

Abstract
Objective:
The aim of this study was to determine whether the genetic background of the disease should be incorporated into treatment decision making.
Background:
Carotid body paragangliomas are rare tumors that often affect patients with genetic mutations of the succinate dehydrogenase complex (SDHx). Despite growing evidence that germ line genetic mutations alter the aggressiveness of paragangliomas, treatment decisions are currently based only on clinical symptoms and tumor size in patients with carotid body paragangliomas.
Methods:
Retrospective analysis of 34 patients with carotid body paragangliomas who underwent genetic testing and surgical treatment. Recurrence was defined by the return of locoregional disease and/or development of distant metastases. Clinical characteristics and genetic testing results were analyzed as predictors of patient outcomes.
Results:
Thirty-four patients underwent 41 primary carotid body paraganglioma resections (median follow-up time of 42 months, range: 1–293). Overall survival was 91.2%. Twelve patients had germ line mutations in SDHB, 17 in SDHD, and 5 carried no known mutation. Surgical resection of larger tumors was associated with higher operative complications (odds ratio: 5.4, P = 0.05). Tumor size at resection was significantly smaller in patients with SDHB mutations than in patients with non-SDHB mutations (2.1 vs 3.3 cm, P = 0.02). Patients with a mutation in the SDHB gene also had significantly worse disease-free survival compared with patients without an SDHB gene mutation (P = 0.03).
Conclusions:
Mutations in the SDHB gene are associated with worse disease- free survival after resection in patients with carotid body paragangliomas despite earlier intervention. This suggests that a more aggressive surgical approach is warranted in patients with SDHB mutations.
Keywords: carotid body tumor, genetics, metastasis, paraganglioma, surgery
Paragangliomas are highly vascular tumors arising from chromaffin cells associated with autonomic paraganglia. Head and neck paragangliomas are rare, comprising only 0.03% of all tumors with an annual incidence of approximately 1 case per 100,000 persons.1,2 Head and neck paragangliomas can arise in several anatomic locations, including jugulotympanic, laryngeal, subclavian, intravagal, and carotid body.3,4 Unlike abdominal and thoracic paragangliomas, head and neck paragangliomas are usually biochemically silent or demonstrate minimal catecholamine secretion.5,6
Carotid body paragangliomas are the most common type of head and neck paraganglioma and can be particularly challenging to manage operatively.7–9 Although carotid body paragangliomas have traditionally been viewed as requiring surgical resection, recent evidence demonstrating that many tumors have relatively low rates of malignancy has led to interest in nonsurgical treatment.10 Effective and precise identification of patients with less aggressive disease could lead to better preoperative decision making and allow patients with indolent disease to avoid the morbidity of surgical resection.11,12
The development of carotid body paragangliomas is often associated with germ line genetic mutations, most commonly involving genes in the succinate dehydrogenase complex (SDHx).9,13,14 Paraganglioma syndromes 1 through 4 are associated with mutations in SDHD, SDHAF2, SDHC, and SDHB, respectively. Mutation of the SDHA gene is also associated with paragangliomas but does not cause one of the traditional paraganglioma syndromes. Phenotypic differences are notable depending on the type of the SDHx mutation. Overall, mutations in SDHB result in more aggressive disease with higher rates of metastasis compared with mutations in SDHD, whereas mutations in SDHD result in a higher incidence of head and neck paragangliomas and a comparatively lower incidence of pheochromocytomas.13–19
Despite these observed differences in disease characteristics based on genetic findings, there are currently limited surgical data in the literature, and thus no guidelines or recommendations on the incorporation of genetic testing results into operative decision making. This study was designed to assess surgical outcomes of patients who underwent resection of hereditary and nonhereditary carotid body paragangliomas and determine whether surgical indications should be altered on the basis of clinical characteristics and/or patient genetics.
