

Abstract
The cellular stress response has a vital role in regulating homeostasis by modulating cell survival and death. Stress granules are cytoplasmic compartments that enable cells to survive various stressors. Defects in the assembly and disassembly of stress granules are linked to neurodegenerative diseases, aberrant antiviral responses and cancer1–5. Inflammasomes are multi-protein heteromeric complexes that sense molecular patterns that are associated with damage or intracellular pathogens, and assemble into cytosolic compartments known as ASC specks to facilitate the activation of caspase-1. Activation of inflammasomes induces the secretion of interleukin (IL)-1β and IL-18 and drives cell fate towards pyroptosis—a form of programmed inflammatory cell death that has major roles in health and disease6–12. Although both stress granules and inflammasomes can be triggered by the sensing of cellular stress, they drive contrasting cell-fate decisions. The crosstalk between stress granules and inflammasomes and how this informs cell fate has not been well-studied. Here we show that the induction of stress granules specifically inhibits NLRP3 inflammasome activation, ASC speck formation and pyroptosis. The stress granule protein DDX3X interacts with NLRP3 to drive inflammasome activation. Assembly of stress granules leads to the sequestration of DDX3X, and thereby the inhibition of NLRP3 inflammasome activation. Stress granules and the NLRP3 inflammasome compete for DDX3X molecules to coordinate the activation of innate responses and subsequent cell-fate decisions under stress conditions. Induction of stress granules or loss of DDX3X in the myeloid compartment leads to a decrease in the production of inflammasome-dependent cytokines in vivo. Our findings suggest that macrophages use the availability of DDX3X to interpret stress signals and choose between pro-survival stress granules and pyroptotic ASC specks. Together, our data demonstrate the role of DDX3X in driving NLRP3 inflammasome and stress granule assembly, and suggest a rheostat-like mechanistic paradigm for regulating live-or-die cell-fate decisions under stress conditions.
Both stress granules and NLRP3 inflammasomes are induced under stress conditions and form cytosolic compartments1,4,7,13–18. However, our understanding of the crosstalk between stress granules and NLRP3 inflammasome activation remains limited. To address this gap in our knowledge, we constructed a system based on a well-established approach that uses sodium arsenite to induce the formation of stress granules1,19. We used lipopolysaccharide (LPS) priming followed by nigericin treatment to induce the activation of the NLRP3 inflammasome in bone-marrow-derived macrophages (BMDMs)20. To decouple the effect of translation inhibition by the induction of stress granules from the potential crosstalk between pro-survival stress granules and pro-cell-death NLRP3 inflammasomes, we induced stress granules in BMDMs by adding sodium (meta)arsenite (hereafter referred to as arsenite) after 4 h of LPS priming. Confocal microscopy imaging of G3BP1, a marker of stress granules, and ASC, an adaptor protein that is a component of the NLRP3 inflammasome, revealed that arsenite induced the formation of stress granules in LPS-primed BMDMs (Fig. 1a). ASC was not recruited to the stress granules, and there was no assembly of inflammasome-driven ASC specks in the cytoplasm (Fig. 1a). Conversely, activation of NLRP3 inflammasomes by nigericin in LPS-primed BMDMs led to the assembly of ASC specks without inducing stress granules (Fig. 1a). To test the effect of stress granules on NLRP3 inflammasome activation, we triggered the assembly of stress granules in LPS-primed BMDMs by treating them with arsenite before adding nigericin. Notably, we observed defects in the assembly of ASC specks in cells that contained stress granules, which suggests that NLRP3 inflammasome activation is inhibited by stress granules (Fig. 1a). Induction of stress granules before treatment with nigericin resulted in a strong reduction, compared to cells in which stress granules had not been induced, in the cleavage of caspase-1 (CASP1) and gasdermin D (GSDMD), as well as a decrease in secretion of the leaderless pro-inflammatory cytokines IL-1β and IL-18—again indicating that NLRP3 inflammasome activation is inhibited by stress granules (Fig. 1b, c). Real-time monitoring of cell death by staining with SYTOX Green dye showed that arsenite-induced stress granules also inhibited the pyroptosis that is driven by the NLRP3 inflammasome, suggesting that a pro-survival cell-fate decision is induced by stress granules (Fig. 1d). To directly determine whether stress granules modulate cell-fate decisions, we monitored the dynamics of pyroptotic death in BMDMs during changes in stress-granule-inducing conditions. Stress granule assembly maintained a pro-survival phenotype in cells even after the removal of the stress-granule-inducing trigger (Fig. 1e). To further confirm the specificity of stress-granule-mediated inhibition of NLRP3 inflammasome activation, we inhibited arsenite-induced stress granule assembly by treating BMDMs with anisomycin; this treatment reduced both stress granule assembly (Extended Data Fig. 1a) and NLRP3 inflammasome activation (Fig. 1f, g). However, using anisomycin followed by arsenite significantly rescued NLRP3 inflammasome activation (Fig. 1f, g) and significantly increased the percentage of cells that contained ASC specks, compared with cells that were treated with arsenite alone (Extended Data Fig. 1b). Several stimuli activate the NLRP3 inflammasome, through mechanisms that either depend on or are independent of potassium efflux. Stress granules inhibited NLRP3 activation that was brought about by a potassium efflux-independent trigger (LPS and imiquimod) and by mechanical damage (LPS and silica) (Fig. 1h, i). Treatment with arsenite also inhibited NLRP3 inflammasome activation in BMDMs primed with Pam3CSK4, β−1, 3-glucan (curdlan) or polyinosinic:polycytidylic acid (poly(I:C)) (Fig. 1j, k, Extended Data Fig. 1c). The release of the cytokines IL-6, KC (also known as CXCL1) and TNF was unaffected by stress granules (Extended Data Fig. 1d). In addition, stress granules did not affect the activation of the AIM2 or NLRC4 inflammasomes (Extended Data Fig. 1e, f), which suggests that the inhibitory effect of stress granules is specific to the NLRP3 inflammasome. These findings indicate that the induction of stress granules by arsenite treatment specifically suppresses NLRP3 inflammasome activation to enable the cells to make a pro-survival cell-fate decision.
Fig. 1 |. Arsenite-induced stress granules inhibit the NLRP3 inflammasome and pyroptosis.

a, Confocal microscopy of BMDMs stimulated with LPS and treated with arsenite (Ar) (1 h), nigericin (N) (45 min), or arsenite (30 min) followed by nigericin (45 min), stained for G3BP1 and ASC. Scale bars, 10 μm. Representative images (n > 3). b, Immunoblot analysis of pro-CASP1 (p45) and cleaved CASP1 (p20), and GSDMD (p50) and cleaved GSDmD (p30). Representative images (n = 3). c, Enzyme-linked immunosorbent assay measurement of IL-1β and IL-18. ****P < 0.0001 (unpaired two-sided t-test; n > 10). d, Measurement of pyroptosis by SYTOX Green staining. ****P ≤ 0.0001 for LPS + N versus LPS + Ar + N (two-way analysis of variance (ANOVA); n = 4). e, Measurement of pyroptosis by SYTOX Green staining in BMDMs stimulated with LPS and treated with arsenite and nigericin, with or without removal of nigericin after 15 min of treatment (N(R)). ****P ≤ 0.0001 for LPS + N(R) versus LPS + Ar + N(R) (two-way ANOVA; n = 4) Data are mean ± s.e.m. (c-e). f, Immunoblot analysis of CASP1 in BMDMs stimulated with LPS and treated with nigericin, arsenite and anisomycin (An) as indicated. Representative images (n > 6). g, Measurement of IL-18 and IL-1β from LPS-stimulated BMDMs treated as indicated. P values (from left to right): ****P < 0.0001, *P = 0.0109, ****P < 0.0001, ***P= 0.0003 (unpaired two-sided t-test; n> 6). h-k, Immunoblot analysis of CASP1 cleavage (representative blots, n > 2) and measurement of IL-1β in LPS-stimulated BMDMs treated with arsenite and imiquimod (Im) (h; n= 4), silica (Si) (i; n= 2), Pam3CSK4 (Pam3) with or without nigericin (j, n = 4), or curdlan (Cn) with or without nigericin (k, n= 2). P values (from left to right): ****P < 0.0001, ****P = 0.0001 (unpaired two-sided t-test). Data are mean ± s.e.m (g-k).
The inhibition of NLRP3 inflammasome activation that was mediated by stress granules was not caused by an arrest of translation, as levels of pro-IL-1β (the inactive cytoplasmic precursor of IL-1β), NLRP3, AsC and CASP1 were unaffected (Extended Data Fig. 1g). We observed no colocalization of G3BP1 and NLRP3, which indicates that NLRP3 was not sequestered in stress granules (Extended Data Fig. 1h). Stress granules inhibited NLRP3 inflammasome activation in BMDMs that lack essential components of the autophagy machinery (BECN1 and ATG5), which suggests that the mechanism of inhibition is autophagy-independent (Extended Data Fig. 1i).
