

Abstract
Background
Feline cryptosporidiosis is an increasing problem, especially in catteries. In humans, close contact with cats could be a potential source of infection although the risk of contracting cryptosporidiosis caused by Cryptosporidium felis is considered to be relatively low. Sequencing of the 60-kDa glycoprotein gene is a commonly used tool for investigation of the genetic diversity and transmission dynamics of Cryptosporidium species. However, until now the sequence of gp60 from C. felis has not been available and genotyping has been limited to less discriminatory markers, such as 18S rRNA, COWP and HSP70.
Methods
We have identified the gp60 orthologue within the genome sequence of C. felis, and used the sequence to design a nested PCR for subtyping purposes. A total of 128 clinical isolates of both feline and human origin, were used to evaluate the marker.
Results
Sequence analysis revealed large variations between the different samples. The C. felis gp60 lack the characteristic serine-tract found in many other cryptosporidian orthologues, instead it has an insertion of variable length (361–742 nt). Also, two cases of suspected zoonotic transmission of C. felis between cats and humans were successfully confirmed.
Conclusions
We have identified the gp60 gene in C. felis and show how this highly variable marker can be used in epidemiological investigations.
Keywords: Cryptosporidiosis, Molecular typing, Zoonotic transmission, 60-kDa glycoprotein, Source tracking, Genetic variability, Epidemiological marker
Background
Cryptosporidium species are significant pathogens that infect a wide range of vertebrate hosts, causing a considerable burden of gastrointestinal disease [1]. Human cryptosporidiosis is mainly caused by Cryptosporidium hominis and Cryptosporidium parvum, the latter also being an important zoonotic pathogen [2]. However, the use of molecular techniques has shown that other species of Cryptosporidium also have zoonotic potential, including, but not limited to, Cryptosporidium meleagridis, Cryptosporidium cuniculus, Cryptosporidium ubiquitum, Cryptosporidium canis and Cryptosporidium felis [2].
The close association between owners and their companion animals has thus a potentially significant role in the zoonotic transmission of parasites [3, 4]. Even though the population-based risk of contracting Cryptosporidium spp. from domestic cats is considered to be relatively low [5, 6], there have been several reports showing human infection with C. felis [2, 7–12]. In 2015, one investigation of a possible zoonotic transmission between a cat and its owner showed that, although the C. felis isolated from both the cat and the owner had identical sequences at several markers, so did all other C. felis samples from unrelated cats [11]. This highlighted the need for a higher-resolution subtyping method for this Cryptosporidium species.
The 60-kDa glycoprotein gene (gp60) is the most commonly used marker for subtyping of Cryptosporidium spp., mainly C. parvum and C. hominis, and has been a useful tool to study sources of infection, genetic diversity and host adaptation. Recently, subtyping methods targeting the gp60 gene of C. meleagridis, C. ubiquitum, C. viatorum, Cryptosporidium skunk genotype and chipmunk genotype I have been developed [13–17]. These additional subtyping tools are needed since the PCR primers designed on C. parvum and C. hominis sequences are not capable of amplifying efficiently the gp60 gene of these divergent Cryptosporidium spp. [18].
Given that C. felis is relatively genetically distant from C. hominis and C. parvum, the gp60 orthologue has previously not been identified. In this study, the gp60 gene of C. felis was identified by massive parallel sequencing of a clinical C. felis sample and PCR primers were developed and used to characterize C. felis isolates retrieved from both humans and cats.
Methods
Samples
DNA extracts from 128 C. felis-positive fecal samples were included in the present study; 93 samples were from humans and 35 from cats (Table 1), and spanned the period 2000 to 2015. Human samples used for screening were collected either by the Public Health Agency of Sweden (eight samples) or as part of national molecular epidemiology in the Cryptosporidium Reference Unit in the UK [19] (85 samples) (Table 1). All the samples were originally identified as containing C. felis by restriction fragment length polymorphism (RFLP) analysis and/or by sequencing of the partial 18S rRNA gene [20]. Samples related to foreign travel were identified either from the submission form or from a routinely administered patient questionnaire (UK) [12], or from information obtained through the national mandatory notifications system (SmiNet) or the local department of Communicable Disease Control (Sweden). Samples from patients travelling to Croatia, Dominican Republic, India, Indonesia, Pakistan, Peru, Spain, Sudan and USA were included in addition to samples from those that had not travelled (Table 1).
