

Abstract
Aging of the nervous system, and the occurrence of age-related brain diseases such as stroke, are associated with changes to a variety of cellular processes controlled by many distinct genes. MicroRNAs (miRNAs), short non-coding functional RNAs that can induce translational repression or site-specific cleavage of numerous target mRNAs, have recently emerged as important regulators of cellular senescence, aging, and the response to neurological insult. Here, we focused on the assessment of the role of miR-34a in stroke. We noted increases in miR-34a expression in the blood of stroke patients as well as in blood and brain of mice subjected to experimental stroke. Our methodical genetic manipulation of miR-34a expression substantially impacted stroke-associated preclinical outcomes and we have in vitro evidence that these changes may be driven at least in part by disruptions to blood brain barrier integrity and mitochondrial oxidative phosphorylation in endothelial cells. Finally, aging, independent of brain injury, appears to be associated with shifts in circulating miRNA profiles. Taken together, these data support a role for miRNAs, and specifically miR-34a, in brain aging and the physiological response to age-related neurological insult, and lay the groundwork for future investigation of this novel therapeutic target.
Keywords: Stroke, Sex, miR-34a, MicroRNA, Blood-brain barrier, Ischemia, aging
Introduction
Polygenetic regulators of neurological function are indicated by the observation that a variety of neurological conditions are not tied specifically to known mutations. As such, an assessment of regulatory factors that could affect a number of cellular processes involved in diseases or injury to the brain is critically needed. One such polygenetic regulatory mechanism involves microRNAs (miRNAs). miRNAs are small, endogenous ∼22 nucleotide RNAs whose biogenesis occurs in all mammalian and plant cells and can play important regulatory roles by targeting messenger RNAs (mRNAs) for cleavage or translational repression. In recent years, there has been a focus on the study of miRNA function, in part to further elucidate the specific mechanisms of miRNA action and study the possible use of miRNAs in the diagnosis and treatment of disease. Importantly, a single miRNA has the potential for targeting many genes simultaneously. Like practically all other cellular processes, genes associated with many features of aging are regulated by miRNAs. Indeed, miRNAs have recently emerged as important regulators of cellular senescence, aging and response to neurological insult. Several reviews describing miRNAs and their role in the aging of the nervous system have been published [1–3].
miRNAs are short non-coding functional RNAs that recognize certain mRNAs based on sequence complementarity resulting in translational repression or site-specific cleavage [4, 5]. Over 1881 human miRs have been reported in miRBase [http://www.mirbase.org/], but miRBase miRNAs are only sequence based, and many of them are yet to be functionally validated, making this number likely overestimated. Individual miRNAs target many gene products and concomitantly down regulate multiple biological pathways by repressing mRNA translation/or increasing its degradation [4, 5]. As such, the expression of novel miRNAs in processes associated with the aging nervous system and the brain’s response to injury may maximize the number of target genes possibly involved in aging and senescence-associated neurological conditions.
One such novel miRNA of interest is miR-34a. The miR-34 family members, which consist of miR-34a, b and c, were discovered computationally and later verified experimentally as a part of the p53 tumor suppressor network [6, 7]. In humans, the miR-34a (chromosome 1p36.22 located within the second exon of its non-coding host gene) and miR-34b/c (chromosome 11q23.1 located within intron 1 and exon 2, respectively) are ubiquitously expressed with high levels in the ovary, prostate and testes, and with intermediate levels in brain, lung, thymus, and kidney [8]. In mice, miR-34a (chromosome 4) and miR-34b/c (chromosome 9) are also ubiquitously expressed, with the highest miR-34a levels in the brain; miR-34b/c are mainly expressed in the lung [9]. It has been reported that the miR-34 family members play important roles as a tumor suppressors [9–12].
We have focused on the assessment of the role of miR-34a in a variety of neurological conditions, especially stroke, for several reasons. Our bioinformatic analysis of murine miRNA database predicts that miR-34a may also target and silence multiple mitochondria associated mRNAs [13]. This indicates that the miR-34 family may play an important role in the regulation of mitochondrial energy production, a potentially important finding given the critical involvement in mitochondria for optimal neurological function. As well, our miR-34a promoter analysis by transcription factor search revealed the presence of p53, NF-kB, fos, c-rel, STAT1, and CREB transcription factor binding response elements in the promotor region of the miR-34a gene [13]. NF-kB, c-rel and STAT1, are known to be activated in response to inflammation [14–16]; CREB, and fos are activated by neuronal excitation [17, 18]; and p53 by oxidative stress [19]. Also, it has been shown that SIRT1, p53 and miR-34a are involved in a positive feedback loop for miR-34a gene transcription in a fashion whereby increased miR-34a suppresses its target SIRT1 expression, thereby resulting in increased p53 transcriptional activity leading to continued up-regulation of miR-34a [20].