METHODS
Patients
The study was approved by the Office of Human Subject Research at the National Institutes of Health. All research participants provided written informed consent. Patient demographics, genetic testing results, radiologic findings, and operative interventions were reviewed in patients diagnosed with head and neck paragangliomas who were evaluated at the National Institutes of Health Warren Magnuson Clinical Center. Patients underwent genetic testing for germ line mutations and deletions in SDHA, SDHB, SDHC, SDHD, SDHAF2, RET, VHL, and TMEM127. These genetic tests were performed in collaboration with the Mayo Clinic in Rochester, Minnesota. Preoperative evaluation and postoperative follow-up consisted of biochemical testing (fractionated plasma catecholamines and metanephrines) and imaging studies [computed tomography (CT), magnetic resonance imaging, 18F-FDG-PET/CT, 18F-DOPA-PET/CT, and 18F-dopamine-PET/CT] as part of the National Institutes of Health clinical protocol. Fifty-three patients with a diagnosis of carotid body paraganglioma were identified. Patients were excluded if they were managed nonoperatively, if operative records of their primary resection were unavailable, or if they developed metastatic paraganglioma from a noncarotid primary tumor before developing a carotid body paraganglioma. This led to a study cohort of 34 patients who underwent a total of 41 primary resections.
Classification of Operative Complications
The major complications analyzed were perioperative cerebrovascular accident and operative mortality. Other complications considered were postoperative sensory disturbances, cranial nerve injuries, Horner syndrome, prolonged hoarseness, and difficulty swallowing. Only complications that had not resolved at postoperative discharge were included in the analysis.
Statistical Analysis
The primary outcome variables analyzed were disease-free survival (DFS) and overall survival. DFS was defined as the duration between complete operative resection and the development of locoregional recurrence or metastatic disease. Metastatic disease was defined as development of paraganglioma tumors at sites normally devoid of chromaffin cells.20–22 Development of a contralateral carotid body tumor or other primary paraganglioma elsewhere in the body was not considered an end point with respect to DFS.
Data analysis was performed including all 41 operations. The association between clinical and genetic test results and DFS was assessed by the Kaplan-Meier method with censored survival. The statistical difference between Kaplan-Meier curves was determined using the log-rank test. Differences between the mean values for 2 groups of continuous variables were analyzed by t test for Gaussian distributions and the Mann-Whitney U test for non-Gaussian distributions. Associations between dichotomous outcomes and clinical variables were determined by the Fisher exact test. All P values are 2-tailed and P value of less than 0.05 was considered statistically significant. Data for continuous variables are presented as mean ± 1 standard deviation with median and range; cohort characteristics are presented as absolute number n, with cohort percentage within parentheses; follow-up and survival intervals are presented as median value, with range within parentheses.
RESULTS
Patient Characteristics
Thirty-four patients developed a total of 45 carotid body paragangliomas. Forty-one of those lesions were resected, with 4 patients having only 1 of bilateral tumors resected at last follow-up. The average age at surgery was 34 years. Nine patients (26.5%) were asymptomatic at the time of diagnosis. Seventeen patients (50%) were positive for an SDHD mutation and 12 patients (35.3%) were positive for an SDHB mutation. The remaining 5 patients (14.7%) were found to have no identifiable genetic mutation. Fifteen patients (44.1%) developed noncarotid paragangliomas during follow-up. Detailed clinical characteristics of the cohort are shown in Table 1.
TABLE 1.
