

Abstract
Native ion mobility-mass spectrometry (IM-MS) is a rapidly growing field for studying the composition and structure of biomolecules and biomolecular complexes using gas-phase methods. Typically, ions are formed in native IM-MS using gentle nanoelectrospray ionization conditions, which in many cases can preserve condensed-phase stoichiometry. Although much evidence shows that large-scale condensed-phase structure, such as quaternary structure and topology, can also be preserved, it is less clear to what extent smaller-scale structure is preserved in native IM-MS. This review surveys computational and experimental efforts aimed at characterizing compaction and structural rearrangements of protein and protein complex ions upon transfer to the gas phase. A brief summary of gas-phase compaction results from molecular dynamics simulations using multiple common force fields and a wide variety of protein ions is presented and compared to literature IM-MS data.
Keywords: native ion mobility-mass spectrometry, electrospray ionization, molecular dynamics, ion structure, protein, gas phase
1. Introduction.
Native ion mobility-mass spectrometry (IM-MS) is an exciting and rapidly growing field of analytical chemistry with a central goal of learning about condensed-phase properties of biomolecules and biomolecular complexes by exploiting the high sensitivity, chemical specificity, and rapidity of gas-phase techniques [1–3]. In native IM-MS, ion shape and size measurements made using IMS are performed in tandem with mass and charge measurement. Because flexible ions, such as polymers or biomolecules, of identical mass and charge can adopt different shapes under different conditions, the IM and MS steps can provide somewhat orthogonal information about an ion’s properties. The measurements are not completely orthogonal in all cases, however, as an ion’s size (measured in IMS as its collision cross section, CCS, which is akin to its orientationally-averaged “shadow”) tends to increase with mass, albeit with different scaling laws for different classes of shapes, e.g., linear versus globular ions [4]. Native IM-MS almost universally uses nanoelectrospray ionization (nESI) to transfer ions from aqueous buffer solutions into the gas phase, as the small size of the nESI capillaries helps to avoid artefactual oligomer formation induced by in-droplet condensation, and nESI conditions can be controlled to minimize heating and structural changes of the complexes upon transfer into the instrument [1, 5, 6].
At the heart of native IM-MS is a long-standing question: to what extent and under what conditions are condensed-phase properties preserved upon transfer into the gas phase [7]? This question ranges in scope from coarse-grained properties such as native oligomeric state and subunit stoichiometry down to atomistic properties such as the number and location of hydrogen bonds, salt bridges, and coordination of particular functional groups. Although a great wealth of information about condensed phase properties of biomolecules has already been learned from native IM-MS, the answer to this question is far from complete and remains a subject of much current research. This review aims to survey the state of the art concerning changes in condensed-phase structure of globular proteins and protein complexes upon transfer to the gas phase using gentle nESI conditions, focusing on experimental native IM-MS and computational modeling approaches.
2.1. What are “native” nESI conditions?
The low-pressure gas-phase environment of a mass spectrometer (and of most IM instrumentation used in native IM-MS) differs significantly from the aqueous condensed phase in a number of important ways. The relative permittivity is close to 1 (some experiments suggest a slightly higher effective relative permittivity for protein ions owing to the polarizability of various functional groups [8]), which intrinsically increases charge-charge and many charge-dipole interaction energies. This difference, and the absence of solvent molecules to favorably solvate them, might be expected to drive “self-solvation” of charged groups by interacting with nearby oppositely-charged or polar groups. With the effects of water solvent also absent in the gas phase, one might anticipate that gas-phase biomolecular ion structure would differ strongly from condensed-phase structure, with self-solvation driving large-scale rearrangement or even “inversion” of condensed-phase structure [7].
How, then, can structures resembling those in the condensed phase be preserved upon ionization and transfer to the gas phase? While small- or even large-scale rearrangements may be thermodynamically favorable, energy barriers for these rearrangements can be intrinsically quite high and may increase further relative to analogous condensed-phase rearrangements due to the absence of solvent and the low relative permittivity of the gas-phase environment. Because high barriers typically require high activation energy or long timescales to overcome, structures resembling those in the condensed phase can become kinetically trapped upon transfer to the gas phase on the millisecond timescale of native IM-MS experiments (see Figure 1) [9–11]. Gas-phase techniques that probe detailed ion structure, including infrared photodissociation spectroscopy [12], indicate that “memory” of solution-phase structure, such as charge sites in small dibasic organic ions [13], can sometimes be preserved in the gas phase under suitably gentle ionization and transfer conditions. Deliberate heating of biomolecular ions in the gas phase via energetic collisions with background gas, by contrast, can cause extensive unfolding driven by electrostatic repulsion of charge sites (“Collision Induced Unfolding”, CIU) [14, 15], which has been used to infer structural information about proteins and protein complexes [14].
Figure 1.

(A-D) Depiction of charging and self-solvation of charge sites (+ and − symbols) for globular protein ions during evaporation of the nanoelectrospray droplet on the nanosecond to picosecond timescale and (E-G) subsequent structural rearrangement and unfolding at longer timescales. Figure copyright 2008 National Academy of the Sciences, reproduced with permission from the National Academy of the Sciences.
Native IM-MS instrumentation offers a number of observables that can be measured to assess whether ions have likely retained native-like structures. Much evidence indicates that globular biomolecules typically adopt a gas-phase charge just below the “Rayleigh limit” charge for an aqueous droplet with the same volume as the biomolecule [4, 16]. The “Charged Residue Model” (CRM) states that charges originally at the surface of this end-stage droplet are then deposited onto the protein, determining its charge state distribution [17, 18], which is relatively low and narrow as compared to denatured or intrinsically disordered protein ions [19]. Conversely, several authors have shown a correlation between observed charge states of protein ions in native MS and the solvent-accessible surface area of their condensed-phase structures [4, 16]. Similarly, experimental CCS for globular protein ions typically scales as roughly the two-thirds power of mass, as expected for roughly spherical objects of similar density (see Figure 2) [4, 20, 21].
Figure 2.

(A) Schematic illustration of condensed-phase protein structure types and (B) typical charge state and CCS distributions in IM-MS experiments. (C) Average charge states for protein ions formed under non-denaturing (native) conditions (open triangles) and denaturing conditions (all other symbols) as a function of mass and (D) relationships between average charge state and condensed-phase surface area for the same ions. A, B reprinted from Current Opinion in Chemical Biology, v. 42, D. Stuchfield and P. Barran, “Unique insights into intrinsically disordered proteins provided by ion mobility mass spectrometry,” pp. 177–185, copyright 2018, with permission from Elsevier. C, D reprinted with permission from Analytical Chemistry, v. 83, L. Testa, S. Brocca, and R. Grandori, “Charge-Surface Correlation in Electrospray Ionization of Folded and Unfolded Proteins,” pp. 6459–6463, copyright 2011 American Chemical Society.
In the remainder of this review, we focus on the use of computational simulations and IM-MS data to understand the degree of compaction and structural rearrangement undergone by native-like, globular protein ions upon transfer to the gas phase from solution.
2.2. What happens to folded protein ions upon transfer to the gas phase?
As described above, charging slightly below the Rayleigh limit is typically observed for many globular protein and protein complexes upon native nESI, but CCSs measured by IM-MS are often smaller than those predicted using condensed-phase structures drawn from x-ray crystallography or NMR experiments or from condensed-phase MD simulations [19]. This difference in the CCS predicted for condensed-phase structures versus experimental native IMMS values varies from ~0–20% for globular proteins and protein complexes ranging in mass from 2.8 kDa (melittin) to 336 kDa (glutamate dehydrogenase hexamer) (see Figure 3). This gas-phase compaction effect has long been attributed to “self-solvation” of charged and polar sidechains at the surface of the ion [9, 22,23]. IM-MS studies of polyalanine peptide ions indicate that self-solvation of the N-terminal charge on protonated ions of these peptides results in destruction of most α-helical content, illustrating the dramatic effects of self-solvation possible in the gas phase [24].