Because a single miRNA targets many genes and concomitantly down regulates multiple biological pathways, we sought to identify the role of miR-34a in stroke outcome.
Post-stroke increase in miR-34a in human subjects
If a miRNA is involved in the sequelae following stroke, one would predict that changes in levels of that miRNA would be seen after stroke. We assessed post-stroke changes in miR-34a in human subject plasma with confirmed ischemic stroke. Subjects who presented to the WVU Medicine Ruby Memorial Hospital were consented and enrolled in a pilot observational study based on their presentation with an ischemic stroke, and blood samples were obtained within 6h of “last seen normal”, then 24 h, 5, 30 and 90 days later. Plasma samples were stored for later assay of miR-34a (and other miRNAs). Thirteen subjects and 15 non-stroke (stroke mimics, controls) were acquired. mir-34a levels post-stroke are shown in Figure 1. At 6h a significant, but highly variable elevation in miR-34a was observed, while at 24h, about half of the subjects showed an increase in miR-34a. Plasma miR-34a levels were elevated significantly in all subjects at 5, 30 and 90 days post-stroke. Collectively, these data indicate that plasma miR-34a is initially highly variable, then persistently increased after stroke.
Figure 1: Analysis of miR-34a expressed in the serum of stroke patients compared to controls.
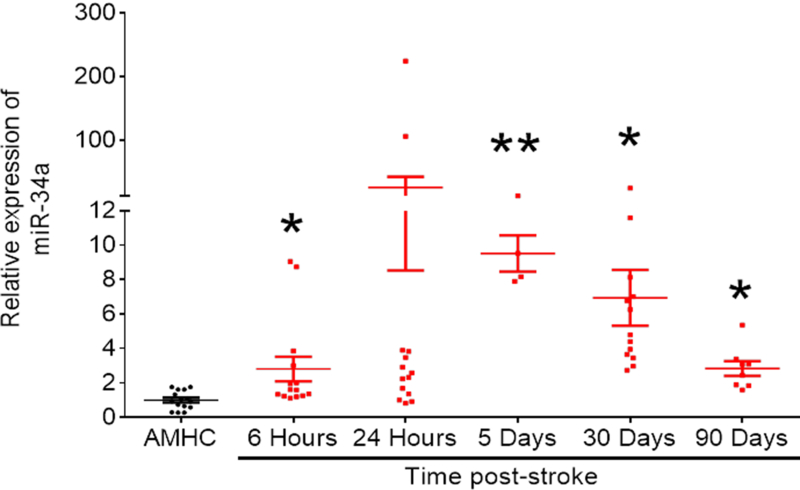
miR-34a was significantly upregulated in the serum of stroke patients at several reperfusion times (6 hours, 5, 30 and 90 days). Data were pooled from both male and female patients. Age matched healthy controls (AMHC) were used. Expression of spike-in C. elegans miR-39 was used to calculate relative fold change in miR-34a expression levels. All reactions were analyzed by qRT-PCR performed in triplicates. Data are expressed as mean ± S.D. Sample sizes were reflected by dot plots; One-way ANOVA followed by post-hoc Tukey’s test. *, p < 0.05; **, p < 0.01. (Lewis, et al., unpublished observations)
Post-stroke increase in miR-34a in mice
We next assessed the time course of miR-34a in brain and serum following transient middle cerebral artery occlusion (tMCAO) model of stroke in mice. There was an increase in serum miR-34a at 6 but not at 24 h post-stroke. Interestingly, the 6 h elevation in miR-34a was associated with an increase in blood-brain barrier (BBB) opening, as evidenced by a profound increase in Evans blue flux into the brain at that time (Figure 2A & B). Finally, the coincidence of the increases in miR-34a in serum with the initial opening of the BBB at 6 h (Figure 2C) led us to assess the potential role of miR-34a in BBB opening and its mechanism (See below).
Figure 2. Time course of miR-34a following ischemia in mouse serum and brain following stroke.
