

Abstract
A major challenge for new antibiotic discovery is predicting the physicochemical properties that enable small molecules to permeate Gram-negative bacterial membranes. We have applied physicochemical lessons from previous work to redesign and improve the antibacterial potency of pyridopyrimidine inhibitors of biotin carboxylase (BC) by up to 64-fold and 16-fold against E. coli and P. aeruginosa, respectively. Antibacterial and enzyme potency assessments in the presence of an outer membrane permeabilizing agent or in efflux-compromised strains indicate that penetration and efflux properties of many redesigned BC inhibitors could be improved to various extents. Spontaneous resistance to the improved pyridopyrimidine inhibitors in P. aeruginosa occurs at very low frequencies between 10−8 to 10−9. However, resistant isolates had alarmingly high MIC shifts (16 to >128-fold) compared to the parent strain. Whole genome sequencing of resistant isolates revealed that either BC target mutations or efflux pump overexpression can lead to the development of high-level resistance.
Graphical Abstract
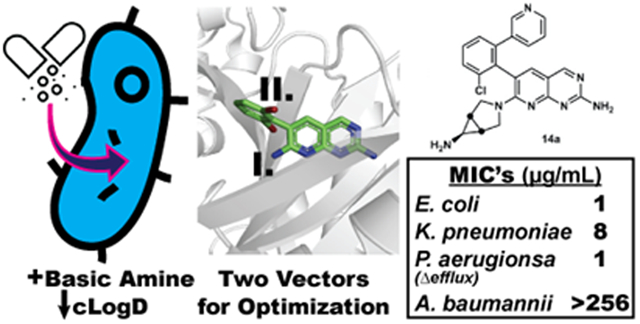
INTRODUCTION
The spread of multi-drug resistant (MDR) bacteria presents a threat to our healthcare system. An analysis from the Centers for Disease Control indicates that approximately 23,000 people die annually in the US from these pathogens,1 and more recent reports suggest this estimate is conservative.2,3 In the future, routine life-saving procedures such as the use of coronary stents, anti-cancer chemotherapy, and organ transplants could carry the risk of life-threating bacterial infections. The situation is particularly alarming for MDR infections caused by the so-called Gram-negative “ESKAPE” pathogens - E. coli, K. pneumoniae, A. baumannii, and P. aeruginosa. This point is illustrated by the recent discovery of a strain of K. pneumoniae from a US ICU patient that is resistant to 26 antibiotics.4 To address this threat from MDR pathogens, a variety of new antibiotic discovery approaches are warranted including optimization of previously discovered compounds with existing, although sometimes low levels, of antibacterial potency.
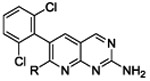
One such previously discovered compound that serves as an enticing starting point for further optimization is the well-characterized biotin carboxylase (BC) inhibitor 1 that was identified in a high-throughput screen against a membrane-compromised, efflux pump-deficient strain of E. coli (tolC, imp).5 BC is a member of the fatty acid biosynthesis pathway (Figure S1) and has attracted attention over the past decade as a promising target for the development of novel, broad-spectrum antibiotics for Gram-negative bacterial infections.6-11 The first committed step in this pathway is catalyzed by an essential cytoplasmic acetyl-CoA carboxylase multienzyme complex12 (see also Supporting Information Text S1), and inhibitors of the BC active site of this complex have shown antibacterial activity against several Gram-negative species.5, 13-17 The BC active site is highly conserved across a number of bacterial pathogens, including P. aeruginosa, E. coli, and H. influenzae.18 While 1 lacks the potency to become a therapeutic agent, it is a promising starting point for medicinal chemistry efforts because of its selective on-target activity, low molecular weight, and available co-crystal structures with BC that enable structure-based molecular design.
Inhibitors of BC must pass through both the outer and inner membranes of the Gram-negative bacteria to reach this cytoplasmic-residing enzyme. The outer membrane bars the passage of many exogenous small molecules. However, porins in the membrane allow some hydrophilic molecules with a size and shape compatible with the diameter of the porin channels to diffuse into the periplasm of the cell.19 Once in the periplasm, molecules must then pass through the phospholipid inner membrane to reach cytoplasmic targets, a process favored by lower polarity. Because the molecular properties of the outer and inner membranes differ, the two barriers act orthogonally to greatly hinder the access of most potential inhibitors to their cytoplasmic target sites. Even when a molecule successfully penetrates both membranes, efflux pumps that actively remove foreign compounds from the cell further reduce the concentration of inhibitor reaching a cytoplasmic target.20,21 These hurdles are major challenges to the development of small molecule inhibitors designed to kill bacteria by acting within the cytoplasm.
To address the challenge of discovering inhibitors that can readily access cytoplasmic targets, retrospective chemoinformatic approaches have been used to evaluate the physicochemical properties of molecules that accumulate effectively in Gram-negative cells.22-24 A pioneering analysis by O’Shea and Moser, showed that, in general, antibiotics with Gram-negative activity are significantly more polar (as measured by relative polar surface area and cLogD values) and have more H-bond acceptor and donor atoms compared to a reference set of drugs from other therapeutic areas.25 For the subset of antibiotics that act against targets in the cytoplasm, the physicochemical differences compared to the reference set were quite diverse, and less pronounced for some classes of antibiotics, but still trended toward having higher overall polarity.24,25 However, a more recent assessment of Gram-negative cell compound accumulation that examined charge and polarity (cLogD) properties separately suggests that polarity alone is not predictive of cell accumulation.26
To illustrate the physicochemical properties of cytoplasm-acting inhibitors that are sufficient for antibacterial potency, we compared a set of exemplar Gram-negative antibiotics (Table 1). These antibiotics have cLogD values ranging from −3.6 to 2.0 and all contain groups that ionize near physiological pH (7.4).
Table 1.