Figure 3.

Plot of fractional compaction of protein and protein complex ions produced by nESI under native conditions measured by IM-MS in He or N2 buffer gas as compared to CCSs for condensed-phase structures computed using Collidoscope [86]. Experimental CCSs from ref. [84] and [85]. Protein Data Bank identifiers for all protein structures are listed in Figure 4.
Simulations of gas-phase compaction of globular protein and protein complex ions have been undertaken through a variety of molecular dynamics (MD) approaches and with a range of force fields (FF) [10, 23–50]. These approaches, along with examples from the literature, are summarized in Table 1. A recent review by Konermann discusses other MD simulation parameters and their impact on results [51]. With simulated structures in hand, ion CCSs in the relevant IM-MS buffer gas can be predicted using a variety of computational tools with tradeoffs in physical realism and computational expense. Reference [52] provides an overview of these CCS calculation tools, including their advantages and disadvantages, and the most common widely-used tools are summarized in Table 2.
Table 1.
Overview of Types of Theory Used to Simulate Gas-Phase Protein Ions.
| Type of Theory | Important Features | References | |
|---|---|---|---|
| “Beads on a String” | Coarse-grained; amino acids represented as beads with no explicit accounting for atomistic structure | [20, 93] | |
| Molecular Dynamics | AMBER | Atomistic; partial charges based on gas-phase electrostatics computed with QM | [25–34, 39] |
| OPLS | Atomistic (united-atom also available); partial charges empirically optimized to experimental liquid vaporization and density data | [30, 34–40, 63] | |
| CHARMM | Atomistic (united-atom also available); polarizable variants available; partial charges based on small-molecule-water interactions computed using QM | [23,24,36,39,41–46, 68] | |
| GROMOS | United-atom; solvated and vacuum variants available; partial charges based on experimental vaporization, density, and solvation free energy data | [30, 34, 47, 67, 69], this article | |
| MOIL | Atomistic; based on AMBER and OPLS | [10,30,34,47,48, 67] | |
| Martini | Coarse-grained; parametrized based on experimental thermodynamics, including lipid bilayer properties | [49, 69] | |
| ESFF | Atomistic; partial charges derived from ab initio computation of electronegativity and hardness | [50] | |
| Quantum Mechanics | Atomistic; rigorous accounting of polarization, hydrogen-bonding, and electron density; extremely computationally demanding for large biomolecules | [34, 92, 94] | |
| Quantum Mechanics/Molecular Mechanics (QM/MM) | Multi-scale; Dynamics and conformational space explored with MD; local details (such as atomic coordinates, hydrogen bonding, electron density of a small region) computed with QM | [25] |
Table 2.
Overview of Programs for Computing Collision Cross Sections of Gas-Phase Ions.
| Computation Suite | Method | Scattering Type | Explicit Trajectories | Temperature/Charge State-Dependent | 3D Geometry-Dependent | Buffer gases |
|---|---|---|---|---|---|---|
| MOBCAL | PA | - | - | − / − | - | He |
| [89] | PA* | - | - | − / − | + | He |
| EHSS | elastic | + | − / − | + | He | |
| TM | elastic | + | + / + | + | He | |
| WebPSA [95] | PSA* | - | - | + / + | + | He/N2 |
| LCPA [96] | LCPA | - | - | + / + | + | He/N2 |
| Sigma [97], | PA* | - | - | − / − | + | He |
| CCSCalc [98], | ||||||
| IMPACT [21] | ||||||
| EHSSRot [99] | EHSS | elastic | + | + / − | + | He |
| IMoS [100] | PA | - | - | − / − | - | He/N2/ |
| (D)TM | elastic (inelastic) | + | + / + | + | Ar/CO2/air | |
| EHSS (TDHSS) | elastic (inelastic) | + | + / + | + | ||
| Collidoscope [86], HPCCS [101], MobCal-MPI [102] | TM | elastic | + | + / + | + | He/N2 |
Features present (+) or absent (−) in various CCS calculation tools. Methods in bold type use 2-dimensional projections rather than explicit 3-dimensional scattering for CCS computations. Asterisks (*) indicate that a geometry-dependent “shape factor” is used to partially account for 3-dimensional scattering. PA = Projection Approximation, EHSS = Exact Hard Spheres Scattering, PSA = Projected Superposition Approximation, LCPA = Local Collision Probability Approximation, TM = Trajectory Method, D indicates inclusion of diffuse scattering (see ref. [52]). Adapted with permission from Ewing, S.A., Donor, M.T., Wilson, J.W., Prell, J.S., Collidoscope: An Improved Tool for Computing Collisional Cross-Sections by the Trajectory Method, Journal of the American Society for Mass Spectrometry 28 (2017) 587–596, copyright 2017 American Society for Mass Spectrometry.
A number of FFs have been developed for widespread use in both condensed-phase and vacuum MD simulations. These include the AMBER, OPLS-AA, CHARMM, and GROMOS FF families, which are optimized to reproduce experimental thermodynamics or results from quantum mechanical computations on small prototypical molecules (see Table 1) [53, 54]. Because MD is by nature only an approximation of quantum mechanics, each FF has unique tradeoffs in physical realism and computational efficiency. For example, the AMBER and CHARMM FF families use partial charges based on gas-phase electrostatics and aqueous solvation energies computed with quantum mechanics, respectively, but both of these FFs are known to have bias toward high helical content in condensed-phase protein simulations [54]. GROMOS and OPLS-AA are optimized to reproduce experimental liquid vaporization and density data but have a known bias toward high β-sheet content (GROMOS more so than OPLSAA) [54]. Reference [55] compares results from ten different FFs for solution-phase MD simulations of peptides.
MD simulations vary in physical realism and accuracy. However, nearly universally, these simulations predict compaction of globular protein and protein complex ions, in qualitative agreement with native IM-MS experiments. Few studies to date have directly compared results from multiple FFs for the same ions [30, 34, 39] or over multiple different ions [35, 36], thus we present an overview of some important case studies.
2.2.1. Ubiquitin.
Ubiquitin (8.6 kDa) has long been a “fruit fly” of native IM-MS research, as it is a typical small, globular protein with known condensed-phase native structure [56, 57] and solution-phase unfolding behavior [58]. Studies comparing ubiquitin ion structure predicted with MD simulations to native IM-MS experiments illustrate the power of using MD simulations to interpret experimental data while also providing important caveats [29, 31, 39]. Chen and Russell studied temperature-dependent unfolding of ubiquitin ions in water (to simulate heating inside the nESI droplet) and in vacuum using an AMBER FF, comparing the MD results to IM-MS data from Clemmer [59] and Bowers [60]. Aqueous ubiquitin6+ ions were found to retain structures similar to the starting crystal structure up to ~375 K in simulations, with extensive unfolding at higher temperatures that resulted in loss of most helical content. By contrast, ubiquitin6+ ions in vacuum MD compacted to a small degree (~3%, with a CCS of ~950 Å2) with accompanying loss of secondary structure, but significant unfolding was not observed for temperatures below 600 K. Above 600 K, a large number of energetically-competitive structures were predicted, with a correspondingly wide spread in CCSs [29].
Structural effects of non-volatile salts on compaction and unfolding of ubiquitin have also been investigated with MD and experiment. Starting with folded structures embedded in aqueous nanodrops containing 16 sodium ions, McAllister et al. used a CHARMM FF to simulate charging and desiccation of ubiquitin6+ (with sodium ion adducts as the source of charge rather than protons) [44]. Both the final charge of the protein and the predicted CCS were found to agree with experimentally measured values, within uncertainty, and were found to be consistent with the CRM and kinetic trapping of native-like structure. Further CHARMM simulations by Bartman et al. indicate that common biological metal ion adducts (such as Na+ and Ca2+) can form multidentate interactions with polar groups in ubiquitin6+ that make it resistant to gas-phase unfolding [61], a result echoed by a report from Wagner et al. that chloride anion adducts can have a similar effect [62]. Bartman et al. caution that the MD-predicted structures for activated, metal-adducted ubiquitin6+ have structures bearing little resemblance to the crystal structure, despite similar calculated and experimental CCSs [61].