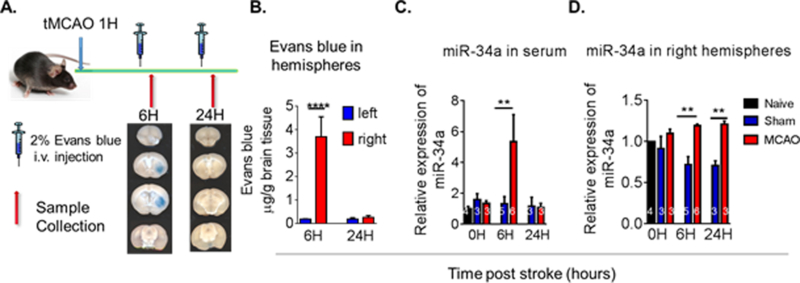
(A) Focal cerebral ischemia was induced by tMCAO for 60 minutes in C57BL/6 male mice and Evan’s blue was injected into the mice (i.v.). Transcardiac perfusion was then performed and brain images acquired as shown by representative coronal brain sections. (B) Quantification of Evan’s blue extravasation in the left and right hemispheres. Data are expressed as mean ± S.D.; n=4/group; One-way ANOVA followed by post-hoc Tukey’s test, ****p<0.0001. (C) At the indicated time post-stroke, tissue samples were collected and miRNA levels in the ischemic brain hemisphere and serum were analyzed by qRT-PCR. In mouse serum following tMCAO, miR-34a levels were increased by 6 hrs post-stroke. (D) miR-34a levels were increased in the brain of tMCAO mice by 6 hrs and at 24 hrs. Data are expressed as mean ± S.D., **p < 0.01. (Naive: N = 2, Sham: N = 3, tMCAO: N = 3) [Jun et al., 44]
Brain concentrations of miR-34a were increased at 6 and 24 h following tMCAO in the ipsilateral brain area (Figure 2D). The source of the post-stroke serum miR-34a is currently unknown, but our in vitro evidence that indicates that miR-34a is rapidly packaged into extracellular vesicles (EVs) and secreted from cells (Russell, unpublished observations) suggests that a brain source is likely. Additionally, in vitro treatment of neurons with TNF-α results in a dose-dependent increase in secreted EV miR-34a, but no change in cellular levels of this miRNA (Russell, unpublished observation). This suggested that at least in neurons, miR-34a is secreted as soon as it is produced. Taken together, our collective findings indicate that in both mice and human subjects, miR-34a responded to stroke with a substantial increase in blood.
Stroke outcome in transgenic mice with miR-34a knockout and over expression
To help determine the role of these changes in miR-34a on stroke outcome, we assessed the effects of miR-34a knockout and its overexpression on infarct volume and neurological deficits after tMCAO. In both studies, wild type (WT) controls and transgenic (Tg) mice were subjected to tMCAO; 24 h later neurological scores were obtained and infarct volume was assessed. Relative to control mice, miR-34a knockout mice (homozygous breeders were purchased from the Jackson Laboratory, stock No. 018279, 3 month old males were used for the study) showed reduced neurological deficit and a 50% reduction in infarct volume (Ren et al., unpublished observations). In contrast, when we made a doxycycline-inducible miR-34a over expression mouse, 4 weeks of induced miR-34a over expression resulted in a 4-fold increase in miR-34a and an exacerbation of infarct volume and neurological deficits (Ren et al., unpublished observations). These data indicate that genetic manipulation of miR-34a can substantially affect stroke outcome in mice.
miR-34a over-expression and blood-brain barrier function
miR-34a can affect stroke outcome through a combination of mechanisms. We have previously demonstrated that miR-34a targets a number of mitochondrial electron transport proteins, and that its overexpression reduces these protein and substantially reduces oxidative phosphorylation (OxPhos) in neurons [13]. We then undertook a series of studies of the effects of miR-34a on cerebral endothelial function, with a focus on BBB permeability and cerebral endothelial mitochondrial function. This was done in view of our observations that inhibition of OxPhos cerebrovascular endothelial cells (CECs) disrupts BBB function [21].
Trans-well cultures of cerebral endothelial cells were used to determine the effects of miR-34a over expression on BBB permeability [21]. miR-34a transfected cells had a significantly greater permeability to a fluorescent dye (MW = 4kDa) than control transfected cells (Figure 3). Additionally, overexpression of miR-34a apparently disrupted tight junctions and resulted in gaps between cells (Figure 3). These data indicate that in vitro, overexpression of miR-34a disrupts the BBB.
Figure 3. Overexpression of miR-34a increases BBB permeability in vitro.
