

Abstract
As a result of the exponential increase in genomic data, discovery of novel ribosomally synthesized and post-translationally modified peptide natural products (RiPPs) has progressed rapidly in the past decade. The lanthipeptides are a major subset of RiPPs. Through genome-mining we identified a novel lanthipeptide biosynthetic gene cluster (lah) from Lachnospiraceae bacterium C6A11, an anaerobic bacterium that is a member of the human microbiota and is implicated in the development of host disease states such as type 2 diabetes and resistance to Clostridium difficile colonization. The lah cluster encodes at least seven putative precursor peptides and multiple post-translational modification (PTM) enzymes. Two unusual class II lanthipeptide synthetases LahM1/M2 and a substrate tolerant S-adenosyl-L-methionine (SAM) dependent methyltransferase LahSB are biochemically characterized in this study. We also present the crystal structure of LahSB in complex with product S-adenosylhomocysteine. This study sets the stage for further exploration of the final products of the lah pathway as well as their potential physiological functions in human/animal gut microbiota.
Keywords: RiPPs, Lanthipeptide, Dehydration, Methyltransferase, PTMs
Graphical Abstract
Two unusual class II lanthipeptide synthetases and a SAM-dependent C-terminal methyltransferase are encoded in a novel lanthipeptide biosynthetic gene cluster (lah) from Lachnospiraceae bacterium C6A11. These enzymes were biochemically characterized, setting the stage for further exploration of the final products and their physiological functions in gut microbiota.
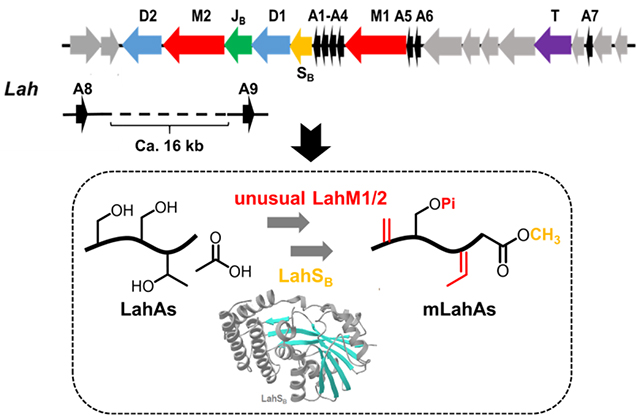
Introduction
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are an expanding class of natural products,[1] the discovery of which has been accelerated by the genome-sequencing advances in the past decade.[2] RiPPs are typically made from a precursor peptide that contains a core peptide where the PTMs take place and a leader peptide that is required for many of the post-translational modification reactions but is removed in one of the later steps of biosynthesis.[3] One of the major classes of RiPPs is the lanthipeptides, which contain the characteristic thioether cross-linked bisamino acids lanthionine and methyllanthionine. They are biosynthesized by dehydration of serine and threonine residues in a precursor peptide, followed by intramolecular Michael-type addition of cysteine thiols to the resulting dehydroalanine (Dha) or dehydrobutyrine (Dhb) residues.[4] For class II lanthipeptides, both reactions are catalyzed by a single enzyme generically called LanM,[5] which uses ATP to first phosphorylate select Ser/Thr residues followed by phosphate elimination to accomplish net dehydration.[6] Recently, we reported a highly substrate tolerant protease domain of the bifunctional transporter LahT encoded in a putative lanthipeptide biosynthetic gene cluster (lah) in Lachnospiraceae bacterium C6A11. This domain called LahT150 efficiently removes leader peptides from a large number of double glycine motif-containing peptides.[7] LahT is a member of the family of ABC-transporter maturation and secretion (AMS) proteins (also called peptidase-containing ATP-binding transporters, PCAT).[8] The lah biosynthetic gene cluster (BGC) in the genome of Lachnospiraceae bacterium C6A11 encodes an intriguing compilation of RiPP PTM enzymes (Figure 1). We identified the lah BGC by BLAST query searching for examples of BGCs that contain multiple precursor peptides based on the flavecin biosynthetic pathway.[7, 9] The lah cluster is unique in that it not only encodes at least seven precursor peptides with diverse core peptides, but also has a highly diverse set of PTM enzymes including two class II lanthipeptide synthetases, two YcaO-like proteins,[10] a dehydrogenase, a protease/transporter, and a methyltransferase. The constellation of multiple precursor substrates along with multiple modification enzymes complicates prediction of the final products since either all enzymes can act on all peptides, or select enzymes may react with only a subset of substrates. The large size of the cluster and the diversity of the precursor peptides was also noted in a previous report without further analysis.[11] Here, we biochemically characterized a SAM-dependent methyltransferase LahSB that methylates the C-terminal carboxyl group of different amino acid residues in a subset of LahA peptides, which constitutes a new PTM in lanthipeptides. In order to rationalize this broad substrate tolerance, we solved the LahSB crystal structure at 2.01 Å resolution. Lastly, we demonstrate that the class II LahM1 enzyme efficiently dehydrates a select set of substrates, but that LahM2 only displays phosphorylation (and not phosphate elimination) activity.
Figure 1.

The lah biosynthetic gene cluster. (A) lah gene cluster. The grey genes between lahA6 and lahT and between lahT and lahA7 encode regulatory elements and ABC transporters; (B) sequences of the nine LahA core peptides. Dehydrated residues (see text) are underlined. (C) LahA leader peptide alignment.