Using affinity purification mass spectrometry, we identified DDX3X—a known component of stress granules—as an interacting partner of NLRP3 under both LPS-primed and inflammasome-activating conditions (Fig. 2a, Extended Data Fig. 2a, Supplementary Table 1) (proteomics data are available through ProteomeXchange with the identifier PXD014828). We confirmed that DDX3X was recruited to the stress granules (Extended Data Fig. 2b). ASC did not colocalize with DDX3X in stress granules—again verifying that ASC is not recruited to stress granules (Extended Data Fig. 2c). Our identification of a physical interaction between NLRP3 and DDX3X led us to speculate that DDX3X might have a role in the stress-granule-mediated inhibition of NLRP3 inflammasomes.
Fig. 2 |. The stress granule component DDX3X interacts with NLRP3 and is required for NLRP3 inflammasome activation.

a, Co-immunoprecipitation (IP) of NLRP3 and DDX3X in BMDMs treated with LPS with or without nigericin. Representative blots (n= 3). b, Immunoblot analysis of CASP1 cleavage in wild-type (WT), Ddx3xfl/fl, Ddx3xfl/flLysMcre and Nlrp3−/− BMDMs treated with LPS and nigericin. Representative blots (n > 3). c, Immunoblots of DDX3X expression and CASP1 cleavage after siRNA-mediated knockdown of Ddx3x in BMDMs treated with LPS with or without nigericin. Representative blots (n = 2). d, Confocal microscopy imaging of ASC specks in BMDMs treated with LPS with or without nigericin, to visualize the subcellular localization of DDX3X, NLRP3 and ASC. Scale bars, 10 μm. Representative images (n = 3). e, Schematic of N-terminal Flag-tagged NLRP3 expression constructs: full-length NLRP3 (Flag-NLRP3-FL), the pyrin domain of NLRP3 (Flag-PYD; amino acids 1–90 of NLRP3), the NACHT domain of NLRP3 (Flag-NACHT; amino acids 91–710 of NLRP3) and the LRR domain of NLRP3 (Flag-LRR; amino acids 711–1034 of NLRP3). f, Immunoblot (IB) analysis of input lysates used for co-immunoprecipitation of Flag-tagged NLRP3 constructs and DDX3X-mCherry. Representative blots (n > 3). g, Immunoblot analysis of immunoprecipitation of DDX3X-mCherry and the indicated NLRP3 constructs. Red asterisk indicates the antibody light chain. Representative blots (n > 3).
We generated mice with a conditional knockout allele of Ddx3x (Ddx3xfl/fl) and deleted the gene from the myeloid compartment by breeding these mice with LysMcre mice (LysM is also known as Lyz2) (Extended Data Fig. 3a-c). Levels of DDX3X protein were markedly decreased in Ddx3xfl/flLysMcre BMDMs (Extended Data Fig. 3c). BMDMs that lacked DDX3X showed similar expression levels of the myeloid cell markers CD11b and F4/80 (Extended Data Fig. 3d), and there was no defect in haematopoietic-cell development (Extended Data Fig. 3e). We observed a significant decrease in the numbers of arsenite-induced stress granules in BMDMs that were derived from Ddx3xfl/flLysMcre mice compared with Ddx3xfl/fl mice (Extended Data Fig. 4a, b).
NLRP3 inflammasome activation was reduced in Ddx3xfl/flLysMcre BMDMs compared with wild-type and Ddx3xfl/fl BMDMs, which suggests that DDX3X itself is required for inflammasome activation (Fig. 2b). We confirmed this result using siRNA-mediated knockdown of Ddx3x in BMDMs (Fig. 2c). NLRP3 inflammasome activation was also defective in peritoneal macrophages that are deficient in DDX3X (Extended Data Fig. 4c). There was less pyroptotic cell death in DDX3X-deficient BMDMs (Extended Data Fig. 4d). In addition, NLRP3 inflammasome activation was defective in Ddx3xfl/flLysMcre BMDMs that were treated with LPS and ATP, Pam3CSK4 and nigericin, and poly(I:C) and nigericin (Extended Data Fig. 4e-g). Together, these results suggest that DDX3X, as well as being essential for stress granule assembly, is critical for activation of the NLRP3 inflammasome.
Activation of the NLRP3 inflammasome and release of IL-1β and IL-18 were strongly suppressed in the absence of DDX3X (Extended Data Fig. 5a). By contrast, activation of the NLRC4, AIM2 and PYRIN inflammasomes was similar in Ddx3xfl/fl and Ddx3xfl/flLysMcre BMDMs (Extended Data Fig. 5b-d). When we overexpressed DDX3X in BMDMs using a doxycycline-inducible promoter, NLRP3 inflammasome activation increased with increased doxycycline concentration (Extended Data Fig. 5e). Constitutive overexpression of DDX3X also rescued NLRP3 inflammasome activation in Ddx3xfl/flLysMcre BMDMs (Extended Data Fig. 5f).
There was no observable difference in the expression of NLRP3, ASC, CASP1, pro-IL-1β and NEK7 between Ddx3xfllf and Ddx3xfllf LysMcre BMDMs (Extended Data Fig. 6a). When coexpressed in HEK293T cells, DDX3X interacted with NLRP3 but not with ASC or CASP1 (Extended Data Fig. 6b-d). DDX3X colocalized with ASC and NLRP3 in ASC specks, which suggests that DDX3X is a component of the inflammasome complex (Fig. 2d). The percentage of cells that contain ASC specks was reduced in DDX3X-deficient BMDMs compared to wild-type cells (Extended Data Fig. 6e), and we recapitulated this result from BMDMs in peritoneal macrophages (Extended Data Fig. 6f, g). These findings support the notion that DDX3X regulates the assembly of the NLRP3 inflammasome by interacting with NLRP3.
We monitored NLRP3 inflammasome activation in BMDMs after treatment with various agents that trigger the formation of stress granules: sorbitol (osmotic shock), thapsigargin (endoplasmic reticulum stress), vincristine (a vinca alkaloid) and paclitaxel. Sorbitol and thapsigargin did not suppress the activation of the NLRP3 inflammasome, whereas vincristine and paclitaxel strongly inhibited activation (Extended Data Fig. 7a–d). Sorbitol and thapsigargin also did not induce stress granules in LPS-primed BMDMs, whereas vincristine and paclitaxel did induce stress granules (Extended Data Fig. 7e). All stressors induced stress granules in the mouse cell line L929 (Extended Data Fig. 7f). Treatment with RK-33 (an inhibitor of DDX3X helicase activity) did not induce stress granules in BMDMs, but efficiently induced them in L929 cells21 (Extended Data Fig. 7e, f). In sum, these findings further indicate that stress granule formation inhibits NLRP3 inflammasome activation.
DDX3X interacted with full-length NLRP3 and with the NLRP3 NACHT domain (also known as the nucleotide-binding domain; NBD) when coexpressed in HEK293T cells (Fig. 2e–g). This suggests that the binding site for DDX3X is located in the NACHT (NBD) domain of NLRP3, and implies that DDX3X has a role in facilitating the oligomerization of NLRP316,18. NLRP3 interacted with full-length, N-terminally truncated and C-terminally truncated versions of DDX3X, as well as with its helicase domain (Extended Data Fig. 8a–f). It is therefore possible that the binding site for NLRP3 resides in the helicase domain of DDX3X (Extended Data Fig. 8c–f). The interaction between DDX3X and NLRP3 might be mediated by intrinsically disordered regions22,23, which are present in the N and C termini and the helicase domain of DDX3X. NLRP3 also contains predicted intrinsically disordered regions in the NACHT domain (Extended Data Fig. 8g, h). Inhibition of DDX3X helicase activity by RK-33 did not affect NLRP3 inflammasome activation (Extended Data Fig. 8i).
Treatment with nigericin induced the oligomerization of NLRP3, DDX3X and ASC when coexpressed in HEK293T cells, and a notable amount of these oligomers was present in the NP-40 insoluble fraction—indicating formation of the inflammasome complex (Extended Data Fig. 9a). Treatment with arsenite disassembled ASC oligomers (Extended Data Fig. 9a). We also monitored endogenous oligomers in BMDMs, with similar findings (Extended Data Fig. 9b). These results show that arsenite-induced stress granules interfere with NLRP3 inflammasome activation.