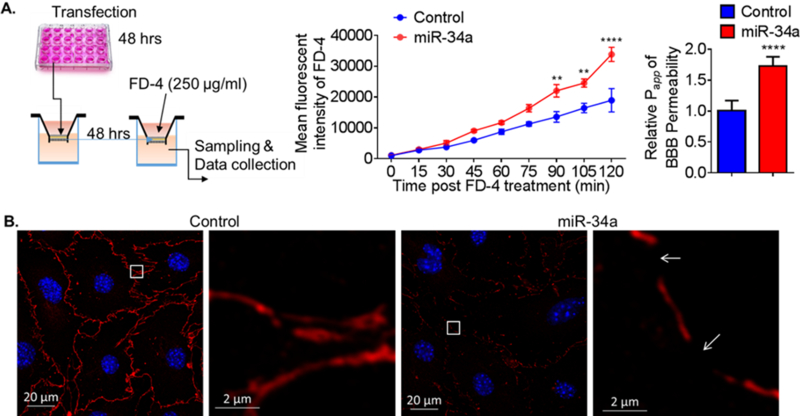
(A) A schematic protocol using fluorescein isothiocyanate–dextran-4 (FD-4) to detect BBB permeability in vitro. FD-4 permeability in CECs that overexpressed miR-34a plasmid (0.017 ng) versus control was presented as real-time rate of FD-4 mean fluorescent intensity (2-way ANOVA followed by post hoc Dunnett’s test; N = 3; **, p < 0.01; ****, p < 0.0001). Calculated apparent permeability coefficient Papp (Student’s t-test; ****, p < 0.0001) is expressed as mean ± SD. (B) Confocal fluorescence images of confluent monolayers confirmed microscopically after transfection with miR-34a plasmid versus control. Fluorescent staining: tight junctions ZO-1 (red), cell nuclei (DAPI, blue). Overexpression of miR-34a apparently disrupted tight junctions and resulted in gaps between cells (white arrows). Results are representative of three independent experiments. [21]
miR-34a over-expression and endothelial cell oxidative phosphorylation
The mechanism by which miR-34a disrupts the BBB could be through its inhibition of mitochondrial OxPhos, in view of our observation of the profound effects of miR-34a overexpression on neuronal OxPhos [13]. As such, we undertook studies to determine if overexpression of miR-34a compromised OxPhos in brain endothelial cells. Overexpression of miR-34a caused a dose- and time dependent reduction in ATP levels (Figure 4) and OxPhos (Figure 5) in brain endothelial cells. Interestingly, of the OxPhos proteins assessed, only cytochrome C (CYC C) was reduced by miR-34a overexpression in this cell type. This is in contrast to our observation of reduction in at least 5 OxPhos proteins with miR-34a overexpression in neurons, representing a cell type-specific effect of this miRNA.
Figure 4. Overexpression of mir-34a reduces mitochondrial function and decreases CYC level in cerebrovascular endothelial cells.
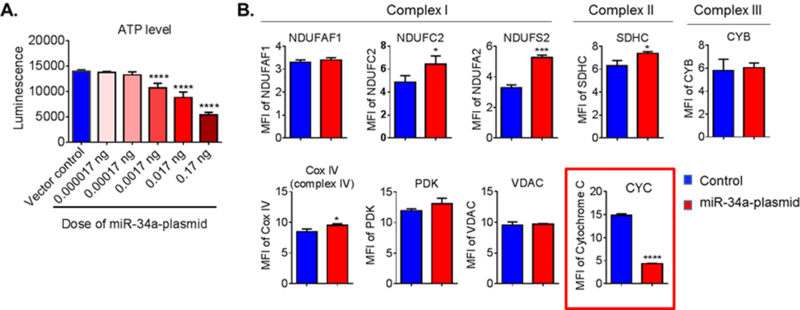
(A) ATP level was measured at 72 h post-transfection. Data are expressed as mean ± SD (N = 5). One-way ANOVA followed by post hoc Tukey’s test. (****, p < 0.0001). (B) Flow cytometry analysis of mitochondrial specific proteins for complex I proteins (NDUFAF1, NDUFC2 and NDUFS2), complex II protein (SDHC), complex III protein (CYB), complex IV protein (CYC C oxidase, Cox IV), CYC, pyruvate dehydrogenase kinase (PDK), and voltage-dependent anion channel protein (VDAC) at 72 h post-transfection. CYC level was significantly lower in the cells that were transfected with the miR- 34a plasmid. Data are presented as mean ± SD (N = 3) and analyzed by Student’s t-test, *, p < 0.05; ***, p < 0.001; ****, p < 0.0001. Results are representative of three independent experiments. [21]
Figure 5. Overexpression of miR-34a reduces OxPhos in cerebrovascular endothelial cells.