Results and Discussion
Bioinformatic survey suggests an intriguing set of PTMs
The lah cluster is the most complex lanthipeptide biosynthetic gene cluster that has been experimentally investigated thus far. Like the prochlorosin systems in cyanobacteria,[12] the lah cluster encodes multiple substrate peptides with diverse core peptides. Seven of the nine putative LanA peptides (LahA1-A7) are encoded within the lah cluster (Figure 1). In addition, two additional putative genes encoding LahA peptides (LahA8 and A9) are located outside of the lah BGC. The distance between the latter two genes and the lah cluster cannot be determined as the genome is not fully assembled and we cannot rule out that these peptides may be modified by different enzymes such as a radical-SAM protein that is encoded nearby. The leader peptides of all nine LahA peptides are conserved and, like the prochlorosins, are members of the Nif11-like protein family[13] ending with a double Gly motif (Figure 1C). Our previous study demonstrated that the LahT150 protease domain recognized this motif for all nine substrates.[7] In contrast, the sequences of the corresponding core peptides exhibit a high degree of sequence divergence and five of the nine peptides distinctly lack Cys residues (Figure 1B). Despite few Cys residues, indicating that at least five (and possibly more) of the nine products are not lanthipeptides, the LahA core peptides contain a number of Ser and Thr residues. Curiously, LahA8 contains neither Ser/Thr nor Cys residues in its predicted core peptide. Whereas all LanM and most Ycao-like proteins act on these three amino acid residues, recent studies on YcaO-proteins have shown thioamidation[2d, 14] and macroamidine-formation[15] activity that does not require β-nucleophile-containing amino acids. By sequence, the leader peptides appear to fall into two groups (LahA1-A5 and LahA6-A9) (Figure 1C). None of the proteins encoded in the lah cluster contains a RiPP recognition element[16] that recognizes the leader peptide in other biosynthetic pathways.[17] Anaerobic cultures of Lachnospiraceae bacterium C6A11 were monitored by mass spectrometry for potential products of the lah cluster, but no such masses were observed.
Two putative class II lanthipeptide synthetases LahM1 and M2 are encoded in the lah cluster (NCBI accession number WP_035625282.1 and WP_081828742.1). Their domain architectures appear to deviate somewhat from those of canonical LanM proteins (Figure 2A) based on Pfam analysis.[18] A prototypical CylM lanthipeptide synthetase contains an approximately 400 amino acid N-terminal domain (Duf4135) and an approximately 350 amino acid LanC-like C-terminal domain. These annotated domains roughly correspond to the dehydratase and cyclase domains within the CylM crystal structure.[19] However, Pfam analysis of both LahM1 and M2 splits the typical dehydratase domain into two parts. The first domain is approximately 200 amino acids in length and the second approximately 100 amino acids. The two domains are connected with a linker of about 50 amino acids that is absent in currently characterized LanM proteins (Figure 2A). It appears that the first domain contains the catalytic machinery for the kinase activity of LanMs and that the second domain corresponds to the kinase activation domain[19] that in canonical LanM enzymes also bears two residues (Arg and Thr) that are critical for phosphate elimination.[20] Inspection of the first domain in an alignment with the canonical LanMs LctM and CylM indicates LahM1 and LahM2 retain the conserved residues Lys159, Asn247, Glu261 (LctM numbering) involved in phosphorylation activity (Figure S1). Both proteins also contain Arg399 in domain 2 that is important for phosphate elimination whereas the key residue Thr405 that is critical for phosphate elimination activity[19–20] is present in LahM1 but mutated to Pro in the LahM2 sequence (Figure 2B). Thus, we predicted that LahM1 could dehydrate its substrate(s) but that LahM2 would only be able to phosphorylate its substrate(s).
Figure 2.

The LahM enzymes differ from canonical LanMs. (A) Pfam domain architecture of various LanM proteins compared to LahM1 and M2. Stars indicate the mutated zinc-binding ligands in the BsjM sequence. (B) Alignment of the conserved active site residues in the dehydration and cyclization domains of canonical LanM enzymes (LctM and CylM), BsjM, and LahM1 and M2. Residues in the dehydration and cyclization domain that differ between the canonical enzymes and the others are shown in red and blue respectively.
The cyclase domains of neither LahM1 nor LahM2 are annotated in a Pfam analysis, and alignment with LctM and CylM shows an absence of the Zn-binding residues His725, Cys781 and Cys836 (LctM numbering) that are conserved in prototypical LanM proteins (Figure 2).[21] A similar scenario of a disrupted Zn-site was observed for BsjM (Figure 2). The gene for this enzyme is clustered with genes for BsjA precursors involved in bicereucin biosynthesis that also predominantly lack Cys residues.[22] The absence of Cys residues in most of the substrates appears therefore to correlate with a cyclase domain lacking the Zn-site that previously has been shown to be essential for cyclization activity of class II lanthipeptide synthetases.[21] Thus, we predicted that neither LahM1 nor LahM2 would be able to form (methyl)lanthionines.
In addition to the two apparent lanthipeptide synthetases, a flavin-dependent oxidoreductase (LahJB) and two YcaO-like proteins are encoded in the BGC (Figure 1). In other systems, LanJB orthologs are responsible for the reduction of dehydro amino acids to their corresponding D-amino acid counterparts,[22–23] whereas YcaO-like proteins install azole, thioamide or amidine structures into RiPPs.[10] The combination of YcaO-type proteins and class II lanthipeptide synthetases is unprecedented in characterized systems. Previously, class I dehydratases and YcaO-proteins were shown to be involved in the biosynthesis of the thiopeptides[24] and the azole and dehydro amino acid containing peptide goadsporin.[25] The two putative YcaO proteins are assigned as LahD1 and D2, following linear azol(in)e-containing peptide nomenclature.[1] They conspicuously lack genes encoding cognate C proteins in the BGC, which typically facilitate substrate recognition through binding the leader peptide on the peptide substrate.[16, 26] Instead, LahD1 and D2 are reminiscent of the stand-alone YcaO proteins involved in bottromycin biosynthesis. Indeed, among various characterized YcaO protein groups bottromycin YcaO proteins have the highest sequence homology with LahD1 (13.7% identity to BotC/BmbD; 11.8% identity to BotCD/BmbE) and D2 (13.7% identity to BotC/BmbD; 11.8% similarity to BotCD/BmbE). Recent studies have presented evidence that the bottromycin YcaO proteins are responsible for the cyclodehydration and macrocyclization of its substrate to provide a macrocyclic amidine.[15]
Finally, a SAM-dependent methyltransferase annotated as LahSB and a peptidase-containing ATP-binding cassette transporter LahT are also encoded in the BGC. The methyltransferase is the second instance that such an enzyme is found in lanthipeptide BGCs after the ubiquitous O-methyltransferases that were recently characterized to convert Asp to isoAsp.[27] These O-methyltransferases were given the general name LanSA (Pfam: PF01135), but since the methyltransferase in the lah cluster belongs to a different methyltransferase family (Pfam: PF13847), we term this protein LahSB (Figure S2).