Analytical ultra-centrifugation analysis revealed that NLRP3, DDX3X and ASC relocalize to complexes of larger molecular weight during inflammasome activation (Fig. 3a). Furthermore, ASC was present in the insoluble fraction, suggesting formation of ASC specks (Fig. 3b). Similar to our observations in oligomer crosslinking experiments, DDX3X and ASC failed to localize to larger complexes in arsenite-treated cells (Fig. 3a). This further confirms that the assembly of a functional NLRP3 inflammasome requires recruitment of both DDX3X and ASC into the complex.
Fig. 3 |. DDX3X promotes NLRP3 inflammasome assembly, and stress granules and the NLRP3 inflammasome compete for DDX3X.

a, Immunoblot analysis of lysates fractionated by analytical ultra-centrifugation to test the oligomerization of NLRP3, DDX3X and ASC in BMDMs stimulated with LPS and treated either with nigericin or with arsenite and nigericin. Representative blots (n = 2). MW, molecular weight. b, Immunoblot analysis of ASC from the insoluble fraction of samples from a under non-reducing (without β-mercaptoethanol; – BME) and reducing (+BME) conditions. Representative blots (n = 2). c, STORM imaging of ASC speck assembly and its spatial organization with DDX3X over time. Scale bars, 5 μm (whole-cell images); 1 μm (magnified images). Representative images (n = 2). d, Plot of the r-index to show the extent of colocalization of ASC and DDX3X with the duration of nigericin treatment (n = 2). R2 was calculated using linear regression analysis; P = 0.0125 (for the significance of the slope being non-zero). Data are mean ± s.e.m. e, f, Immunoblot analysis of CASP1 cleavage in LPS-primed BMDMs to which arsenite was added at various time points (30 and 15 min before adding nigericin, simultaneously with nigericin and 15 and 30 min after adding nigericin), without (e) or with (f) pre-treatment with anisomycin. Representative blots (n = 3). g, h, Structured illumination microscopy of BMDMs to visualize the subcellular localization of DDX3X, G3BP1 and ASC in LPS-primed BMDMs treated with nigericin alone (g) or with arsenite and nigericin (h). Scale bars, 3 μm. Representative images (n = 3).
We performed a time-course experiment for inflammasome activation and measured the extent to which DDX3X and ASC colocalized using stochastic optical reconstruction microscopy (STORM) (Fig. 3c). The colocalization of DDX3X and ASC (as measured by the r-index, a measure developed to analyse colocalization in super-resolution data; X. Liu et al., unpublished protocol) increased with the duration of nigericin treatment, suggesting that DDX3X progressively associates with the NLRP3 inflammasome complex (Fig. 3d). These findings suggest that DDX3X is a critical factor for NLRP3 inflammasome assembly.
In a time-course competition experiment between stress granule assembly and inflammasome activation, LPS-primed BMDMs were treated with arsenite 30 and 15 min before, simultaneously with, or 15 and 30 min after the addition of nigericin. The timing of the arsenite treatment strongly correlated with the extent of NLRP3 inflammasome inhibition (Fig. 3e). Treatment with anisomycin (to inhibit stress granule assembly) before the addition of nigericin ameliorated the inhibition of NLRP3 that was mediated by arsenite-induced stress granules (Fig. 3f). The release of IL-1β and IL-18 followed a similar trend to that of CASP1 cleavage (Extended Data Fig. 9c, d). We fitted a sigmoidal curve to the cytokine data and found that R2 was greater than 0.8 for all conditions for IL-1β, and greater than 0.68 for IL-18. This suggests a strong correlation between the timing of stress granule formation and the amount of IL-1β and IL-18 released (Extended Data Fig. 9c, d). Anisomycin facilitated increased secretion of IL-1β and IL-18, suggesting that stress granule inhibition increased NLRP3 activation (Extended Data Fig. 9c, d).
The time-course analysis of stress granule formation and NLRP3 inflammasome assembly suggests that stress granule assembly and NLRP3 inflammasome activation are mutually exclusive, kinetically controlled processes that can regulate one another by competing for a common essential factor. Loss of DDX3X disrupted the assembly of both stress granules and ASC specks, indicating that DDX3X is required for both processes and might be the common factor. To test the effect of stress granule induction on the localization of DDX3X, we performed structured illumination microscopy with LPS-primed BMDMs that were treated either with nigericin alone or with arsenite and nigericin (Fig. 3g, h). In cells treated with nigericin alone, DDX3X colocalized in ASC specks (Fig. 3g); in cells treated with arsenite and nigericin, DDX3X colocalized with G3BP1 in the stress granules (Fig. 3h). Loss of G3BP1 (which is also a component of stress granules) had only a minimal effect on NLRP3 inflammasome activation (Extended Data Fig. 10a). This suggests that DDX3X is the common essential factor for which stress granules and NLRP3 inflammasomes compete. This competition enables the cells to make a live-or-die cell-fate decision that is based on DDX3X availability.
Injection of arsenite induced stress granules in mice in vivo (Fig. 4a), and DDX3X was required for stress granule assembly (Fig. 4a, b). Arsenite injection led to a decrease in the recruitment of CD11b-positive myeloid cells to the peritoneal cavity in an LPS-induced model of peritonitis (Fig. 4c). Consistent with decreased NLRP3 inflammasome activation, the amount of IL-1β in the serum and peritoneal fluid was also decreased in mice that were treated with arsenite (Fig. 4d, e). By contrast, arsenite injection did not consistently reduce levels of other inflammatory cytokines (Extended Data Fig. 10b, c). Intraperitoneal injection of LPS into Ddx3xfl/flLysMcre mice led to a decrease in IL-1β in the serum and peritoneal fluid compared with control mice (Fig. 4f). Collectively, these data show that the formation of stress granules or loss of DDX3X compromises NLRP3 inflammasome activation in vivo.
Fig. 4 |. Induction of stress granules and loss of DDX3X in the myeloid compartment lead to a decrease of IL-β in serum and peritoneal fluid.

a, Confocal microscopy imaging of peritoneal CD45+ cells to visualize in vivo stress granules in Ddx3xfl/fl and Ddx3xfl/flLysMcre mice that were treated with PBS or arsenite. Scale bars, 10 μm. Representative images (n = 2). b, Quantification of CD45+ cells that contain stress granules, from the peritoneal cavity of Ddx3xfl/fl and Ddx3xfl/flLysMcre mice that were injected with arsenite or PBS. ****P < 0.0001 (unpaired two-sided t-test). Data represent two biologically independent experiments (n = 10 frames). c, Quantification of the numbers and percentage of CD11b+ myeloid cells in the peritoneal cavity. P values (from left to right): ***P = 0.0001, ***P = 0.0005 (unpaired two-sided t-test; n = 7). d, e, Levels of IL-1β in the serum (d) and peritoneal fluid (PF) (e) of mice that were injected with LPS with or without prior arsenite injection in the peritoneum. P values: **P = 0.0026 (d), P = 0.09 (e) (unpaired two-sided t-test; n = 7). f, Levels of IL-1β in the serum and peritoneal fluid of Ddx3xfl/fl and Ddx3xfl/flLysMcre mice that were injected with LPS in the peritoneum. P values (from top to bottom): **P = 0.0073, *P = 0.0123 (unpaired two-sided t-test; n ≥ 6). Data are mean ± s.e.m. (b-f). g, Schematic of the interplay between the inflammasome and stress granules that is involved in cell-fate decisions. DDX3X promotes NLRP3 inflammasome activation and the pro-death cell-fate decision probably by interacting with the NLRP3 NACHT domain through its helicase (Heli) domain. Induction of stress granules causes the sequestration of DDX3X (along with 40S ribosomal subunits and translation initiation factors (eIFs)), thus making it unavailable for NLRP3 inflammasome activation and thereby allowing the cells to make a pro-survival cell-fate choice.
In conclusion, we have identified a mechanism for regulating cell-fate decisions in stressed cells. In this paradigm, pyroptosis that is mediated by the NLRP3 inflammasome depends on the availability of DDX3X molecules, and DDX3X is also required for the pro-survival assembly of stress granules (Fig. 4g). Owing to its importance in both NLRP3 inflammasome activation and stress granule assembly, DDX3X sits at a crossroads between inflammation and stress adaptation, and acts in effect as a live-or-die checkpoint in the stressed cells. DDX3X could therefore represent an attractive target for designing drugs for the therapeutic modulation of stress responses and NLRP3 inflammasome activation.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-019-1551-2
METHODS
DDX3X.
DDX3X is a highly conserved DEAD-box family protein, the loss of which disrupts stress granule assembly24–26. Ddx3x is X-chromosome-linked, with a paralogue present on the Y-chromosome (Ddx3y) that has over 90% similarity. DDX3X has roles in translational control, WNT signalling, viral RNA sensing and interferon-mediated gene expression25,27–29. It is frequently mutated in cancers and has been a target for designing anti-cancer drugs21,30. DDX3X is expressed in all cell types, and DDX3Y is predominantly expressed in male germline and neuronal cells31,32. Genetic deletion of DDX3X in mice is embryonically lethal33,34.