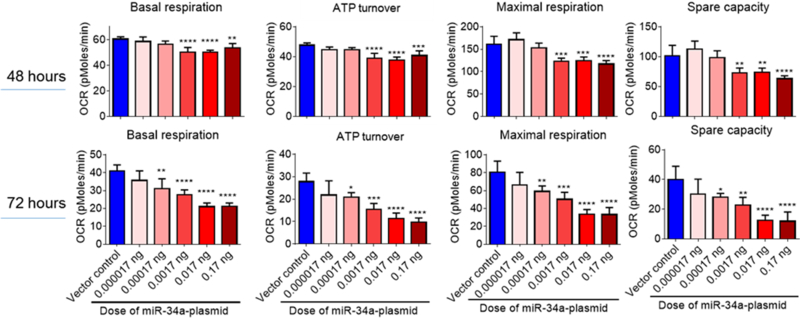
Basal respiration, mitochondrial ATP turnover, maximal respiration, and spare capacity were calculated from the bioenergetics functional assay at post-transfection 48 & 72 hours. Data are expressed as mean ± S.D. (N = 4). 1-way ANOVA followed by post hoc Tukey’s test. (* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). [21]
Aging and miR-34a levels
Finally, we conducted a small pilot study of miRNA levels in serum of aging Fischer 344 rats to determine if age is associated with an expected increase in miRNAs that may compromise brain function and contribute to the well-recognized association between age and stroke severity [22]. In marked contrast to our expectations, age was in general associated with a reduction in miRNAs (Figure 6). An age-related decline in most miRNAs, including miR-34a, was significant for miR-15a and −145 and trended towards significance for miR-let-7f. As well, as Ns in this study were small (Ns = 3–5/group), Cohen’s f effect size calculations conducted on the remaining miRNAs revealed mostly strong effects (miRs-34a, −146, −210, let-7f, and −124). Using this information, a priori power analyses revealed that should the experiment be repeated and similar trends be observed, significant age-related decreases in miRNA expression are likely to be obtained for these miRNAs with Ns/group of between 6–11 rodents, suggesting that changes in peripheral levels of these miRNAs may play an important role in the aging process. From a broader perspective, this observation of an age-related decrease in miRNA expression may be the reflection of a compensatory protective response with aging, or could be due to a survival effect, where animals with elevated miRNAs are eliminated from the population during aging due to premature death from sporadic disease. Further studies aim at assessing these miRNAs in larger cohorts of animals and the comparison of morbid animals with healthy animals would be needed.
Figure 6. Effects of age on serum miR-34a in female Fischer 344 rats.
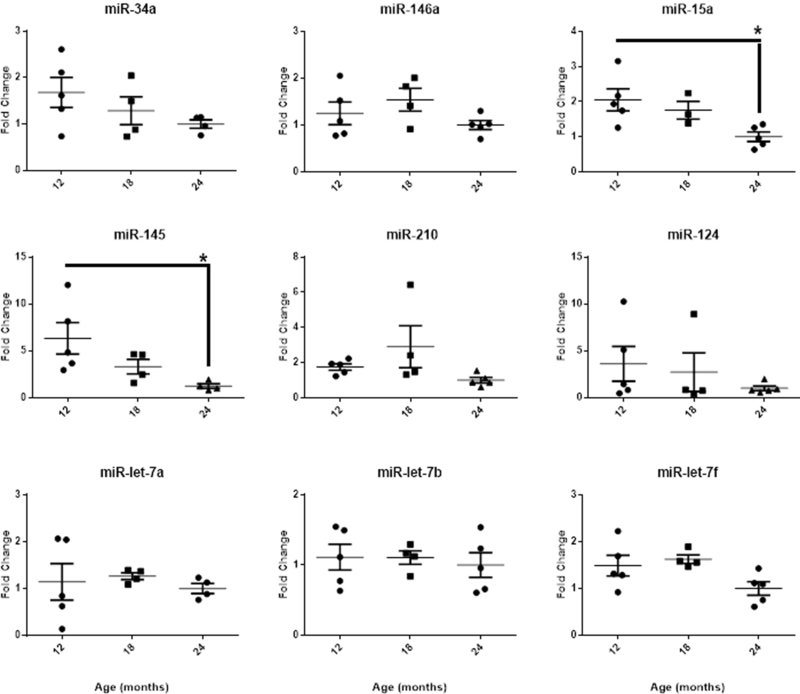
Fischer 344 rats were staged for estrous cycle state and sampled at 12, 18 and 24 months of age for serum concentrations of miRNAs. Reported are mean ± SEM serum miRNA fold change values at each age normalized to 24 month old group. While an Age main effect was significant for miR-15a [F(2,10) = 5.32, p < 0.05] and −145 [F(2,10) = 4.59, p < 0.05] with two-group post-hoc comparisons revealing a decrease in fold change between 12 months and 24 months of age, visual inspection of the graph indicates age-related declines in miR-34a, −146a, −15a, −210, −124, and -let-7f. One-way ANOVA followed by post-hoc Tukey’s test. *, p < 0.05. Significant outliers were excluded from individual miRNA analyses; one subject euthanized prior to the completion of the experiment was excluded from all analyses. (Engler-Chiurazzi et al, unpublished observations).
Discussion
Most miRNAs are increased after stroke
With few exceptions, the miRNAs assessed to date increase after stroke. This suggests that their promotor regions are activated by one or more transcription factors that are responsive to stroke. This is not surprising, because response elements for transcription factors that respond to neuroinflammation, neuronal activity and oxidative stress are present on many of these miRNAs. Two major questions then emerge from this observation. Are increases in miRNAs beneficial or detrimental to stroke outcome? Is one miRNA more important than others to stroke outcome? These questions are discussed below.
Are increases in miRNAs beneficial or detrimental to stroke outcome?
