sodium concentration to urine torsemide concentration
- USG
urine specific gravity
1. INTRODUCTION
Congestive heart failure (CHF) most often occurs secondary to severe degenerative mitral valve disease or dilated cardiomyopathy in dogs and is a common clinical problem in veterinary medicine.1, 2 Medical management typically includes diuretics, the inodilator, pimobendan, and inhibitors of the renin‐angiotensin‐aldosterone system (RAAS), depending on disease stage.3 Diuretics are considered essential for removal of CHF fluid, but publications on the relative efficacy of diuretics in clinical canine patients are sparse. Typically, dogs with CHF succumb to their disease because of continued CHF signs, despite escalating dosages of medications, or because of medication adverse effects. Diuretic resistance is thought to play a role in heart failure progression in humans, but little information is available on this phenomenon in dogs. Knowledge of the existence, importance, and underlying causes for diuretic unresponsiveness could guide treatment modifications specifically directed at restoration of diuresis and improvement of quality and duration of life.
Diuretic resistance is identified in up to 17% of people with CHF and also is suspected to occur in dogs.4, 5 Although reports of diuretic resistance are lacking in veterinary medicine, the American College of Veterinary Internal Medicine (ACVIM) considers a role for diuretic failure in the description of refractory heart disease (HD), termed Stage D.3 Diuretic resistance is negatively associated with prognosis in people with CHF; hence, early recognition is important.6, 7 Diuretic dosage does not always correlate positively with diuretic response, which may be suboptimal in naturally occurring CHF.8, 9 Potential causes of suboptimal diuretic responsiveness include inadequate gastrointestinal drug absorption, decreased renal tubular transport, loop of Henle cell hypertrophy, and RAAS activation.6 In humans, diuretic resistance is defined as persistent CHF despite an appropriate furosemide dosage; fractional excretion of sodium (FE Na) <0.2%; failure of adequate natriuresis after furosemide administration; or urine sodium:urine potassium ratio (uNa:uK) <1.0.5, 10 Some studies in people with CHF also have associated poor diuretic response with deceased spot urine sodium concentration (uNa), hypochloremia, and low urine sodium:urine furosemide ratio (uNa:uFur), the latter indicating pharmacodynamic unresponsiveness to the diuretic.7, 10, 11, 12, 13 Some of these variables have been correlated to urine volume in dogs receiving IV furosemide, but, to our knowledge, they have not been evaluated in dogs receiving PO diuretics.14 Identifying the mechanism(s) for loss of diuretic responsiveness in dogs could direct preventative strategies or treatments to restore effective diuresis in dogs that suffer from uncontrolled CHF.
Our hypothesis was that dogs with naturally occurring HD of different stages (ACVIM Stages B1, B2, C, and D) could be differentiated using specific indicators of renal function, electrolyte homeostasis, RAAS activation, and indices of diuretic efficacy in dogs. One objective was to prospectively measure these variables in dogs with different and progressive HD stages. A second objective was to determine if any of these variables are associated with controlled versus refractory CHF to provide mechanistic insight into potential causes of diuretic resistance in dogs. Such insights may provide stage‐specific therapeutic interventions to restore diuresis and improve quality of life for dogs with refractory CHF.
2. MATERIALS AND METHODS
This study was approved by the Institutional Animal Care and Use Committee (#14‐142‐0) at North Carolina State University Veterinary Hospital, and owner consent was obtained. Two groups of dogs were studied. Group 1 was utilized to evaluate renal variables, electrolytes, and indicators of diuretic efficacy in dogs with different HD stages (ACVIM stages B1, B2, C, and D—see definition below).3 Group 2 was utilized to evaluate serum RAAS components in dogs with CHF (stages C and D). Dogs with naturally occurring HD as a result of degenerative mitral valve disease or dilated cardiomyopathy were included in both groups, as were dogs with right‐sided, left‐sided, or biventricular CHF. Exclusion criteria were the presence of congenital HD, a requirement for hospitalization with parenteral diuretic therapy, and inability to obtain paired urine and serum samples for group 1 dogs. Enrollment for dogs requiring hospitalization was delayed for at least 1 week after discharge.
2.1. Evaluation of renal variables, electrolytes, and diuretic efficacy in dogs with HD stages B1, B2, C, and D (group 1)
2.1.1. Data collection (group 1)
Clinical variables recorded were signalment, cardiac disease diagnosis, body condition score, body weight, ACVIM HD stage, medications, and dosages. Medication dosages were indexed for body weight (kilograms) and expressed as a daily dosage. Torsemide dose was reported as actual dosage (mg/kg/day) as well as proposed “furosemide‐equivalent dosage” (mg/kg/day × 10).8, 15 Systolic blood pressure was recorded as the mean of 5 measurements, using a Doppler unit (Parks Medical Electronic, Las Vegas, Nevada) on a hind leg with the dog in lateral recumbency using a cuff size of 30%‐40% of the leg diameter.
2.1.2. Heart disease staging (group 1)
Determination of HD stage was made using clinical information at the time of the visit (eg, thoracic radiographs, echocardiogram, assessment of medications, and clinical status) in conjunction with the ACVIM HD classification.3 This study included dogs from preclinical stages B1, B2, and CHF stages C and D. Stage B1 dogs were dogs with HD but without cardiac enlargement or clinical signs. Stage B2 dogs were dogs with HD and secondary cardiac enlargement but without clinical signs. Stage C dogs were dogs with HD and CHF controlled using standard medications and dosages according to published guidelines (furosemide <12 mg/kg/day, pimobendan 0.5‐0.6 mg/kg/day, and an angiotensin‐converting enzyme (ACE) inhibitor 0.5‐1 mg/kg/day).3 For the purpose of this study, we defined stage D using ACVIM HD Staging Guidelines3 and a previous publication.16 We defined refractory CHF, as CHF requiring escalated drug dosage(s) to address (successfully or not) uncontrolled CHF. The dosages used as cutoffs for refractory CHF in our practice were (1) PO furosemide >12 mg/kg/day or (2) the transition to PO torsemide to address uncontrolled CHF, or (3) PO pimobendan >0.9 mg/kg/day, in conjunction with at least 4 mg/kg/day furosemide. Higher dosage of pimobendan to address mainstem bronchus compression was not allowed as the sole diagnostic criterion for stage D. Congestive heart failure was defined as the presence of radiographically documented cardiogenic pulmonary edema, pleural effusion, or ascites.