Study Cohort Clinical Characteristics
| N | |
|---|---|
| Sex | |
| Male/female | 14/20 |
| Age at diagnosis, yr | |
| Mean ± standard deviation | 34.5 ± 14.6 |
| Median, range | 33, 9–72 |
| Tumor characteristics | |
| Total resected tumors | 41 (91.1%) |
| Right-sided | 25 (61.0%) |
| Left-sided | 16 (39.0%) |
| Bilateral | 11 (32.4%) |
| Total unresected tumors | 4 (8.9%) |
| Noncarotid body primary paragangliomas* | 15 (44.1%) |
| Abdominal paraganglioma | 5 (14.7%) |
| Pheochromocytoma | 3 (8.8%) |
| Thoracic paraganglioma | 3 (8.8%) |
| Other head and neck paragangliomas | 6 (17.6%) |
| Presenting symptoms | |
| Neck mass | 13 (38.2%) |
| Palpitations/dizziness/syncope | 10 (29.4%) |
| Hypertension | 6 (17.4%) |
| Ear pain/tinnitus | 3 (8.8%) |
| Other† | 8 (23.5%) |
| Genotype | |
| SDHB | 12 (35.3%) |
| SDHD | 17 (50.0%) |
| No known mutation | 5 (14.7%) |
| Postoperative therapy‡ | |
| External beam radiation | 4 (11.8%) |
| Cyclophosphamide, vincristine, and dacarbazine | 2 (5.9%) |
| I-131 MIBG | 1 (2.9%) |
| Experimental chemotherapy | 2 (5.9%) |
All patients developed noncarotid paragangliomas at the same time or after their carotid disease was identified. Two patients had more than 1 additional paraganglioma.
Four patients complained of diaphoresis, 2 with neck pain, 1 with fatigue, and 1 with tongue sluggishness.
Six patients received nonsurgical therapy after developing recurrent disease. Two patients received multiple treatment modalities (1 131I-MIBG and experimental MBq-Octreotide; the other, cyclophosphamide, vincristine, and dacarbazine; XRT, and experimental 90Y-DOTATOC).
Operative Outcomes
Ten patients (24.4%) suffered a total of 14 complications. There were 2 major complications (4.9%), with both involving patients having right-sided middle cerebral artery embolic cerebrovascular accidents. One of these patients had complete resolution of all neurologic deficits at 3-month follow-up. Additional complications included 6 patients with prolonged cranial nerve deficits, 1 patient with Horner syndrome, 1 patient with dysphagia, and 4 patients with hoarseness. A detailed breakdown of operative outcomes is listed in Table 2.
TABLE 2.
Operative Characteristics and Follow-up
| Tumor Characteristics | |
|---|---|
| Resected tumor size, cm (N = 41) | 2.7 ± 1.2 (range, 0.6–6.0) |
| Unresected tumor size, cm (N = 4) | 1.5 ± 0.6 (range, 1.0–2.5) |
| Grossly invasive at resection | 3 (7.3%) |
| Biochemically active at resection | 1 (2.4%) |
| Operative complications (N = 41 operations) | |
| Total number of complications* | 10 (24.3) |
| Major | 2 |
| Cerebrovascular accident | 2 (4.9%) |
| Operative mortality | 0 |
| Minor | 12 |
| Cranial nerve injury† | 6 (14.6%) |
| Prolonged hoarseness | 4 (9.8%) |
| Horner syndrome | 1 (2.4%) |
| Dysphagia | 1 (2.4%) |
| Follow-up, mo | |
| Median | 42 ± 75.3 |
| Range | 1–293 |
| Disease recurrence and metastasis | |
| Locoregional recurrence | 6 (17.6%) |
| Distant metastasis | 6 (17.6%) |
| Patient status at most recent follow-up | |
| Alive | 31 (91.2%) |
| Deceased‡ | 3 (8.8%) |
There were 14 total complications, with 4 operations having more than 1 complication.
Two cranial nerve VII, 1 cranial nerve X, and 3 cranial nerve XII.
All 3 patients had disease-related mortality.
The average size of a resected tumor was 2.7 cm (range: 0.6–6.0 cm), and complete resection was achieved in all operations. Patients who developed postoperative complications had significantly larger tumors than those without complications (P = 0.02, Fig. 1). The average tumor size at the time of operation was 2.1 cm in patients with germ line SDHB mutations, 3.1 cm in patients with germ line SDHD mutations, and 3.5 cm inpatients without germ line mutations. When stratified by germ line mutation status, patients with SDHB mutations had significantly smaller tumors than patients without SDHB mutations (P = 0.02, Fig. 2).
FIGURE 1.

Tumor size in operations with and without complications. The dashed line at 2.0 cm represents common tumor size indication for surgical resection. Maximum tumor diameter defined as maximum dimension radiologically.