LahM1 is selective for LahA3 and LahA5
Since both LanM and YcaO-type proteins typically act on unmodified RiPP precursor peptides, a priori it is difficult to predict whether the enzymes would act on the same substrate making a natural hybrid RiPP or whether each type of modification would be discrete to a particular set of substrates. For the first scenario, it would also be unclear which enzyme(s) would act first. In this initial study we focused our attention on the two lanthipeptide synthetases and performed co-expression studies with the substrates in Escherichia coli, a strategy that has been highly successful for a large number of lanthipeptides.[9, 28] Co-expression of lahA1, A2, A4, A6 and A7 with either lahM1 or M2 resulted in peptides with masses corresponding to unmodified peptide. In contrast, co-expression of lahA5 with lahM1 resulted in three-fold dehydrated peptide, and co-expression of lahA3 with lahM1 resulted in two-fold dehydrated peptide (Figure S3, S4). The clean conversion of LahA3 and LahA5 suggests that the enzyme is fully active and that the other peptides are not substrates for LahM1. All previous co-expression studies of lanthipeptides have provided the correct post-translationally modified products and hence the observed activities are likely the native reactions. In order to investigate the possibility that the expression level of LahM1 might have been insufficient for the modification of LahA1, A2, A4, A6, and A7, purification of LahM1 was undertaken. The expression and purification of a His6-maltose-binding protein fusion[29] of LahM1 (His6-MBP-LahM1) using Ni2+-affinity chromatography was successful (Figure S5). In vitro assays of LahA3 and LahA5 with purified LahM1 resulted in the same modification as that observed in vivo (compare Figures 3, 4 with Figures S3, S4). Thus, we conclude that these two peptides are substrates for LahM1 but the other LahA peptides are not. Although the determinants of enzyme activity are unclear from only examining the LahA sequences, some possible correlations based on the sites of dehydration are discussed below.
Figure 3.

In vitro assays of LahM1 and LahM2 with LahA3. MALDI-TOF MS spectra of (A) His-LahA3 incubated with heat-inactivated enzyme; (B) His-LahA3 incubated with MBP-LahM1; (C) His-LahA3 incubated with MBP-LahM2; and, (D) His-LahA3 incubated with MBP-LahM1 and MBP-LahM2. (E) Tandem ESI-MS fragmentation of LahM1- and LahT150-modified LahA3 core peptide. Masses of the ions shown are listed in Table S2.
Figure 4.

In vitro assays of LahM1 and LahM2 with LahA5. MALDI-TOF MS spectra of (A) His-LahA5 incubated with heat-inactivated enzyme; (B) His-LahA5 incubated with MBP-LahM1; (C) His-LahA5 incubated with MBP-LahM2; and, (D) His-LahA5 incubated with MBP-LahM1 and MBP-LahM2. (E) Tandem ESI-MS fragmentation of LahM1- and GluC-modified LahA5 core peptide. Masses of the ions shown are listed in Table S3.
In contrast to the observations with LahM1, but consistent with the sequence analysis demonstrating the absence of a critical Thr residue for phosphate elimination, LahM2 only demonstrated phosphorylation activity with LahA5, and no activity with LahA3 (Figures 3 and 4). These observations raised the possibility that perhaps the elimination activity of LahM1 might be used to act on peptides phosphorylated by LahM2, or that modification by either enzyme would be a requirement for activity by the second LanM. Therefore, co-expression studies of LahA5 and LahA3 with both the LahM1 and LahM2 proteins were performed, but these did not yield additional modification beyond that observed in the co-expressions with just the individual LahM enzymes. Similar findings were observed in vitro with purified enzymes (Figures 3D and 4D).
To determine the positions of the dehydrated residues in LahA3 and LahA5, the leader peptide was proteolytically removed using the LahT150 protease domain resulting in liberation of the LahA3/A5 core peptide. Alternatively, because the core peptides of LahA3 and LahA5 do not contain a Glu residue, for convenience commercially available endoproteinase Glu-C was employed for some subsequent experiments as indicated. Purified LahM1-modified LahA3 (mLahA3) and LahA5 (mLahA5) core peptides were subjected to electrospray ionization (ESI) quadrupole time-of-flight (TOF) tandem mass spectrometric analysis (ESI-Q-TOF MS-MS). The results illustrated that in the mLahA3 core peptide Ser13 and Thr17 were dehydrated to Dha and Dhb, respectively, while in mLahA5 Ser12, Ser18 and Thr15 were dehydrated (Figures 3E and 4E). The Ser/Thr residues that escape dehydration are near the N- and C-termini of the core peptide, suggesting a putative positional dependence of LahM1. No obvious sequence motif(s) can be detected to explain the observed selectivity, but all Ser/Thr residues that are acted on by LahM1 are flanked by two aliphatic residues and are located in the middle of the peptide (i.e. at a defined distance from the leader peptide). Almost all Ser/Thr residues that are not dehydrated are either flanked by at least one polar residue, are located at the start or end of the core peptide, or have a different leader peptide from LahA3/5 (i.e. LahA and A7). The observation that LahM1 does not act on any of the Cys containing peptides, along with the observed disrupted zinc-binding site, suggests that the Cys residues in these peptides may be substrates for the YcaO-proteins. This prediction as well as the proposed molecular basis for site- and peptide selectivity need to beinvestigated in future studies.