Radical-Directed Dissociation experiments by Ly and Julian on ubiquitin ions formed from denaturing solutions were used to provide experimentally measured distance constraints for MD simulations of ion structure [38]. Intriguingly, they found that constrained ubiquitin 4+ and 6+ charge states adopted structures in OPLS-AA MD simulations that differ significantly from the crystal structure and from unconstrained simulated structures, even though they possess CCSs within the range of those measured experimentally. These results provide an important additional caveat to interpreting MD-simulated structures with CCSs consistent with experimental values as definitive evidence that MD structures do in fact represent the experimental ion population, even when they are compact.
2.2.2. Cytochrome c.
Cytochrome c (12 kDa), which contains a covalently-bound heme group, is another “fruit fly” of IM-MS and has served as a benchmark for gas-phase MD simulations of relevance to native IM-MS [9, 10, 41, 48]. Steinberg et al. performed MD simulations of solvent evaporation from and gas-phase collapse of native cytochrome c [10, 48], building off of earlier CHARMM MD and IM-MS work by Jarrold [41]. Using the MOIL FF (a hybrid of AMBER and OPLS FFs), Steinberg et al. found that cytochrome c6+, starting with its aqueous NMR structure and surrounded by 182 water molecules cools rapidly via loss of some, but not all, water molecules over a period of ~100 ps [48]. Little significant change in the structure of the protein was observed in any case, and the authors concluded that evaporative cooling prevents complete desolvation or restructuring of the ion in the absence of collisions or absorption of blackbody radiation on the picosecond timescale. A subsequent constant-energy MOIL FF study, starting with a completely desolvated cytochrome c7+ ion, predicted a number of structural changes [10]. Charged sidechains were observed to self-solvate within 0–20 ps, and the typical number of salt bridges (SBs) was found to increase over ~10 ps from 6 to 17.2. The average number of hydrogen bonds (HBs) involving positively-charged sidechains increased dramatically from 0 to 11.7 on a similar timescale, but the number of HBs involving negatively-charged sidechains increased only slightly from 5 to 6.3. Charged sidechains moved to decrease the net dipole by ~1/3 during the first 10 ps. No significant breaking of non-covalent bonds was observed during the 4.2 ns simulations, and thus the final structure possessed a similar backbone conformation to the aqueous NMR structure. The MD-predicted integrity of the folded structure was found consistent with experimental native electron capture dissociation experiments [10].
Fascinating IM-MS experiments by Warnke et al., in which charged lysine sidechains of cytochrome c were capped with large crown ether ligands during the nESI process, indicate that self-solvation of charged sidechains in ordinary native IM-MS may actually prevent some collapse of ion structure [22]. Native charge states of cytochrome c7–8+ adopted successively lower CCSs as the number of adducted crown ethers increased from 1 to 5, despite the large size of the ligands. The authors interpreted these results to mean that, in the absence of self-solvation of charged surface residues by polar groups on the protein, the interior of the ion collapses significantly on transfer to the gas phase.
2.2.3. Retention and loss of condensed-phase structure in peptides and small proteins.
Melittin (2.8 kDa) is a 26-amino acid membrane protein from bee venom, with a condensed-phase structure consisting of two α-helices joined by a proline “kink” [26]. This structure is known to be more stable in less polar condensed-phase environments (e.g., methanol [63] or lipid membranes [64]) than in water. Contrasting with results for singly protonated polyalanine peptides with 3–20 amino acids [24] and for Trp-cage1+ (2.2 kDa) [40], melittin3+ formed from acidified 1:1water:methanol was found with a combination of mass-analyzed ion kinetic energy (MIKE) experiments and CHARMM FF MD to retain its helical structure in the gas phase [23]. The authors attributed the retention of structure to the “highly oriented network of hydrogen bonds along the polypeptide backbone” and noted that, with no water surrounding the ions to compete with self-solvation, the intrinsic H-bond network is left undisturbed. Florance et al. performed AMBER FF MD simulated annealing computations between 0 and 800 K to identify structure families consistent with IM-MS data for melittin3–4+ formed by nESI from aqueous solutions containing 0–100% methanol [26]. Computed CCSs correlated with the number (1–3) of α-helical regions in the simulated structures. The MD results supported assignment of a range of partially helical structures to the ions, and the authors noted that there was “slight evidence for solvent memory,” with more helical structures being formed in higher-fraction methanol solutions.
Further support for retention of native-like folds in small, globular native protein ions comes from infrared multiple-photon dissociation spectroscopy results for gas-phase myoglobin (16.7 kDa) and β-lactoglobulin (18 kDa) ions. Seo et al. report that myoglobin8+ and β-lactoglobulin8+ ions retain their highly α-helix- and β-sheet-rich structures, respectively, upon nESI [65]. Driven by repulsion between positively-charged sidechains, β-lactoglobulin ions with higher charge states adopt progressively greater α-helix content. Such studies are expected in the future to provide important targets for MD simulations and assist in interpretation of IM-MS data.
2.2.4. Protein complexes.
Larger proteins and protein complexes, which contain many more degrees of freedom than peptides and small proteins, can be considerably more computationally expensive to simulate with MD and often intractable with high-level QM computations. Ruotolo et al. demonstrated a simple coarse-grained approach to study gas-phase collapse of trp RNA-binding attenuation protein (TRAP) 11-mer assemblies (~90 kDa), in which each protein in the assembly is treated as a sphere [66]. Their simulations indicated that lower native charge states of the assemblies have experimental CCSs consistent with retention of planar ring structures, but higher native charge states collapse ~14% to close-packed, roughly spherical assemblies, with some evidence for additional partially compacted structures. These results illustrate the sensitivity with which native IM-MS can be used to study large-scale gas-phase structural rearrangement, even with simple computational models.
OPLS-AA FF MD simulations in combination with native IM-MS experiments by Hall et al. further examined desolvation-induced compaction as well as gas-phase collision-induced compaction of serum amyloid P component pentamer (SAP, 125 kDa), avidin tetramer (64 kDa), transthyretin tetramer (TTR, 56 kDa), and TRAP 11-mer [35]. Under minimal activation conditions, SAP18–30+ assemblies were found to have experimental CCSs slightly larger (~70 nm2) than those computed for 300 K 18+ ions with native-like ring structures (~68 nm2). Low charge states were predicted by MD to collapse up to ~7% by elimination of the ring’s central cavity upon heating by several hundred K, qualitatively consistent with observed ~10% compaction of these charge states upon collisional activation in IM-MS experiments.
Friemann et al. studied changes in detergent micelle-embedded transmembrane protein β-barrel regions using a GROMOS FF and found that the micelle shields the transmembrane region from structural collapse upon transfer to vacuum [67]. By contrast, hairpin loops extending outside the micelle were found to collapse. They concluded that membrane proteins embedded in micelles are “not very sensitive to the vacuum environment,” a property that may prove highly beneficial in studying their condensed-phase structures using gas-phase measurements. The Robinson group has explored lipid binding to native membrane protein complexes embedded in detergent micelles and “Nanodisc” lipid bilayers and compared them to MD simulations to identify lipid binding preferences [68–70]. In their experiments, all but a few lipids are stripped from the native protein ions to reveal those that closely associate with the protein. The remarkable agreement between simulated and experimentally determined lipid binding preferences, which have been extended to quantitative measurements of lipid binding thermodynamics by the Laganowsky group [71–73], provides indirect evidence that tightly bound lipids do not move significantly during nESI or the gas-phase stripping process.