2.1.3. Procedures and sample tests (group 1)
A 3 mL blood sample was obtained by peripheral venipuncture at the time of the appointment. After centrifugation, serum was removed and aliquoted into 3 separate tubes. One aliquot was immediately analyzed for blood urea nitrogen (BUN), creatinine, and electrolyte (sodium, potassium, chloride) concentrations using an automated analyzer (Roche Cobas C501, Indianapolis, Indiana) at the clinical pathology laboratory of North Carolina State University Veterinary Hospital. A second aliquot was sent to an outside laboratory for symmetric dimethylarginine (SDMA) analysis (IDEXX, Westbrook, Maine), and a third aliquot was stored at −80°C until analysis of either furosemide or torsemide concentrations at the North Carolina State University Veterinary Pharmacology Laboratory, as previously described using a validated reverse‐phase high‐pressure liquid chromatography and fluorescence detection method.17, 18 A voided urine sample (5‐10 mL) was obtained at the time of the appointment, and urine specific gravity (USG) was measured. The urine was centrifuged and the supernatant divided into 3 aliquots. One aliquot was immediately analyzed for creatinine and electrolytes (sodium, potassium, chloride). The other aliquots were stored at −80°C until analysis. Urine furosemide or torsemide was measured as described above for serum using the second aliquot, and the third aliquot was evaluated for urine aldosterone and creatinine concentrations at a veterinary diagnostic laboratory (Diagnostic Center for Population and Animal Health, Michigan State University, Lansing, Maine). Urine aldosterone was measured with a commercially available radioimmunoassay kit (Siemens Medical Diagnostic Solutions, Los Angeles, California) following the manufacturer's instructions as previously described, and urine creatinine was measured using a standard colorimetric assay.19
2.1.4. Calculations and assessments (group 1)
Renal function was assessed using serum creatinine, BUN and SDMA concentrations. The secretion of diuretic medication into the renal tubule was assessed using the ratio of serum to urinary diuretic concentrations (sFur:uFur for furosemide and sTor:uTor for torsemide).20, 21 The FE Na, the percentage of filtered sodium excreted in the urine, was calculated using the formula: FE Na = 100 x ([uNa × serum creatinine]/[serum sodium × urine creatinine]).7 The uNa concentration was converted from mmol/L to mg/dL using the molecular weight of sodium (23 g/mol) to allow unitless indexing to urinary diuretic concentration (furosemide or torsemide, uNa:uFur, or uNa:uTor). This index was used to assess pharmacodynamic effects of the diuretics.7, 22
Urine aldosterone amount (μg) was indexed to urine creatinine amount (grams; urine aldosterone to creatinine ratio [UAldo:C]) as an indicator of RAAS activity.19 The ratio of uNa:uK was calculated as a proposed marker of diuretic resistance and an indicator of aldosterone's effect on electrolyte excretion.23, 24
2.2. Evaluation of serum RAAS components in CHF (HD stages C and D) (group 2)
2.2.1. Procedures
Serum was collected as described above and stored at −80°C until analysis. Clinical patient information and HD staging were performed as described above.
2.2.2. Equilibrium analysis of RAAS components (group 2)
The equilibrium concentrations of 6 different RAAS angiotensin peptide metabolites and aldosterone in canine serum samples were quantified by liquid chromatography‐mass spectrometry/mass‐spectroscopy (LC‐MS/MS), performed at a service provider laboratory (Attoquant Diagnostics, Vienna, Austria), using previously validated and described methods.25, 26, 27 Briefly, serum conditioning for equilibrium analysis was performed at 37°C followed by stabilization by the addition of an enzyme inhibitor cocktail (Waters, Milford, Massachusetts), as described previously.25, 26, 27 Previous results have shown similar qualitative outcomes when comparing the quantification of circulating (stabilized immediately at blood drawing) and equilibrium angiotensin peptide concentrations.25, 26, 27 Stabilized equilibrated serum samples were further spiked with stable isotope labeled internal standards for each angiotensin metabolite as well as with the deuterated internal standard for aldosterone (aldosterone D4) at a concentration of 200 pg/mL. The samples then underwent C‐18‐based solid‐phase extraction and were subjected to LC‐MS/MS analysis using a reversed‐phase analytical column operating in line with a Xevo TQ‐S triple quadruple mass spectrometer. Internal standards were used to correct for peptide and steroid recovery of the sample preparation procedure for each analyte in each individual sample. Analyte levels were reported in pM and are calculated considering the corresponding response factors determined in appropriate calibration curves in original sample matrix, on condition that integrated signals exceeded a signal‐to‐noise ratio of 10. The lower limit of quantification was 3.0 pM for angiotensin I, 2.0 pM for angiotensin II, 3.0 pM for angiotensin 1‐7, 2.0 pM for angiotensin 1‐5, 2.5 pM for angiotensin III, 2.0 pM for angiotensin IV, and 20.0 pM for aldosterone.
The activity of circulating ACE was determined by measuring the angiotensin II formation rate in samples diluted with phosphate‐buffered saline, after spiking samples with angiotensin I and ex vivo incubation at 37°C in the presence and absence of the ACE inhibitor lisinopril (Sigma‐Aldrich, Munich, Germany) at 10 μmoL/L. Aminopeptidase inhibitor and Z‐Pro‐prolinal (Sigma‐Aldrich, Munich, Germany; both 10 μmoL/L) were added to all samples for substrate (angiotensin I) and product (angiotensin II) stabilization. Angiotensin quantification (angiotensin I and angiotensin II) was performed as described earlier, and specific ACE activity was calculated by determining the inhibitor sensitive fraction (solvent minus inhibitor) of angiotensin II formation ([pg/mL]/h).
2.2.3. Calculations (group 2)
The ratio of angiotensin I‐7 to angiotensin II was calculated, because a low ratio is an indicator of RAAS activation and worsening heart failure signs, as reported in people.25 Angiotensin I and angiotensin II were summed as a surrogate for plasma renin concentration.26
2.3. Statistical analysis
Statistical analysis was performed using commercially available software (GraphPad Prism 8, San Diego, California). Data values that were below the lower limit of assay quantification were reported as half the lower limit for statistical analysis.28, 29 Data sets were divided by American College of Veterinary Internal Medicine stage and tested for normality using the D'Agostino and Pearson tests. Parametric data were reported as means and standard deviation, whereas nonparametric data were reported as medians and interquartile range. Parametric data among all ACVIM stages were analyzed using 1‐way analysis of variance testing done using Holm‐Sidak's multiple comparison test if indicated and nonparametric data were analyzed using Kruskal Wallis test with Dunn's multiple comparison test if indicated. Adjusted P values are reported for significant findings. Two‐way, unpaired t‐tests were used to compare parametric data specific to diuresis for ACVIM stages C and D and Mann‐Whitney test was used if data was nonparametric. Torsemide variables were not compared statistically among groups or correlated to PO dose because of the small sample sizes. Correlations between PO furosemide daily doses and serum furosemide concentrations were explored using Spearman's correlation because data was nonnormally distributed. Receiver operating characteristic curves were developed to determine sensitivity and specificity of variables that differentiated stage C from D dogs. Data for group 1 dogs were analyzed with and without dogs with dilated cardiomyopathy; results were not different and so all dogs are reported together. Significance was set at P < .05.
3. RESULTS
3.1. Evaluation of renal variables, electrolytes, and diuretic efficacy in dogs with HD stages B1, B2, C, and D (group 1)
The first group consisted of 149 dogs of which 9 were stage B1, 62 were stage B2, 62 were stage C, and 16 were stage D (Table 1). Stage D dogs represented 21% of CHF dogs in this study. The majority of dogs with CHF received furosemide (n = 71). The minority received torsemide instead of furosemide (n = 7), with 6 of these classified as stage D. Most CHF dogs received an ACE inhibitor (51 of 62 stage C dogs and 15 of 16 stage D dogs) and spironolactone (26 of 62 stage C dogs and 16 of 16 stage D dogs). Enalapril was administered to 90% of dogs that received an ACE inhibitor, whereas benazepril was administered to 10%. All dogs with CHF received pimobendan. No stage C dogs and 2 of 16 stage D dogs received potassium supplementation. A minority of dogs received nonsteroidal anti‐inflammatory medications (0 stage B1, 4 stage B2, 1 stage C, 0 stage D) or prednisone (0 stage B1, 2 stage B2, 2 stage C, 0 stage D).