Examples of cytoplasm-targeted Gram-negative agents with physicochemical properties of interest
| Compound | Structurec | MW (g/mol) |
cLogDd | Charge classe |
|---|---|---|---|---|
| Levofloxacin | 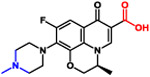 |
361 | −2.0 | Zwitterionic |
| Trimethoprim | 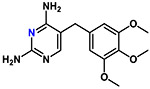 |
290 | 1.1 | Basic |
| Sulfamethoxazole | 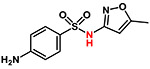 |
253 | −1.1 | Acidic |
| Chloramphenicol | 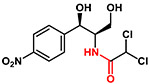 |
323 | 1.0 | Acidic |
| Tetracycline | 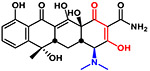 |
444 | −3.6 | Zwitterionic |
| Nitrofurantoin | 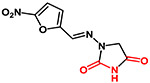 |
238 | −0.27 | Acidic |
| ACHN-975a | 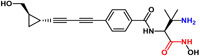 |
369 | 2.0 | Zwitterionic |
| Triusb | 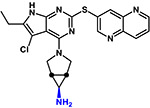 |
420 | 1.3 | Basic |
| 1 | 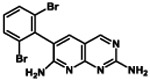 |
395 | 3.3 | Neutral |
Comparing the property values in Table 1 reveals that BC inhibitor 1 with a cLogD value of 3.3 is significantly less polar than the proven antibacterials. In addition and perhaps most importantly, 1 is the only compound in Table 1 that does not contain an ionizable group. These comparisons suggest that the potency of 1 could be improved by increasing its polarity and installing ionizable groups with pKa values in the physiological pH range. Further support for this approach was reported recently by the Hergenrother group, which showed that the addition of a primary amine to a molecule with a pKa in the physiological pH range can significantly enhance cellular accumulation.26,29
Herein, we report a structure-guided approach that was used to modulate the physicochemical properties of 1 to favor membrane penetration and efflux avoidance while simultaneously maintaining or improving BC target affinity, resulting in a significant overall improvement in antibacterial potency.
CHEMISTRY
Design Strategy.
The previously determined structure of 1 bound to E. coli BC suggested two vectors allowing for physicochemical optimization while also permitting the retention, or even improvement of, BC target inhibition. The first vector points from the amine at the 7-position of the pyridopyrimidine ring toward two conserved glutamic acid residues that coordinate Mg2+ when substrate ATP is bound (Figure 1A). We hypothesized that this vector would provide an opportunity to place an ionizable amine group that would reduce the cLogD value of the compound while also engaging the glutamic acid residues via H-bonding and/or electrostatic interactions. The second vector points off the dihalo-aromatic ring through a small slot in the active site surface toward solvent (Figure 1B). We begin by focusing our attention on the first vector at the 7-position of the aromatic ring system and subsequently return to the second vector for further optimization. In the final phase of the campaign compounds consisting of derivatizations at both vectors are presented.
Figure 1.

Compound 1 (carbon, green; nitrogen, blue; bromine, dark red; all rendered as sticks) co-crystallized with E. coli BC at 2.1 Å as reported in Ref. 5 (PDB code 2V58). (A) View of compound 1 in the BC active site showing the 7-position vector that points toward two proximal glutamate residues as illustrated by the two arrows. (B) View of the solvent accessible surface representation, in transparent grey, of E. coli BC co-crystallized with 1. The second vector points off the dihalo-aromatic ring of 1 through a small slot in the active site surface toward solvent. Images prepared using PyMOL.
Synthesis.
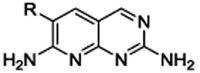
Simple 2,7-diaminopyridopyrimidines such as 1 and 4a-e are readily synthesized in one step from aryl acetonitriles as shown in Scheme 1.
Scheme 1.

Synthesis of 6-aryl pyridopyrimidines
Reagents and conditions: (a) Base, solvent see Experimental Section.
To install an amine at the 7-position, we developed a novel synthetic route that allowed for differentiation of the 2- and 7- positions as illustrated in Scheme 2. The route begins with a condensation between aryl acetonitrile 2a and pyrimidine 5, wherein the eventual amine at the 2- position is masked as a thiomethyl group. Following condensation, the 7-position is converted to pyridone 7 by nitration and hydrolysis. Chlorination with POCl3 provided 8 with a handle for installation of various amines by simple SnAr displacement. The sulfide was converted to an amine by oxidation to the sulfoxide and displacement with ammonia to provide analogs 12a-b. If required, final Boc deprotection was affected with TFA, to afford inhibitors 13c-h. This route could also be modified to allow for late-stage diversification via Suzuki reaction, as illustrated in Scheme 3, for the synthesis of compounds 14a–e.
Scheme 2.

Synthesis of Compounds 12a, b, and 13c-h
aReagents and conditions: (a) NaH, DMF 45%; (b) NaNO2, trifluoroacetic acid, 90%; (c) POCl3, DMF (cat), 95 °C, 98%; (d) 9a-h, MeCN, 1,4-dioxane, 80 °C; (e) mCPBA or Davies oxazirdine, DCM; (f) NH3, MeOH, THF, 65 °C; (g) Trifluoroacetic acid, DCM.
Scheme 3.

Synthesis of Compounds 14a–d
Reagents and conditions: (a) NaH, DMF; (b) NaNO2, trifluoroacetic acid; (c) POCl3, DMF (cat), 95 °C; (d) 9h, MeCN, 1,4-dioxane, 80 °C; (e) mCPBA or Davies oxazirdine, DCM; (f) NH3, MeOH, THF, 65 °C; (g) Trifluoroacetic acid, DCM; (h) R(BOH)2, Pd(dppf)Cl2, Na2CO3, dioxane, water, 80 °C.
RESULTS AND DISCUSSION
Structure-activity relationships for derivatives off the pyridopyrimidine 7-position.
Compound 4a was chosen as the parent molecule for this first campaign because it is identical to 1 except for the substitution of the dibromo ring with a dichloro ring, which results in a slightly lower cLogD. In addition, the IC50 value measured for 4a is within 2-fold of the IC50 value measured here for 1, indicating that the ring substitution does not have a large impact on BC target-inhibition. As expected based on the potency reported for 1, compound 4a has limited microbiological activity against wild type (WT) ESKAPE pathogens A. baumannii, E. coli, and K. pneumoniae, and no activity against P. aeruginosa was detected (MIC >256μg/mL) (Table 2).
Table 2.