2.2.5. Explicit modeling of nESI droplet evaporation and ion charging process.
During the last decade or so, efforts to accurately simulate desiccation of biomolecules within ESI droplets and concomitant acquisition of charge have been undertaken by several groups [36, 37, 44, 45, 51, 67, 74–77]. In addition to the aforementioned simulations by Steinberg et al. [48], the Konermann and Consta groups have applied MD simulations to charged droplets containing biomolecules to learn about ionization mechanisms, ion compaction upon desiccation, and heating-induced unfolding and dissociation. These computations can be especially sensitive to choice of water model, treatment of electrostatics, temperature control, and simulation length, among other user-determined variables [51]. These simulations provide insight into native charge distributions of proteins and protein complexes produced by nESI [44], droplet evaporation dynamics [75–77], as well as the role of charge hopping and charge-charge repulsion in both the ionization and collision-induced dissociation processes [36].
3.1. Comparison of MD results with different force fields for ion compaction upon transfer to vacuum.
As discussed above, electrostatics, hydrogen bonding, and other local molecular properties and interactions can play cooperative or competitive roles in determining both solution and gas-phase structure for proteins and protein ions. Because MD FFs differ in their treatment of these properties and interactions [53], different FFs may in principle lead to different conclusions about both local and global protein ion structure even if computed CCSs are similar. Despite the wealth of information about gas-phase native-like protein ions gained from the comparison of MD and experimental IM-MS results discussed above, results from different FFs have only rarely been compared for the same ions, and typically only for one or perhaps a small number of separate proteins. One advantage of comparing results from different FFs is that biases of specific FFs as well as commonalities can be identified. To contribute to this discussion, we present here a brief comparison of vacuum MD ion compaction results for 5 different common FFs (AMBER94 [78], OPLS-AA/L [79], CHARMM27 [80, 81], GROMOS96 43a2 [82], and GROMOS96 54b7 [83]) and 17 different natively-charged proteins with well-characterized experimental CCSs in helium and nitrogen that serve as calibration standards for native IM-MS experiments [84, 85].
3.1.1. MD simulation method.
Initial structure for all 17 proteins were taken from the Protein Data Bank (PDB) structures indicated in Figure 4A, with any missing residues appended using PyMol. All MD simulations were conducted with GROMACS v. 2016.4. After a brief (1 ns) relaxation of any added residues in explicit water solvent, all water molecules were deleted, and low-energy protomers for these structures with native charge states were identified using the Charge Placement algorithm in Collidoscope, leaving SB structures from the MD simulations intact [86]. Subsequent fixed-charge-site vacuum MD simulations for the protonated ions consisted of a short vacuum relaxation step followed by a 5 ns production run at 300 K with a Berendsen thermostat. CCSs for typical final structures were computed using He or N2 buffer gas in Collidoscope with the Trajectory Method. Variations of this procedure were conducted for alcohol dehydrogenase tetramer to assess the sensitivity of the results to vacuum simulation length (5, 50, and 500 ns) and velocity seeding, and GROMOS96 43a2 heat ramp studies (from 300 to 600 K in 25 K increments every 5 ns) were conducted for melittin, insulin monomer, and ubiquitin. These variant methods were found to result in only minor differences (typically no more than ~1%) in computed CCSs from the above-described method, with the exception of the heat ramp studies, which predicted up to 4% variability of the CCS between 300 and 600 K.
Figure 4.

(A) Structures of protein and protein complex ions from Fig. 3 before (green mesh) and after (blue solid) MD simulation of gas-phase compaction using GROMOS96 43a2 FF (see text). Protonated ions simulated (with Protein Data Bank identifiers indicated below structures) are melittin4+ (2MLT), insulin3+ (3E7Y), ubiquitin5+ (1UBQ), insulin dimer5+ (5BTS), cytochrome c7+ (1HRC), β-lactoglobulin monomer7+ (3BLG), insulin hexamer10+ (4EY9), transthyretin tetramer15+ (1F41), avidin tetramer16+ (1AVE), bovine serum albumin15+ (4F5S), concanavalin A tetramer21+ (3CNA), serum amyloid P component pentamer24+ (1SAC), alcohol dehydrogenase tetramer24+ (4W6Z), pyruvate kinase tetramer32+ (1F3W), serum amyloid P component decamer33+ (2A3W), glutamate dehydrogenase hexamer40+ (3JCZ). (B) Plot of average percent difference between experimental CCS data from ref. [84] and [85] and CCSs computed for MD-compacted ions shown in A using Collidoscope for each of the 5 FFs tested (see text). (C) Schematic depiction of typical degree of surface (red) and interior (dark blue) compaction predicted by MD simulations using 5 different FFs for ions represented in A and B. Embedded circles represent the typical size (small: 5–12 Å diameter, medium: 12–25 Å, and large: ≥ 25 Å) of cavities in the ions that are fully eliminated (dark blue), sometimes eliminated (light blue), or not eliminated (white) during the MD simulations.
3.1.2. Maximal degree of compaction predicted from MD simulations.
All five FFs tested produced structures that were on average compacted relative to the PDB structures, as measured by computed CCS. The GROMOS FFs resulted in the greatest global compaction relative to the PDB structures (up to 20%), whereas the other three force fields compacted ions only up to 9–10% (see Table 3). To provide a coarse-grained picture of ion compaction, ion “surface” was defined as the set of all residues with at least 30% water solvent accessibility as determined with SwissPDBViewer, and the remainder of the ion was defined as the “interior”. At the global structural level, all five FFs compacted the ion surface relative to the PDB structure, resulting in a smaller number of surface residues and a larger number of polar contacts involving charged residues initially at the surface. However, there were notable differences in the degree to which the interior of the ions were compacted by each FF (see Table 3 and Figure 4), as determined by using the computed CCS of the interior as a measure of its size. Thus, differences in global compaction for the five FFs were largely attributed to the extent of compaction of the interior of the ions.
Table 3.
Summary of Results from MD Simulations of Ion Compaction on Transfer to Gas Phase.
| Force Field | Global | Surface | Interior | Secondary Structure | Cavities/Grooves | Non-covalent Interactions | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Maximum % Compaction (He/N2) | Mean Change in Number of Surface Residues | Mean Number Polar Contacts for Charged Sidechains | Mean Change in Interior CCS, He | Mean Change in Interior CCS, N2 | Mean Change in α-Helical Content | Mean Change in β-Sheet Content | Diameter 5–12 Å → 25 Å | Mean Number of HB (% NHBmax) |
Mean Number of SBs | |
| (1.3 for condensed-phase structures) | (43 for condensed-phase structures) | (8 for condensed-phase structures) | ||||||||
| GROMOS96 43a2 | 20/18 | −42% | 4.1 | −1.2% | −6.3% | −4.1% | −0.1% |  |
49 | 9 |
| GROMOS96 54b7 | 14/12 | −28% | 1.5 | +0.3% | −2.0% | −1.6% | −1.8% |  |
49 | 12 |
| AMBER94 | 10/10 | −28% | 3.7 | +2.9% | −1.1% | −0.9% | −2.1% |  |
48 | 15 |
| OPLS-AA/L | 9/9 | −25% | 3.7 | +2.0% | −0.4% | −6.8% | −1.0% |  |
44 | 11 |
| CHARMM27 | 9/9 | −20% | 3.7 | +3.6% | +1.0% | −2.7% | −0.1% | 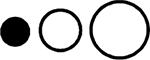 |
45 | 14 |
Maximum % compaction refers to largest % compaction as measured by computed CCS (condensed-phase − collapsed)/condensed-phase among all 17 protein and protein complex ions studied (see Figure 4). Methods for determining number of non-covalent interactions described in text. Shading of cavities same as in Figure 4.
3.1.3. Changes in secondary structure and number of hydrogen bonds and salt bridges.
All five FFs resulted in minor loss of α-helical content (up to 7% for OPLS-AA/L) and very minor loss of β-sheet content (up to 2% for AMBER94). These results were largely consistent with known tendencies of these FFs (see above) [54]. Using Marklund’s algorithm for determining the “maximum possible” number of hydrogen bonds for each ion (NHBmax) [87] and PyMol to determine the number of hydrogen bonds actually present in each structure (NHB), NHB/NHBmax was found on the whole to increase for each FF in vacuum, with the greatest increase for the GROMOS FFs. By contrast, the change in the number of SBs (determined using PyMol) increased among the five FFs but exhibited no clear dependence on mass.