Biochemical Characterization of LahSB
Co-expression of LahA1, A6 or A7 with LahSB (NCBI accession number WP_051646488.1) in E. coli resulted in masses corresponding to unmodified peptide (Figures S5–S7). In contrast, co-expression of lahA2-A5 with lahSB resulted in products corresponding to a mass addition of one methyl group, indicating that LahSB is selectively active on LahA peptides independently of other PTM enzymes. Subsequently, the activity was confirmed in vitro using MBP-tagged recombinant LahSB (Figure S6) with LahA2-A5 as substrates (Figure 5B, Figures S7–9), demonstrating consistent results with the observations from co-expression. Furthermore, LahM1-modified LahA3 and LahA5 as well as the GluC-treated mLahA3 and mLahA5 peptides were also substrates for LahSB during in vitro assays (Figure 5D and S10–13). Tandem MS/MS analysis of LahSB-modified mLahA5 and mLahA3 peptides revealed that a methyl group was installed at the C-terminus of both peptides (Figures 5E, S14, S15). Based on all the data above, LahSB activity is independent of the leader peptide and prior dehydration. Collectively the peptides that are substrates contain Ile, Val, and Met as C-terminal residues, suggesting that LahSB is tolerant with respect to the amino acid residue that it methylates. At present, the factors that determine substrate specificity are not entirely clear. A kinetic investigation of LahSB using a synthetic 15-mer peptide FLGSAIVAASSAGAV corresponding to the C-terminus of LahA3 revealed a Km of 103 ± 10 μM and kcat of 60.9 ± 0.3 s−1 (Figure 5F). C-terminal methylation has not been previously observed in lanthipeptides, but it was reported for cyanobactins, another group of RiPPs.[30] Modification of the C-terminus of RiPPs likely contributes beneficially to environmental stability by providing protection from carboxypeptidases. C-terminal methylation adds to previous protecting group strategies reported for lanthipeptides such as C-terminal decarboxylation.[31] LahSB might therefore have potential utility for synthetic biology as a methyl ester-introducing element.
Figure 5.

MALDI-TOF mass spectra of (A) His-LahA5 reacted with heat-inactivated MBP-LahSB; (B) MBP-LahSB reacted with His-LahA5; (C) GluC-treated mLahA5 MBP-LahSB reacted with reacted with heat-inactivated MBP-LahSB; (D) GluC-treated mLahA5 MBP-LahSB reacted with reacted with MBP-LahSB. Masses of the ions shown are listed in Table S4. (E) Tandem MS-MS spectrum of LahSB-modified GluC-treated mLahA5. Masses of the ions shown are listed in Table S5. (F) Dependence of LahSB activity on substrate (LahA3-15-mer peptide) concentration. The data were fit to a nonlinear regression using the Michaelis-Menten equation. The plot was generated using GraphPad Prism. Reported values are means ± S.D. (n=3).
Overall structure of LahSB
In order to gain insights into the substrate scope, LahSB was purified as described in the Experimental Section and subjected to crystallization. The LahSB structure was determined at a resolution of 2.01 Å using diffraction data collected from crystals of SeMet labeledprotein. Relevant data collection and refinement statistics may be found in Table S6. The overall structure of LahSB is representative of other members of class I methyltransferases, and consists of a core Rossman-like fold. The domain architecture is composed of a central seven-stranded β sheet with the β7 strand running antiparallel to strands β1 through β6 (Figure 6A). This central β-sheet is surrounded by α–helices on both sides. A DALI search against the Protein Data Bank showed the S-methyltransferase TmtA (PDB ID 5EGP; Figure S16) to be the closest characterized protein with an RMSD of 4.2 Å.[32] TmtA is a self-resistance protein that methylates gliotoxin in the producing fungus Aspergillus fumigatus. Density corresponding to a molecule of bound S-adenosyl homocysteine (SAH) can be seen at the presumptive active site, where the hydroxyl groups of the ribose are hydrogen bonding with Asp72 (Figure 6B). Asp98 interacts with the amino group of the adenine moiety. The main chain oxygen atoms of His115 and Gly49 interact with the amino group from the methionine moiety in SAH while Arg21 and Thr116 interacts with the carboxyl group. Gly49 is part of the GXGXG motif known to make contact with the amino acid moiety of SAM. Efforts to obtain a co-crystal structure of LahSB with one of its peptide substrates were not successful. However, a surface representation of the structure reveals a large cavity adjacent to the bound SAH (Figure 6C). Docking studies with the last five residues of LahA3 shows that the cavity can accommodate a linear conformation of the peptide, where the carboxy terminus would be appropriately situated for methyl transfer from SAM (Figure 6D). The edge of this cavity is defined by residues Phe11, Phe16, and Phe149, which provide a hydrophobic pocket where the C-terminal residue of the substrate would be expected to bind. This pocket may account for the selectivity of LahSB for only a subset of the precursor peptides.
Figure 6.

Structure of the LahSB methyltransferase. (A) Overall architecture of the enzymes with helices and strands numbered in order. (B) Difference Fourier map (F0-FC) showing SAH binding site (C) Surface diagram of a AutoDock Vina model of the last five amino acids of LahA3 (sticks) bound to the presumptive substrate-binding cavity. (D) Close-up view of the active site with LahA3 modeled in as in C
Conclusions
By genome mining we identified an unusual gene cluster that combines hallmarks from other RiPP biosynthetic pathways. Like the thiopeptides,[33] polytheonamides,[34] and bottromycins,[35] the lah cluster combines genes encoding enzymes that are characteristic for different classes of RiPPs, in this case class II lanthipeptide synthetases and YcaO-type enzymes. In the other examples, all of the enzymes act on a single substrate peptide (or a small set of peptides closely related by sequence). Because the lah cluster encodes at least seven and possibly as many as nine precursor peptides with very different predicted core peptide sequences, it is not clear whether the biosynthetic enzymes act on all or a subset of substrates. Our results with the lanthipeptide synthetase LahM1 and the methyltransferase LahSB suggest the latter. As such, the lah cluster may encode an unusual example of a multi-peptide system[36] in which two or more products act synergistically to exert a particular biological function. Usually the peptides in multi-peptide systems are structurally related and belong to the same class of natural products (e.g. two-component lantibiotics). In this case the individual components might be post-translationally modified by a different subset of enzymes and belong to different RiPP classes. A somewhat related system is the two-peptide antimicrobial peptide bicereucin, in which one component contains a lanthionine ring and the second component does not have any (methyl)lanthionines.[22] However, bicereucin is still made by a single LanM synthetase. One alternative scenario that we cannot currently rule out is that the peptides that are not substrates for the enzymes investigated in this work might be encoded by cryptic or pseudogenes.