Table 1.
Clinical data are presented for group 1 dogs in each ACVIM Heart disease stage
| Variable | ACVIM heart disease stage | Adjusted P value for post hoc significance if indicated | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage B1 (n = 9) | Stage B2 (n = 62) | Stage C (n = 62) | Stage D (n = 16) | P value | B1 versus B2 | B1 versus C | B1 versus D | B2 versus C | B2 versus D | C versus D | |
| Age (years) | 11.1 (9.1‐12.1) | 10.1 (8.6‐11.8) | 10.3 (8.0‐12.2) | 10.5 (9.8‐12.0) | .7 | ||||||
| Weight (kg) | 10.5 (7.6‐14.9) | 9.9 (7.6‐13.3) | 8.1 (4.8‐18.8) | 6.9 (6.2‐10.9) | .2 | ||||||
| Underlying disease | 9 DMVD 0 DCM | 57 DMVD 5 DCM | 49 DMVD 13 DCM | 16 DMVD 0 DCM | |||||||
| Systolic Blood Pressure (mmHg) | 140 (125‐170) | 140 (125‐145) | 120 (110‐140) | 110 (100‐133) | .002 | .17 | .03 | .005 | .13 | .03 | .17 |
| Furosemide dosage (mg/kg/day) | n = 61; 3.5 (2.9‐4.5) | n = 10; 8.1 (6.0‐8.9) | <.0001 | ||||||||
| Torsemide dosage (mg/kg/day) | n = 1; 0.68 | n = 6; 1.3 (1.2‐1.5) | ND | ||||||||
| ACE inhibitor dosage (mg/kg/day) | n = 39; 0.83 (0.72‐1.0) | n = 51; 0.85 (0.71‐1.0) | n = 15; 0.98 (0.83‐1.2) | .02 | .8 | .03 | .02 | ||||
| Pimobendan dosage (mg/kg/day) | n = 25; 0.57 (0.50‐0.74) | n = 62; 0.73 (0.6‐1.0) | n = 16; 1.9 (1.4‐3.1) | <.0001 | .04 | <.0001 | <.0001 | ||||
| Spironolactone dosage (mg/kg/day) | n = 26; 1.8 (1.6‐2.4) | n = 16; 2.7 (2.1‐3.9) | <.0001 | ||||||||
| Furosemide equivalent dosage (mg/kg/day) | n = 62; 3.53 (2.93‐4.50) | n = 16; 8.90 (7.18‐12.83) | <.0001 | ||||||||
Notes: Overall P value is shown for comparison between stage C and D and for between all groups. Adjusted P value is shown when multiple comparison testing was performed due to overall significance. Data are reported as medians and interquartile ranges.
Abbreviations: ACVIM, American College of Veterinary Internal Medicine, DMVD, degenerative mitral valve disease; DCM, dilated cardiomyopathy; ND, not done.
Median diuretic, ACE inhibitor, pimobendan, and spironolactone dosages were significantly higher in stage D dogs compared to stage C dogs (Table 1). Median pimobendan dosage also was significantly higher in stage C dogs compared to stage B2 dogs (P = .04). No differences in age (P = .7), weight (P = .2) or sex distribution (P = 0.7) were found among HD stages. The systolic blood pressure was significantly lower in CHF stages (C and D) compared to preclinical stages (B1 and B2) but was not different between stages C and D or between stages B1 and B2 (Table 1).
Biochemical data for the first group of dogs (Table 2) indicated that renal variables (BUN, creatinine, and SDMA) were significantly higher and USG significantly lower in dogs with CHF (stages C and D) compared to dogs with preclinical disease (stages B1 and B2), but these variables did not differentiate stage C from D or stage B1 from B2. Serum electrolyte concentrations (sodium, chloride, and potassium) were significantly lower for stage D dogs as compared to stages B2 and C dogs. In addition, serum chloride concentrations were significantly different among stages B1, C, and D, and serum potassium concentrations were significantly different between stages B1 and D (Table 2). Areas under curves and receiver operating characteristic data for serum electrolyte concentrations are shown in Table 3. Serum chloride concentration showed the best receiver operating characteristic curve in this population (Figure 1); a serum chloride concentration <103.5 mmol/L predicted stage D with a sensitivity of 81% and specificity of 76%. All stage D dogs were hypochloremic.
Table 2.
Biochemical data are presented for group 1 dogs in each ACVIM heart disease stage
| Variable | Reference range | ACVIM staging by current consensus | Adjusted P value for post hoc significance if indicated | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage B1 (n = 9) | Stage B2 (n = 62) | Stage C (n = 62) | Stage D (n = 16) | P value | B1 versus B2 | B1 versus C | B1 versus D | B2 versus C | B2 versus D | C versus D | ||
| BUN (mg/dL) | 6‐26 | 15.0 (11.0‐21.5) | 17.5 (14.0‐24) | 27.0 (21‐37.3) | 31.0 (20.5‐45.5) | <.0001 | 1.0 | .001 | .002 | <.0001 | .0003 | 1.0 |
| Creatinine (mg/dL) | 0.7‐1.5 | 0.70 (0.45‐1.00) | 0.75 (0.60‐0.93) | 1.10 (0.80‐1.40) | 1.10 (1.00‐1.20) | <.0001 | 1.0 | .02 | .03 | <.0001 | .001 | 1.0 |
| SDMA (μg/dL) | 0‐14 | 10.0 (7.5‐10.0) | 10.0 (8.0‐13.0) | 12.0 (10.0‐16.0) | 13.5 (11.3‐17.0) | <.0001 | 1.0 | .02 | .003 | .003 | .002 | 1.0 |
| Urine specific gravity | 1.015‐1.045 | 1.026 (1.019‐1.040) | 1.032 (1.019‐1.044) | 1.011 (1.009‐1.016 | 1.010 (1.007‐1.013 | <.0001 | 1.0 | .007 | .002 | <.0001 | <.0001 | 1.0 |
| Serum sodium (mmol/L) | 140‐156 | 147.0 (145.5‐148.5) | 147.5 (147.0‐149.0) | 148.0 (146.0‐150.0) | 146.5 (144.3‐147.0) | .008 | 1.0 | 1.0 | 1.0 | 1.0 | .03 | .006 |
| Serum chloride (mmol/L) | 108‐122 | 109.0 (107.0‐111.5) | 109.0 (107.0‐111.0) | 106.0 (103.8‐108.0) | 102.0 (99.3‐103.0) | <.0001 | 1.0 | .03 | <.0001 | <.0001 | <.0001 | .007 |
| Serum potassium (mmol/L) | 4‐5.3 | 4.70 (4.40‐5.15) | 4.70 (4.400‐5.03 | 4.50 (4.20‐4.70) | 4.20 (3.70‐4.53) | <.0001 | .71 | .25 | .003 | .08 | <.0001 | .005 |
Notes: Overall P value is shown as well as the adjusted P value when multiple comparison testing was performed due to overall significance. Data are reported as medians and interquartile ranges.
Abbreviations: ACVIM, American College of Veterinary Internal Medicine, BUN, blood urea nitrogen.
Table 3.