3.1.4. Collapse of cavities and grooves.
Many of the proteins and protein complexes studied here possess sizeable cavities or grooves in their condensed-phase structures. The largest cavities (~25 Å in diameter) were not completely collapsed at the end of the simulations by any of the FFs, whereas all FFs resulted in collapse of small cavities and grooves with diameters ranging from ~5–12 Å. For cavities and grooves with diameters in between these extremes (in β-lactoglobulin dimer and concanavalin A tetramer), significant differences were observed between the FFs. The GROMOS FFs collapsed these intermediate-sized cavities, whereas CHARMM27 collapsed neither, and AMBER94 and OPLS-AA/L completely collapsed only one of the two cavities. Taken together with the results described above, this assessment of gas-phase collapse leads to the schematic depiction of compaction represented in Figure 4C for the ions investigated with these five FFs.
3.1.5. Comparison of MD results to native IM-MS CCS data.
A comparison of average differences in computed and experimental CCSs in both He and N2 buffer gas is shown in Figure 4B. Both GROMOS FFs outperformed the others in these simple simulations in reproducing experimental CCSs in both buffer gases. As seen in Figure 4B, the average difference in CCSs between simulated structures and experiment was 0 ± 4% and −4 ± 3% for GROMOS96 43a2 in N2 and He buffer gas, respectively. Results from GROMOS96 54b7 were slightly better for He buffer gas (2 ± 4%), especially for ions below ~36 kDa in mass. The other three FFs (AMBER94, OPLS-AA/L, and CHARMM27) resulted in less average compaction, with the average difference between simulation and experiment being more than one standard deviation away from 0 (see Figure 4B). These trends were confirmed by MD simulation of three additional native-like membrane protein complex ions (multi-antimicrobial extrusion protein, 50 kDa; aquaporin Z tetramer, 99 kDa; and ammonia channel B trimer, 127 kDa) [88], which have an average He CCS difference of 0 ± 4% between simulation and experiment using GROMOS96 43a2 and somewhat higher differences for the other FFs. Based on the results from this simple and relatively low-expense computational method, we recommend use of GROMOS96 43a2 for similar simulations to predict experimental CCSs in N2 or He buffer gas. We anticipate that slight reparametrization of He Lennard-Jones parameters in Collidoscope (and other CCS calculation programs that use the same parameters, including MOBCAL [89]) based on these results may further improve accuracy of He CCS prediction in native IM-MS [86].
4. Summary and Outlook.
As the above survey of the literature shows, the last two decades of research into the structure of biomolecular ions upon transfer from solution into the gas phase indicates that many ions can retain native-like structure, including secondary structure, upon native nESI through the timescale of IM-MS experiments. Although important exceptions, especially for smaller ions such as peptides, indicate that conversion to more stable gas-phase structures with accompanying loss of native-like structure can sometimes occur, the vast majority of results are very promising for native IM-MS work aimed at inferring solution-phase structure from gas-phase data. Both IM-MS data and MD simulations using a variety of FFs indicate that native-like ions compact by several percent upon desiccation and self-solvation of charge sites, although detailed results can be discrepant between FFs. This highlights the need for comparison between FFs in MD simulations, and we hope that the example given in section 3 of this review illustrates a path forward for more reliable interpretation of IM-MS data by use of MD simulation results.
Very recent advances in interpretation of IM-MS data, including simulation of entire experimental CCS distributions [90], are likely to provide a more complete, holistic picture of ion structure by simultaneously matching large sets of data for individual ions rather than single CCS values. Combining these approaches with structural “fingerprinting” via collision-induced unfolding [14] as well as with H/D exchange experiments [50], novel dissociation methods [91], ion spectroscopy [12, 92], and other experimental methods should provide yet more structural constraints for MD simulations. The recent inclusion of nanoscale water droplet environments [45, 75] and mobile charges [36] in MD simulations represents a step forward in realism, and more accurate parametrization of water models for nanoscale droplets is expected to provide unparalleled insight into native ion compaction and structure.
Highlights.
Molecular dynamics simulations provide insight into ion desolvation and collapse
Self-solvation of charged sidechains drives ion surface collapse
Much condensed-phase structure can be preserved up to microsecond timescale
Common molecular dynamics force fields predict different degrees of compaction of ion interior and perform differently as compared to experimental IM-MS data
Acknowledgements.
This work was supported by the National Institutes of Health (grant numbers R21AI125804–2 [to J.S.P.] and T32 GM007759 [to A.D.R.]). A.D.R. is an ARCS Scholar supported by the ARCS Oregon Chapter.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests Statement.
The authors declare no competing interests.
References
- [1].Heck AJR, van den Heuvel RHH, Investigation of Intact Protein Complexes by Mass Spectrometry, Mass Spectrom. Rev 23 (2004) 368–389. [DOI] [PubMed] [Google Scholar]
- [2].Konijnenberg A, Butterer A, Sobott F, Native Ion Mobility-Mass Spectrometry and Related Methods in Structural Biology, Biochim. Biophys. Acta, Proteins Proteomics 1834 (2013) 1239–1256. [DOI] [PubMed] [Google Scholar]
- [3].Gabelica V, Marklund E, Fundamentals of Ion Mobility Spectrometry, Curr.Opin. Chem. Biol 42 (2018) 51–59. [DOI] [PubMed] [Google Scholar]
- [4].Testa L, Brocca S, Grandori R, Charge-Surface Correlation in Electrospray Ionization of Folded and Unfolded Proteins, Anal. Chem 83 (2011) 6459–6463. [DOI] [PubMed] [Google Scholar]
- [5].Benesch JLP, Ruotolo BT, Simmons DA, Robinson CV, Protein Complexes in the Gas Phase: Technology for Structural Genomics and Proteomics, Chem. Rev 107 (2007) 3544–3567. [DOI] [PubMed] [Google Scholar]
- [6].Wheeler LC, Donor MT, Prell JS, Harms MJ, Multiple Evolutionary Origins of Ubiquitous Cu2+ and Zn2+ Binding in the S100 Protein Family, PLOS ONE 11 (2016) e0164740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wolynes PG, Biomolecular Folding in Vacuo!!!(?), Proc. Natl. Acad. Sci. U. S. A 92 (1995) 2426–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schnier PD, Gross DS, Williams ER, Electrostatic Forces and Dielectric Polarizability of Multiply Protonated Gas-Phase Cytochrome C Ions Probed by Ion/Molecule Chemistry, J. Am. Chem. Soc 117 (1995) 6747–6757. [Google Scholar]