Mice.
The Ddx3x allele was modified in mice using CRISPR-Cas9 gene editing. Pronuclear-stage zygotes were injected with two single-guide RNAs (sgRNAs) that target introns 6 and 14 of Ddx3x with Cas9 mRNA transcripts, and homology- directed repair oligonucleotides (DDX3X-I6-LoxP-HDR and DDX3X-I14-LoxP-HDR) designed to incorporate loxP sites together with EcoRI restriction sites to facilitate targeting and genotyping. Cre recombination of the modified Ddx3x allele (Ddx3xfl/fl) resulted in deletion of exons 7–14. Injected embryos were surgically transplanted into the oviducts of pseudo-pregnant CD1 females, and newborn mice bearing the exon 7–14 deletion were identified by amplifying a 1.1-kb fragment with primers that flank the two break sites. Nlrp3−/−(ref. 7), Aim2−/− (ref. 35), Nlrc4−/− (ref. 36) and Pyrin−/− (ref. 37) mice have been described previously. All mice were bred at St. Jude Children’s Research Hospital. Mouse studies were conducted in accordance with protocols approved by the St. Jude Animal Care and Use Committee and in compliance with all relevant ethical guidelines. Mice (6–8 weeks old) from the same cage were randomly selected for different treatments. Littermate controls were used, wherever possible. Experiments were performed with both females and males. Investigators were not blinded, as none of the reported experiments required subjective decision making. Key experiments were repeated by at least three independent researchers.
Cell culture.
Primary BMDMs were grown for 6 days in DMEM (Thermo Fisher Scientific, 11995–073) supplemented with 10% FBS (Biowest, S1620), 30% L929- conditioned medium and 1% penicillin and streptomycin (Sigma Aldrich). BMDMs were seeded in antibiotic-free medium in 12-well plates at a density of 1 × 106 cells per well and incubated overnight before stimulation with inflammasome ligands. HEK293T and L929 cell lines were acquired from ATCC, which thoroughly authenticates cell lines. Cell lines tested negative for mycoplasma contamination.
Stimulation of BMDMs.
To study the activation of the canonical NLRP3 inflammasome, BMDMs were primed for 4 h with 100 ng ml−1 ultrapure LPS from Salmonella minnesota R595 (InvivoGen, tlrl-smlps) and stimulated for 30–45 min with 20 μM nigericin (Cayman Chemical, 11437). For DNA transfection, each reaction consisted of 2 μg poly(dA:dT) (InvivoGen, tlrl-patn) resuspended in PBS and mixed with 0.5 μl Xfect polymer in Xfect Reaction Buffer (Clontech Laboratories, 631318). After 10 min, DNA complexes were added to BMDMs in Opti-MEM (Thermo Fisher Scientific, 31985–070); this was incubated for 3–5 h. For bacterial protein transfection, 0.5 μg ultrapure flagellin from Salmonella enterica subsp. enterica serovar Typhimurium (InvivoGen, tlrl-epstfla-5) was resuspended in PBS and mixed with 20 μl DOTAP (Roche, 11202375001) for each reaction. The reaction mixture was incubated for 40 min then added to BMDMs in 500 μl Opti-MEM.
To induce stress granule formation, BMDMs were first primed with LPS for 4 h, then treated with 50 μM sodium (meta)arsenite (Sigma-S7400) for 30 min before ligands were added to activate the inflammasomes. To inhibit stress granule formation, 25 μg ml−1 anisomycin (Cayman Chemical, 11308) was added to the LPS-primed BMDMs 20 min before the arsenite treatment. To induce stress granules in analyses of BMDMs and L929 cells, 400 μM sorbitol38 (Alfa Aesar, 36404), 2 μg ml−1 thapsigargin (Cayman Chemical, 10522), 750 μM vincristine39 (Cayman Chemical, 11764), 400 μM paclitaxel (Cayman Chemical, 10461) and 5 μM RK-33 (Selleckchem, S8246) were added for 30 min before nigericin treatment or 1 h when used without nigericin. Cell culture supernatants were collected for enzyme-linked immunosorbent assays (ELISAs) after each stimulation.
Immunoblotting analysis.
Immunoblotting was performed as previously described40. In brief, cells were washed with cold PBS and lysed in RIPA buffer and sample-loading buffer containing SDS and 2-mercaptoethanol. BMDM cell lysates and culture supernatants were combined in caspase lysis buffer (containing protease inhibitors, 5% NP-40 and 25 mM DTT) and sample-loading buffer (containing SDS and 2-mercaptoethanol) for immunoblot analysis of CASP1. Proteins were separated on 8–12% polyacrylamide gels and transferred onto PVDF membranes. Membranes were blocked in 5% skimmed milk then incubated with primary antibodies and secondary horseradish peroxidase (HRP)-conjugated antibodies. The primary antibodies were anti-CASP1 (AdipoGen, AG-20B-0042), anti-NLRP3 (AdipoGen, AG-20B-0014), anti-NLRP3 (Cell Signaling Technology (CST), 15101), anti-ASC (AdipoGen, AG-25B-006-C100), anti-IL-1|3 (R&D Systems, AF-401-NA), anti-DDX3X (Bethyl Laboratories, A300–474A), anti- NEK7 (Abcam, ab133514), anti-G3BP1 (Proteintech, 27299-I-AP), anti-GSDMD (Abcam, ab209845), anti-mCherry (Abcam, ab167453), anti-Flag (Sigma, F1804) and anti-GAPDH (CST, 5174). Membranes were then washed and incubated with the appropriate HRP-conjugated secondary antibody (Jackson Immuno Research Laboratories anti-rabbit (111-035-047), anti-mouse (315-035-047) or anti-goat (705-035-003)) for 1 h. Proteins were visualized using Luminata Forte Western HRP Substrate (Millipore, WBLUF0500).
Co-immunoprecipitation.
For immunoprecipitation, BMDMs were lysed in NP-40 lysis buffer (1% NP-40, 150 mM NaCl, 50 mM HEPES), and lysates were cleared by centrifugation at maximum speed for 15 min. Whole-cell lysates were incubated with 4 μg of the indicated primary antibodies on a rocking platform for 4–5 h at 4 °C. Protein A/G PLUS-Agarose (Santa Cruz) beads were washed, incubated for 30 min with 3% BSA and then added to the samples. Incubation was continued for an additional 2 h on the rocking platform. The agarose beads were collected by centrifugation and washed three times with the NP-40 lysis buffer. After washes, immunoprecipitates were eluted in sample buffer and used for immunoblotting analysis. For mass spectrometry analysis, the same protocol was used, except 2 μg of antibody was used. Two biologically independent samples were used. The eluate was loaded in a 10-well 4–20% Bis-Tris gel (BioRad) and allowed to run for 5 min, until all of the sample had entered the gel. The gel was washed with deionized water twice and stained with simple blue stain (Thermo Fisher Scientific, LC6060). The bands were excised, and in-gel digestion was performed with a mixture of trypsin and LysC. The peptides were separated by in-line reversed phase liquid chromatography and analysed on a Q-Exactive mass spectrometer. RAW files were analysed using the MaxQuant software package with the Andromeda search engine41. The mouse proteome was downloaded from the Uniprot database. Normalized spectral counts generated by MaxQuant were used for quantification. An empirical constant value of 0.1 was imputed for missing values and was used for statistical analysis and fold change calculations.
Reconstitution of DDX3X expression in BMDMs.
DDX3X-YFP was obtained from GeneCopoeia in the pReceiver-Lv123 expression vector. DDX3X-mCherry was cloned in the pCW57.1 vector (Addgene, plasmid 41393) using Gibson assembly. Lentiparticles for transducing primary BMDMs were generated in HEK293T cells using packaging plasmids pMD2.G and psPAX2 in DMEM supplemented with 10% FBS. Lentiviruses were collected 48 h after transfection, supplemented with 10% FBS and stored at −80 °C. Primary BMDMs were transduced with lentiparticles in the presence of 6 μg ml−1 polybrene on day 2 of the differentiation protocol. BMDMs were seeded on day 5 in 12-well tissue-culture-treated plates subjected to doxycycline treatment (250 ng ml−1 or 1 μg ml−1) for 48 h before treatment with LPS and nigericin.
Immunoprecipitation studies to probe for domain specificity of NLRP3 and DDX3X.