MIC values, IC50 values, and physicochemical properties of interest for compounds elaborated at the 7-position
 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| -R |  |
 |
 |
||||||||
| Organisma | 4a | 12a | 12b | 13c | 13d | 13e | 13f | 13g | 13h | ||
| MIC (mg L−1) | A. baumannii | 256 | >256 | >64 | >256 | >256 | >256 | >256 | >128 | 128 | |
| E. coli | 32 | 64 | 32 | 16 | 32 | 64 | 16 | 32 | 2 | ||
| K. pneumoniae | 64 | 256 | >64 | 256 | 256 | >256 | 64 | 128 | 8 | ||
| P. aeruginosa | >256 | >256 | >64 | >256 | >256 | >256 | >256 | >128 | >256 | ||
| E. coli +PMBN | 1 | 4 | 2 | 8 | 4 | 8 | 2 | 4 | 0.25 | ||
| E. coli (ΔtolC)b | 0.5 | 0.5 | 0.25 | 1 | 2 | 1 | 0.5 | 1 | 0.12 | ||
| E. coli (ΔtolC) +PMBN | 0.25 | 0.12 | 0.12 | 0.25 | 1 | 0.25 | 0.12 | 0.5 | 0.06 | ||
| P. aeruginosa (ΔmexAB, etc.)c | 16 | 256 | >64 | 256 | 256 | >64 | 64 | 64 | 8 | ||
| fold- change | E. coli +/−PMBN | 32 | 16 | 16 | 2 | 8 | 8 | 8 | 8 | 8 | |
| E. coli +/−efflux | 64 | 128 | 128 | 16 | 16 | 64 | 32 | 32 | 16 | ||
| E. coli (ΔtolC) +/−PMBN | 2 | 4 | 2 | 4 | 2 | 4 | 4 | 2 | 2 | ||
| P. aeruginosa +/−efflux | >16 | >1 | - | >1 | >1 | - | >4 | >2 | >32 | ||
| IC50 P. aeruginosa BC (nM) | 320 | 170 | 140 | 190 | 440 | 83 | 64 | 320 | 33 | ||
| MW (g/mol) | 307 | 403 | 390 | 405 | 407 | 405 | 389 | 403 | 387 | ||
| pKad | 4.8 | (3.2) | 5.0 | 7.6 | 7.9 | 8.7 | 9.6 | 10.1 | 8.0 | ||
| clogD (pH 7.4) | 2.8 | 2.9 | 3.5 | 2.3 | 1.6 | 0.6 | −0.1 | 0.8 | 2.7 | ||
The strains of each bacterial species were as follows: A. baumannii (wild type) ATCC 19606; E. coli (wild type) DM4100; K. pneumoniae (wild type) ATCC 43816; P. aeruginosa (wild type) PAO1.
The E. coli ΔtolC strain contains a targeted knockout of the efflux pump component TolC.
The P. aeruginosa ΔmexAB, etc. strain contains targeted knockouts of efflux pumps MexAB-OprM, MexCD-OprJ, and MexEF-OprN, and the efflux pump components MexXY are expected to be compromised by the absence of OprM.
Calculated pKa values are in parentheses.
To test the ability of 4a to penetrate the Gram-negative outer membrane, we conducted MIC assays in the presence and absence of the permeabilizing agent polymyxin B nonpeptide (PMBN). At the concentration used (8 μg/mL), PMBN does not have an antibacterial effect but is expected to render the outer membrane more permeable by interacting with and disrupting the negatively charged lipopolysaccharide (LPS) molecules that otherwise maintain the outer membrane order.30 When PMBN is included, the MIC value of compound 4a against E. coli is reduced 32-fold from 32 to 1 μg/mL. This significant enhancement of potency in the presence of PMBN suggests that when the outer membrane is compromised, a greater proportion of 4a is able to reach the BC active site in the cytoplasm. By contrast, ACHN-975 (Table 1), which also targets a cytoplasmic enzyme but has physicochemical properties that may be more conducive to membrane penetration, is only 2- to 4-fold more potent in the presence of PMBN (data not shown) suggesting that a greater proportion of ACHN-975 readily crosses the outer membrane.
To test the contribution of efflux to the limited potency of 4a, an E. coli (ΔtolC) mutant was tested in the MIC assay. With this key efflux component deleted, the potency of 4a is strengthened by 64-fold (32 to 0.5 μg/mL), suggesting that WT E. coli uses a TolC-containing efflux pump to dramatically reduce the intracellular concentration of 4a. Similar results were observed when comparing the MIC values of 4a against WT and efflux-compromised strains of P. aeruginosa (Table 2).
When PMBN was included with compound 4a the MIC for E. coli ΔtolC strain is 0.25 μg/mL, the lowest value observed for all of the strains and conditions tested. Because the efflux-compromised and WT strains are expected to have the same levels of LPS in their outer membranes, we initially reasoned that PMBN should reduce the MIC by the same proportion for both strains. Instead, the presence of PMBN induced only a 2-fold reduction in MIC against the efflux-compromised strain (0.5 to 0.25 μg/mL), compared to a 32-fold reduction against the WT strain (32 to 1 μg/mL). This disconnect suggests that when the outer membrane and efflux mechanisms are both compromised, other properties of the bacterial cell and inhibitor, such as the BC enzyme affinity and/or the ability of the inner membrane to exclude the inhibitor, become limiting to the observed MIC potency.
Structural elaboration of 4a at the 7-position of the pyridopyrimidine ring yielded compounds 12a-b and 13c-h shown in Table 2. To test our structural expectation that groups at this position could be readily accommodated by the BC enzyme, we measured the IC50 values against purified P. aeruginosa BC. The results in Table 2 show that the IC50 values measured are all similar or better compared to 4a, demonstrating that the 7-position modification does not disrupt BC inhibition and in one case (13h) improves inhibition by nearly 10-fold.
In addition to having similar BC affinity compared to 4a, the compounds 12a and 12b, also have similar cLogD and pKa values. Thus, it was expected that these compounds would perform similarly to 4a in the MIC panel. The MIC data for 12a and 12b in Table 2 reflect these expectations, with similarly low potency against WT strains and similarly strong MIC shifts observed in the presence of PMBN and against the efflux mutants. These observations are consistent with the premise that compounds with high cLogD values and no ionizable groups at physiological pH values have difficulty entering cells and avoiding efflux.