Area under the receiver operating characteristic (ROC) curve and 95% confidence intervals (CI) are shown for serum electrolytes for group 1 dogs
| Variable | Area under the ROC curve | 95% CI | P value | Cutoff value (mmol/L) | Sensitivity (%) | Specificity (%) |
|---|---|---|---|---|---|---|
| Serum sodium (mmol/L) | 0.75 | 0.63‐0.86 | .003 | <147.5 | 88 | 61 |
| Serum chloride (mmol/L) | 0.81 | 0.71‐0.91 | .0001 | <103.5 | 81 | 76 |
| Serum potassium (mmol/L) | 0.71 | 0.55‐0.87 | .01 | <4.3 | 75 | 68 |
Figure 1.
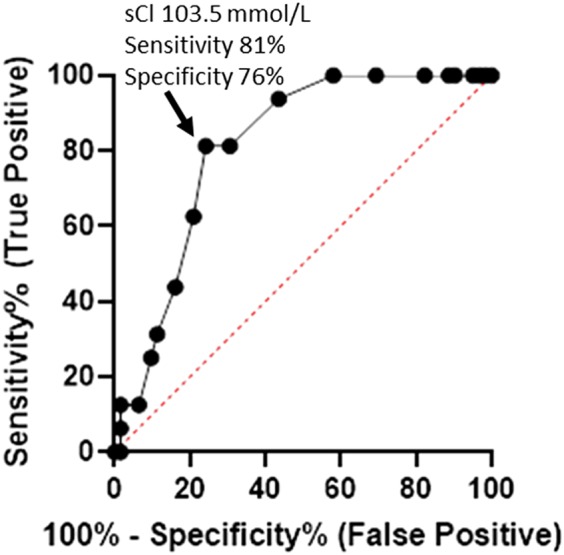
Receiver operating characteristic curve for serum chloride for 150 dogs (Group 1). A serum chloride concentration less than 103.5 mmol/L predicted stage D with a sensitivity of 81% and specificity of 75% (likelihood ratio 3.4). The line of identity is shown as the dotted line bisecting the graph. sCl, serum chloride concentration
Biochemical data reflective of response to diuretic medications were different between preclinical stages and CHF stages but did not differentiate stages C and D (Table 4). Fractional excretion of sodium and chloride were significantly higher and FE of potassium significantly lower in dogs with CHF (stages C and D, receiving cardiac treatments, including diuretics), compared to those with preclinical disease (stages B1 and B2, not receiving diuretics). Only 1 of 16 stage D dogs had an FE Na <0.2% and only 4 of 16 stage D dogs had a uNa:uK ratio <1.0, both of which are proposed cutoff values associated with diuretic resistance in human beings.10, 24 One of these dogs met both criteria. In fact, stage D dogs as a group had numerically (but not statistically) higher uNa:uK, compared to stage C dogs, because of less potassium excretion (Table 4). Stage D dogs that met diuretic resistance criteria used in humans, using FE Na or uNa:uK, had significantly lower serum sodium concentrations (142.3 ± 3.8 mmol/L) and serum chloride concentrations (98.0 ± 3.2 mmol/L) compared to stage D dogs that did not meet these criteria (sodium, 146.5 ± 1.6 mmol/L, P = .08; chloride, 102.5 ± 2.2 mmol/L, P = .007). Serum potassium concentrations were not significantly different between dogs that met or did not meet diuretic resistance criteria (P = .7).
Table 4.
Biochemical and urine data associated with diuretic response are presented for group 1 dogs in each ACVIM heart disease stage
| Variable | ACVIM heart disease stage | Adjusted P value for post hoc significance if indicated | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| B1 (n = 9) | B2 (n = 62) | C (n = 62) | D (n = 16) | P value | B1 versus B2 | B1 versus C | B1 versus D | B2 versus C | B2 versus D | C versus D | |
| Fractional excretion sodium (%) | 0.19 (0.12‐0.86) | 0.45 (0.23‐0.66) | 0.93 (0.42‐1.55) | 0.91 (0.69‐1.53) | <.0001 | 1.0 | .03 | .04 | .0004 | .02 | 1.0 |
| Fractional excretion chloride (%) | 0.38 (0.21‐1.15) | 0.55 (0.30‐0.89) | 22.5 (17.36‐32.52) | 18.18 (13.86‐29.67 | <.0001 | 1.0 | <.0001 | .0002 | <.0001 | <.0001 | 1.0 |
| Fractional excretion potassium (%) | 8.83 (6.21‐19.59) | 14.74 (11.37‐19.59) | 1.20 (0.39‐2.34) | 0.92 (0.53‐1.58) | <.0001 | 1.0 | .0003 | .001 | <.0001 | <.0001 | 1.0 |
| uNa (mmol/L) | 64.(36‐143) | 100 (58‐167) | 46 (30‐73) | 42 (17‐63) | <.0001 | 1.0 | .8 | .4 | <.0001 | .0001 | 1.0 |
| uNa:uFur (μg/mL):(μg/mL) | n = 56; 95.12 (47.80‐168.40) | n = 10; 65.33 (30.03‐100.80) | .13 | ||||||||
| uNa:uTor (μg/mL):(μg/mL) | n = 1; 465.9 | n = 6; 85.60 (46.17‐267.00) | ND | ||||||||
| sFur:uFur (μg/mL):(μg/mL) | n = 56; 0.018 (0.012‐0.031) | n = 9; 0.015 (0.010‐0.024) | .4 | ||||||||
| sTor:uTor (μg/mL):(μg/mL) | n = 1; 1.67 | n = 6; 1.47 (0.78‐1.92) | ND | ||||||||
| Urine Aldo:Creatinine | 1.00 (0.63‐1.45) | 1.13 (0.77‐1.92) | 1.55 (0.96‐3.85) | 3.06 (1.29‐5.95) | .005 | 1.0 | .4 | .03 | .3 | .02 | .4 |
| Serum furosemide (μg/mL) | n = 59; 0.18 (0.10‐0.28) | N = 9; 0.22 (0.15‐0.46) | .2 | ||||||||
| Serum torsemide (μg/mL) | n = 1; 6.5 | n = 6; 10.85 (8.22‐14.93) | |||||||||
| uNa:uK (mmol/L):(mmol/L) | 0.75 (0.43‐1.20) | 0.92 (0.55‐1.49) | 1.15 (0.60‐1.96) | 1.65 (1.03‐2.05) | .07 | ||||||
Notes: Overall P value is shown for comparison between Stage C and D and for comparison between all groups. Adjusted P value is shown when multiple comparison testing was performed due to overall significance. Data are reported as medians and interquartile ranges.
Abbreviations: ACVIM, American College of Veterinary Internal Medicine; ND, not done; sFur:uFur, serum furosemide concentration to urine furosemide concentration; sTor:uTor, serum torsemide concentration to urine torsemide concentration; uNa, urine sodium concentration; uNa:uFur, urine sodium concentration to urine furosemide concentration; uNa:uK, urine sodium concentration to urine potassium concentration; uNa:uTor, urine sodium concentration to urine torsemide concentration.
The activity of RAAS was evaluated using uNa:uK (activation indicated by lower value) and UAldo:C (activation indicated by higher value).23, 24 The uNa:uK was higher numerically but not statistically in stage D, as compared to stage C dogs (P = .07 among all stages; Table 4). Although UAldo:C was higher in dogs with stage D CHF compared to dogs with stages B1 (P = .03) and B2 (P = .02), this ratio did not differentiate stage C from stage D statistically (P = .4; Table 4).