The pReceiver-M56-DDX3X-mCherry expression clone was purchased from GeneCopoeia. DDX3X-ΔNterm (residues 156–662), DDX3X-ΔCterm (residues 1–573) and DDX3X-helicase (residues 156–573) constructs were generated in the same vector background. NLRP3 expression constructs pCDNA3-N-Flag-NLRP3, pCDNA3-N-Flag-NLRP3 1–90, pCDNA3-N-Flag-NLRP3 91–710 and pCDNA3-N-Flag-NLRP3 711–1034 were purchased from AddGene. To interrogate domain specificity of DDX3X and NLRP3, HEK293T cells were transiently transfected with 4 μg of the full-length DDX3X or NLRP3 construct in combination with 4 μg of one of the domain constructs of NLRP3 or DDX3X using Xfect (Takara) or Lipofectamine-2000 (Invitrogen) in Opti-MEM (Gibco). Opti-MEM was replaced with DMEM with 10% FBS 8 h after transfection, and 24–30 h after transfection, cells were lysed with NP-40 lysis buffer (1% NP-40, 150 mM NaCl, 50 mM Tris and protease-phosphatase inhibitor cocktail). Whole-cell lysates were centrifuged to separate the insoluble fraction, and the soluble fraction was further subjected to immunoprecipitation experiments. Antibodies for mCherry (Abcam), Flag-M2 (Sigma) and NLRP3 (AdipoGen/CST) were used to detect DDX3X and NLRP3 on immunoblots.
Separation of soluble and insoluble NP-40 detergent fractions.
Primary BMDMs or HEK293T cells transiently transfected with DDX3X-mCherry, NLRP3 and ASC expression clones were subjected to lysis with NP-40 lysis buffer containing 1 mM DSP (dithiobis(succinimidyl propionate)) crosslinker (CovaChem). Whole-cell lysates were centrifuged at 10,000 r.p.m. for 15–20 min to separate insoluble fraction (pellet) from soluble fraction. Insoluble lysates were washed once and further dissolved in NP-40 lysis buffer. Both soluble and insoluble lysates were mixed with 4 × sample buffer with or without BME and subjected to SDS-PAGE to analyse the oligomeric status of complexes.
AUC to fractionate whole-cell lysates.
After treatments, cells were lysed using NP-40 lysis buffer containing 1 mM DSP crosslinker, followed by centrifugation to separate soluble and insoluble NP-40 fractions. The soluble lysate fraction was concentrated using a 10-kDa cut-off centricon (Millipore). A 15–45% glycerol gradient containing NP-40 lysis buffer was constructed by layering solutions at decreasing percentages from 45% to 15% glycerol (13 layers, 98 μl each; total volume: 1.30 ml; height: 3.0 cm) in an 11 × 34−mm centrifuge tube. Concentrated soluble lysate (50 μl) was layered on top of the gradient followed by 50 μl silicon oil to prevent evaporation. The tube was then placed in a pre-cooled bucket and centrifuged for 5 h at a rotor speed of 55,000 r.p.m. at 4 °C in an Optima TLX preparative ultracentrifuge using a swinging bucket TLS-55 rotor (Beckman Coulter). Deceleration was performed without braking, and the tube was immediately placed on ice. Microfractionation of the tube contents was carried out using a BRANDEL automated micro-fractionator equipped with the FR-HA 1.0 block assembly (Brandel). The tube was placed in the receptacle, and fractions were removed from the upper surface of the solution by stepwise elevation of the receptacle by a precise increment of height. A total of 27 fractions were collected in a 96-well plate; each fraction was approximately 45 μl in volume, and the bottom fraction was 125 μl. The fractions were then further processed for SDS-PAGE.
Flow cytometry.
Two monoclonal antibodies were used for flow cytometry analyses: anti-CD11b (M1/70; Affymetrix eBioscience, 48-0112-82) and anti-F4/80 (BM8; BioLegend, 123109). Both antibodies were diluted 1:300. Flow cytometry data were acquired on a FACSCalibur flow cytometer (BD) and analysed with FlowJo software (FlowJo and Illumina).
Immunofluorescence staining and imaging.
After stimulation, BMDMs were fixed for 15 min at room temperature in 4% paraformaldehyde (ChemCruz, sc-281692). Non-specific binding was blocked with 10% normal goat serum (Sigma). Cells were incubated with the following antibodies overnight at 4 °C or for 2 h at room temperature: anti-ASC (AdipoGen, AG-25B-006-C100; diluted 1:250), anti-DDX3X (Bethyl Laboratories, A300–474A; diluted 1:100), anti-ASC (Millipore, 04–147; diluted 1:100), rabbit anti-NLRP3 (CST, 15101; diluted 1:100), rat anti-NLRP3 (R&D systems, MAB7578; diluted 1:100) and anti-G3BP1 (Proteintech, 27299-I-AP; diluted 1:250). The secondary antibodies used were Alexa Fluor 568-conjugated anti-rabbit IgG (Life Technologies, A11036; diluted 1:250), Alexa Fluor 568-conjugated anti-rat IgG (Life Technologies, A11077; diluted 1:250) and Alexa Fluor 488-conjugated anti-rabbit IgG (Life Technologies, A11034; diluted 1:250). Cells were counterstained with DAPI mounting medium (Vecta Labs), and images were acquired on a Marianas (Intelligent Imaging Innovations) or Leica SP8 (Leica Microsystems) confocal microscope. Confocal images were rendered using the vendor software. For structured illumination microscopy, cells were stained with DAPI and mounted in Prolong Gold mounting medium (Thermo Fisher Scientific, P10144). Images were acquired with a Zeiss Elyra PS.1 microscope (Carl Zeiss Microscopy) with a plan apochromatic 63X, 1.4 NA objective and Andor EMCCD. Images were first processed with the Zen 2012 Black software then rendered in Imaris (Bitplane). For in vivo stress-granule detection, 1 ml thioglycollate was injected into the peritoneal cavity of the mice. Three days after thioglycollate injection, 1 mg kg−1 arsenite or 200 μL PBS was injected. One hour after arsenite injection, mice were killed, and the peritoneal cavity was washed with 3 ml of fixative (0.1% glutaraldehyde, 2% paraformaldehyde, 0.1% T-100, 1% DMSO). Cells in the fixative were spun down, permeabilized, stained and immobilized on microscopy-compatible multi-well plates coated with 5 μg ml−1 anti-CD45 antibody (clone 30-F11, NovusBio, NB100-77417). Images were acquired on a Marianas and rendered using vendor-supplied software.
STORM.
BMDMs were seeded 24 h before stimulation. Cells were primed with LPS for 4 h, then nigericin was added to activate the NLRP3 inflammasome, and the cells were incubated at 37 °C for 15–45 min. Cells were fixed with 4% paraformaldehyde and processed for STORM imaging. The antibodies used for labelling were anti-DDX3 (Bethyl Laboratories, A300–474A), anti-ASC (Millipore, 04–147) and anti-tubulin (Thermo Fisher Scientific, clone YOL1/34; diluted 1:1,000). STORM acquisition of 10 fields per condition was performed using a Nikon N-STORM system consisting of an inverted TiE stand, a 100 X, 1.45 NA objective, a DU-897 EMCCD camera, an Agilent laser launch and appropriate optics, as previously reported42. Images of single-molecule reconstructions were exported for downstream analysis by Imaris software. Coordinate (x, y) positions of individual molecules were exported for analysis with a new statistical measure for analysing colocalization in super-resolution images (r-index; X. Liu et al., unpublished protocol). With this method, a value of 1 indicates colocalization, a value of 0 indicates complete randomness and a value of − 1 indicates exclusion.
Real-time cell-death analysis.
Real-time cell-death assays were performed using a two-colour IncuCyte Zoom incubator imaging system (Essen Biosciences). In brief, BMDMs were seeded in 24-well tissue-culture vessels (250,000 cells per well) in the presence of 20 nM SYTOX Green (Thermo Fisher Scientific, S7020), a cell-impermeable DNA-binding fluorescent dye that rapidly enters dying cells after membrane permeabilization. The resulting images were analysed using the software package supplied with the IncuCyte imager, which enables a precise analysis of the number of SYTOX Green-positive cells present in each image. Experiments were conducted using a minimum of three separate wells for each experimental condition and a minimum of four image fields per well. Dead-cell events were acquired via SYTOX Green counting and plotted using GraphPad Prism software.
Prediction of intrinsically disordered regions.
The Predictor of Natural Disordered Regions (PONDR) and the PSIPRED43 servers were used to predict secondary structures and disordered regions in DDX3X and NLRP3. Ordered and disordered regions predicted by PONDR and PSIPRED were overlaid for highlighting intrinsically disordered regions in DDX3X and NLRP3.
Generation of G3bp1-knockout BMDMs using CRISPR-Cas9.