To test the physicochemical hypothesis further, compound 12b was modified with the addition of a nitrogen atom with a pKa value near physiological pH, resulting in secondary amine-containing compound 13c. In addition to becoming ionizable, 13c also has a modest decrease in cLogD. Importantly, the BC affinity of 13c remains relatively constant compared to the parent compound so that any changes in potency can more likely be attributed to changes in the two important physicochemical properties. Table 2 shows that 13c does not have a large increase in antibacterial potency against WT strains. However, the presence of PMBN improves the potency of 13c by only 2-fold, suggesting that it crosses the outer membrane more easily than compounds 4a and 12a-b, which have MIC shifts of 16- to 32-fold in the presence of PMBN. Likewise, the 16-fold shift observed with 13c against the E. coli efflux-compromised strain compared to WT strain is much lower than the 64- to 128-fold shifts seen with compounds 4a and 12a-b. The lower fold-change suggests that compound 13c also avoids efflux more readily than the neutral compounds with higher cLogD values. Despite these apparent cellular penetration improvements and maintenance of BC target affinity, the lack of corresponding improvement against WT strains suggests that 13c has lost potency due to some other difference(s) compared to 4a and 12a-b. While currently untested, one hypothesis is that 13c has a reduced ability to cross the Gram-negative inner membrane relative to 4a and 12a-b. The direct measurement of the accumulation of compound inside the cell could help test this hypothesis.
Further derivatives of 13c were designed to increase the pKa and lower the cLogD, again with the expectation that such changes would improve outer membrane penetration and efflux avoidance. Contrary to this expectation, these derivatives (13d-g in Table 2) all have higher MIC shifts in the presence of PMBN compared to 13c (8-fold versus 2-fold, respectively). In addition, all had equal or larger MIC shifts against the efflux compromised strain compared to 13c. These observations suggest that an unknown counteracting property of these derivatives reduce their ability to penetrate the outer membrane and avoid efflux relative to 13c. One possible explanation is that the basicity of the amines of 13d-g is too high since they all have pKa values greater than 13c. If true, this would suggest there is a narrow pKa range for this scaffold to achieve optimal cellular penetration. When the majority of the compound population is positively charged at physiological pH, as is the case when the pKa is >8, the membrane penetration and efflux avoidance properties of the compounds may get worse despite relatively low cLogD values. While this model also remains untested, we focused our further medicinal chemistry efforts in this series on making compounds with amine pKa values less than 8.
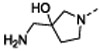
After further rounds of elaboration at the 7-position, the most potent compound against the WT strains identified 13h, which contained an amino azabicyclohexane group with a pKa value of 8. This compound had similar physicochemical properties as compounds 13d-g and yielded similar MIC shifts in the presence of PMBN and against the efflux compromised strain. However, 13h achieved the lowest MIC values against the WT strains of interest (except against P. aeruginosa, which remained above the measurable range), presumably due to its approximately 10-fold improvement in BC target affinity compared to the parent 4a. The resiliency of the WT P. aeruginosa strain was not unexpected as this species is known to have a less permeable membrane and a more extensive efflux system compared to the other ESKAPE pathogens tested here.31-35
Structure-activity relationships of slot position derivatives.
In an effort to further improve the potency of parent compound 4a, we turned our attention to the second vector of interest pointing off the dihalo-aromatic ring (Figure 1B). Structural elaboration at this position occurred in two phases. The first phase involved the addition of various aryl groups off of the dibromo aromatic ring that were designed to fit within the small surface slot of the BC active site (Figure 1B). These modifications did not contain groups capable of ionizing near physiological pH nor did they result in large reductions to the cLogD so it was not expected that these compounds would have enhanced membrane penetration or efflux avoidance.
These new compounds were tested against our MIC panel to assess cellular entry properties and overall potency (Table 3). In accordance with expectations, this set of compounds had large MIC shifts when PMBN was present, similar to parent compounds 1 and 4a. These compounds were also much more potent against the E. coli efflux-compromised strain relative to the WT strain, with MIC shifts ranging from 64- to >512-fold. These observations support the prediction that this set of compounds would retain relatively poor outer membrane permeability and high efflux potential because of unoptimized physicochemical properties. However, we note that compound 4c has a cLogD value a full unit lower than parent 1, yet the MIC fold-shift for this compound suggests no improvement in either outer membrane penetration or efflux avoidance. While this reduction in cLogD value may not be large enough to yield predictable penetration and efflux avoidance improvements, it may also be that introduction of polarity at this particular position on the molecule is ineffective for reasons not captured by the simple expectation based solely on bulk physicochemical properties. Given that 4c has the same cLogD as 13c, which did not show large MIC shifts in the presence of PMBN or when efflux was compromised, it may be that the presence of an ionizable amine, as on 13c and not 4c, is the more important property in determining cellular accumulation compared to the cLogD parameter.
Table 3.
MIC values, IC50 values, and physicochemical properties of interest for compounds elaborated at the slot-position
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| -R | 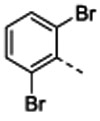 |
 |
 |
 |
 |
 |
||
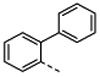
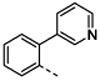
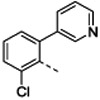
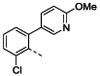
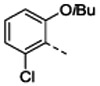
| Organisma | 1 | 4b | 4c | 4d | 4e | 4f | ||
| MIC (mg L−1) | A. baumannii | 128 | >256 | >256 | >256 | >256 | >128 | |
| E. coli | 32 | >256 | 256 | 8 | 8 | 32 | ||
| K. pneumoniae | 64 | >256 | 256 | 32 | >16 | >128 | ||
| P. aeruginosa | >256 | >256 | >256 | >256 | >256 | >128 | ||
| E. coli +PMBN | 0.5 | 2 | 4 | 0.5 | 0.25 | 1 | ||
| E. coli (ΔtolC)b | 0.5 | 0.5 | 2 | 0.12 | 0.06 | 0.25 | ||
| E. coli (ΔtolC) +PMBN | 0.12 | 0.25 | 0.5 | 0.06 | 0.03 | ≤0.06 | ||
| P. aeruginosa (ΔmexAB, etc.)c | 16 | >256 | 16 | 4 | 8 | 32 | ||
| fold- change | E. coli +/−PMBN | 64 | >128 | 64 | 16 | 32 | 32 | |
| E. coli +/−efflux | 64 | >512 | 128 | 64 | 128 | 128 | ||
| E. coli (ΔtolC) +/−PMBN | 4 | 2 | 4 | 2 | 2 | >4 | ||
| P. aeruginosa +/−efflux | >16 | - | >16 | >64 | >32 | >4 | ||
| IC50 P. aeruginosa BC (nM) | 180 | 79 | 130 | 36 | 32 | 93 | ||
| MW (g/mol) | 395 | 313 | 314 | 349 | 379 | 344 | ||
| pKad | 4.8 | (4.7) | (4.9) | (4.4) | (3.8) | (3.9) | ||
| clogD (pH 7.4) | 3.3 | 3.2 | 2.3 | 2.9 | 2.6 | 2.9 | ||
The strains of each bacterial species were as follows: A. baumannii (wild type) ATCC 19606; E. coli (wild type) DM4100; K. pneumoniae (wild type) ATCC 43816; P. aeruginosa (wild type) PAO1.