FIGURE 2.

Tumor size by the presence of SDHB mutation status. The non-SDHB group includes patients with SDHD mutations and patients without any known germ line mutation. Maximum tumor diameter defined as maximum dimension radiologically.
Postoperative Survival
Although surgical resection was the primary method of disease control in all 34 patients, 6 patients (17.6%) did receive postoperative medical therapy because of recurrent disease (Table 1). Median follow-up was 42 months (range: 1–293 months), with an overall survival of 91.2% at last follow-up and 100% at median follow-up. Twelve patients (35.3%) developed recurrent or metastatic disease after a median of 126 months (range: 1–293), with 6 patients developing tumors at the site of previous resection and 6 patients developing distant metastases (Fig. 3A). Four patients developed pulmonary metastasis, with 3 of those 4 also having bony metastases. The fifth patient had multiple mediastinal metastases, whereas the sixth had an isolated iliac lesion. Three of the 6 patients developing metastatic disease had other primary paragangliomas (1 pheochromocytoma, 1 abdominal paraganglioma, and 1 cardiac paraganglioma).
FIGURE 3.
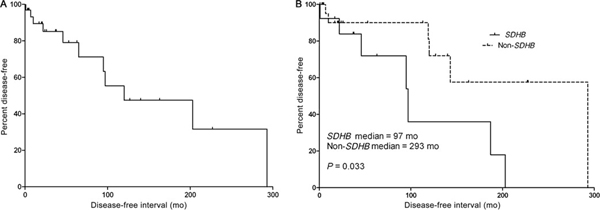
Disease-free survival in patients with carotid body paragangliomas. A, Disease-free survival in patients who underwent surgical resection of their carotid body paragangliomas. Twelve patients developed recurrent or metastatic disease after a median of 126-month follow-up. B, Disease-free survival stratified by the presence of SDHB mutation status. The non-SDHB group includes patients with SDHD mutations and patients without any known mutation.
Among the clinical, genetic, and demographic parameters analyzed, germ line SDHB mutation status was the only variable significantly associated with disease-free interval (P = 0.03, Fig. 3B). When patients with synchronous noncarotid body paragangliomas were excluded, there was still a trend toward worse DFS in patients with SDHB mutations (P = 0.09). Although not statistically significant, patients with SDHB mutations were more likely to develop recurrence (50% in SDHB vs 22.7% in non-SDHB, P = 0.10). There was no difference in overall survival between SDHB and non-SDHB groups (91.7% vs 91.0%). Clinical factors not associated with recurrence included sex (P = 0.57), age (P = 0.80), number of tumors at presentation (P = 0.65), and the presence of noncarotid body paragangliomas (P = 0.40).
DISCUSSION
In this study, we analyzed the clinical factors and genetic testing results associated with DFS and overall survival in a cohort of 34 patients who underwent 41 primary resections of carotid body paragangliomas. Operative complication rates were relatively low, with fewer than 25% of patients affected by any of the defined complications and only 2 patients suffering major complications (cerebrovascular accident). Operative complications were higher in patients with large tumors. Patients with germ line SDHB mutations also had shorter DFS after surgical resection than patients without SDHB mutations. However, there was no association between germ line SDHx mutation status and overall survival. We also found no association between age at diagnosis, number of head and neck paragangliomas at diagnosis, or the presence of abdominal or thoracic paragangliomas with patient outcome.
The most important finding in this study is the relatively poor DFS in patients with SDHB mutations after surgical resection of smaller tumors. The lower DFS observed in this cohort is consistent with data indicating that germ line SDHB mutations are associated with more aggressive disease.13–17 However, it is remarkable that patients with germ line SDHB mutations had a lower DFS despite the fact that the tumors were resected at a smaller tumor size than patients without an SDHB mutation (Fig. 2). The statistical significance of the tumor size difference at the time of operation is unlikely to reflect disease biology or tumor growth rate but rather it is likely due to active screening and a more aggressive surgical approach in patients with a preidentified SDHB mutations. Although the size discrepancy certainly reflects a clinical selection bias, it is nonetheless notable that the earlier intervention in these patients did not result in improved DFS. Thus, these data suggest that delaying resection until a predetermined size (2 cm or larger) does not provide optimal disease control in patients with known SDHB mutations. On the basis of the results of this study, we have altered our surgical approach in patients with carotid body paragangliomas, which incorporates the genetic testing results as outlined in Figure 4.