Lentiviral particles that express two different guide RNAs (gRNAs) that target G3bp1 were prepared by transfecting HEK293T cells with gRNA-expressing construct, pPAX2 and pMD2 plasmids. Lentiviruses were collected 48 h after transfection, supplemented with 10% FBS and stored at −80 °C. In-house-generated immortalized BMDMs that express the Cas9 protein were transduced with gRNA lentiparticles in combination with 6–8 μg ml−1 polybrene. Then, 24 h after transduction, lentivirus-containing medium was replaced with fresh DMEM with FBS and incubated for another 24 h. Cells were subjected to puromycin selection (2.5–3 μg ml−1) to isolate G3bp1-knockout immortalized BMDMs. After puromycin selection, G3bp1 - knockout immortalized BMDMs and wild-type-dCas9-immortalized BMDMs were subjected to NLRP3 activation stimuli as described above.
siRNA knockdown of Ddx3x.
The SMART siRNA pool for Ddx3x (Dharmacon, M-043597-01-0005) was used; it contains four different siRNAs, siRNA-1: GCAAGCA AAGGGCGUUAUA; siRNA-2: GGUUAGACUUCUGCAAAUA; siRNA-3: GAAAGAGGUGGAAAUAGUC; siRNA-4: ACUAAAGGAUUCUAUGCA. A total of 5 nmol was dissolved in nuclease-free water at 50 μM, and 0.5 μl siRNA was added to 1 × 107 BMDMs. Electroporation was performed using the neon transfection system (Invitrogen), with parameters −1500 V, 1 pulse and 1-ms width. After electroporation, BMDMs were immediately transferred into 12-well plates with a seeding density of 1 × 106 cells per well. Forty-eight hours after transfection, BMDMs were treated with LPS and nigericin to activate NLRP3.
LPS peritonitis.
Wild-type mice were injected with 1 mg kg−1 arsenite prepared in sterile PBS intraperitoneally Two hours after arsenite injection, the mice were injected with 50 mg kg−1 LPS (Escherichia coli O111:B4, Sigma-Aldrich) intraperitoneally. Mice were killed 4 h after LPS injection to collect serum and peritoneal fluid for cytokine measurements and fluorescence-activated cell sorting analysis.
Statistics and reproducibility.
GraphPad Prism 7.0 software was used for data analysis. Statistical significance was determined by unpaired two-tailed t-test and by one-way and two-way ANOVA. Unless otherwise noted, the mean was used to represent central tendency, and error bars represent the standard error of the mean (s.e.m.). A ROUT test was used to exclude outliers, in which Q = 0.1 was used as a cut-off. Unless otherwise noted, every experiment was done with at least three biologically independent replicates. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P ≤ 0.0001. For ELISA data, values from multiple experiments were pooled together, which might have led to overlap of values between panels. For quantification of pyroptosis based on SYTOX Green staining, mean and standard deviations were calculated in the vendor-supplied software, and data were imported to GraphPad Prism for graphing. Representative western blots and microscopy images are shown from at least three biologically independent replicates that showed similar results. No statistical methods were used to predetermine sample size and the experiments were not randomized.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Extended Data
Extended Data Fig. 1 |. Specific inhibition of the NLRP3 inflammasome is not due to differences in levels of core inflammasome component proteins, NLRP3 sequestration in stress granules or induction of autophagy.

a, Confocal microscopy imaging of LPS-primed BMDMs treated with arsenite (50 μM) or with anisomycin (25 μg ml−1) and arsenite. Scale bars, 10 μm. Representative images (n = 2).b, Quantification of ASC+ cells stimulated with LPS and treated with nigericin, arsenite and anisomycin as indicated. ***P = 0.0002 (unpaired two-sided t-test). Data are mean ± s.e.m. (n > 70 frames). c, Immunoblot analysis of pro-CASP1 (p45) and cleaved CASP1 (p20) in BMDMs treated with poly(I:C) (P(I:C)); poly(I:C) and arsenite; poly(I:C) and nigericin; or poly(I:C), arsenite and nigericin. Representative blots (n = 2 biologically independent experiments). d, Quantification of inflammatory cytokines in BMDMs treated with medium (Mock), LPS, LPS and arsenite, or arsenite. P values for LPS versus LPS + Ar: IL-6, P = 0.8561; KC, P = 0.9522; TNF, P = 0.2599. Data are mean ± s.e.m. (n = 4). e, f, Immunoblot analysis of CASP1 cleavage in BMDMs treated with the AIM2-activating ligand poly(dA:dT) (pA:T) (e) or the NLRC4-activating ligand flagellin (Fla) (f). Representative blots (n = 3 biologically independent experiments each). g, Immunoblot analysis of protein levels of core NLRP3 inflammasome components after LPS priming, arsenite-mediated induction of stress granules and anisomycin-mediated inhibition of stress granules. Representative blots (n = 3 biologically independent experiments). h, Confocal microscopy imaging of BMDMs treated with LPS alone or with LPS and arsenite to visualize NLRP3 localization. G3BP1 was used as a marker of stress granules. Scale bars, 10 μm (whole-cell images); 5 μm (magnified images). Representative images (n = 2 biologically independent experiments). i, Immunoblot analysis of CASP1 cleavage in autophagy-deficient BMDMs that lack Becn1 and Atg5, stimulated with LPS and treated with nigericin, arsenite and anisomycin as indicated. Representative blots (n = 2 biologically independent experiments).
Extended Data Fig. 2 |. DDX3X colocalizes with G3BP1 in stress granules in BMDMs treated with arsenite.
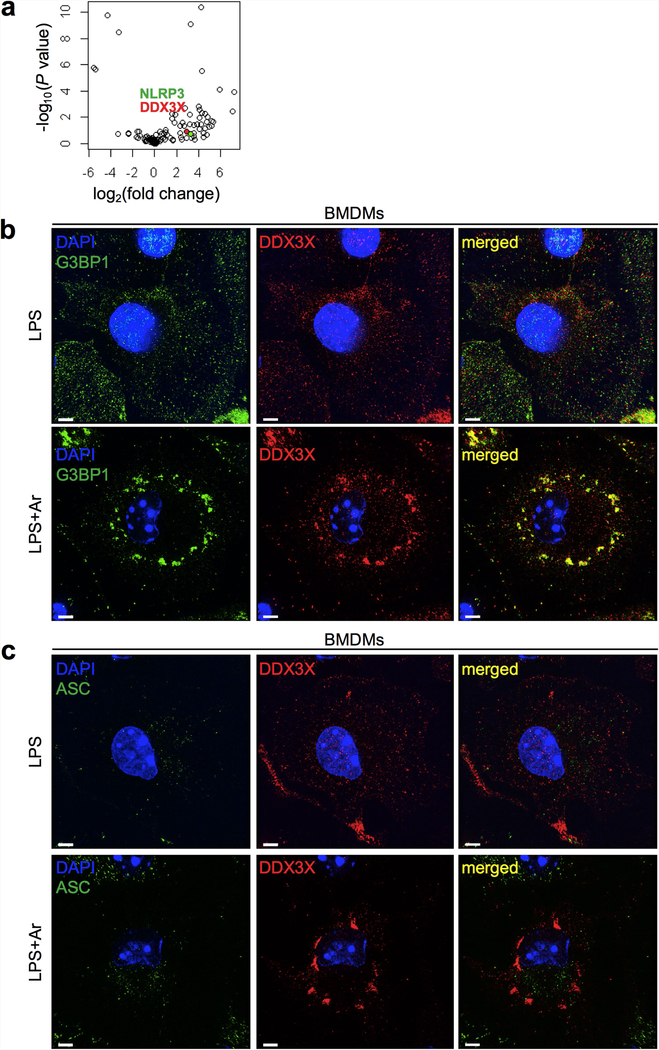
a, Volcano plot of the affinity purification mass spectrometry analysis of the NLRP3 interactome, with NLRP3 (green) and DDX3X (red) highlighted. Two biologically independent replicates each of samples treated with LPS and samples treated with LPS and nigericin were pooled together for analysis. Data can be accessed using ProteomeXchange with the identifier PXD014828. b, c, Structured illumination microscopy of LPS-primed BMDMs treated with or without arsenite to visualize DDX3X, the stress granule marker G3BP1 (b) and ASC (c). Scale bars, 3 μm. Representative images (n = 2 each).
Extended Data Fig. 3 |. Strategy for generating the Ddx3x gene knockout and characterization of immune cells.

a, Genomic locations of loxP sites in the Ddx3x locus. b, Genotyping PCR for the 5′ and 3′ loxP sites. Representative gel (n = 1). L, molecular-weight ladder. c, Immunoblot analysis of the levels of DDX3X protein in BMDMs. Data are from three different mice with each genotype. d, Flow cytometry analysis of ex vivo differentiated BMDMs stained for CD11b and F4/80. e, Quantification of the numbers of different immune-cell types from the blood of the indicated mouse strains. RBC, red blood cells. Data are mean ± s.e.m. (n = 3).