Table 1.
Origin and history of Cryptosporidium felis DNA extracts investigated in the study
| Host | Collection | Origin | n | Travel history |
|---|---|---|---|---|
| Cat | Sweden | Domestic | 11 | |
| Cat | Denmark | Domestic and feral | 24 | |
| Human | Sweden | Domestic | 6 | |
| Human | Sweden | Travel-related | 2 | India, Indonesia |
| Human | UK | Domestic or travel history unknown | 73 | |
| Human | UK | Travel-related | 12 | Croatia, Dominican Republic, USA India, Pakistan, Peru, Spain, Sudan, |
Abbreviation: n, number of samples
Feline samples were obtained from the National Veterinary Institute (NVI) and the Public Health Agency in Sweden and from the Technical University of Denmark, National Veterinary Institute (DTU-VET) in Denmark. At NVI the samples were collected from routine diagnosis of gastroenteritis while the samples at the Public Health Agency were collected as part of a study or because zoonotic transmission was suspected. The Danish samples originated from domestic and feral cats from veterinary clinics, cat shelters and breeders in the Copenhagen Metropolitan Region. The samples were collected as part of an epidemiological study of cryptosporidiosis and giardiasis in cats. A total of 24 samples were included from Denmark and 11 from Sweden (Table 1). All of these samples had previously been tested positive for Cryptosporidium by immunofluorescence microscopy and C. felis had been detected in some of the samples by PCR and sequencing of the 18S and/or HSP70 loci (Enemark et al. unpublished). All samples included in the present study were confirmed positive for C. felis by PCR and sequencing of 18S rRNA locus [20].
Subtyping marker
Initially, a draft genome sequence of C. felis, obtained from a clinical isolate from a Swedish household cat with diarrhea (unpublished data), was screened for gp60 using tBLASTx homology searches, with Cryptosporidium orthologues as seed sequences. The C. felis draft genome sequence comprised 8.74 Mbp in 109 contigs. However, this approach generated very weak hits. Instead we aligned amino acid sequences of gp60 orthologues using Clustal X, and the resulting alignment was searched to identify the most conserved motif. A homology search using amino acid sequence FVMWFGEGTPVATLKCGGY identified a homologous motif, FTVWFDGGIPITTIGCG (e = 0.57), embedded in a predicted coding sequence of 1899 nt, 633 aa. Using the complete 1899 nt to perform a homology search of sequences from various species of Cryptosporidium yielded a best hit to an uncharacterized Cryptosporidium sequence (GenBank: AJW72319) (47% identity E = 6e−53) followed by a gp60 ortholog in C. viatorum (GenBank: AQY61281, 46% identity E = 3e−51). The C. felis gp60 candidate sequence was aligned to a set of orthologues using Clustal X and the most conserved part of the alignment were used to design primers for nested PCR.
Primer design
A conserved region of the gp60 gene was selected to design primer sets for nested PCR. PCR amplification was performed using the primers GP60CF_F1 (5′-TTT CCG TTA TTG TTG CAG TTG CA-3′) and GP60CF_R1 (5′-ATC GGA ATC CCA CCA TCG AAC-3′) for primary reactions, and GP60CF_F2 (5′-GGG CGT TCT GAA GGA TGT AA-3′) and GP60CF_R2 (5′-CGG TGG TCT CCT CAG TCT TC-3′) for secondary reactions with PCR products expected ~ 1200 bp and 900 bp respectively based on the genome sequence of C. felis (unpublished data).
PCR screening
Nested PCR was carried out in a total volume of 20 μl of PCR mixture, containing Maxima Hot Start 2× PCR master mix (Thermo Fisher Scientific, Waltham, USA), 0.5 μM of each primer, and 1 to 3 μl of extracted DNA. For the primary PCR 0.4 μl of bovine serum albumin (20 mg/ml) (Thermo Fisher Scientific) was added to the mix. For the secondary PCR, 1 μl of the primary PCR product was used. Reaction conditions were initial denaturation of 95 °C for 4 min followed by 35 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 1.5 min, followed by a final extension step at 72 °C for 7 min.
DNA sequence analysis
Positive products of the secondary gp60 PCR were purified using ExoSAP-IT (Thermo Fisher Scientific) and Sanger sequenced bidirectionally (BigDye chemistry; Applied Biosystems, Waltham, USA) using the primers designed for secondary PCR. Sequences obtained were manually edited and analyzed using BioEdit sequence alignment editor (version 7.0.9.0). Subsequently, nucleotide sequences were aligned and compared with the full C. felis gp60 gene sequences using CLUSTALW algorithm [21].