The E. coli ΔtolC strain contains a targeted knockout of the efflux pump component TolC.
The P. aeruginosa ΔmexAB, etc. strain contains targeted knockouts of efflux pumps MexAB-OprM, MexCD-OprJ, and MexEF-OprN, and the efflux pump components MexXY are expected to be compromised by the absence of OprM.
Calculated pKa values are in parentheses.
To assess the effect of the structural changes off the dibromo aromatic ring on target BC inhibition, IC50 values were measured using purified P. aeruginosa BC. As these derivatives were designed to sterically fit into a small slot extending from the BC active site but not make new specific interactions, it was expected that the IC50 values of the elaborated compounds would be quite similar to parent compound 1. As expected, the IC50 values reported in Table 3 are approximately equal to or lower by up to 6-fold of 1, indicating that the introduction of additional ring systems at this position do not cause major disruptions to target inhibition. Wary of the potential for the additional aromatic ring to reduce solubility, it was discovered that certain aliphatic groups, such as isobutyl 4f, could also fit the slot position and afford activity against the enzyme.
For the final phase of the structural elaboration off the dihalo-aromatic ring we chose a starting scaffold with the best amine-containing group identified during the derivatization campaign at the 7-position of the pyridopyrimidine ring, 13h. The first two related compounds in the series, 14a and 14b (Table 4), maintained the physicochemical properties of the parent compound 13h and showed an improvement in BC inhibition, with IC50 values recorded at the lower limit of detection of ≤15 nM – an improvement of ≥2-fold relative to the parent 13h. This BC target inhibition improvement could be responsible for the small MIC improvements observed against the WT strains (with the exception of A. baumannii). No substantially different MIC shifts were observed in the presence of PMBN or when efflux was compromised, as expected given that 14a and 14b have pKa and cLogD values similar to the parent comparator 13h and all compounds contain an ionizable amine.
Table 4.
MIC values, IC50 values, and physicochemical properties of interest for compounds elaborated in the final phase of the medicinal chemistry campaign
 |
|||||||
|---|---|---|---|---|---|---|---|
| -R | 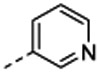 |
 |
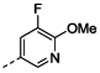 |
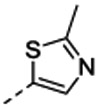 |
|||
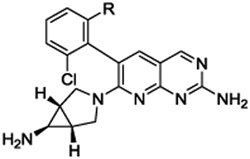
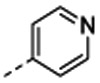
| Organisma | 14a | 14b | 14c | 14d | 14e | ||
| MIC (mg L−1) | A. baumannii | >256 | 128 | 32 | 128 | 32 | |
| E. coli | 1 | 1 | 1 | 0.5 | 2 | ||
| K. pneumoniae | 8 | 4 | 4 | 2 | 8 | ||
| P. aeruginosa | 256 | 128 | 128 | 128 | >64 | ||
| E. coli +PMBN | 0.12 | 0.12 | 0.06 | 0.03 | 0.06 | ||
| E. coli (ΔtolC)b | 0.03 | 0.03 | 0.015 | 0.008 | 0.03 | ||
| E. coli (ΔtolC) +PMBN | 0.008 | 0.015 | 0.008 | 0.004 | 0.015 | ||
| P. aeruginosa (ΔmexAB, etc.)c | 1 | 2 | 2 | 1 | 8 | ||
| fold- change |
E. coli +/−PMBN | 8 | 8 | 16 | 16 | 32 | |
| E. coli +/−efflux | 32 | 32 | 64 | 64 | 64 | ||
| P. aeruginosa +/−efflux | 256 | 64 | 64 | 128 | >8 | ||
| IC50 P. aeruginosa BC (nM) | ≤15 | ≤15 | ≤15 | ≤15 | ≤15 | ||
| MW (g/mol) | 430 | 430 | 478 | 450 | 425 | ||
| pKad | 8.4 | (8.1) | (8.1) | (8.1) | 8.1 | ||
| cLogD (pH 7.4) | 2.5 | 2.5 | 2.4 | 2.6 | 2.8 | ||
The strains of each bacterial species were as follows: A. baumannii (wild type) ATCC 19606; E. coli (wild type) DM4100; K. pneumoniae (wild type) ATCC 43816; P. aeruginosa (wild type) PAO1; S. aureus (wild type) ATCC 29213.
The E. coli ΔtolC strain contains a targeted knockout of the efflux pump component TolC.
The P. aeruginosa ΔmexAB, etc. strain contains targeted knockouts of efflux pumps MexAB-OprM, MexCD-OprJ, and MexEF-OprN, and the efflux pump components MexXY are expected to be compromised by the absence of OprM.
Calculated pKa values are in parentheses.
The next compounds in the series, 14c-14e, all maintained BC IC50 values below 15 nM and achieved the most potent microbiological activity observed in the series. As these compounds also maintained the general physicochemical properties of the starting scaffold, the MIC shifts observed in the presence of PMBN or against the efflux compromised strain were similar to the shifts observed with the parent compound.
Structural Biology.
To assess the structural consequences of the 7-position modifications, the X-ray co-crystal structure of 13f with the WT BC enzyme from E. coli was solved at 2.06 Å (see Table S1 for crystallographic statistics). Figure 2A shows that the pyridopyrimidine ring scaffold of 13f occupies the same position as the previously reported structure of 1.5 Thus, neither the dibromo- to dichloro-substitution nor the modification off the 7-position altered the scaffold binding position in the BC active site pocket.
Figure 2.