The seven substrate peptides with Nif11-type leader peptides are also reminiscent of the prochlorosins in which one class II lanthipeptide synthetase modifies thirty[12a] (or up to 80)[12b] peptides with highly diverse core sequences. But unlike the prochlorosin system, the majority of the LahA peptides do not contain Cys, and the LahM proteins do not have the characteristic Zn2+ coordinated by three Cys ligands implicated in conferring high substrate tolerance to ProcM-like enzymes for the cyclization of diverse core peptide sequences.[37] Finally, although we show that the methyltransferase LahSB acts independently of LahM activity, we cannot exclude the possibility that LahM1/M2 substrate recognition and therefore activity are predicated on the presence of other PTMs. A strict sequence of PTM installation has been observed previously for some RiPP biosynthetic pathways such as the thiopeptides,[24a] select lanthipeptides,[38] microcin C7,[39] and the bottromycins.[40] However, in other systems such as the cyanobactins, the biosynthetic enzymes have demonstrated much plasticity and can act in a variety of orders.[41] Although we cannot rule out the possibility of strict order for the lah system, we find it unlikely that LahM1/M2 would act on a large number of previously posttranslationally modified LahA peptides. In particular, the high sequence divergence of their core peptides makes it hard to envision how LahM1/M2 would accept a specific pre-installed structure the way for instance the TbtB enzyme recognizes a highly specific hexazole-containing peptide during thiomuracin biosynthesis.[24b] The answers to these questions lie likely in the activity of the two YcaO proteins and the dehydrogenase, which will be the focus of future studies.
Experimental Section
Protein cloning, expression and purification:
The gene encoding either LahSB, LahM1, or LahM2 was first amplified using genomic DNA of Lachnospiraceae bacterium C6A11 as template and the corresponding primers (Table S1) and cloned into the multiple cloning site of the pET28a-MBP vector[23a] linearized by BamHI using Gibson assembly to generate pETMBP-LahSB, pETMBP-LahM1 and pETMBP-LahM2 (for details, see Supplementary Methods). E. coli BL21 (DE3) cells were transformed with the corresponding constructs and plated on a Luria Broth (LB) agar plate containing 50 mg/L kanamycin. A single colony was picked and grown in 15 mL of LB with 50 mg/L kanamycin at 37 °C for 15 h, and the resulting culture was used to inoculate 3 L of LB medium. Cells were cultured at 37 °C until the OD600 reached 0.6 and cooled on ice for 30 min. Subsequently IPTG was added to a final concentration of 0.3 mM. The cells were cultured at 16 °C for another 18 h before harvesting. The cell pellets were resuspended in buffer A (20 mM Tris, 1 M NaCl, pH 7.8 at 25 °C) and lysed using a high pressure homogenizer (Avestin, Inc.). The sample was centrifuged at 23,700×g for 30 min. The supernatant was passed through 0.45-μm syringe filters (Fisherbrand®) and loaded onto a 5 mL HisTrap immobilized metal affinity chromatography (IMAC) column pre-charged with Ni2+ and equilibrated with buffer A. The column was attached to an ÄKTA fast protein liquid chromatography (FPLC) system (GE Healthcare) and washed with up to 25% buffer B (20 mM Tris, 1 M NaCl, 500 mM imidazole, pH 7.8 at 25 °C) in buffer A at a flow rate of 1.5 mL/min. Then the protein was eluted using a gradient of 25-100% buffer B over 45 min. UV absorbance at 280 nm was monitored and fractions were collected and analyzed by SDS-PAGE (Bio-Rad). The fractions containing the desired proteins were combined and exchanged back to buffer A using a PD-10 desalting column (GE Healthcare) and subsequently concentrated using an Amicon Ultra-15 Centrifugal Filter Unit (Millipore). Furthermore, a gel filtration step with a HiLoad 16/60 column containing Superdex200 resin (GE Healthcare) was employed. The column was eluted with 120 mL (2 CV) storage buffer (10% glycerol, 20 mM Tris, 500 mM NaCl, pH 7.8) and the fractions were concentrated, aliquoted and frozen in liquid nitrogen and then stored at −80 °C.
Peptide expression and purification:
E. coli BL21 (DE3) cells were transformed with pRSFDuet-LahAs-LahSB/M1/M2 (for construction, see Supplementary Methods) and plated on a Luria Broth (LB) agar plate containing 50 mg/L kanamycin. A single colony was picked and grown in 15 mL of terrific broth (TB) amended with 10 mM MgCl2 and 50 mg/L kanamycin at 37 °C for 15 h, and the resulting culture was used to inoculate 1.5 L of TB medium containing 50 mg/L kanamycin with 10 mM MgCl2. Cells were cultured at 37 °C until the OD600 reached 0.6 and cooled on ice for 30 min. Subsequently IPTG was added to a final concentration of 0.7 mM. The cells were cultured at 18 °C for another 18 h before harvesting. The cell pellets were resuspended at room temperature in LanA start buffer (20 mM NaH2PO4, pH 7.5 at 25 °C, 500 mM NaCl, 0.5 mM imidazole, 20% glycerol) and lysed using a high pressure homogenizer (Avestin, Inc.). The sample was centrifuged at 23,700×g for 30 min, and the supernatant was kept. The pellets were then resuspended in LanA buffer 1 (6 M guanidine hydrochloride, 20 mM NaH2PO4, pH 7.5 at 25 °C, 500 mM NaCl, 0.5 mM imidazole) and lysed again. The insoluble portion was removed by centrifugation at 23,700×g for 30 min, and the soluble portion was kept. Both soluble portions were passed through 0.45-μm syringe filters (Fisherbrand®), and the His6-tagged modified peptides were purified by IMAC as previously described[42]. The eluted fractions were desalted by preparative reversed phase (RP) HPLC using a Waters Delta-pak C4 column (15 μm; 300 Å; 25 mm × 100 mm). The desalted peptides were lyophilized and stored at −20 °C.