Serum furosemide concentrations were not significantly different between stages C and D dogs despite the higher PO furosemide dosages for stage D dogs (Tables 1 and 4). Although numerically lower, uNa:uFur (P = .13) and sFur:uFur (P = .4) ratios were not statistically different for stage D dogs compared to stage C dogs (Table 4). The median PO furosemide dosage for stage D dogs was approximately 6‐fold higher than the median PO torsemide dosage, yet the median serum torsemide concentration was approximately 50‐fold higher than the median serum furosemide concentration. The sTor:uTor ratio for stage D dogs was approximately 100‐fold higher than the sFur:uFur ratio, indicating a lower secretory rate into the renal tubule, yet the urine diuretic concentrations were similar.
A significant, but weak, correlation was found between serum furosemide concentrations and daily PO furosemide dosages (P = .003, r = .359; Table 5). This relationship was further explored by examining dosages ≤6 mg/kg/day versus dosages >6 mg/kg/day, because there was minimal overlap of stage C and D dogs at this cutoff. The correlation between dosage and serum concentration remained significant and weak with furosemide dosages ≤6 mg/kg/day (P = .02, r = .320) but was lost with furosemide dosages >6 mg/kg/day (P = .6). Correlations were not explored for torsemide because of the low numbers of dogs receiving this diuretic.
Table 5.
Correlation of PO administered furosemide daily doses with serum diuretic concentrations for group 1 dogs
| Correlation with serum furosemide concentrations | P value | r value |
|---|---|---|
| All furosemide dosages (n = 68) | .003 | .359 |
| Furosemide dosages ≤ 6 mg/kg/day (n = 57) | .02 | .32 |
| Furosemide dosages > 6 mg/kg/day (n = 11) | .6 | −.156 |
3.2. Evaluation of RAAS metabolites in CHF HD stages C and D (group 2)
Group 2 consisted of 22 dogs of which 17 were stage C and 5 were stage D (Table 6). The majority of dogs with CHF received furosemide (n = 20) with the minority receiving torsemide (n = 2). All dogs received pimobendan and an ACE inhibitor, and most dogs received spironolactone (15 of 17 stage C and 5 of 5 stage D). No dogs received potassium supplementation. Median furosemide, pimobendan, and ACE inhibitor dosages were higher in stage D dogs compared to stage C dogs, but median spironolactone dosage was not different between stages (Table 6). No difference in age (P = .2), weight (P = .3) or sex distribution (P = .9) was found among CHF stages.
Table 6.
Clinical and renin‐angiotensin‐aldosterone data are presented for group 2 dogs in ACVIM heart disease stages C and D
| Variable | ACVIM Stage C (n = 17) median (IQR) | ACVIM Stage D (n = 5) median (IQR) | P value | ACVIM Stage C mean (SD) | ACVIM Stage D mean (SD) |
|---|---|---|---|---|---|
| Age (years) | 12.0 (10.4‐13.1) | 11.1 (8.4‐11.8) | .2 | 11.9 (2.2) | 10.4 (1.9) |
| Weight (kg) | 6.62 (4.94‐15.30) | 14.8 (7.29‐48.50) | .3 | ||
| Furosemide dosage (mg/kg/day) | n = 16; 3.50 (2.46‐4.69) | n = 4; 7.69 (6.63‐14.54) | 0.003 | ||
| Torsemide dosage (mg/kg/day) | n = 1; 0.39 | n = 1; 2.36 | ND | ||
| Pimobendan dosage (mg/kg/day) | 0.69 (0.55‐1.14) | 2.03 (1.43‐3.73) | .002 | ||
| ACE‐inhibitor dosage (mg/kg/day) | 0.92 (0.76‐1.03) | 1.01 (0.94‐1.63) | .01 | 0.91 (0.18) | 1.23 (0.37) |
| Spironolactone dosage (mg/kg/day) | N = 15; 2.06 (1.64‐2.65) | N = 5; 2.14 (2.10‐4.37) | .3 | ||
| Ang I (1‐10) (pM) | 1628 (881‐2129) | 2986 (1933‐5372) | .002 | 1414 (715) | 3519 (2081) |
| Ang II (1‐8) (pM) | 25.2 (14.85‐61.00) | 19.20 (12.75‐26.2) | .3 | ||
| Ang 1‐7 (pM) | 359 (145‐555) | 821 (286‐1210) | .01 | 356 (194) | 763 (527) |
| Ang III (2‐8) (pM) | 1.25 (1.25‐1.25) | 1.25 (1.25‐1.25) | 0.999 | ||
| Ang 1‐5 (pM) | 6.20 (1.45‐18.20) | 7.00 (2.55‐42.5) | .4 | ||
| Ang IV (3‐8) (pM) | 3.2 (1.0‐5.4) | 4.9 (1.0‐6.3) | .8 | ||
| Aldosterone (pM) | 50.9 (14.8‐295.6) | 136.0 (39.3‐486.9) | .5 | ||
| Ang I‐7/Ang II (1‐8) | 13.5 (6.5‐16.7) | 42.2 (15.8‐74.4) | .01 | ||
| Ang I (1‐10) + Ang II (1‐8) (renin surrogate) | 1640 (903‐2157) | 2995 (1951‐5399) | .002 | 1459 (726) | 3539 (2088) |
| Endogenous ACE activity [(pg Ang II/mL solvent)/hour] | 2861 (2559‐3273) | 1747 (1271‐2017) | .004 | 2937 (719) | 1696 (424) |
Notes: Data are reported as medians and interquartile ranges as well as means and standard deviations if normally distributed.
Abbreviations: ACE, angiotensin‐converting enzyme; ACVIM, American College of Veterinary Internal Medicine; Ang, angiotensin; IQR, interquartile range; ND, not done; SD, standard deviation.
Angiotensin I (P = .002) and angiotensin 1‐7 (P = .01) were higher in stage D dogs compared to stage C dogs (Table 6). The remainder of the RAAS components, including aldosterone, was not different among stages (Table 6, Figure 2). Endogenous ACE activity was lower in stage D dogs (P = .004). The sum of angiotensin I and angiotensin II, a surrogate for plasma renin activity, was higher in stage D dogs (P = .002). The ratio of angiotensin 1‐7 to angiotensin II was higher in stage D dogs (P = .01).
Figure 2.