Extended Data Fig. 4 |. Lack of DDX3X leads to defects in both stress granule assembly and NLRP3 inflammasome activation.

a, Confocal microscopy of Ddx3xfl/fl and Ddx3xfl/flLysMcre BMDMs stimulated with LPS and arsenite to visualize localization of the stress granule marker G3BP1 and DDX3X. Scale bars, 10 μm. b, Quantification of the number of stress granules per cell. Each data point represents the average number of stress granules per cell in each field of view. ***P = 0.0003 (unpaired two-sided t-test). Data are mean ± s.e.m. (n = 10). c, Immunoblot analysis of CASP1 cleavage in peritoneal macrophages stimulated with LPS with or without nigericin. Representative blots (n = 2 biologically independent experiments). d, Pyroptotic cell death as measured by the number of SYTOX Green+ cells. Cells were stimulated with LPS and treated with nigericin or with nigericin and arsenite. ****P ≤ 0.0001 (two-way ANOVA) for LPS + N-treated Ddx3xfl/fl versus LPS + N-treated Ddx3xfl/fl LysMcre BMDMs; LPS + N-treated versus LPS + Ar + N-treated Ddx3xfl/fl BMDMs; and LPS + N-treated versus LPS + Ar + N-treated Ddx3xfl/fl LysMcre BMDMs. Data are mean ± s.e.m. (n = 4). e–g, Immunoblot analysis of CASP1 cleavage in BMDMs treated with LPS or LPS and ATP (e), Pam3CSK4 or Pam3CSK4 and nigericin (f), or poly(I:C) or poly(I:C) and nigericin (g). Representative blots (n = 2 biologically independent experiments each).
Extended Data Fig. 5 |. Loss of DDX3X in BMDMs leads to the specific inhibition of NLRP3 inflammasome activation.

a–d, CASP1 cleavage in stimulated and unstimulated BMDMs and IL-1β and IL-18 release from BMDMs stimulated to activate the NLRP3, NLRC4, AIM2 and PYRIN inflammasomes by using LPS and nigericin (a) (ELISA, n > 10); flagellin (b) (ELISA, n = 2); poly(dA:dT) (c) (ELISA, n = 2); and Clostridium difficile toxin B (TcdB) (d) (ELISA, n = 2). P values in a (from left to right): *P = 0.0170, *P = 0.0116 (unpaired two-sided t-test); n.d., not detected. ELISA data are mean ± s.e.m. Representative blots (n = 3 biologically independent experiments each). e, Immunoblot analysis of CASP1 cleavage in wild-type BMDMs expressing a doxycycline-inducible DDX3X–mCherry construct and treated with LPS and nigericin. Representative blots (n = 2 biologically independent experiments). f, Immunoblot analysis of CASP1 cleavage in wild-type BMDMs and Ddx3xfl/flLysMcre BMDMs that constitutively express DDX3X–YFP from a cytomegalovirus promoter, treated with LPS and nigericin. Representative blots (n = 2 biologically independent experiments).
Extended Data Fig. 6 |. DDX3X interacts with NLRP3 but not ASC and CASP1, and its loss does not lead to a decrease in the levels of core components of the NLRP3 inflammasome.

a, Immunoblot analysis of the levels of NLRP3, ASC, CASP1, pro-IL-1β, NEK7, DDX3X and GAPDH proteins in Ddx3xfl/fl, Ddx3xfl/flLysMcre and Nlrp3−/− BMDMs after LPS priming. Representative blots (n = 3 biologically independent experiments). b, Immunoblot analysis of NLRP3 and DDX3X immunoprecipitation from HEK293T cells transfected with Flag–NLRP3 and DDX3X–mCherry expression constructs. S.E., short exposure; L.E., long exposure. Representative blots (n = 3 biologically independent experiments). c, Immunoblot analysis of ASC and DDX3X immunoprecipitates from HEK293T cells transfected with ASC and DDX3X–mCherry expression constructs. Representative blots (n = 3 biologically independent experiments). d, Immunoblot analysis of CASP1 and DDX3X immunoprecipitates from HEK293T cells transfected with CASP1 and DDX3X–mCherry expression constructs. Representative blots (n = 2 biologically independent experiments). e, Quantification of the percentage of cells that contain ASC specks in Ddx3xfl/fl and Ddx3xfl/fl LysMcre BMDMs. ****P ≤ 0.0001 (unpaired two-sided t-test). Data are mean ± s.e.m. (n = 48). f, Quantification of the percentage of cells that contain ASC specks in wild-type and Ddx3xfl/flLysMcre peritoneal macrophages (PMs). ****P ≤ 0.0001 (unpaired two-sided t-test). Data are mean ± s.e.m. (n = 48). g, Confocal microscopy imaging of wild-type and Ddx3xfl/flLysMcre peritoneal macrophages treated with LPS and nigericin to visualize ASC and DDX3X. Scale bar, 10 μm.
Extended Data Fig. 7 |. Induction of stress granules by various stressors.
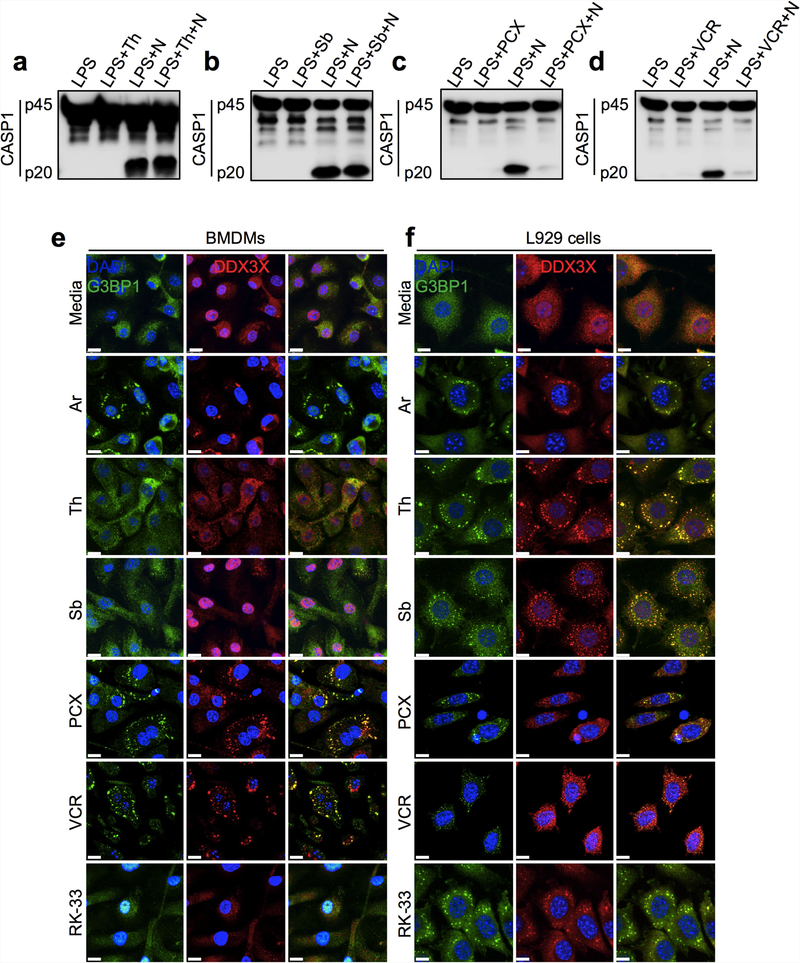
a–d, Immunoblot analysis of CASP1 cleavage in LPS-primed BMDMs. Representative blots (n = 3 biologically independent experiments each). a, BMDMs were treated with 2 μg ml−1 thapsigargin (Th) for 1 h before NLRP3 inflammasome activation by nigericin. b, BMDMs were treated with 400 μM d-sorbitol (Sb) for 30 min before NLRP3 inflammasome activation by nigericin. c, BMDMs were treated with 400 μM paclitaxel (PCX) for 30 min before NLRP3 inflammasome activation by nigericin. d, BMDMs were treated with 750 μM vincristine (VCR) for 30 min before NLRP3 inflammasome activation by nigericin. e, f, Confocal microscopy images to visualize the induction of stress granules in BMDMs (e) and L929 cells (f) using 50 μM arsenite, 2 μg ml−1 thapsigargin, 400 μM d-sorbitol, 400 μM paclitaxel, 750 μM vincristine and 5 μM RK-33. Cells were incubated with stressors for 1 h and stained with the stress granule marker G3BP1 and with DDX3X. Representative images (n = 3 biologically independent experiments each).