Our observations of post-stroke increases in miR-34a should be put in the context of observations made with other miRNAs. Since the first demonstration of stroke-induced miRNA changes [23, 24], analyses of the interaction between ischemic pathology and miRNAs has yielded valuable insights into the brain injury progression and towards the development of biomarker panels and novel therapeutic targets. In human stroke patients, microarray assays of blood or cerebrospinal fluid levels of miRNAs have been found to undergo dramatic changes as compared to healthy controls [25], non-ischemic controls with vascular risk factors [26], or patients with other non-acute ischemic stroke neurological disorders [27]. Like our observation with miR-34a, increases in miR-let-7b, miR-30a, and miR-126 persist for several weeks to months beyond the acute recovery period [28]. Further, at least some of the altered miRNAs, whose biological roles have been implicated in cardiovascular and endothelial function, angiogenesis, hypoxia, inflammation/immune system response, and cellular restoration pathways, are associated with clinical outcome [24, 25].
Whether the post-stroke increase in miRNAs is detrimental or beneficial is open to debate. For instance, levels of a brain-specific miRNA, miR-124, are increased in plasma following permanent MCAO in rats [29] but reduced in brains of rats subjected to tMCAO [30]. Inhibition of brain miR-124 reduced infarct size and improved neurological outcomes in both mice [31] and rats [30]. Yet, interestingly, in mice experiencing tMCAO, brain miR-124 levels increased following stroke and when levels were methodically manipulated, overexpression was associated with neuroprotective effects and improved functional consequences [32, 33], suggesting potential interactive effects of species and ischemic model on stroke-induced changes in brain miRNA levels.
miR-181 has also been shown to change following stroke and modulation of these levels has yielded beneficial impacts. Indeed, in male rats subjected to bilateral carotid artery occlusion, miR-181a levels were increased relative to sham animals; treatment of stroke animals with a miR-181a antagomir (an oligonucleotide antagonist) increased hippocampal Bcl-2 levels and decreased neuronal loss [34]. However, among mice subjected to 20 min tMCAO, stroke reduced levels of miR-181b; reduced miR-181b expression reduced cell death and improved neurological scores [35]. One reason for this discrepancy could relate to the brain region sampled as mice subjected to 60 min tMCAO had increased levels of miR-181a, -b, -c, and –d in the ischemic core but decreased levels in the penumbra. However, given that the above studies differed in the species evaluated and the experimental stroke model employed, the contribution of region sampled is not entirely clear; future evaluations specifically addressing this question are warranted.
In the context of stroke, miR-29 appears to be associated with cell survival/death pathways. Indeed, increased cell death was associated with increased brain levels of miR-29b at 24 hours following 90min tMCAO in rats [36]. Similarly, miR-29c overexpression reduced anti-apoptotic proteins, increased infarct, and negatively impacted neurological outcomes [37]. This effect may be species specific as interestingly, in mice, miR-29b [38] and -c [39] levels are reduced in the post-stroke brain and overexpression reduces lesion volume, edema, BBB disruption, and cell death. Added clarity is needed regarding these conflicting findings.
Finally, given that age is a significant non-modifiable risk factor for stroke (Boehme et al., 2017) and that we noted an age-related change in serum miRNA levels, the consideration of biological variables in the context of miRNA response to stroke is key. Indeed, Selvamani and colleagues [40] noted age- and sex-dependent effects of stroke on ischemic outcome measures as well as miRNA expression; at least acutely, the variance in miRNA expression changes were attributed to age while at further post-stroke time points, sex tended to account for these changes. Other recent studies have also noted sex differences in miRNA response to brain insult [41, 42], highlighting the importance of addressing these factors in research that is only just beginning to be explored in depth.
In spite of these limitations, these collective findings suggest important roles for several unique miRNAs in the pathology of stroke. More investigation will contribute to the understanding of the precise nature of miRNA expression changes in the context of injury and hopefully identify novel therapeutic targets in addition to biomarkers for disease progression.
Is miR-34a special?
Does miR-34a hold a special place in stroke, given that the genetic depletion of several miRNAs has been shown to improve stroke outcome? The answer is complex and relates to the complexity of the sequelae of stroke. Clearly, numerous cell death pathways are activated with ischemia and targeting any one of them is expected to improve stroke outcome. As such, various miRNAs could improve stroke outcome by targeting one or more of these signaling pathways.