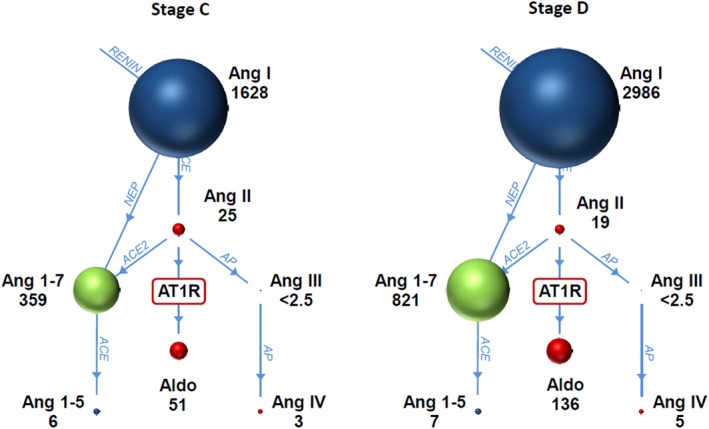
Renin‐angiotensin‐aldosterone system graphs for 22 dogs with stage C and stage D CHF. Median values for each of the angiotensin metabolites and aldosterone are shown underneath each name. The size of the ball is proportional to the value. Stage D dogs had greater angiotensin I and angiotensin 1‐7 concentrations compared to stage C dogs. ACE, angiotensin‐converting enzyme; ACE2, angiotensin‐converting enzyme 2; Aldo, aldosterone; Ang 1‐5, angiotensin 1‐5; Ang 1‐7, angiotensin 1‐7; Ang I, angiotensin 1; Ang II, angiotensin II; Ang III, angiotensin III; Ang IV, angiotensin IV; AP, aminopeptidase; AT1R, angiotensin II receptor type I; NEP, neprilysin
4. DISCUSSION
We evaluated biochemical variables, RAAS components, and proposed markers of diuretic efficacy for their ability to differentiate dogs of clinically determined separate and progressive HD stages. Some findings, such as higher medication dosages in more advanced stages, were expected based on disease stage definitions. Whether this was because of clinician directed dosage increases, weight loss, or a combination of these factors was not determined from this study. Other findings were expected based on disease pathophysiology and the effect of cardiac medications (eg, decrease in systolic blood pressure and USG, increase in BUN, creatinine, and SDMA concentrations for CHF stages, compared to preclinical stages). The differences in these variables between preclinical and CHF stages corroborated results of previous studies30, 31, 32 but did not differentiate controlled CHF (stage C) from refractory CHF (stage D).
Stage D dogs made up 21% of the CHF population in our study. This percentage is similar to reports of people with CHF that show diuretic resistance, but we do not know if all (or any) of these dogs had diuretic resistance.5 Laboratory variables, including FE Na, uNa, uNa:uK, and uNa:Fur, have been proposed as markers of diuretic resistance, associated with refractory CHF, in people. However, in our study, these variables did not differentiate dogs with stage D from stage C CHF. Furthermore, only 4 of 16 stage D dogs (5.1% of the total CHF population) met published definitions of diuretic resistance for FE Na or uNa:uK, used in people.5, 6
Serum electrolyte (sodium, chloride, and potassium) concentrations were significantly lower in stage D dogs compared to stage B2 and stage C dogs. The sensitivity and specificity of hypochloremia for differentiating stage D from stage C HD is a clinically important finding of our study. Additionally, both serum chloride and sodium concentrations were significantly lower in stage D dogs that met criteria used in human medicine to identify diuretic resistance (FE Na <0.2% and uNa:uK <1), compared to stage D dogs that did not meet these criteria. However, only serum chloride concentrations were below the reference range. The observation that more marked hypochloremia was present in the 4 stage D dogs with FE Na or uNa:uK results suggesting diuretic resistance supports a role for serum chloride concentrations in the identification of dogs with diuretic resistance. Hypochloremia may be caused by high diuretic dosages in refractory CHF because chloride loss is intrinsic to the mechanism of loop diuretics. Importantly, hypochloremia also could be a marker for, and driver of, diuretic resistance because chloride regulates renal tubular sodium transporters.12 Furthermore, renal chloride wasting, independent of sodium loss, has been correlated to higher renin concentrations and diuretic resistance in people.12 Improved diuretic responsiveness in people has been demonstrated with restoration of normal serum chloride concentrations.12, 33 Antidiuretic hormone may have contributed to serum electrolyte dilution and lowered electrolyte concentrations but was not evaluated in this study.34
Fractional excretion of sodium and chloride was higher in CHF stages C and D, compared to preclinical stages (B1 and B2) but did not differentiate between stages C and D. This higher excretion in stages C and D was attributed to diuretic administration.17 Supportive of this conclusion, we observed increased potassium excretion in CHF stages. Loop diuretics produce potassium wasting, which is countered by the administration of ACE inhibitors and spironolactone in CHF. Despite the administration of these potassium‐sparing agents, serum potassium concentrations were lower in progressive HD stages, especially stage D compared to stage C. Lower serum potassium concentrations may have been caused by lower dietary intake or progressively greater loss in response to higher diuretic dosages in refractory CHF but, based on other findings, were not consistent with inadequate RAAS suppression.
In the entire CHF population (C and D stages), a weak but significant correlation of furosemide dosages with serum concentrations was found for dosages ≤6 mg/kg/day, but no significant correlation was found at dosages >6 mg/kg/day. The weak correlation at low dosages suggests that other factors may impact serum concentrations. There is high biologic variability in furosemide absorption from the gastrointestinal tract, possibly exacerbated by advanced CHF, despite the use of high dosages.20, 35 This observation may encourage increased use of torsemide, as an alternative, because it is a potent loop diuretic with better and more consistent absorption after PO dosing.8, 21, 36
Variables, including uNa:uFur, sFur:uFur, and sFur concentrations were used to assess furosemide pharmacodynamics after absorption from the gastrointestinal tract. The lack of difference in these variables between stages C and D indicated that furosemide secretion into the renal tubule remains stable even in refractory CHF, and results were consistent with those of previous studies.21, 22 Both drugs are highly protein bound, but torsemide is a weak acid with a higher pKa than furosemide, which is a strong acid (low pKa).21 Therefore torsemide has a lower affinity for organic anion transporters, which results in a slower but more prolonged secretion into the tubular lumen and a more sustained diuretic effect than furosemide.21
Evaluation of UAldo:C and uNa:uK in group 1 provided insights into RAAS activation, which is 1 of several suspected causes of diuretic resistance. The UAldo:C was higher in progressive HD stages but was only significantly different when preclinical stages (B1 and B2) were compared to stage D (Table 4). The frequent administration of spironolactone to dogs with CHF in our study complicates this interpretation because spironolactone interferes with aldosterone's binding to receptor sites, resulting in increased blood and urine aldosterone concentrations.37 The ratio of uNa:uK, however, reflects the action of aldosterone on urine electrolyte excretion, and results from group 1 dogs suggested similar aldosterone influence in both stage C and D dogs.24
The RAAS was further evaluated by measuring angiotensin metabolites, aldosterone, and ACE activity in 22 additional dogs with stage C and D CHF (group 2). The RAAS was suppressed in both stage C and D, but the profile indicated more RAAS inhibition in stage D dogs. Angiotensin I and angiotensin 1‐7 were higher and ACE activity lower in stage D dogs compared to stage C dogs. This finding theoretically could be attributed to a significant, but only slightly higher ACE inhibitor dosage for stage D dogs (1.01 mg/kg/day) compared to stage C dogs (0.92 mg/kg/day; P = .01). The higher angiotensin I concentrations in stage D dogs were attributed to increased renin production with CHF and diuretic treatment, ACE inhibition, low renal tubular chloride concentrations, or some combination of these.12, 26 This conclusion is supported by finding that the sum of angiotensin I and angiotensin II concentrations, a surrogate for plasma renin concentration, was higher in stage D dogs.26