- [9].Breuker K, McLafferty FW, Stepwise Evolution of Protein Native Structure with Electrospray into the Gas Phase, 10−12 to 102 S, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 18145–18152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Steinberg MZ, Elber R, McLafferty FW, Gerber RB, Breuker K, Early Structural Evolution of Native Cytochrome C after Solvent Removal, ChemBioChem 9 (2008) 2417–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bakhtiari M, Konermann L, Protein Ions Generated by Native Electrospray Ionization: Comparison of Gas Phase, Solution, and Crystal Structures, J. Phys. Chem. B 123 (2019) 1784–1796. [DOI] [PubMed] [Google Scholar]
- [12].Polfer NC, Infrared Multiple Photon Dissociation Spectroscopy of Trapped Ions, Chem. Soc. Rev 40 (2011) 2211–2221. [DOI] [PubMed] [Google Scholar]
- [13].Steill JD, Oomens J, Gas-Phase Deprotonation of P-Hydroxybenzoic Acid Investigated by Ir Spectroscopy: Solution-Phase Structure Is Retained Upon Esi, J. Am. Chem. Soc 131 (2009) 13570–13571. [DOI] [PubMed] [Google Scholar]
- [14].Dixit SM, Polasky DA, Ruotolo BT, Collision Induced Unfolding of Isolated Proteins in the Gas Phase: Past, Present, and Future, Curr. Opin. Chem. Biol 42 (2018) 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Donor MT, Mroz AM, Prell JS, Experimental and Theoretical Investigation of Overall Energy Deposition in Surface-Induced Unfolding of Protein Ions, Chem. Sci 10 (2019) 4097–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kaltashov IA, Mohimen A, Estimates of Protein Surface Areas in Solution by Electrospray Ionization Mass Spectrometry, Anal. Chem 77 (2005) 5370–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fernandez J de la Mora, Electrospray Ionization of Large Multiply Charged Species Proceeds Via Dole’s Charged Residue Mechanism, Anal. Chim. Acta 406 (2000) 93–104. [Google Scholar]
- [18].Dole M, Mack LL, Hines RL, Mobley RC, Ferguson LD, Alice MB, Molecular Beams of Macroions, J. Chem. Phys 49 (1968) 2240–2249. [Google Scholar]
- [19].Jurneczko E, Barran PE, How Useful Is Ion Mobility Mass Spectrometry for Structural Biology? The Relationship between Protein Crystal Structures and Their Collision Cross Sections in the Gas Phase, Analyst 136 (2011) 20–28. [DOI] [PubMed] [Google Scholar]
- [20].Donor MT, Ewing SA, Zenaidee MA, Donald WA, Prell JS, Extended Protein Ions Are Formed by the Chain Ejection Model in Chemical Supercharging Electrospray Ionization, Anal. Chem 89 (2017) 5107–5114. [DOI] [PubMed] [Google Scholar]
- [21].Marklund Erik G., Degiacomi Matteo T., Robinson Carol V., Baldwin Andrew J., Justin L.P. Benesch, Collision Cross Sections for Structural Proteomics, Structure 23 (2015) 791–799. [DOI] [PubMed] [Google Scholar]
- [22].Warnke S, Von Helden G, Pagel K, Protein Structure in the Gas Phase: The Influence of Side-Chain Microsolvation, J. Am. Chem. Soc 135 (2013) 1177–1180. [DOI] [PubMed] [Google Scholar]
- [23].Kaltashov IA, Fenselau C, Stability of Secondary Structural Elements in a Solvent-Free Environment: The A Helix, Proteins 27 (1997) 165–170. [DOI] [PubMed] [Google Scholar]
- [24].Hudgins RR, Mao Y, Ratner MA, Jarrold MF, Conformations of Glynh+ and Alanh+ Peptides in the Gas Phase, Biophys. J 76 (1999) 1591–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li J, Lyu W, Rossetti G, Konijnenberg A, Natalello A, Ippoliti E, Orozco M, Sobott F, Grandori R, Carloni P, Proton Dynamics in Protein Mass Spectrometry, J. Phys. Chem. Lett 8 (2017) 1105–1112. [DOI] [PubMed] [Google Scholar]
- [26].Florance HV, Stopford AP, Kalapothakis JM, McCullough BJ, Bretherick A, Barran PE, Evidence for A-Helices in the Gas Phase: A Case Study Using Melittin from Honey Bee Venom, Analyst 136 (2011) 3446–3452. [DOI] [PubMed] [Google Scholar]
- [27].Kulesza A, Marklund EG, MacAleese L, Chirot F, Dugourd P, Bringing Molecular Dynamics and Ion-Mobility Spectrometry Closer Together: Shape Correlations, Structure-Based Predictors, and Dissociation, J. Phys. Chem. B 122 (2018) 8317–8329. [DOI] [PubMed] [Google Scholar]
- [28].Pagel K, Natan E, Hall Z, Fersht AR, Robinson CV, Intrinsically Disordered P53 and Its Complexes Populate Compact Conformations in the Gas Phase, Angew. Chem., Int. Ed 52 (2013) 361–365. [DOI] [PubMed] [Google Scholar]
- [29].Chen S-H, Russell DH, How Closely Related Are Conformations of Protein Ions Sampled by Im-Ms to Native Solution Structures?, J. Am. Soc. Mass Spectrom 26 (2015) 1433–1443. [DOI] [PubMed] [Google Scholar]
- [30].Krone MG, Baumketner A, Bernstein SL, Wyttenbach T, Lazo ND, Teplow DB, Bowers MT, Shea J-E, Effects of Familial Alzheimer’s Disease Mutations on the Folding Nucleation of the Amyloid B-Protein, J. Mol. Biol 381 (2008) 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Segev E, Wyttenbach T, Bowers MT, Gerber RB, Conformational Evolution of Ubiquitin Ions in Electrospray Mass Spectrometry: Molecular Dynamics Simulations at Gradually Increasing Temperatures, Phys. Chem. Chem. Phys 10 (2008) 3077–3082. [DOI] [PubMed] [Google Scholar]
- [32].Bernstein SL, Liu D, Wyttenbach T, Bowers MT, Lee JC, Gray HB, Winkler JR, A-Synuclein: Stable Compact and Extended Monomeric Structures and Ph Dependence of Dimer Formation, J. Am. Soc. Mass Spectrom 15 (2004) 1435–1443. [DOI] [PubMed] [Google Scholar]
- [33].Cole HL, Kalapothakis JMD, Bennett G, Barran PE, MacPhee CE, Characterizing Early Aggregates Formed by an Amyloidogenic Peptide by Mass Spectrometry, Angew. Chem., Int. Ed 49 (2010) 9448–9451. [DOI] [PubMed] [Google Scholar]
- [34].Marchese R, Grandori R, Carloni P, Raugei S, A Computational Model for Protein Ionization by Electrospray Based on Gas-Phase Basicity, J. Am. Soc. Mass Spectrom 23 (2012) 1903–1910. [DOI] [PubMed] [Google Scholar]
- [35].Hall Z, Politis A, Bush MF, Smith LJ, Robinson CV, Charge-State Dependent Compaction and Dissociation of Protein Complexes: Insights from Ion Mobility and Molecular Dynamics, J. Am. Chem. Soc 134 (2012) 3429–3438. [DOI] [PubMed] [Google Scholar]
- [36].Popa V, Trecroce DA, McAllister RG, Konermann L, Collision-Induced Dissociation of Electrosprayed Protein Complexes: An All-Atom Molecular Dynamics Model with Mobile Protons, J. Phys. Chem. B 120 (2016) 5114–5124. [DOI] [PubMed] [Google Scholar]
- [37].Konermann L, Molecular Dynamics Simulations on Gas-Phase Proteins with Mobile Protons: Inclusion of All-Atom Charge Solvation, J. Phys. Chem. B 121 (2017) 8102–8112. [DOI] [PubMed] [Google Scholar]
- [38].Ly T, Julian RR, Elucidating the Tertiary Structure of Protein Ions in Vacuo with Site Specific Photoinitiated Radical Reactions, J. Am. Chem. Soc 132 (2010) 8602–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Meyer T, de la Cruz X, Orozco M, An Atomistic View to the Gas Phase Proteome, Structure 17 (2009) 88–95. [DOI] [PubMed] [Google Scholar]