FIGURE 4.

Management algorithm for carotid body paragangliomas based on genetic testing results. CT indicates computed tomography; MRI, magnetic resonance imaging.
We believe that the present results contribute significantly to operative planning. Current surgical decision making in asymptomatic patients is based largely on tumor size, and previous data have demonstrated that size and growth rate were the best predictors of future tumor behavior.10,11 However, no study has stratified patients on the basis of the presence of genetic mutations to see whether size remains the most important clinical variable for patient outcome. Our data suggest that the genetic background of a carotid body paraganglioma should be the primary driver in the clinical management of patients with these tumors. Furthermore, larger carotid body paragangliomas are more likely to cause morbidity associated with local extension, whereas our data indicate that smaller paragangliomas are associated with fewer complications. This indicates that aggressive resection of carotid body paragangliomas in patients with germ line SDHB mutations, regardless of size or clinical symptoms, would result in safer operations and that the earlier intervention is justified to reduce the risk of disease recurrence.
There are several limitations to this study. There is no nonoperative control group of patients to compare patient outcome, largely because most patients undergo surgical treatment on the basis of symptoms and the risk of malignancy. Our results, however, suggest that a clinical trial comparing active surveillance with surgical treatment may be warranted in patients without germ line SDHB mutations. Second, 15 patients (44.1%) developed noncarotid paragangliomas during follow-up, which could be a confounding factor because of the possibility that metastatic disease developed from a noncarotid primary tumor. This is a difficulty when studying paraganglioma syndromes, but we did exclude patients whose disease burden was predominantly related to other paraganglioma sites. In addition, the presence of noncarotid body paragangliomas was not associated with reduced DFS, and when patients with synchronous noncarotid body tumors were excluded, there was still a trend toward worse DFS (due to local recurrence, P = 0.10) inpatients with SDHB mutations. The clinical applicability of these data is dependent on the patient having a known germ line SDHB mutation before developing a carotid body paraganglioma. Although this will make early resection difficult or impossible for the index case of paraganglioma syndrome type 4 in a given family, subsequent screening will allow for earlier surgical intervention in at-risk family members. As genetic testing continues to expand and more families are identified, resection at subclinical tumor sizes will become increasingly feasible and should be known in any patient suspected to have a carotid body paraganglioma before treatment decisions are made. Finally, although our findings are based on a relatively small cohort, this study represents one of the largest series in the literature because of the rarity of carotid body paragangliomas. The clinical applicability of the small cohort is further enhanced by the uniform genetic testing performed for common susceptibility genes and comprehensive anatomic and functional imaging studies at presentation and follow-up.
CONCLUSIONS
In summary, our results indicate that patients with carotid body paragangliomas in the setting of germ line SDHB mutations are more likely to develop locoregional recurrence or metastatic disease than patients with non-SDHB mutations. These outcomes are observed despite the fact that, on average, patients with SDHB mutations had surgical interventions for smaller tumors. Our data challenge the notion that only tumor size should drive operative decision making in the absence of symptoms and indicate that patients with SDHB mutations should be surgical candidates even when only small carotid body tumors are detected. Conversely, more conservative management based on symptoms, tumor size, and absence of invasive imaging tumor characteristics may be justified in patients without germ line SDHB mutations.
Acknowledgments
Disclosure: Supported by the intramural research program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
The authors declare no conflicts of interest.