The cause for differences in RAAS activation between dogs in CHF stages C and D (indicated by more renin activity [angiotensin I + angiotensin II] and angiotensin 1‐7, with less ACE activity in stage D CHF) is undetermined. Possible explanations include 1 or more of the following: functional differences associated with ACE gene polymorphisms; an undescribed endogenous substance affecting converting enzymes in the RAAS cascade; recruitment of neprilysin‐mediated production of angiotensin 1‐7 from either angiotensin I or angiotensin 1‐9 (the latter produced from ACE2‐mediated conversion from angiotensin 1 as recently described in dogs) or differences in diuretic dosages or adverse effects with enhanced stimulation of renin release in refractory CHF.27, 38, 39
Our results support the use of RAAS suppressive treatment with ACE inhibitors and spironolactone, as administered in this study, but do not support the routine use of additional RAAS inhibition in stage D dogs with refractory CHF. Angiotensin 1‐7 is a beneficial, vasodilatory angiotensin metabolite whose production by neprilysin is promoted when the ACE pathway is blocked by ACE inhibitors.27 The RAAS profile in group 2 stage D dogs appears to be favorable. This conclusion is supported by higher uNa:uK, lower ACE activity, and higher angiotensin 1‐7 to angiotensin II ratios in stage D dogs, indicating successful RAAS suppression.25
Our study had several limitations that may influence the interpretation. A gold standard currently does not exist for identification of refractory CHF patients. One of our objectives was to identify variables that could separate stage C from stage D dogs, so we relied upon a recently published classification method16 and the 2009 ACVIM staging scheme. These definitions and differentiators of stages C and D may have flaws. An updated ACVIM consensus statement was published after completion of our study, but this document does not add substantially to the definition of CHF stages.40 Furthermore, CHF stages change progressively, and not all stage D dogs are equivalent. Because ours was a clinical study, we could not control for differences in diet among dogs, and there may have been differences in dietary amounts of electrolytes. These differences may have affected CHF control, RAAS activation, and blood test results. Samples were collected during the later morning or early afternoon, 4‐6 hours after medication administration, and timing varied within this window. Although timing of sampling may have impacted correlation results, it was similar to a previous report.26 We were not able to group dogs into ACE polymorphism genotypes.38 Additionally, our study evaluated circulating ACE, whereas tissue‐bound ACE or other non‐ACE enzymes also may be clinically important in these dogs. Finally, the small number of dogs in stages B1 and D likely decreased the statistical power for detecting significance in differences among groups for some variables.
5. CONCLUSIONS
Our study provides new information to strengthen guidelines for the identification and possibly treatment of dogs with refractory (stage D) CHF. Serum chloride concentrations <103.5 mmol/L accurately identified stage D, which supports the use of serum chloride concentrations in the definition of refractory CHF. Very few dogs in our study met proposed criteria in people for diuretic resistance, and these dogs were more hypochloremic than other stage D dogs. Inhibition of the RAAS with ACE inhibitors and spironolactone appeared to improve potassium retention and increase beneficial RAAS metabolites in dogs with stage C and D CHF. Further study will be required to determine whether more ACE inhibition in stage D dogs is a marker for advanced disease, higher medication dosages, or, conversely, prolonged survival. Earlier use of torsemide, instead of increasingly higher dosages of furosemide, to achieve diuresis in refractory CHF should be considered. This conclusion is supported by the evidence of a lower secretory rate of torsemide and lack of significant PO furosemide dosage to serum concentration correlation with higher doses of furosemide. A lower cutoff furosemide dosage for defining stage C versus D also might bear consideration in future attempts to stage HD in dogs.
CONFLICT OF INTEREST DECLARATION
Dr. Adin has received funding from CEVA Animal Health. Dr. Atkins has received funding and has consulted for CEVA Animal Health, Boehringer‐Ingelheim, and Vetoquinol. Drs. Papich supervises the Clinical Pharmacology Laboratory at North Carolina State University. Drs. Vaden and Kurtz do not have disclosures related to this study.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
This study was approved by the IACUC (#14‐142‐0) at North Carolina State University Veterinary Hospital and owner consent was obtained.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
ACKNOWLEDGMENT
The authors acknowledge Allison Klein for assistance with data collection. Publication of this article was funded in part by the University of Florida Open Access Publishing Fund. This project was supported by the American Kennel Club Canine Health Foundation. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the views of the Foundation.
Adin D, Kurtz K, Atkins C, Papich MG, Vaden S. Role of electrolyte concentrations and renin‐angiotensin‐aldosterone activation in the staging of canine heart disease. J Vet Intern Med. 2020;34:53–64. 10.1111/jvim.15662
This article was published online on 26 November 2019. An Error was subsequently identified in Funding information. This notice is included in the online version to indicate that has been corrected 26 December 2019.
Funding information American Kennel Club Canine Health Foundation, Grant/Award Number: 02456; University of Florida Open Access Publishing Fund
REFERENCES
- 1. Borgarelli M, Buchanan JW. Historical review, epidemiology and natural history of degenerative mitral valve disease. J Vet Cardiol. 2012;14(1):93‐101. [DOI] [PubMed] [Google Scholar]
- 2. Wess G, Domenech O, Dukes‐McEwan J, Häggström J, Gordon S. European Society of Veterinary Cardiology screening guidelines for dilated cardiomyopathy in Doberman Pinschers. J Vet Cardiol. 2017;19(5):405‐415. [DOI] [PubMed] [Google Scholar]
- 3. Atkins C, Bonagura J, Ettinger S, et al. ACVIM Consensus Statement_Guidelines for diagnosis and treatment of canine chronic valvular heart disease. J Vet Intern Med. 2009;23(1):1142‐1150. [DOI] [PubMed] [Google Scholar]
- 4. Oyama MA, Peddle GD, Reynolds CA, Singletary GE. Use of the loop diuretic torsemide in three dogs with advanced heart failure. J Vet Cardiol. 2011;13(4):287‐292. [DOI] [PubMed] [Google Scholar]
- 5. Doering A, Jenkins CA, Storrow AB, et al. Markers of diuretic resistance in emergency department patients with acute heart failure. Int J Emerg Med. 2017;10(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. ter Maaten JM, Valente M a E, Damman K, Hillege HL, Navis G, Voors A a. Diuretic response in acute heart failure‐pathophysiology, evaluation, and therapy. Nat Rev Cardiol. 2015;12(3):184‐192. [DOI] [PubMed] [Google Scholar]