Nucleotide as well as amino acid sequences were aligned using T-coffee alignment software [22] and short sequences were removed to not affect the result of the tree. The final analysis involved 102 nucleotide sequences. Ends of all sequences were trimmed, resulting in an alignment covering 1291 bases. A phylogenetic model selector as implemented in MEGA 6 [23] was used to test which model best fitted the data. A phylogenetic tree was inferred in MEGA 6 by using the Maximum Likelihood method based on the Tamura 3-parameter model [24]. A discrete Gamma distribution was used to model evolutionary rate differences among sites (1 category (+G, parameter = 0.1703) [24]). The rate variation model allowed for some sites to be evolutionarily invariable (+I, 75.8281% sites). The robustness of the phylogeny was tested with 1000 bootstrap replicates. In order to avoid over-interpretation, only confidence levels for well-supported nodes are shown.
To illustrate the difference between Cryptosporidium species within the gp60, an amino acid alignment using BioEdit sequence alignment editor (7.0.9.0) was made comparing reference sequences of C. hominis, C. parvum, C. viatorum, C. meleagridis, C. fayeri, C. ubiquitum, Cryptosporidium chipmunk genotype I and C. felis. All the sequences were downloaded from the NCBI GenBank database with the exception of C. felis in which case the full gp60 sequences (SWEFEL1) generated by massive parallel sequencing were used.
Nucleotide sequence accession numbers
Nucleotide sequences of C. felis subtype variants identified have been deposited in the GenBank database under the accession numbers MH240831-MH240912 (Additional file 1: Table S1).
Results
Cryptosporidium felis gp60 gene and sample characteristics
A comparison of the region harbouring the C. felis gp60 orthologue revealed synteny to C. parvum and C. hominis, further strengthening the hypothesis that a true gp60 orthologue had been identified. To characterize the C. felis gp60 gene, 128 C. felis positive DNA samples from different locations and origins were analyzed (Table 1). A total of 102 samples were successfully amplified and sequenced (80 human samples and 22 cat samples) whereof 82 unique sequences were obtained. Negative amplification attempts were most likely due to extensive DNA degradation in old fecal samples or those not stored at optimal conditions. Samples positive by nested gp60 PCR showed fragment lengths ranging from 896 bp to 1285 bp, as predicted, but differences in length could not be linked to host or origin of samples. All C. felis sequences lacked the characteristic serine-tract of many gp60 orthologues identified in cryptosporidia, instead an insertion (Fig. 1, highlighted in blue), of variable length (361–742 nt), with no homology to the serine tracts was present. As previously reported in the gp60 of C. viatorum [16] some randomly occurring double peaks were observed in 10/102 (10%) samples.
Fig. 1.

Features of the gp60 gene of C. felis. Amino acid alignment illustrating the differences in the gp60 between Cryptosporidium species. The insertion present in C. felis is highlighted in blue and the serine repeats region characteristic of other Cryptosporidium species is highlighted in green. Cryptosporidium ubiquitum lacks the typical S repeat, the region is highlighted in yellow
In a general overview, more C. felis samples from humans were from men (n = 44) than women (n = 36). The age of the patients ranged from 1 to 83 years-old (median: 26 years-old; mean: 27 years-old). When it comes to the cats the age ranged from below 0.5 to 10 years-old and the majority were domestic cats. A few were stray or feral cats of which the age was unknown. Four samples from cats representing a feline outbreak in a breeding establishment (F1) were included (Enemark et al. unpublished), and so were samples from both cat and owner in two zoonotic cases (Z1 and Z2). (Additional file 1: Table S1)
Phylogeny
Nucleotide and amino acid sequences were used to calculate phylogenies using the Maximum Likelihood method based on the Tamura 3-parameter model. However, the data did not yield any fully-resolved, well-supported trees. Three well supported (bootstrap ≥ 80) clusters were identified, B–D (Fig. 2). Two of the supported clusters (C and D) and one additional cluster (A, Fig. 2) consisted of sequences isolated from human cases of cryptosporidiosis only, and one cluster (B) was mixed, comprising sequences from a suspected zoonotic transmission between a cat and its’ owner. Cluster A comprised 15 sequences from human cases, 14 from the UK and one travel-related, from the Dominican Republic; the sex distribution in this cluster was slightly dominated by female patients (n = 9). Cluster B comprised 13 sequences isolated from both cats and humans, with the majority being male (7/10). Cluster C contained six sequences, all isolated from UK patients. These sequences represent four male and two females.