Alternatively, it is well know that a single mRNA can be targeted by hundreds of miRNAs, and as such, many miRNAs may target an essential causative signaling pathway in neuronal cell death after stroke. The latter case is less likely, since inhibiting one miRNA should not have a robust effect on stroke outcome. Nonetheless, there are many examples of inhibition of a single miRNA that robustly reduces stroke outcome. This would argue that miRNAs targeting different cell death pathways is more likely.
miR-34a is of great interest to us because of the essential role that mitochondria play in cell death [43]. In neurons, miR-34a targets and reduces numerous glycolysis and OxPhos proteins [13]; as shown in Fig 4 and 5; in cerebral endothelial cells, a more selective suppression of CYC C is seen, but is associated with substantial OxPhos reductions [21]. Our studies suggest that the associated mitochondrial dysfunction is integrally involved in opening of the BBB after stroke [21]. It remains to be seen whether miR-34a exerts influence on the numerous other aspects of the ischemic neuropathological cascade, including cell death, glucose metabolism, etc.
Remaining issue with miRNAs and stroke
There are a number of issues that remain to be resolved with respect to miRNAs and stroke outcome. First, we do not know if the miRNAs inhibit mRNA within the cell in which they are produced, in neighboring cells, or between organs. Second, we do not yet know which cell type produces the dynamic raise in miRNAs in blood after stroke – neurons, endothelial cells, glial cells, immune cell, etc. The answer to these questions may lead us to the specific cell type to be targeted to protect the brain from the devastating effects of stroke. Finally, the aforementioned issue of the role in individual miRNAs versus multiple miRNAs in stroke outcome needs to be resolved, because this will have critical influence on our ability to target specific miRNAs for acute stroke therapy.
Acknowledgements:
This work was supported by NIH Grants P20 GM109098, P01 AG027956, U54 GM104942, T32 AG052375, AHA grant 16SDG31170008 and the WVU Research Office.
Footnotes
Declarations of interest: none
References
- 1.Harries LW, MicroRNAs as Mediators of the Ageing Process. Genes (Basel), 2014. 5(3): p. 656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jung HJ and Suh Y, MicroRNA in Aging: From Discovery to Biology. Curr Genomics, 2012. 13(7): p. 548–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarkar SNR, A. E.; Engler-Chiurazzi EB; Porter KN; Simpkins JW, MicroRNAs and the genetic nexus of brain aging, neuroinflammation, neurodegeneration, and brain trauma. Aging and Disease In press. 10.14336/AD.2018.0409 [DOI] [PMC free article] [PubMed]
- 4.Ambros V, The functions of animal microRNAs. Nature, 2004. 431(7006): p. 350–5. [DOI] [PubMed] [Google Scholar]
- 5.McManus DD and Ambros V, Circulating MicroRNAs in cardiovascular disease. Circulation, 2011. 124(18): p. 1908–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He L, et al. , A microRNA component of the p53 tumour suppressor network. Nature, 2007. 447(7148): p. 1130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, et al. , MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res, 2009. 69(19): p. 7569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei JS, et al. , The MYCN oncogene is a direct target of miR-34a. Oncogene, 2008. 27(39): p. 5204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bommer GT, et al. , p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol, 2007. 17(15): p. 1298–307. [DOI] [PubMed] [Google Scholar]
- 10.Cole KA, et al. , A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol Cancer Res, 2008. 6(5): p. 735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lujambio A, et al. , A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A, 2008. 105(36): p. 13556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen Z, et al. , MicroRNA-34a affects the occurrence of laryngeal squamous cell carcinoma by targeting the antiapoptotic gene survivin. Med Oncol, 2012. 29(4): p. 2473–80. [DOI] [PubMed] [Google Scholar]
- 13.Sarkar S, et al. , Expression of MicroRNA-34a in Alzheimer’s Disease Brain Targets Genes Linked to Synaptic Plasticity, Energy Metabolism, and Resting State Network Activity. Brain research, 2016. 1646: p. 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin P, et al. , Anti-inflammatory and anti-amyloidogenic effects of a small molecule, 2,4-bis(p-hydroxyphenyl)-2-butenal in Tg2576 Alzheimer’s disease mice model. J Neuroinflammation, 2013. 10: p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HK, et al. , Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell, 2003. 112(5): p. 631–43. [DOI] [PubMed] [Google Scholar]
- 16.Sarnico I, et al. , NF-kappaB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J Neurochem, 2009. 108(2): p. 475–85. [DOI] [PubMed] [Google Scholar]
- 17.Impey S, et al. , Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron, 1998. 21(4): p. 869–83. [DOI] [PubMed] [Google Scholar]
- 18.Morgan JI and Curran T, Role of ion flux in the control of c-fos expression. Nature, 1986. 322(6079): p. 552–5. [DOI] [PubMed] [Google Scholar]