- 7. Singh D, Shrestha K, Testani JM, et al. Insufficient natriuretic response to continuous intravenous furosemide is associated with poor long‐term outcomes in acute decompensated heart failure. J Card Fail. 2014;20(0 1):392‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hori Y, Takusagawa F, Ikadai H, Uechi M, Hoshi F, Higuchi S. Effects of oral administration of furosemide and torsemide in healthy dogs. Vet Res. 2007;68(10):1058‐1063. [DOI] [PubMed] [Google Scholar]
- 9. Testani JM, Brisco MA, Turner JM, et al. Loop diuretic efficiency: a metric of diuretic responsiveness with prognostic importance in acute decompensated heart failure. Circ Hear Fail. 2015;27(4):215‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. ter Maaten JM, Valente MAE, Metra M, et al. A combined clinical and biomarker approach to predict diuretic response in acute heart failure. Clin Res Cardiol. 2016;105(2):145‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferreira JP, Girerd N, Medeiros PB, et al. Spot urine sodium excretion as prognostic marker in acutely decompensated heart failure: the spironolactone effect. Clin Res Cardiol. 2016;105(6):489‐507. [DOI] [PubMed] [Google Scholar]
- 12. Hanberg JS, Rao V, Ter Maaten JM, et al. Hypochloremia and diuretic resistance in heart failure. Circ Hear Fail. 2016;9(8):1‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumar D, Bagarhatta R. Fractional excretion of sodium and its association with prognosis of decompensated heart failure patients. J Clin Diagnostic Res. 2015;9(4):OC01‐OC03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adin D, Atkins C, Papich MG. Pharmacodynamic assessment of diuretic efficacy and braking in a furosemide continuous infusion model. J Vet Cardiol. 2018;20(2):92‐101. [DOI] [PubMed] [Google Scholar]
- 15. Uechi M, Matsuoka M, Kuwajima E, et al. The effects of the loop diuretics furosemide and torasemide on diuresis in dogs and cats. J Vet Med Sci. 2003;65(10):1057‐1061. 10.1292/jvms.65.1057. [DOI] [PubMed] [Google Scholar]
- 16. Beaumier A, Rush JE, Yang VK, Freeman LM. Clinical findings and survival time in dogs with advanced heart failure. J Vet Intern Med. 2018;32(3):944‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adin D, Atkins C, Papich M, et al. Furosemide continuous rate infusion diluted with 5% dextrose in water or hypertonic saline in normal adult dogs: a pilot study. J Vet Cardiol. 2017;19(1):44‐56. [DOI] [PubMed] [Google Scholar]
- 18. Johansson AM, Gardner SY, Levine JF, et al. Pharmacokinetics and pharmacodynamics of furosemide after oral administration to horses. J Vet Intern Med. 2004;18(5):739‐743. . [DOI] [PubMed] [Google Scholar]
- 19. Lantis AC, Ames MK, Werre S, et al. The effect of enalapril on furosemide‐activated renin–angiotensin–aldosterone system in healthy dogs. J Vet Pharmacol Ther. 2015;38:513‐517. [DOI] [PubMed] [Google Scholar]
- 20. Hirai J, Miyazaki H, Taneike T. The pharmacokinetics and pharmacodynamics of furosemide in the anesthetized dog. J Vet Pharmacol Ther. 1992;15:231‐239. [DOI] [PubMed] [Google Scholar]
- 21. Sogame Y, Okano K, Hayashi K, Uchida T, Tsuda Y. Urinary excretion profile of torasemide and its diuretic action in dogs. J Pharm Pharmacol. 1996;48(4):375‐379. [DOI] [PubMed] [Google Scholar]
- 22. Rose HJ, Pruitt AW, Dayton PG, McNay JL. Relationship of urinary furosemide excretion rate to natiuretic effect in experimental azotemia. J Pharmacol Exp Ther. 1976;199:490‐497. [PubMed] [Google Scholar]
- 23. Brandish PE, Chen H, Szczerba P, Hershey JC. Development of a simplified assay for determination of the antimineralocorticoid activity of compounds dosed in rats. J Pharmacol Toxicol Methods. 2008;57(2):155‐160. [DOI] [PubMed] [Google Scholar]
- 24. Eudy RJ, Sahasrabudhe V, Sweeney K, et al. The use of plasma aldosterone and urinary sodium to potassium ratio as translatable quantitative biomarkers of mineralocorticoid receptor antagonism. J Transl Med. 2011;9(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Basu R, Poglitsch M, Yogasundaram H, Thomas J, Rowe BH, Oudit GY. Roles of angiotensin peptides and recombinant human ACE2 in heart failure. J Am Coll Cardiol. 2017;69(7):805‐819. [DOI] [PubMed] [Google Scholar]
- 26. Pavo N, Goliasch G, Wurm R, et al. Low‐and high‐renin heart failure phenotypes with clinical implications. Clin Chem. 2018;64(3):597‐608. [DOI] [PubMed] [Google Scholar]
- 27. Domenig O, Manzel A, Grobe N, et al. Neprilysin is a mediator of alternative renin‐angiotensin‐system activation in the murine and human kidney. Sci Rep. 2016;6:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keizer RJ, Jansen RS, Rosing H, et al. Incorporation of concentration data below the limit of quantification in population pharmacokinetic analyses. Pharmacol Res Perspect. 2015;3:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ogden TL, Court M, Way M, De D. Handling results below the level of detection. Ann Occup Hyg. 2010;54(3):255‐256. [DOI] [PubMed] [Google Scholar]
- 30. Pouchelon JL, Atkins CE, Bussadori C, et al. Cardiovascular‐renal axis disorders in the domestic dog and cat: a veterinary consensus statement. J Small Anim Pract. 2015;56(9):537‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nicolle AP, Chetboul V, Allerheiligen T, et al. Azotemia and glomerular filtration rate in dogs with chronic valvular disease. J Vet Intern Med. 2007;21(5):943‐949. [DOI] [PubMed] [Google Scholar]
- 32. Martinelli E, Locatelli C, Bassis S, et al. Preliminary investigation of cardiovascular–renal disorders in dogs with chronic mitral valve disease. J Vet Intern Med. 2016;30(5):1612‐1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ter Maaten JM, Damman K, Hanberg JS, et al. Hypochloremia, diuretic resistance, and outcome in patients with acute heart failure. Circ Hear Fail. 2016;9(8):1‐9. [DOI] [PubMed] [Google Scholar]
- 34. Goldsmith SR, Bart BA, Piña IL. Neurohormonal imbalance: a neglected problem—and potential therapeutic target—in acute heart failure. Curr Probl Cardiol. 2018;43(7):294‐304. [DOI] [PubMed] [Google Scholar]
- 35. Brater DC. Clinical pharmacology of loop diuretics in health and disease. Eur Heart J. 1992;13(suppl G):10‐14. [DOI] [PubMed] [Google Scholar]
- 36. Dodion L, Willems JL. Study of the elimination kinetics of torasemide, a novel loop diuretic, in renal insufficiency. Eur J Clin Pharmacol. 1986;31(1 Supplement):49‐51. [DOI] [PubMed] [Google Scholar]
- 37. Guyonnet J, Elliott J, Kaltsatos V. A preclinical pharmacokinetic and pharmacodynamic approach to determine a dose of spironolactone for treatment of congestive heart failure in dog. J Vet Pharmacol Ther. 2010;33(3):260‐267. [DOI] [PubMed] [Google Scholar]
- 38. Meurs KM, Stern JA, Atkins CE, et al. Angiotensin‐converting enzyme activity and inhibition in dogs with cardiac disease and an angiotensin‐converting enzyme polymorphism. J Renin Angiotensin Aldosterone Syst. 2017;18(4):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larouche‐Lebel É, Loughran KA, Oyama MA, et al. Plasma and tissue angiotensin‐converting enzyme 2 activity and plasma equilibrium concentrations of angiotensin peptides in dogs with heart disease. J Vet Intern Med. 2019;33:1571‐1584. 10.1111/jvim.15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keene BW, Atkins CE, Bonagura JD, et al. ACVIM consensus guidelines for the diagnosis and treatment of myxomatous mitral valve disease in dogs. J Vet Intern Med. 2019;33:1‐14. 10.1111/jvim.15488. [DOI] [PMC free article] [PubMed] [Google Scholar]