In vitro assays of LahSB:
The activity of LahSB in vitro was monitored in the presence of 100 μM of LahA or variants (modified, unmodified, full-length and core peptide) and 20 μM of His-tagged-MBP-LahSB in buffer (50 mM HEPS pH 7.8) and 1 mM S-adenosyl methionine (SAM). Aliquots of 100 μL were desalted via C18 ZipTip (EMD Millipore) according to the manufacturer’s instructions, and the peptide was eluted using a saturated solution of sinapinic acid in 60% aq. MeCN. The eluent was analyzed by MALDI-TOF MS.
In vitro assays of LahM1 and LahM2:
To reconstitute the activity of His-MBP-LahM1 and His-MBP-LahM2 in vitro, 20 μM of linear LahA peptides were supplied in a reaction vessel with 4 mM MgCl2, 4 mM ATP, 2 mM DTT and 50 mM HEPES (pH 7.5), followed by the addition of the LahM to a final concentration of 0.5 μM. Reactions were incubated at room temperature for 4 h. Control reactions were set up with all other components with heat-inactivated LahM1 and/or LahM2. Each sample was zip-tipped and analyzed by MALDI-TOF MS.
Determination of Kinetic Parameters for LahSB:
Kinetic analysis of LahSB was performed using a coupled spectrophotometric assay using the Cayman Methyltransferase Fluorometric Assay Kit (Catalog No. 700150, Cayman Chemical) following the manufacturer’s instruction and with assay conditions that ensured the saturation of S-adenosylmethionine, coupling enzyme, and varying the concentration of the substrate peptide. A LahSB concentration of 1 μM was used, which ensured that the time-dependent activity observed was within the linear range in the time frame observed. Three replicates were run per sample, and resorufin standards were used to create a standard curve. The kinetic parameters (Km and kcat) were calculated using GraphPad Prism 7 software (GraphPad Software Inc.) using standard settings for non-linear regression curve fitting to the Michaelis–Menten equation.
Crystallization of LahSB:
Following the removal of the MBP tag (Figure S6B) and further purification using size exclusion chromatography (buffer of 20 mM Hepes pH 7 and 100 mM KCl), LahSB was concentrated to 10 mg/ml using Amicon centrifugal filters. Crystallization conditions were initially determined using commercial screens. Crystals of LahSB were grown using the hanging drop method in conditions containing 0.2 M sodium malonate and varying concentrations (16-22%) of PEG3350. Crystals were obtained at 9 oC after 2 days, and were rapidly soaked in precipitant solution supplemented with 30% glycerol or ethylene glycol prior to vitrification by direct immersion in liquid nitrogen. Anomalous scattering data, collected on crystals of SeMet-substituted, was used to determine crystallographic phases. Data were indexed and scaled using HKL2000.[43] Selenium sites were identified using HySS, and subsequently refined in autoSHARP.[44] The model was fitted using COOT[45] and refined with REFMAC5.[46] The structure has been deposited in the Protein Data Bank (ID 6UAK).
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R37 GM 058822 to W.A.V. and RO1 GM 079038 to S.K.N.) and the Natural Sciences and Engineering Research Council of Canada (NSERC) (to J.Z.A). We thank Dr. William Kelly (AgResearch, New Zealand) for providing Lachnospiraceae bacterium C6A11 and the Hungate1000 project and Joint Genomes Institute for sequencing of its genome. We also thank Jefferson Chan (UIUC) for use of a fluorometer for the LahSB kinetic study.
Footnotes
Additional experimental methods are provided in the Supporting information via the link at the end of the document.
References
- [1].Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA, Nat. Prod. Rep 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Letzel AC, Pidot SJ, Hertweck C, BMC Genomics 2014, 15, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hetrick KJ, van der Donk WA, Curr. Opin. Chem. Biol 2017, 38, 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Poorinmohammad N, Bagheban-Shemirani R, Hamedi J, Antonie van Leeuwenhoek 2019 [DOI] [PubMed] [Google Scholar]; d) Santos-Aberturas J, Chandra G, Frattaruolo L, Lacret R, Pham TH, Vior NM, Eyles TH, Truman AW, Nucleic Acids Res. 2019, 47, 4624–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Oman TJ, van der Donk WA, Nat. Chem. Biol 2010, 6, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Plat A, Kuipers A, Rink R, Moll GN, Curr. Protein Pept. Sci 2013, 14, 85–96. [DOI] [PubMed] [Google Scholar]
- [4].a) Bierbaum G, Sahl HG, Curr. Pharm. Biotechnol 2009, 10, 2–18 [DOI] [PubMed] [Google Scholar]; b) Cotter PD, Hill C, Ross RP, Curr. Protein Pept. Sci 2005, 6, 61–75 [DOI] [PubMed] [Google Scholar]; c) Repka LM, Chekan JR, Nair SK, van der Donk WA, Chem. Rev 2017, 117, 5457–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Siezen RJ, Kuipers OP, de Vos WM, Antonie van Leeuwenhoek 1996, 69, 171–184 [DOI] [PubMed] [Google Scholar]; b) Xie L, Miller LM, Chatterjee C, Averin O, Kelleher NL, van der Donk WA, Science 2004, 303, 679–681. [DOI] [PubMed] [Google Scholar]