BC co-crystal structures of new pyridopyrimidine inhibitors overlaid with previously identified parent compound 1 co-crystallized with E. coli BC (shown in green; PDB 2V58). (A) Compound 13f (carbon, magenta; nitrogen, blue; chlorine, light green; all rendered as sticks) co-crystallized with E. coli BC at 2.06 Å (magenta ribbon cartoon; PDB code 6OI9). The core pyridopyrimidine scaffold of 13f is found in the identical position as that of parent compound 1. The 7-position moiety extending off 13f points toward the conserved glutamate residues placing its proximal amine group nitrogen atom 3.16 and 2.17 Å in mol A and 2.97 and 2.65 Å in mol B away from the nearest oxygen atoms of the carboxylate groups of Glu288 and Glu276, respectively. (B) Compound 14a (carbon, light blue; nitrogen, blue; chlorine, light green; all rendered as sticks) co-crystallized with the H. influenzae BC at 2.52 Å (light blue-ribbon cartoon; PDB code 6OI8). The T-loop (Ref. 36) in the H. influenzae crystal structure is presumably disordered, as observed previously in other BC crystal structures (Ref. 37) and not represented by the ribbon cartoon. The core pyridopyrimidine scaffold of 14a is found in the identical position as that of parent compound 1. The 7-position moiety extending off 14a points toward the conserved glutamate residues placing its proximal amine group nitrogen atom 3.29 and 2.91 Å away from the nearest oxygen atoms of the carboxylate groups of Glu288 and Glu276, respectively. (C) The vector of 14a that extends off the dihalo-aromatic ring is positioned, as expected, within the small slot in the active site surface as shown by the partially transparent solvent accessible surface representation in grey. Images prepared using PyMOL.
Consistent with our hypothesis based on the structure of 1, the 7-position modification of 13f extends further into the interior of the BC enzyme placing the distal amine within H-bonding distance to Glu288 and Glu276. Consistent with this possibility, the IC50 results show a 5-fold improvement in the inhibition of 13f compared to 4a, possibly reflecting the binding contribution of the new H-bond interactions to the conserved glutamate residues that are formed by 13f; however, mutation studies involving both glutamate residues would be needed to more definitively assign the binding affinity consequences of the 7-position modification.
To investigate the structural consequences of modifications off the dihalo ring, the crystal structure of 14a in complex with WT BC enzyme from H. influenzae was solved at 2.52 Å (Figure 2B and C; see Table S1 for crystallographic statistics). The active site of the H. influenzae BC enzyme is nearly identical to the other Gram-negative species of interest.5,18 The structure shows that the pyridopyrimidine ring scaffold of 14a occupies the same position as 13f and the previously reported structure of 1 demonstrating that the pyridyl modification off the dihalo ring does not alter the binding conformation of the core ring system. This modification is positioned, as intended, within the slot region of the BC active site, potentially making packing interactions to the residues in the walls of the slot (Figure 2C). The structure also shows that the 7-position moiety of 14a extends toward the conserved Glu288 and Glu276 residues positioning the primary amine of 14a within H-bonding distance to the two acid residues (Figure 2B). Thus, both modifications found on 14a had the intended structural consequences.
Bacterial Cytological Profiling.
The optimization of antibacterial compounds may be compromised by off-target activity that would complicate interpretation of structure-activity relationships.38 To test that the improved antibacterial potency of the novel BC inhibitors was due to on-target activity, Bacterial Cytological Profiling was used. Briefly, this technique uses high resolution fluorescence microscopy to compare the effects on cytological structure of novel antibiotics to a set of reference control antibiotics with known targets.39,40 Compounds that inhibit different pathways generate reproducible and distinct cytological signatures that can be used to infer the mechanism of action of unknown compounds.39-44 When E. coli cells were treated with the previously reported BC inhibitor 14 at ~3-fold the MIC for 2 hours they formed short chains of 3 to 8 cells, a phenotype that is characteristic of lipid biogenesis inhibitors (Figure 3). Two novel inhibitors, 13f and 14e, were tested in the profiling assay. It was found that both showed cytological profiles very similar to each other and to parent compound 1 suggesting that they inhibit the same target in vivo.
Figure 3. Cells treated with BC inhibitors produce chains of cells.

Cells were treated with either DMSO or compound at the indicated concentrations for 2 hours. The top panel shows FM4-64 stained cell membranes (red) and the bottom panel shows both cell membranes and DNA stained with DAPI (blue). Scale bar equals 1 micron.
Selectivity characterization.
Compound 1 is a member of a class of compounds that were originally identified as inhibitors of VEGFR2 and FGFR1 eukaryotic protein kinases.45 However, in the original report,5 1 was shown to have acceptable selectivity for bacterial BC over these and 28 other eukaryotic protein kinases. To test if the potency improvements of the derivative compounds synthesized in the current work came at the expense of losses in selectivity, two particularly promising compounds, 14a and 14e, were subjected to a similar panel of kinases. Compounds were profiled at a single concentration of 25 μM in the Z’-LYTE kinase activity assay at Invitrogen. The results are shown in Table 5. Compared to parent compound 1, the novel compounds retain excellent and even improved selectivity against this panel of kinases.
Table 5.
Percent inhibition of eukaryotic kinases at a compound concentration of 25 μM
| compound | |||
|---|---|---|---|
| kinase | 1 | 14a | 14e |
| ABL1 | 97 | 38 | 30 |
| FYN | 93 | 12 | 12 |
| SRC | 93 | 8 | 8 |
| BTK | 92 | 16 | 32 |
| FGFR1 | 78 | 57 | 60 |
| EGFR (ErbB1) | 58 | 65 | 6 |
| MAP4K4 (HGK) | 52 | 21 | 24 |
| NTRK1 (TRKA) | 47 | 47 | 40 |
| MAPK1 (ERK2) | 26 | 1 | 6 |
| MAPK14 (p38 alpha) Direct | 24 | 1 | 5 |
| MET (cMet) | 21 | 7 | 15 |
| AURKA (Aurora A) | 20 | 9 | 8 |
| PAK4 | 20 | 25 | 23 |
| CDK1/cyclin B | 18 | 4 | 4 |
| CSNK1D (CK1 delta) | 16 | 3 | 3 |
| CHEK2 (CHK2) | 13 | 20 | 67 |
| MAPKAPK2 | 10 | 26 | 21 |
| SGK (SGK1) | 7 | 32 | 32 |
| INSR | 6 | 3 | 3 |
| GSK3B (GSK3 beta) | 6 | 4 | 7 |
| IKBKB (IKK beta) | 5 | 5 | 8 |
| AKT1 (PKB alpha) | 4 | 6 | 7 |
| CSNK2A1 (CK2 alpha 1) | 4 | 0 | 1 |
| PDK1 Direct | 3 | 0 | 0 |
| PRKCZ (PKC zeta) | 0 | 5 | −8 |
| CHEK1 (CHK1) | −1 | 28 | 42 |
| IKBKE (IKK epsilon) | −7 | 3 | 0 |
Determination of resistance liabilities.