Extended Data Fig. 8 |. Identification of the region of DDX3X required for its interaction with NLRP3, prediction of disordered regions using PONDR and PSIPRED, and effect of DDX3X helicase activity inhibition on NLRP3 inflammasome activation.

a, Schematic of C-terminal mCherry-tagged DDX3X domain-deletion expression constructs. b, Immunoblot analysis of the input lysates used for immunoprecipitation of Flag–NLRP3-FL and DDX3X–mCherry constructs. Representative blots (n = 2 biologically independent experiments). c–f, Immunoblot analysis of immunoprecipitates with Flag–NLRP3-FL and mCherry-tagged DDX3X domain-deletion expression constructs: full-length DDX3X (c), DDX3X with an N-terminal deletion (d), DDX3X with a C-terminal deletion (e) and the DDX3X helicase domain (f). Red asterisk indicates a non-specific band. Representative blots (n = 2 biologically independent experiments each). g, Overlay of the predicted disordered regions in NLRP3 from PONDR and PSIPRED. h, Overlay of the predicted disordered regions in DDX3X from PONDR and PSIPRED. i, Immunoblot analysis of CASP1 cleavage in BMDMs treated with LPS; LPS and nigericin; LPS and RK-33; LPS, RK-33 and arsenite; LPS, arsenite and nigericin; LPS, RK-33 and nigericin; and LPS, RK-33, arsenite and nigericin. Representative blots (n = 2 biologically independent experiments).
Extended Data Fig. 9 |. Stress granule-mediated inhibition of oligomerization of NLRP3 and ASC and induction of NLRP3 aggregation.

a, Immunoblot analysis of crosslinked and un-crosslinked (BME-treated) lysates of HEK293T cells expressing mouse ASC, DDX3X and NLRP3. Representative blots (n = 3). WCE, whole-cell extracts. b, Immunoblot analysis of crosslinked and non-reduced (that is, without BME treatment) BMDM lysates. Representative blots (n = 3). c, d, Secretion of inflammasome-dependent cytokines IL-1β (c) and IL-18 (d) from BMDMs treated with arsenite for different durations before the addition of nigericin to activate the NLRP3 inflammasome. Arsenite was added 30 or 15 min before, simultaneously with or 15 or 30 min after nigericin. Samples were collected after 75 min of nigericin treatment. For the anisomycin-treated samples, anisomycin was given 20 min before the start of the earliest arsenite treatment (−30 min). A sigmoid curve was fitted to the data to show the correlation between the timing of arsenite addition and the release of inflammasome-dependent cytokines. The goodness of fit was determined by the R2 value. P values (for the column factor that represents anisomycin treatment): **P = 0.0010 (c) and **P = 0.0066 (d) by two-way ANOVA. Data are mean ± s.e.m. (n = 3).
Extended Data Fig. 10 |. Effects of the loss of G3BP1 on NLRP3 inflammasome activation and intraperitoneal injection of arsenite on the release of inflammatory cytokines.

a, Immunoblot analysis of CASP1 cleavage, total G3BP1 and GAPDH in wild-type and dCas9-expressing immortalized BMDMs transduced with two different gRNAs that target G3bp1. Representative blots (n = 3). b, c, Quantification of inflammatory cytokines in an LPS-induced peritonitis model with or without intraperitoneal injection of arsenite, in serum (b) and in peritoneal fluid (c). P values: **P = 0.0051 (IP-10 in serum), **P = 0.0024 (RANTES in peritoneal fluid) by unpaired two-sided t-test. Data are mean ± s.e.m. (n ≥ 6).
Supplementary Material
Acknowledgements
We thank all the members of the Kanneganti laboratory for their comments and suggestions; F. Phillips and N. Lantz for their help with generating BMDMs and with mouse husbandry; K. A. Laycock and R. Tweedell for scientific editing of the manuscript; K. Kodali and V. Pagala for their help with affinity purification mass spectrometry analysis; and S. Miller and S. Sakurada for helping with the CRISPR-Cas9-mediated gene disruption experiment. psPAX2 (Addgene plasmid 12260) and pMD2.G (Addgene plasmid 1225) were gifts from D. Trono. T-D.K. is supported by NIH grants AI101935, AI124346, AR056296 and CA163507 and by the American Lebanese Syrian Associated Charities; the St. Jude Children’s Research Hospital Cell and Tissue Imaging Center is supported by St. Jude Children’s Research Hospital and by National Cancer Institute grant P30 CA021765–35; R.J.G. is supported by Cancer Research UK, the Mathile Family Foundation, Cure Search, the Sohn Foundation and NIH grants P01CA96832 and R0CA1129541.
Footnotes
Data availability
The proteomics data generated and analysed during the current study are available in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD014828. Raw data for figures are available as Source Data to the relevant figure. All other datasets generated and analysed during the current study are available from the corresponding authors on reasonable request. Any requests for data or materials should, in the first instance, be addressed to T.-D.K.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Competing interests The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41586-019-1551-2.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Protter DSW & Parker R Principles and properties of stress granules. Trends Cell Biol. 26, 668–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolozin B Regulated protein aggregation: stress granules and neurodegeneration. Mol. Neurodegener. 7, 56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White JP & Lloyd RE Regulation of stress granules in virus systems. Trends Microbiol. 20, 175–183 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson P & Kedersha N RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 10, 430–436 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Anderson P, Kedersha N & Ivanov P Stress granules, β-bodies and cancer. Biochim. Biophys. Acta 1849, 861–870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinon F, Burns K & Tschopp J The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Kanneganti T-D et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Mariathasan S et al. Cryopyrin activates the inflammasome in response to toxins and ATP Nature 440, 228–232 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Karki R & Kanneganti T-D Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 19, 197–214 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malireddi RKS et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 215, 1023–1034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Man SM & Kanneganti T-D Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 16, 7–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venegas C et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Muñoz-Planillo R et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanneganti T-D & Lamkanfl M K+ drops tilt the NLRP3 inflammasome. Immunity 38, 1085–1088 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Groß CJ et al. K+ efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity 45, 761–773 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Lu A et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franklin BS et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 15, 727–737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franklin BS, Latz E & Schmidt FI The intra- and extracellular functions of ASC specks. Immunol. Rev. 281, 74–87 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Kedersha N et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169, 871–884 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Man SM et al. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell 167, 382–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bol GM et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol. Med. 7, 648–669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin Y & Brangwynne C P Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Aditi F, Folkmann AW & Wente SR Cytoplasmic hGlelA regulates stress granules by modulation of translation. Mol. Biol. Cell 26, 1476–1490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilliker A, Gao Z, Jankowsky E & Parker R The DEAD-box protein Dedl modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol. Cell 43, 962–972 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih J-W et al. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 441, 119–129 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Cruciat C-M et al. RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt-β-catenin signaling. Science 339, 1436–1441 (2013) [DOI] [PubMed] [Google Scholar]
- 28.Soulat D et al. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 27, 2135–2146 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stunnenberg M, Geijtenbeek TBH & Gringhuis SI DDX3 in HIV-1 infection and sensing: a paradox. Cytokine Growth Factor Rev. 40, 32–39 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Robinson G et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ditton HJ, Zimmer J, Kamp C, Rajpert-De Meyts E & Vogt PH The AZFa gene DBY(DDX3Y) is widely transcribed but the protein is limited to the male germ cells by translation control. Hum. Mol. Genet. 13, 2333–2341 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Vakilian H et al. DDX3Y, a male-specific region of Y chromosome gene, may modulate neuronal differentiation. J. Proteome Res. 14, 3474–3483 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Chen C-Y et al. Targeted Inactivation of murine Ddx3x: essential roles of Ddx3x In placentation and embryogenesis. Hum. Mol. Genet. 25, 2905–2922 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Li Q et al. DDX3X regulates cell survival and cell cycle during mouse early embryonic development. J. Biomed. Res. 28, 282–291 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones JW et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl Acad. Sci. USA 107, 9771–9776 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franchi L et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol. 7, 576–582 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Van Gorp H et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl Acad. Sci. USA 113, 14384–14389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wheeler JR, Matheny T, Jain S, Abrisch R & Parker R Distinct stages in stress granule assembly and disassembly. eLife 5, e18413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szaflarski W et al. Vinca alkaloid drugs promote stress-induced translational repression and stress granule formation. Oncotarget 7, 30307–30322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karki R et al. IRF8 regulates transcription of Naips for NLRC4 inflammasome activation. Cell 173, 920–933 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyanova S, Temu T & Cox J The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protocols 11, 2301–2319 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Kesavardhana S et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 214, 2217–2229 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchan DWA & Jones DT The PSIPRED Protein Analysis Workbench:20 years on. Nucleic Acids Res. 47 (W1), W402–W407 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.