- [6].Chatterjee C, Miller LM, Leung YL, Xie L, Yi M, Kelleher NL, van der Donk WA, J. Am. Chem. Soc 2005, 127, 15332–15333. [DOI] [PubMed] [Google Scholar]
- [7].Bobeica SC, Dong S, Huo L, Mazo N, McLaughlin MIH, Jimenéz-Osés G, Nair SK, van der Donk WA, eLife 2019, 8, e42305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Håvarstein LS, Diep DB, Nes IF, Mol. Microbiol 1995, 16, 229–240 [DOI] [PubMed] [Google Scholar]; b) Lin DY, Huang S, Chen J, Nature 2015, 523, 425–430. [DOI] [PubMed] [Google Scholar]
- [9].Zhao X, van der Donk WA, Cell Chem. Biol 2016, 23, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Burkhart BJ, Schwalen C, Mann G, Naismith JH, Mitchell DA, Chem. Rev 2017, 117, 5389–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Azevedo AC, Bento CB, Ruiz JC, Queiroz MV, Mantovani HC, Appl. Environ. Microbiol 2015, 81, 7290–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, van der Donk WA, Proc. Natl. Acad. Sci. U.S.A 2010, 107, 10430–10435 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cubillos-Ruiz A, Berta-Thompson JW, Becker JW, van der Donk WA, Chisholm SW, Proc. Natl. Acad. Sci. USA 2017, 114, E5424–E5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Haft DH, Basu MK, Mitchell DA, BMC Biol. 2010, 8, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Dong SH Liu A, Mahanta N, Mitchell DA, Nair SK, ACS Cent. Sci 2019, 5, 842–851 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nayak DD, Mahanta N, Mitchell DA, Metcalf WW, eLife 2017, 6, e29218. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kawahara T, Izumikawa M, Kozone I, Hashimoto J, Kagaya N, Koiwai H, Komatsu M, Fujie M, Sato N, Ikeda H, Shin-Ya K, J. Nat. Prod 2018 [DOI] [PubMed] [Google Scholar]; d) Frattaruolo L, Lacret R, Cappello AR, Truman AW, ACS Chem. Biol 2017, 12, 2815–2822 [DOI] [PubMed] [Google Scholar]; e) Kjaerulff L, Sikandar A, Zaburannyi N, Adam S, Herrmann J, Koehnke J, Müller R, ACS Chem. Biol 2017, 12, 2837–2841. [DOI] [PubMed] [Google Scholar]
- [15].a) Crone WJK, Vior NM, Santos-Aberturas J, Schmitz LG, Leeper FJ, Truman AW, Angew. Chem., Int. Ed 2016, 55, 9638–9642 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schwalen CJ, Hudson GA, Kosol S, Mahanta N, Challis GL, Mitchell DA, J. Am. Chem. Soc 2017, 139, 18154–18157 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Franz L, Adam S, Santos-Aberturas J, Truman AW, Koehnke J, J. Am. Chem. Soc 2017, 139, 18158–18161. [DOI] [PubMed] [Google Scholar]
- [16].Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA, Nat. Chem. Biol 2015, 11, 564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Ortega MA, Hao Y, Zhang Q, Walker MC, van der Donk WA, Nair SK, Nature 2015, 517, 509–512 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koehnke J, Mann G, Bent AF, Ludewig H, Shirran S, Botting C, Lebl T, Houssen WE, Jaspars M, Naismith JH, Nat. Chem. Biol 2015, 11, 558–563 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Grove TL, Himes PM, Hwang S, Yumerefendi H, Bonanno JB, Kuhlman B, Almo SC, Bowers AA, J. Am. Chem. Soc 2017, 139, 11734–11744 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Evans RL 3rd, Latham JA, Xia Y, Klinman JP, Wilmot CM, Biochemistry 2017, 56, 2735–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, Finn RD, Nucleic Acids Res. 2019, 47, D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dong SH, Tang W, Lukk T, Yu Y, Nair SK, van der Donk WA, eLife 2015, 4, e07607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].You YO, van der Donk WA, Biochemistry. 2007, 46, 5991–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Paul M, Patton GC, van der Donk WA, Biochemistry. 2007, 46, 6268–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huo L, van der Donk WA, J. Am. Chem. Soc 2016, 138, 5254–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Yang X van der Donk WA, J. Am. Chem. Soc 2015, 137, 12426–12429; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lohans CT, Li JL, Vederas JC, J. Am. Chem. Soc 2014, 136, 13150–13153. [DOI] [PubMed] [Google Scholar]
- [24].a) Hudson GA, Zhang Z, Tietz JI, Mitchell DA, van der Donk WA, J. Am. Chem. Soc 2015, 137, 16012–16015 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Z, Hudson GA, Mahanta N, Tietz JI, van der Donk WA, Mitchell DA, J. Am. Chem. Soc 2016, 138, 15511–15514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Hayashi S, Ozaki T, Asamizu S, Ikeda H, Omura S, Oku N, Igarashi Y, Tomoda H, Onaka H, Chem. Biol 2014, 21, 679–688 [DOI] [PubMed] [Google Scholar]; b) Ozaki T, Kurokawa Y, Hayashi S, Oku N, Asamizu S, Igarashi Y, Onaka H, ChemBioChem 2016, 17, 218–223. [DOI] [PubMed] [Google Scholar]