One concern with inhibitors targeting a single enzyme is that single point mutations in the target enzyme may lead to high-level resistance. Due to the high bacterial burden and error-prone replication, it is feasible for bacteria to sample every possible point mutation during the course of an infection. If a single-point mutation can disrupt inhibitor binding without drastically impairing enzyme function then the infection will become resistant [for example see Ref. 46]. Efflux-mediated resistance is more common than target-based resistance, but upregulation of a single efflux system usually has a moderate impact on potency.
To determine whether high-level resistance to our inhibitors could arise in P. aeruginosa, we selected for spontaneous resistant mutants in the efflux-compromised P. aeruginosa strain. The lack of genes encoding the three major antibiotic efflux systems increases the likelihood of identifying rare mutations in other genes. However, it should be noted that P. aeruginosa encodes additional efflux systems that can contribute to resistance.
We selected for mutants on agar plates containing 4-, 8-, or 16-fold the MIC of compounds 1, 14a, or 14e. The spontaneous mutation frequency of these compounds in this strain background was similar, ranging from 2×10−9 for 14e to 9×10−9 for 14a at 8-fold above the MIC. These frequencies are low and similar to the frequencies observed for compound 1 previously.5 We note however that spontaneous mutation frequencies would likely be much higher for a WT P. aeruginosa strain carrying the full suite of efflux systems as these additional components can also mutate to more efficiently efflux lethal compounds.
Several resistant colonies of the efflux-compromised P. aeruginosa strain were found to harbor mutations in the BC coding sequence. These mutations encoded changes to amino acids Leu278 and Ile437, which are located in the BC active site according to a structure of the P. aeruginosa enzyme in complex with a substrate analog (PDB: 2VQD).18 Both residues are highly conserved across BC species including E. coli and H. influenzae.5,18 The I437N and I437T mutations identified here have also been observed previously when efflux-compromised strains of E. coli and H. influenzae were grown in the presence of 1.5 The L278F mutation observed here in P. aeruginosa was not identified in either the E. coli or H. influenzae enzymes.5 However, several other active site mutations were observed in E. coli and H. influenzae that were not observed in the current study, possibly because not enough resistant colonies were screened to exhaustively identify all possible mutants. If this were the case, additional screening would eventually reveal the same set of resistance conferring mutants for all BC species. Alternatively, the different sets of active site mutations observed in the BC enzymes from different species may reflect the influence of subtle BC structural differences that result in truly different sets of possible resistance-conferring mutations.
All three P. aeruginosa BC active site mutations isolated in this study have markedly stronger effects on the potency of 14a compared to 14e (Table 6). The L278F mutation raised the MIC of 14a by 256-fold above the MIC of the parent strain, while both I437 mutations raised the MIC 128-fold. In contrast, 14e was less affected by all three mutations, showing 8-fold MIC shifts above the parent strain.
Table 6.
MIC changes conferred by spontaneous P. aeruginosa mutations
| MIC (mg L−1) | fold-change from WT | |||||
|---|---|---|---|---|---|---|
| Strain | 1 | 14a | 14e | 1 | 14a | 14e |
| WT backgrounda | 16 | 1 | 8 | (1) | (1) | (1) |
| BC L278F | 128 | 256 | 64 | 8 | 256 | 8 |
| BC I437N | >128 | 128 | 64 | >8 | 128 | 8 |
| BC I437T | >128 | 128 | 64 | >8 | 128 | 8 |
| MexT F201I | >128 | 2 | 8 | >8 | 2 | 1 |
| MexT P202L | >128 | 2 | 8 | >8 | 2 | 1 |
| MexT V226L | >128 | 1 | 8 | >8 | 1 | 1 |
| INV(muxA-mexZ) | >128 | 32 | 32 | >8 | 32 | 4 |
P. aeruginosa ΔmexAB-oprM, ΔmexCD-oprJ, ΔmexEF-oprN [PAM1626 from Ref. 47]
Examination of the 14a H. influenzae BC X-ray co-crystal structure overlaid with the previously reported P. aeruginosa BC with substrate analog co-crystal structure18 offers a speculative model for how the resistance-conferring mutations might disrupt inhibitor binding but not significantly impact substrate turnover (Figure 4). In the H. influenzae structure, the pyridopyrimidine ring moiety of the 14a ligand extends further toward Leu278 than the corresponding adenine group of the substrate analog. Because of this difference, mutation to the larger phenylalanine amino acid could sterically hinder 14a binding while leaving substrate and transition state binding unaffected. By contrast, the structural overlap of the inhibitor and substrate near the 437 position appears quite extensive in the structural alignment shown in Figure 4. As the resistance-conferring mutations at this position introduce more polarity, it may be that electrostatic forces play a large role in affording the differential effect on substrate/transition state versus inhibitor binding. Further catalytic and inhibition studies as well as additional X-ray crystal structures would be needed to understand the precise consequences of the resistance-conferring mutations. These future studies may also reveal why 14e MIC potency is not as largely affected by the resistance-conferring mutations as the other inhibitors.
Figure 4.

Alignment of the P. aeruginosa BC enzyme (shown in yellow ribbon; PBD 2VQD) co-crystallized with substrate analog AMPCP (carbon, yellow; nitrogen, blue; phosphate, orange; oxygen, red; all rendered as sticks) and the H. influenzae BC enzyme (shown in light blue ribbon) co-crystallized with 14a (carbon, light blue; nitrogen, blue; chlorine, light green; all rendered as sticks; PDB code 6OI8). Resistance-conferring mutants were found at residue positions 278 and 437, which in the WT enzymes shown here are leucine and isoleucine, respectively (shown as sticks).