Fig. 2.
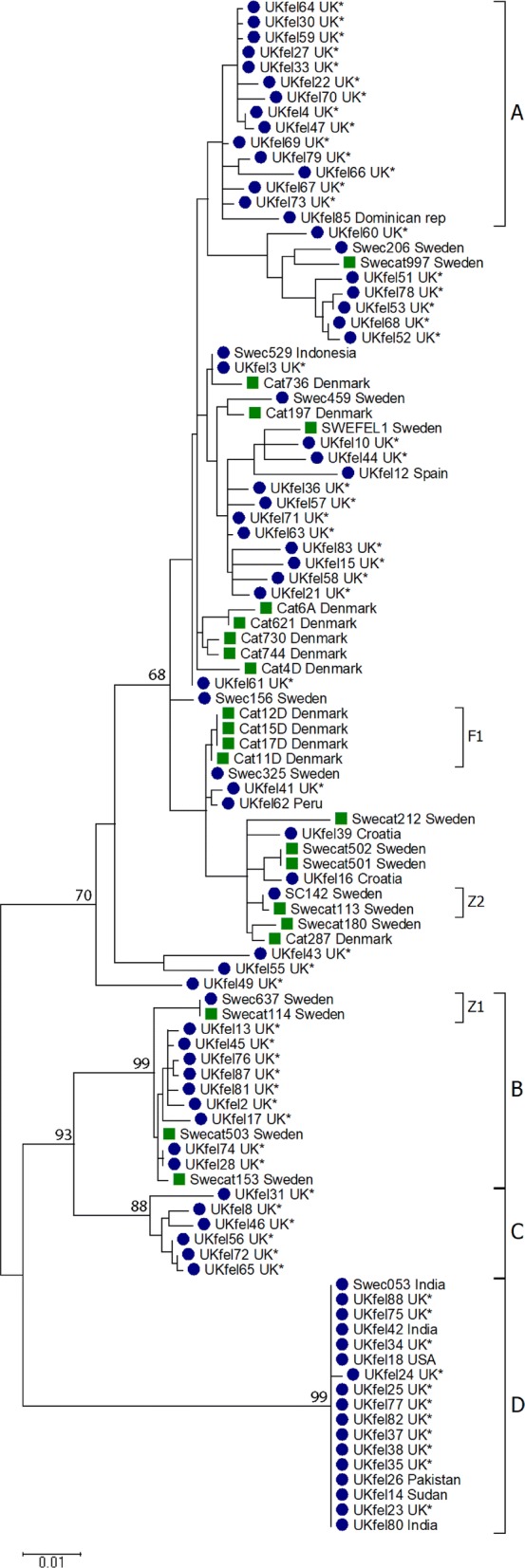
Phylogenetic analysis of 102 gp60 sequences from C. felis isolated from humans and cats. Blue dots indicate sample from human and green squares indicate sample from cat. All cases with known travel history have been indicated while samples with unknown travel history are marked with an asterisk
Cluster D comprised 17 sequences from human hosts. More cases were male (n = 16) than female (n = 1) [23] and stemmed mainly from the UK. However, there were also several cases that had travelled to India or Pakistan in Cluster D. The sequences within Cluster D had almost no variation (0–3 nt) and this was the most distinct group in our dataset.
Connected cases
All four cat sequences from the outbreak in a Danish cattery grouped together in the phylogenetic tree (Fig. 2, F1). Three of the isolates, from an adult cat and two kittens, were identical, whereas an isolate from a third kitten differed by 37 nucleotides (of which 36 nucleotides represents indels and one a SNP).