- 19.Gambino V, et al. , Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell, 2013. 12(3): p. 435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamakuchi M, Ferlito M, and Lowenstein CJ, miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A, 2008. 105(36): p. 13421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bukeirat M, et al. , MiR-34a regulates blood-brain barrier permeability and mitochondrial function by targeting cytochrome c. J Cereb Blood Flow Metab, 2016. 36(2): p. 387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boehme AK, Esenwa C, and Elkind MS, Stroke Risk Factors, Genetics, and Prevention. Circ Res, 2017. 120(3): p. 472–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeyaseelan K, Lim KY, and Armugam A, MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke, 2008. 39(3): p. 959–66. [DOI] [PubMed] [Google Scholar]
- 24.Tan KS, et al. , Expression profile of MicroRNAs in young stroke patients. PLoS One, 2009. 4(11): p. e7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, et al. , Circulating MicroRNAs as Novel Potential Biomarkers for Early Diagnosis of Acute Stroke in Humans. Journal of Stroke and Cerebrovascular Diseases, 2014. 23(10): p. 2607–2613. [DOI] [PubMed] [Google Scholar]
- 26.Jickling GC, et al. , microRNA expression in peripheral blood cells following acute ischemic stroke and their predicted gene targets. PLoS One, 2014. 9(6): p. e99283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sorensen SS, et al. , miRNA expression profiles in cerebrospinal fluid and blood of patients with acute ischemic stroke. Transl Stroke Res, 2014. 5(6): p. 711–8. [DOI] [PubMed] [Google Scholar]
- 28.Long G, et al. , Circulating miR-30a, miR-126 and let-7b as biomarker for ischemic stroke in humans. BMC Neurol, 2013. 13: p. 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weng H, et al. , Plasma miR-124 as a biomarker for cerebral infarction. Biomed Res, 2011. 32(2): p. 135–41. [DOI] [PubMed] [Google Scholar]
- 30.Zhu F, et al. , MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J Mol Neurosci, 2014. 52(1): p. 148–55. [DOI] [PubMed] [Google Scholar]
- 31.Liu XR, et al. , MicroRNA-124-Mediated Regulation of Inhibitory Member of Apoptosis-Stimulating Protein of p53 Family in Experimental Stroke. Stroke, 2013. 44(7): p. 1973–1980. [DOI] [PubMed] [Google Scholar]
- 32.Doeppner TR, et al. , MicroRNA-124 protects against focal cerebral ischemia via mechanisms involving Usp14-dependent REST degradation. Acta Neuropathol, 2013. 126(2): p. 251–65. [DOI] [PubMed] [Google Scholar]
- 33.Sun Y, et al. , MicroRNA-124 protects neurons against apoptosis in cerebral ischemic stroke. CNS Neurosci Ther, 2013. 19(10): p. 813–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moon JM, Xu L, and Giffard RG, Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J Cereb Blood Flow Metab, 2013. 33(12): p. 1976–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Z, et al. , Downregulation of miR-181b in mouse brain following ischemic stroke induces neuroprotection against ischemic injury through targeting heat shock protein A5 and ubiquitin carboxyl-terminal hydrolase isozyme L1. J Neurosci Res, 2013. 91(10): p. 1349–62. [DOI] [PubMed] [Google Scholar]
- 36.Shi GD, et al. , Upregulated miR-29b promotes neuronal cell death by inhibiting Bcl2L2 after ischemic brain injury. Experimental Brain Research, 2012. 216(2): p. 225–230. [DOI] [PubMed] [Google Scholar]
- 37.Huang LG, et al. , MicroRNA-29c Correlates with Neuroprotection Induced by FNS by Targeting Both Birc2 and Bak1 in Rat Brain after Stroke. CNS Neurosci Ther, 2015. 21(6): p. 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, et al. , MicroRNA-29b is a therapeutic target in cerebral ischemia associated with aquaporin 4. J Cereb Blood Flow Metab, 2015. 35(12): p. 1977–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khanna S, et al. , Loss of miR-29b following acute ischemic stroke contributes to neural cell death and infarct size. J Cereb Blood Flow Metab, 2013. 33(8): p. 1197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selvamani A, et al. , Circulating miRNA profiles provide a biomarker for severity of stroke outcomes associated with age and sex in a rat model. Clin Sci (Lond), 2014. 127(2): p. 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selvamani A and Sohrabji F, Mir363–3p improves ischemic stroke outcomes in female but not male rats. Neurochem Int, 2017. 107: p. 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stary CM, et al. , Inhibition of miR-181a protects female mice from transient focal cerebral ischemia by targeting astrocyte estrogen receptor-alpha. Mol Cell Neurosci, 2017. 82: p. 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duarte FV, Palmeira CM, and Rolo AP, The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes (Basel), 2014. 5(4): p. 865–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jun SS, S. N.; Tennant C; Hu H; Quintana DD; Doll DN; Ren X; Barr T; Simpkins JW Jun S, Sarkar SN, Tennant C, Hu H, Quintana DD, Doll DN, Ren X, Barr T, Simpkins JW. Time-course response of miR-34a, miR-132 and miR-146 to ischemic stroke. in Society for Neuroscience 2014. Washington D.C. [Google Scholar]