- [26].Dunbar KL, Melby JO, Mitchell DA, Nat. Chem. Biol 2012, 8, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Acedo JZ, Bothwell IR, An L, Trouth A, Frazier C, van der Donk WA, 2019, submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Nagao J, Harada Y, Shioya K, Aso Y, Zendo T, Nakayama J, Sonomoto K, Biochem. Biophys. Res. Commun 2005, 336, 507–513 [DOI] [PubMed] [Google Scholar]; b) Shi Y, Yang X, Garg N, van der Donk WA, J. Am. Chem. Soc 2011, 133, 2338–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Caetano T, Krawczyk JM, Mosker E, Süssmuth RD, Mendo S, Chem. Biol 2011, 18, 90–100 [DOI] [PubMed] [Google Scholar]; d) Lin Y, Teng K, Huan L, Zhong J, Microbiol. Res 2011, 166, 146–154 [DOI] [PubMed] [Google Scholar]; e) okesli A, Cooper LE, Fogle EJ, van der Donk WA, J. Am. Chem. Soc 2011, 133, 13753–13760 [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Shi Y, Bueno A, van der Donk WA, Chem. Commun 2012, 48, 10966–10968 [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Tang W, van der Donk WA, Nat. Chem. Biol 2013, 9, 157–159 [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Wang J, Zhang L, Teng K, Sun S, Sun Z, Zhong J, Appl. Environ. Microbiol 2014, 80, 2633–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Kuthning A, Mösker E, Süssmuth RD, Appl. Microbiol. Biotechnol 2015, 99, 6351–6361 [DOI] [PubMed] [Google Scholar]; j) Garg N, Tang W, Goto Y, van der Donk WA, Proc. Natl. Acad. Sci. U. S. A 2012, 109, 5241–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Huo L, Ökesli A, Zhao M, van der Donk WA, Appl. Environ. Microbiol 2017, 83, e02698–02616 [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Chen E, Chen Q, Chen S, Xu B, Ju J, Wang H, Appl. Environ. Microbiol 2017, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Smith TE, Pond CD, Pierce E, Harmer ZP, Kwan J, Zachariah MM, Harper MK, Wyche TP, Matainaho TK, Bugni TS, Barrows LR, Ireland CM, Schmidt EW, Nat. Chem. Biol 2018, 14, 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Ongey EL, Giessmann RT, Fons M, Rappsilber J, Adrian L, Neubauer P, Front. Microbiol 2018, 9, 1688. [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Hegemann JD, van der Donk WA, J. Am. Chem. Soc 2018, 140, 5743–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Diguan C, Li P, Riggs PD, Inouye H, Gene. 1988, 67, 21–30 [DOI] [PubMed] [Google Scholar]; b) Kapust RB, Waugh DS, Protein Sci. 1999, 8, 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sardar D, Hao Y, Lin Z, Morita M, Nair SK, Schmidt EW, J. Am. Chem. Soc 2017, 139, 2884–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sit CS, Yoganathan S, Vederas JC, Acc. Chem. Res 2011, 44, 261–268. [DOI] [PubMed] [Google Scholar]
- [32].Duell ER, Glaser M, Le Chapelain C, Antes I, Groll M, Huber EM, ACS Chem. Biol 2016, 11, 1082–1089. [DOI] [PubMed] [Google Scholar]
- [33].a) Wieland Brown LC, Acker MG, Clardy J, Walsh CT, Fischbach MA, Proc. Natl. Acad. Sci. U.S.A 2009, 106, 2549–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kelly WL, Pan L, Li C, J. Am. Chem. Soc 2009, 131, 4327–4334 [DOI] [PubMed] [Google Scholar]; c) Morris RP, Leeds JA, Naegeli HU, Oberer L, Memmert K, Weber E, Lamarche MJ, Parker CN, Burrer N, Esterow S, Hein AE, Schmitt EK, Krastel P, J. Am. Chem. Soc 2009, 5946–5955 [DOI] [PubMed] [Google Scholar]; d) Liao R, Duan L, Lei C, Pan H, Ding Y, Zhang Q, Chen D, Shen B, Yu Y, Liu W, Chem. Biol 2009, 16, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Freeman MF, Gurgui C, Helf MJ, Morinaka BI, Uria AR, Oldham NJ, Sahl HG, Matsunaga S, Piel J, Science 2012, 338, 387–390. [DOI] [PubMed] [Google Scholar]
- [35].a) Huo L, Rachid S, Stadler M, Wenzel SC, Müller R, Chem. Biol 2012, 19, 1278–1287 [DOI] [PubMed] [Google Scholar]; b) Crone WJK, Leeper FJ, Truman AW, Chem. Sci 2012, 3, 3516–3521 [Google Scholar]; c) Hou Y, Tianero MD, Kwan JC, Wyche TP, Michel CR, Ellis GA, Vazquez-Rivera E, Braun DR, Rose WE, Schmidt EW, Bugni TS, Org. Lett 2012, 14, 5050–5053 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gomez-Escribano JP, Song L, Bibb MJ, Challis GL, Chem. Sci 2012, 3, 3522–3525. [Google Scholar]
- [36].Garneau S, Martin NI, Vederas JC, Biochimie 2002, 84, 577–592. [DOI] [PubMed] [Google Scholar]
- [37].Yu Y, Mukherjee S, van der Donk WA, J. Am. Chem. Soc 2015, 137, 5140–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].An L, Cogan DP, Navo CD, Jiménez-Osés G, Nair SK, van der Donk WA, Nat. Chem. Biol 2018, 14, 928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Severinov K, Nair SK, Future Microbiol. 2012, 7, 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sikandar A, Franz L, Melse O, Antes I, Koehnke J, J. Am. Chem. Soc 2019, 141, 9748–9752. [DOI] [PubMed] [Google Scholar]
- [41].Gu W, Sardar D, Pierce E, Schmidt EW, J. Am. Chem. Soc. 2018, 140, 16213–16221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li B, Cooper LE, van der Donk WA, Methods Enzymol. 2009, 458, 533–558. [DOI] [PubMed] [Google Scholar]
- [43].Minor W, Cymborowski M, Otwinowski Z, Chruszcz M, Acta Crystallogr. D Biol. Crystallogr 2006, 62, 859–866. [DOI] [PubMed] [Google Scholar]
- [44].Vonrhein C, Blanc E, Roversi P, Bricogne G, Methods Mol. Biol 2007, 364, 215–230. [DOI] [PubMed] [Google Scholar]
- [45].Emsley P, Cowtan K, Acta Crystallogr. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- [46].Murshudov G, Skubák P, Lebedev A, Pannu N, Steiner R, Nicholls R, MD W, Long F, Vagin A, Acta Crystallogr. D Biol. Crystallogr 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.