REFERENCES
- 1.Schipper J, Boedeker CC, Maier W, et al. [Paragangliomas in the head-/neck region. I: classification and diagnosis]. HNO. 2004;52:569–574; quiz 575. [DOI] [PubMed] [Google Scholar]
- 2.Lee JH, Barich F, Karnell LH, et al. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer. 2002;94:730–737. [DOI] [PubMed] [Google Scholar]
- 3.Glenner GG, Grimley PM. Tumors of the Extra-Adrenal Parnganglion System (Including Chemoreceptors). Washington: Armed Forces Institute of Pathology; for sale by the American Registry of Pathology, Armed Forces Institute of Pathology; 1974. [Google Scholar]
- 4.Kleihues P, Cavenee WK, International Agency for Research on Cancer. Pathology and Genetics of Tumours of the Nervous System. Lyon: IARC Press; 2000. [Google Scholar]
- 5.Schwaber MK, Glasscock ME, Nissen AJ, et al. Diagnosis and management of catecholamine secreting glomus tumors. Laryngoscope. 1984;94:1008–1015. [DOI] [PubMed] [Google Scholar]
- 6.Erickson D, Kudva YC, Ebersold MJ, et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. 2001;86:5210–5216. [DOI] [PubMed] [Google Scholar]
- 7.Maier W, Marangos N, Laszig R. Paraganglioma as a systemic syndrome: pitfalls and strategies. J Laryngol Otol. 1999;113:978–982. [DOI] [PubMed] [Google Scholar]
- 8.Pellitteri PK, Rinaldo A, Myssiorek D, et al. Paragangliomas of the head and neck. Oral Oncol. 2004;40:563–575. [DOI] [PubMed] [Google Scholar]
- 9.Boedeker CC. Paragangliomas and paraganglioma syndromes. GMS Curr Top Otorhinolaryngol Head Neck Surg. 2011;10:Doc03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langerman A, Athavale SM, Rangarajan SV, et al. Natural history of cervical paragangliomas: outcomes of observation of 43 patients. Arch Otolaryngol Head Neck Surg. 2012;138:341–345. [DOI] [PubMed] [Google Scholar]
- 11.Jansen JC, van den Berg R, Kuiper A, et al. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer. 2000;88:2811–2816. [PubMed] [Google Scholar]
- 12.de Flines J, Jansen J, Elders R, et al. Normal life expectancy for paraganglioma patients: a 50-year-old cohort revisited. Skull Base. 2011;21:385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. [DOI] [PubMed] [Google Scholar]
- 14.Boedeker CC, Neumann HP, Maier W, et al. Malignant head and neck paragangliomas in SDHB mutation carriers. Otolaryngol Head Neck Surg. 2007;137:126–129. [DOI] [PubMed] [Google Scholar]
- 15.Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63:5615–5621. [PubMed] [Google Scholar]
- 16.Matro J, Giubellino A, Pacak K. Current and future therapeutic approaches for metastatic pheochromocytoma and paraganglioma: focus on SDHB tumors. Horm Metab Res. 2013;45:147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Hulsteijn LT, Dekkers OM, Hes FJ, et al. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: a systematic review and meta-analysis. J Med Genet. 2012;49:768–776. [DOI] [PubMed] [Google Scholar]
- 18.Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab. 2007;3:92–102. [DOI] [PubMed] [Google Scholar]
- 19.Fruhmann J, Geigl JB, Konstantiniuk P, et al. Paraganglioma of the carotid body: treatment strategy and SDH-gene mutations. Eur J Vasc Endovasc Surg. 2013;45:431–436. [DOI] [PubMed] [Google Scholar]
- 20.O’Riordain DS, Young WF Jr, Grant CS, et al. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg. 1996;20:916–921; discussion 922. [DOI] [PubMed] [Google Scholar]
- 21.Wangberg B, Muth A, Khorram-Manesh A, et al. Malignant pheochromocytoma in a population-based study: survival and clinical results. Ann N Y Acad Sci. 2006;1073:512–516. [DOI] [PubMed] [Google Scholar]
- 22.Zarnegar R, Kebebew E, Duh QY, et al. Malignant pheochromocytoma. Surg Oncol Clin N Am. 2006;15:555–571. [DOI] [PubMed] [Google Scholar]