Within our dataset, two zoonotic transmissions between cat and owner were further confirmed; in both cases the owner and cat grouped together in the phylogenetic tree (Fig. 2). In one of the cases the transmission of infection was suspected but never established (Fig 2, Z1), while in the other case, the transmission from cat to owner was established by both epidemiological and molecular investigations [11]. In zoonotic case 1 (Z1) the sequences retrieved from both the cat and owner were identical, but differed significantly from all other sequences within our dataset (Cluster B, Fig 2). The sequences retrieved from the cat and owner in zoonotic case 2 (Z2) also differed from all other sequences obtained, but also from each other by one nucleotide in the gp60 sequence; where all other sequences in our dataset displayed an adenine (A) in position 833, the sequence retrieved from the cat had a guanine (G). This single nucleotide polymorphism resulted in an amino acid change. However, careful inspection of C. felis NGS data from this cat (which was the donor of the sample from which the genome sequence used to identify the gp60 sequence was derived) showed that one of 62 high quality sequence reads had an A in that position. This clearly shows that observed variation and dominant variant within a single C. felis isolate shifted when transmitted from one host (cat) to another host (human) (Fig. 3).
Fig. 3.

a In zoonotic case 2 (Z2) one nucleotide in the gp60 sequence clearly showed how a dominant variant of C. felis shifted to another when transmitted from one host (cat) to another (human). The nucleotide in position 833 differed between the cat (G) and the owner (A). Massive parallel sequencing of the cat sample, covering the named position 62 times, showed that one of the 62 reads indeed had the adenine (A) found in the human sample. b All other cats and humans in the study had A in this specific position
Nomenclature
The subtype family of C. felis gp60 is designated XIX. Since the gene is so variable and most of the sequences are unique, and no serine tract is present, no specific subtype nomenclature has been established.
Discussion
The gp60 gene is currently the most commonly used genetic marker for subtyping of Cryptosporidium spp., often using the number of, and variation in, repeats in the serine tract present in most species explored in combination with the adjacent sequence. The serine tract is lacking in the genome of C. felis and C. ubiquitum [14] as well as in the recently described C. suis gp60 sequences (GenBank: MH187875 and MH187874). The variability among Cryptosporidium gp60 sequences indicates that the use of universal primers for gp60 amplification is inappropriate.
We have identified the gp60 orthologue in C. felis and used a novel, variable region for subtyping (Fig. 1). The gp60 orthologue in C. felis is highly variable and most samples generated fragments that vary greatly in size which renders weak phylogenies and thus gives poorly supported trees under all algorithms tested. However, the marker is very useful to confirm relatedness between samples, as for example in source tracing and suspected zoonotic transmission. This need, to have a distinct subtyping tools for this purpose, was highlighted in the previous report on a possible zoonotic transmission of C. felis from a cat to its owner [11]. In this type of case, the diverged nature of the C. felis gp60 orthologue is useful to trace zoonotic transmissions, since the large variability makes the likelihood of false positive connections low.
Comparing the human and feline isolates stemming from the established zoonotic transmission from a cat to its owner revealed one single nucleotide substitution (A to G). However, for this particular feline isolate, we have NGS data at a 62-fold coverage of the locus of the substitution. Although the majority of reads displayed this feline isolate-specific adenine, one high quality read matched the sequence found in the human-derived isolate. In the DNA sequence generated from Sanger sequencing of some PCR products covering this region there were double peaks in this exact position, further strengthening our observation. Samples with Cryptosporidium parasites are more likely to contain mixed populations of oocysts rather than a clonal population, and should be looked on as mixed infections. The variability in an infection may change when the population moves from one host to another which was apparent in the case described herein.
We have detected a large variability among the gp60 sequences in our dataset. However, there were also some conserved groups. The most striking example is a cluster (Cluster D, Fig. 2) of 17 almost identical sequences, obtained from humans in both Sweden and the UK, many of whom had recently travelled abroad. More strikingly for the cases in this highly conserved group is the overrepresentation of men (16/17 cases), which is unusual as women are generally overrepresented among adults with cryptosporidiosis [19]. Another intriguing observation linked to this cluster is the median age of the group, 37 years-old, which is atypical for a mostly pediatric infection [25]. Furthermore, this group represents the cluster most distinct from the other gp60 sequences obtained. Altogether our data suggest that these 17 sequences represent a separate, conserved variant. It would be interesting to study other genetic markers among samples from this group.
Conclusions
We have identified the gp60 gene in C. felis and show how this highly variable marker can be used to supplement epidemiological investigations and aid in source tracking. Due to occasionally occurring double peaks in these sequences it is imperative to have a close look at chromatograms before drawing conclusions.
Supplementary information
Additional file 1: Table S1. Sample origin and GenBank accession number as well as sex and age of each patient.