- [40].Patriksson A, Adams CM, Kjeldsen F, Zubarev RA, van der Spoel D, A Direct Comparison of Protein Structure in the Gas and Solution Phase: The Trp-Cage, J. Phys. Chem. B 111 (2007) 13147–13150. [DOI] [PubMed] [Google Scholar]
- [41].Jarrold MF, Unfolding, Refolding, and Hydration of Proteins in the Gas Phase, Acc. Chem. Res 32 (1999) 360–367. [Google Scholar]
- [42].Devine PWA, Fisher HC, Calabrese AN, Whelan F, Higazi DR, Potts JR, Lowe DC, Radford SE, Ashcroft AE, Investigating the Structural Compaction of Biomolecules Upon Transition to the Gas-Phase Using Esi-Twims-Ms, J. Am. Soc. Mass Spectrom 28 (2017) 1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Baumketner A, Bernstein SL, Wyttenbach T, Bitan G, Teplow DB, Bowers MT, Shea J-E, Amyloid B-Protein Monomer Structure: A Computational and Experimental Study, Protein Sci 15 (2006) 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].McAllister RG, Metwally H, Sun Y, Konermann L, Release of Native-Like Gaseous Proteins from Electrospray Droplets Via the Charged Residue Mechanism: Insights from Molecular Dynamics Simulations, J. Am. Chem. Soc 137 (2015) 12667–12676. [DOI] [PubMed] [Google Scholar]
- [45].Duez Q, Metwally H, Konermann L, Electrospray Ionization of Polypropylene Glycol: Rayleigh-Charged Droplets, Competing Pathways, and Charge State-Dependent Conformations, Anal. Chem 90 (2018) 9912–9920. [DOI] [PubMed] [Google Scholar]
- [46].Saikusa K, Fuchigami S, Takahashi K, Asano Y, Nagadoi A, Tachiwana H, Kurumizaka H, Ikeguchi M, Nishimura Y, Akashi S, Gas-Phase Structure of the Histone Multimers Characterized by Ion Mobility Mass Spectrometry and Molecular Dynamics Simulation, Anal. Chem 85 (2013) 4165–4171. [DOI] [PubMed] [Google Scholar]
- [47].Borysik AJ, Kovacs D, Guharoy M, Tompa P, Ensemble Methods Enable a New Definition for the Solution to Gas-Phase Transfer of Intrinsically Disordered Proteins, J. Am. Chem. Soc 137 (2015) 13807–13817. [DOI] [PubMed] [Google Scholar]
- [48].Steinberg MZ, Breuker K, Elber R, Gerber RB, The Dynamics of Water Evaporation from Partially Solvated Cytochrome C in the Gas Phase, Phys. Chem. Chem. Phys 9 (2007) 4690–4697. [DOI] [PubMed] [Google Scholar]
- [49].Fegan SK, Thachuk M, Suitability of the Martini Force Field for Use with Gas-Phase Protein Complexes, J. Chem. Theory Comput 8 (2012) 1304–1313. [DOI] [PubMed] [Google Scholar]
- [50].Valentine SJ, Clemmer DE, Temperature-Dependent H/D Exchange of Compact and Elongated Cytochrome C Ions in the Gas Phase, J. Am. Soc. Mass Spectrom 13 (2002) 506–517. [DOI] [PubMed] [Google Scholar]
- [51].Konermann L, Metwally H, McAllister RG, Popa V, How to Run Molecular Dynamics Simulations on Electrospray Droplets and Gas Phase Proteins: Basic Guidelines and Selected Applications, Methods 144 (2018) 104–112. [DOI] [PubMed] [Google Scholar]
- [52].Prell JS, Chapter One - Modelling Collisional Cross Sections, in: Donald WA, Prell JS, Comprehensive Analytical Chemistry, Elsevier, Amsterdam, 2019, pp. 1–22. [Google Scholar]
- [53].Guvench O, MacKerell AD Jr., Comparison of Protein Force Fields for Molecular Dynamics Simulations, in: Kukol A, Molecular Modeling of Proteins, Humana Press, Totowa, NJ, 2008, pp. 63–88. [DOI] [PubMed] [Google Scholar]
- [54].Smith MD, Rao JS, Segelken E, Cruz L, Force-Field Induced Bias in the Structure of Aβ21–30: A Comparison of Opls, Amber, Charmm, and Gromos Force Fields, J. Chem. Inf. Model 55 (2015) 2587–2595. [DOI] [PubMed] [Google Scholar]
- [55].Cino EA, Choy W-Y, Karttunen M, Comparison of Secondary Structure Formation Using 10 Different Force Fields in Microsecond Molecular Dynamics Simulations, J. Chem. Theory Comput 8 (2012) 2725–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Vijay-Kumar S, Bugg CE, Cook WJ, Structure of Ubiquitin Refined at 1.8 Å Resolution, J. Mol. Biol 194 (1987) 531–544. [DOI] [PubMed] [Google Scholar]
- [57].Weber PL, Brown SC, Mueller L, Sequential Proton Nmr Assignments and Secondary Structure Identification of Human Ubiquitin, Biochemistry 26 (1987) 7282–7290. [DOI] [PubMed] [Google Scholar]
- [58].Ibarra-Molero B, Makhatadze GI, Sanchez-Ruiz JM, Cold Denaturation of Ubiquitin, Biochim. Biophys. Acta-Protein Struct. Molec. Enzym 1429 (1999) 384–390. [DOI] [PubMed] [Google Scholar]
- [59].Koeniger SL, Clemmer DE, Resolution and Structural Transitions of Elongated States of Ubiquitin, J. Am. Soc. Mass Spectrom 18 (2007) 322–331. [DOI] [PubMed] [Google Scholar]
- [60].Wyttenbach T, Bowers MT, Structural Stability from Solution to the Gas Phase: Native Solution Structure of Ubiquitin Survives Analysis in a Solvent-Free Ion Mobility–Mass Spectrometry Environment, J. Phys. Chem. B 115 (2011) 12266–12275. [DOI] [PubMed] [Google Scholar]
- [61].Bartman CE, Metwally H, Konermann L, Effects of Multidentate Metal Interactions on the Structure of Collisionally Activated Proteins: Insights from Ion Mobility Spectrometry and Molecular Dynamics Simulations, Anal. Chem 88 (2016) 6905–6913. [DOI] [PubMed] [Google Scholar]
- [62].Wagner ND, Kim D, Russell DH, Increasing Ubiquitin Ion Resistance to Unfolding in the Gas Phase Using Chloride Adduction: Preserving More “Native-Like” Conformations Despite Collisional Activation, Anal. Chem 88 (2016) 5934–5940. [DOI] [PubMed] [Google Scholar]
- [63].Bazzo R, Tappin MJ, Pastore A, Harvey TS, Carver JA, Campbell ID, The Structure of Melittin, Eur. J. Biochem 173 (1988) 139–146. [DOI] [PubMed] [Google Scholar]
- [64].Vogel H, Comparison of the Conformation and Orientation of Alamethicin and Melittin in Lipid Membranes, Biochemistry 26 (1987) 4562–4572. [DOI] [PubMed] [Google Scholar]
- [65].Seo J, Hoffmann W, Warnke S, Bowers MT, Pagel K, von Helden G, Retention of Native Protein Structures in the Absence of Solvent: A Coupled Ion Mobility and Spectroscopic Study, Angew. Chem., Int. Ed 55 (2016) 14173–14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ruotolo BT, Giles K, Campuzano I, Sandercock AM, Bateman RH, Robinson CV, Evidence for Macromolecular Protein Rings in the Absence of Bulk Water, Science 310 (2005) 1658–1661. [DOI] [PubMed] [Google Scholar]
- [67].Friemann R, Larsson DSD, Wang Y, van der Spoel D, Molecular Dynamics Simulations of a Membrane Protein-Micelle Complex in Vacuo, J. Am. Chem. Soc 131 (2009) 16606–16607. [DOI] [PubMed] [Google Scholar]
- [68].Bechara C, Noell A, Morgner N, Degiacomi MT, Tampe R, Robinson CV, A Subset of Annular Lipids Is Linked to the Flippase Activity of an Abc Transporter, Nat. Chem 7 (2015) 255–262. [DOI] [PubMed] [Google Scholar]