Acknowledgements
We would like to thank Martin Norling at the SciLifeLab bioinformatic platform NBIS for help with the bioinformatic analyses and Tobias Lilja for help with the phylogenetic analysis. The Technical University of Denmark, National Veterinary Institute is acknowledged for providing the C. felis isolates from Danish cats.
Abbreviations
- gp60
60-kDa glycoprotein
- 18S
18S ribosomal RNA gene
- COWP
Cryptosporidium oocyst wall protein
- HSP70
heat-shock protein 70
- PCR
polymerase chain reaction
- Z1
zoonotic case 1
- Z2
zoonotic case 2
- F1
feline outbreak 1
Authors’ contributions
LR-L performed experiments, analyzed results and was a major contributor in writing of the manuscript. JB and KT were major contributors in the conceptualization, experimental design, analysis of results and writing of the manuscript. KE and RMC collected and performed species determination on all UK samples. HLE performed species determination on all samples from Denmark. All authors read and approved the final manuscript.
Funding
This work, including KT and JB, was funded by a grant from the Swedish Civil Contingencies Agency (Grant Number: 2012-172). LR was funded by the Mexican National Council of Science and Technology (CONACyT, Mexico).
Availability of data and materials
All data generated or analysed during this study are included in this published article. All newly generated sequences were deposited in the GenBank database under the accession numbers MH240831-MH240912.
Ethics approval and consent participate
The study was conducted in accordance with the Swedish Disease Act (2004:168), and special approval for samples collected during individual studies had been obtained from the Ethical Review Board at Karolinska Institutet, Stockholm, Sweden. The DNA from oocysts collected in the UK were shared for public health monitoring and quality assurance purposes and ethical approval was not required. Feline samples were sent to the National Veterinary Institute, Uppsala, Sweden, for diagnostic purposes and ethical approval is not required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Laura Rojas-Lopez, Email: laura.rojas@icm.uu.se.
Kristin Elwin, Email: kristin.elwin@wales.nhs.uk.
Rachel M. Chalmers, Email: rachel.chalmers@wales.nhs.uk
Heidi L. Enemark, Email: heidi.enemark@vetinst.no
Jessica Beser, Email: jessica.beser@folkhalsomyndigheten.se.
Karin Troell, Email: karin.troell@sva.se.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13071-020-3906-9.
References
- 1.Xiao L, Fayer R, Ryan U, Upton SJ. Cryptosporidium taxonomy: recent advances and implications for public health. Clin Microbiol Rev. 2004;17:72–97. doi: 10.1128/CMR.17.1.72-97.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan U, Fayer R, Xiao L. Cryptosporidium species in humans and animals: current understanding and research needs. Parasitology. 2014;141:1667–1685. doi: 10.1017/S0031182014001085. [DOI] [PubMed] [Google Scholar]
- 3.Sotiriadou I, Pantchev N, Gassmann D, Karanis P. Molecular identification of Giardia and Cryptosporidium from dogs and cats. Parasite. 2013;20:8. doi: 10.1051/parasite/2013008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito Y, Itoh N, Kimura Y, Kanai K. Molecular detection and characterization of Cryptosporidium spp. among breeding cattery cats in Japan. Parasitol Res. 2016;115:2121–2123. doi: 10.1007/s00436-016-4984-7. [DOI] [PubMed] [Google Scholar]
- 5.Glaser CASS, Reingold A, Newman TB. Association between Cryptosporidium infection and animal exposure in HIV-infected individuals. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;17:79–82. doi: 10.1097/00042560-199801010-00012. [DOI] [PubMed] [Google Scholar]