- [69].Pliotas C, Dahl ACE, Rasmussen T, Mahendran KR, Smith TK, Marius P, Gault J, Banda T, Rasmussen A, Miller S, Robinson CV, Bayley H, Sansom MSP, Booth IR, Naismith JH, The Role of Lipids in Mechanosensation, Nat. Struct. Mol. Biol 22 (2015) 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Marty MT, Hoi KK, Gault J, Robinson CV, Probing the Lipid Annular Belt by Gas-Phase Dissociation of Membrane Proteins in Nanodiscs, Angew. Chem.-Int. Edit 55 (2016) 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cong X, Liu Y, Liu W, Liang X, Russell DH, Laganowsky A, Determining Membrane Protein–Lipid Binding Thermodynamics Using Native Mass Spectrometry, J. Am. Chem. Soc 138 (2016) 4346–4349. [DOI] [PubMed] [Google Scholar]
- [72].Harvey SR, Liu Y, Liu W, Wysocki VH, Laganowsky A, Surface Induced Dissociation as a Tool to Study Membrane Protein Complexes, Chem. Commun 53 (2017) 3106–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Liu Y, Cong X, Liu W, Laganowsky A, Characterization of Membrane Protein–Lipid Interactions by Mass Spectrometry Ion Mobility Mass Spectrometry, J. Am. Soc. Mass Spectrom 28 (2017) 579–586. [DOI] [PubMed] [Google Scholar]
- [74].Ahadi E, Konermann L, Modeling the Behavior of Coarse-Grained Polymer Chains in Charged Water Droplets: Implications for the Mechanism of Electrospray Ionization, J. Phys. Chem. B 116 (2012) 104–112. [DOI] [PubMed] [Google Scholar]
- [75].Consta S, In Oh M, Kwan V, Malevanets A, Strengths and Weaknesses of Molecular Simulations of Electrosprayed Droplets, J. Am. Soc. Mass Spectrom 29 (2018) 2287–2296. [DOI] [PubMed] [Google Scholar]
- [76].Oh MI, Malevanets A, Paliy M, Frenkel D, Consta S, When Droplets Become Stars: Charged Dielectric Droplets Beyond the Rayleigh Limit, Soft Matter 13 (2017) 8781–8795. [DOI] [PubMed] [Google Scholar]
- [77].Consta S, Oh MI, Sharawy M, Malevanets A, Macroion–Solvent Interactions in Charged Droplets, J. Phys. Chem. A 122 (2018) 5239–5250. [DOI] [PubMed] [Google Scholar]
- [78].Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA, A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules, J. Am. Chem. Soc 117 (1995) 5179–5197. [Google Scholar]
- [79].Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL, Evaluation and Reparametrization of the Opls-Aa Force Field for Proteins Via Comparison with Accurate Quantum Chemical Calculations on Peptides, J. Phys. Chem. B 105 (2001) 6474–6487. [Google Scholar]
- [80].MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins, J. Phys. Chem. B 102 (1998) 3586–3616. [DOI] [PubMed] [Google Scholar]
- [81].Mackerell AD Jr., Feig M, Brooks CL III, Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations, J. Comput. Chem 25 (2004) 1400–1415. [DOI] [PubMed] [Google Scholar]
- [82].Schuler LD, Van Gunsteren WF, On the Choice of Dihedral Angle Potential Energy Functions for N-Alkanes, Mol. Simulat 25 (2000) 301–319. [Google Scholar]
- [83].Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, van Gunsteren WF, Definition and Testing of the Gromos Force-Field Versions 54a7 and 54b7, Eur. Biophys. J 40 (2011) 843–856. [DOI] [PubMed] [Google Scholar]
- [84].Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT, Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology, Anal. Chem 82 (2010) 9557–9565. [DOI] [PubMed] [Google Scholar]
- [85].Salbo R, Bush MF, Naver H, Campuzano I, Robinson CV, Pettersson I, Jørgensen TJD, Haselmann KF, Traveling-Wave Ion Mobility Mass Spectrometry of Protein Complexes: Accurate Calibrated Collision Cross-Sections of Human Insulin Oligomers, Rapid Commun. Mass Spectrom 26 (2012) 1181–1193. [DOI] [PubMed] [Google Scholar]
- [86].Ewing SA, Donor MT, Wilson JW, Prell JS, Collidoscope: An Improved Tool for Computing Collisional Cross-Sections with the Trajectory Method, J. Am. Soc. Mass Spectrom 28 (2017) 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].van der Spoel D, Marklund EG, Larsson DSD, Caleman C, Proteins, Lipids, and Water in the Gas Phase, Macromol. Biosci 11 (2011) 50–59. [DOI] [PubMed] [Google Scholar]
- [88].Allison TM, Landreh M, Benesch JLP, Robinson CV, Low Charge and Reduced Mobility of Membrane Protein Complexes Has Implications for Calibration of Collision Cross Section Measurements, Anal. Chem 88 (2016) 5879–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF, Structural Information from Ion Mobility Measurements: Effects of the Long-Range Potential, J. Phys. Chem 100 (1996) 16082–16086. [Google Scholar]
- [90].Bleiholder C, Structure Elucidation from Ion Mobility-Mass Spectrometry Data: Are Detailed Structures Amenable?, Advancing Mass Spectrometry for Biophysics and Structural Biology Meeting, Ann Arbor, MI, 2017. (conference paper). [Google Scholar]
- [91].Zhou M, Wysocki VH, Surface Induced Dissociation: Dissecting Noncovalent Protein Complexes in the Gas Phase, Acc. Chem. Res 47 (2014) 1010–1018. [DOI] [PubMed] [Google Scholar]
- [92].Nagornova NS, Guglielmi M, Doemer M, Tavernelli I, Rothlisberger U, Rizzo TR, Boyarkin OV, Cold-Ion Spectroscopy Reveals the Intrinsic Structure of a Decapeptide, Angew. Chem., Int. Ed 50 (2011) 5383–5386. [DOI] [PubMed] [Google Scholar]
- [93].Williams ER, Proton Transfer Reactivity of Large Multiply Charged Ions, J. Mass Spectrom 31 (1996) 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Roy TK, Nagornova NS, Boyarkin OV, Gerber RB, A Decapeptide Hydrated by Two Waters: Conformers Determined by Theory and Validated by Cold Ion Spectroscopy, J. Phys. Chem. A 121 (2017) 9401–9408. [DOI] [PubMed] [Google Scholar]
- [95].Bleiholder C, Wyttenbach T, Bowers MT, A Novel Projection Approximation Algorithm for the Fast and Accurate Computation of Molecular Collision Cross Sections (I). Method, Int. J. Mass Spectrom 308 (2011) 1–10. [Google Scholar]
- [96].Bleiholder C, A Local Collision Probability Approximation for Predicting Momentum Transfer Cross Sections, Analyst 140 (2015) 6804–6813. [DOI] [PubMed] [Google Scholar]
- [97].Wyttenbach T, von Helden G, Batka JJ, Carlat D, Bowers MT, Effect of the Long-Range Potential on Ion Mobility Measurements, J. Am. Soc. Mass Spectrom 8 (1997) 275–282. [Google Scholar]
- [98].Paizs B, A Divide-and-Conquer Approach to Compute Collision Cross Sections in the Projection Approximation Method, Int. J. Mass Spectrom 378 (2015) 360–363. [Google Scholar]
- [99].Shvartsburg AA, Mashkevich SV, Siu KWM, Incorporation of Thermal Rotation of Drifting Ions into Mobility Calculations: Drastic Effect for Heavier Buffer Gases, J. Phys. Chem. A 104 (2000) 9448–9453. [Google Scholar]
- [100].Larriba-Andaluz C, Fernandez-Garcia J, Ewing MA, Hogan CJ, Clemmer DE, Gas Molecule Scattering & Ion Mobility Measurements for Organic Macro-Ions in He Versus N-2 Environments, Phys. Chem. Chem. Phys 17 (2015) 15019–15029. [DOI] [PubMed] [Google Scholar]
- [101].Zanotto L, Heerdt G, Souza PCT, Araujo G, Skaf MS, High Performance Collision Cross Section Calculation—Hpccs, J. Comput. Chem 39 (2018) 1675–1681. [DOI] [PubMed] [Google Scholar]
- [102].Ieritano C, Crouse J, Campbell JL, Hopkins WS, A Parallelized Molecular Collision Cross Section Package with Optimized Accuracy and Efficiency, Analyst (2019) doi: 10.1039/C8AN02150C. [DOI] [PubMed] [Google Scholar]