- 6.Hunter PR, Hughes S, Woodhouse S, Syed Q, Verlander NQ, Chalmers RM, et al. Sporadic cryptosporidiosis case-control study with genotyping. Emerg Infect Dis. 2004;10:1241–1249. doi: 10.3201/eid1007.030582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caccio S, Pinter E, Fantini R, Mezzaroma I, Pozio E. Human infection with Cryptosporidium felis: case report and literature review. Emerg Infect Dis. 2002;8:85–86. doi: 10.3201/eid0801.010269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pieniazek NJ, Bornay-Llinares FJ, Slemenda SB, da Silva AJ, Moura IN, Arrowood MJ, et al. New Cryptosporidium genotypes in HIV-infected persons. Emerg Infect Dis. 1999;5:444–449. doi: 10.3201/eid0503.990318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cieloszyk J, Goni P, Garcia A, Remacha MA, Sanchez E, Clavel A. Two cases of zoonotic cryptosporidiosis in Spain by the unusual species Cryptosporidium ubiquitum and Cryptosporidium felis. Enferm Infecc Microbiol Clin. 2012;30:549–551. doi: 10.1016/j.eimc.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 10.de Lucio A, Merino FJ, Martinez-Ruiz R, Bailo B, Aguilera M, Fuentes I, et al. Molecular genotyping and sub-genotyping of Cryptosporidium spp. isolates from symptomatic individuals attending two major public hospitals in Madrid, Spain. Infect Genet Evol. 2016;37:49–56. doi: 10.1016/j.meegid.2015.10.026. [DOI] [PubMed] [Google Scholar]
- 11.Beser J, Toresson L, Eitrem R, Troell K, Winiecka-Krusnell J, Lebbad M. Possible zoonotic transmission of Cryptosporidium felis in a household. Infect Ecol Epidemiol. 2015;5:28463. doi: 10.3402/iee.v5.28463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elwin K, Hadfield SJ, Robinson G, Chalmers RM. The epidemiology of sporadic human infections with unusual cryptosporidia detected during routine typing in England and Wales, 2000–2008. Epidemiol Infect. 2012;140:673–683. doi: 10.1017/S0950268811000860. [DOI] [PubMed] [Google Scholar]
- 13.Guo Y, Cebelinski E, Matusevich C, Alderisio KA, Lebbad M, McEvoy J, et al. Subtyping novel zoonotic pathogen Cryptosporidium chipmunk genotype I. J Clin Microbiol. 2015;53:1648–1654. doi: 10.1128/JCM.03436-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li N, Xiao L, Alderisio K, Elwin K, Cebelinski E, Chalmers R, et al. Subtyping Cryptosporidium ubiquitum, a zoonotic pathogen emerging in humans. Emerg Infect Dis. 2014;20:217–224. doi: 10.3201/eid2002.121797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stensvold CR, Beser J, Axen C, Lebbad M. High applicability of a novel method for gp60-based subtyping of Cryptosporidium meleagridis. J Clin Microbiol. 2014;52:2311–2319. doi: 10.1128/JCM.00598-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stensvold CR, Elwin K, Winiecka-Krusnell J, Chalmers RM, Xiao L, Lebbad M. Development and application of a gp60-based typing assay for Cryptosporidium viatorum. J Clin Microbiol. 2015;53:1891–1897. doi: 10.1128/JCM.00313-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan W, Alderisio K, Roellig DM, Elwin K, Chalmers RM, Yang F, et al. Subtype analysis of zoonotic pathogen Cryptosporidium skunk genotype. Infect Genet Evol. 2017;55:20–25. doi: 10.1016/j.meegid.2017.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao L, Feng Y. Molecular epidemiologic tools for waterborne pathogens Cryptosporidium spp. and Giardia duodenalis. Food Waterborne Parasitol. 2017;8–9:14–32. doi: 10.1016/j.fawpar.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chalmers RM, Elwin K, Thomas AL, Guy EC, Mason B. Long-term Cryptosporidium typing reveals the aetiology and species-specific epidemiology of human cryptosporidiosis in England and Wales, 2000 to 2003. Euro Surveill. 2009;14:19086. doi: 10.2807/ese.14.02.19086-en. [DOI] [PubMed] [Google Scholar]
- 20.Xiao L, Morgan UM, Limor J, Escalante A, Arrowood M, Shulaw W, et al. Genetic diversity within Cryptosporidium parvum and related Cryptosporidium species. Appl Environ Microbiol. 1999;65:3386–3391. doi: 10.1128/AEM.65.8.3386-3391.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 23.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol. 1992;9:678–687. doi: 10.1093/oxfordjournals.molbev.a040752. [DOI] [PubMed] [Google Scholar]
- 25.Caccio SM, Chalmers RM. Human cryptosporidiosis in Europe. Clin Microbiol Infect. 2016;22:471–480. doi: 10.1016/j.cmi.2016.04.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Sample origin and GenBank accession number as well as sex and age of each patient.
Data Availability Statement
All data generated or analysed during this study are included in this published article. All newly generated sequences were deposited in the GenBank database under the accession numbers MH240831-MH240912.