Additional resistant mutants were subjected to whole-genome sequencing to identify the underlying resistance mechanism. Several mutants were found to have mutations conferring single amino acid changes in MexT, which is a transcriptional regulator affecting many cellular processes.48-50 The effect of mexT mutation was highly compound-specific. Many BC inhibitors, including 14a and 14e, were unaffected by any mexT mutation (Table 6). However, the potency of 1 was reduced >8-fold by any of the three individual mexT mutations. The mexT mutants also had elevated resistance to ciprofloxacin and levofloxacin (data not shown), suggesting a general efflux-mediated mechanism is likely responsible for the observed phenotypes.
Sequencing also uncovered a large (647 kb) chromosomal inversion disrupting the coding sequences of two efflux-related genes: mexZ, a repressor of mexXY, and muxA, which encodes the periplasmic membrane fusion component of the MuxABC-OpmB efflux system. The disruption presumably upregulates MexXY production while at the same time abrogates the function of MuxABC-OpmB, but these hypotheses were not tested experimentally. The muxA-mexZ inversion affected all tested BC inhibitors, reducing their potency 4- to 32-fold, and also reduced the potency of ciprofloxacin and levofloxacin (data not shown), supporting the premise that resistance is efflux-mediated. The lack of mutations in other pathways suggests that efflux and BC coding mutations are the major resistance mechanisms for this set of compounds.
CONCLUSIONS
In this report, the physicochemical properties of starting compound 1 were rationally optimized to improve Gram-negative cellular accumulation and BC target inhibition resulting in the overall enhancement of antibacterial activity. Reducing the cLogD value and installing a primary amine with a pKa value in the physiological pH range resulted in compounds such as 14a and 14e, which improved potency up to 64-fold against WT E. coli, 32-fold against WT K. pneumoniae, and 16-fold against an efflux-compromised strain of P. aeruginosa.
While these MIC improvements demonstrate the potential of BC inhibitors as antibacterial agents, several challenges remain. First, the understanding of the parameters affecting Gram-negative penetration and efflux is still incomplete. For example, 13h showed large MIC and cell penetration improvements compared to the parent compound, despite having an almost identical cLogD value. This observation lends further support to the conclusion drawn by more recent studies that suggest cLogD is not as predictive of gram-Negative cellular accumulation as was once proposed.26 In addition, 14a, one of the most potent compounds, still had rather large decreases in MIC in the presence of PMBN and against efflux-compromised strains, indicating that cell penetration and efflux are still significant barriers to the potency of this compound, despite having “optimal” properties. These results suggest that there are other, as of yet, undiscovered properties that likely contribute to penetration and efflux avoidance. Recent work has begun to provide evidence that these other properties could likely include a molecule’s globularity and number of rotatable bonds.26,29 In addition, the optimization strategy used here was largely ineffective at improving potency against WT A. baumannii and P. aeruginosa. The unique membrane composition and efflux mechanisms of these species may require different or additional molecular properties to be satisfied before adequate antibacterial potencies can be achieved.
A second challenge illustrated by this work is the inherent resistance liability associated with targets encoded by a single-copy gene like BC. All clinically relevant antibiotics overcome this challenge by inhibiting targets encoded by multiple gene copies such as the ribosome (aminoglycosides, macrolides, etc.) or multiple related targets such as penicillin binding proteins (beta-lactams) and topoisomerases (quinolones) or by binding to non-protein targets (vancomycin, polymyxins, etc.). The large MIC shifts observed herein from BC active-site mutations illustrate the ease by which single amino acid changes can disrupt inhibitor binding without compromising catalytic activity enough to jeopardize cell survival. In principle, if an inhibitor mimicked the molecules along the catalytic pathway extensively enough, such single point mutations could not arise. Designing such inhibitors has proved challenging but compounds such as 14e with reduced MIC shifts in the presence of BC active site mutations represent a promising starting point for further optimization.
EXPERIMENTAL SECTION
Chemistry.
All chemicals were purchased from commercial suppliers and used as received. Flash chromatography was carried out with prepacked silica cartridges from either ISCO or SiliCycle on an ISCO Companion chromatography system using gradient elution. NMR spectra were recorded on an Oxford 400 MHz spectrometer and referenced to tetramethylsilane. Preparative HPLC was performed on a Luna C18 5 μm column (50 mm 21 mm), eluting with mixtures of water acetonitrile. All final compounds were purified to >95% chemical purity, as assayed by HPLC with UV detection at λ = 254 and 210 nm.
Analytical LCMS was conducted on an Agilent 1200 series UPLC coupled with an Agilent G1946D Mass spectrometer and an Agilent 1200 DAD UV detector. All methods utilized the following parameters: Capillary voltage: 2500V; Fragmentor/Gain: 100; Gain: 1.0; Drying gas flow: 12.5 L/min; Gas Temperature: 350 °C; Nebulizer Pressure: 60 psi; Scan Range: 100-1000 amu; Eluent A: 0.1% TFA in water; Eluent B: 0.1% TFA in acetonitrile; Flow: 0.8 mL/min; Column: Agilent SB-C18 RRHD 1.8 μm; 2.1 × 50 mm; Column temperature: 35 °C; LC retention times are based on the following methods:HPLC Method 1: Gradient: 2-50% eluent B over 2.0 minutes; HPLC Method 2: Gradient: 2-99% eluent B over 2.0 minutes; HPLC Method 3: Gradient: 2-30% eluent B over 2.0 minutes
2-[2-(3-pyridyl)phenyl]acetonitrile (2c): General procedure A: 2-(2-bromophenyl)acetonitrile (100 mg, 0.51 mmol), 3-pyridylboronic acid (130 mg, 1.0 mmol), potassium fluoride (60 mg, 1.0 mmol) were combined in 1,4-dioxane (3 mL) and methanol (0.90 mL) under nitrogen. Pd2(dba)3 (9.3 mg, 0.010 mmol) and dicyclohexyl-[2-(2,6-dimethoxyphenyl)phenyl]phosphane (SPhos) (4.2 mg, 0.010 mmol) were added and the reaction was heated at 95 °C overnight. After cooling to room temperature, the reaction was filtered and the filtrate was concentrated and used in the next step without further purification (97 mg, 98%). LCMS (ESI) [M+H]+ = 195.0.
2-[2-chloro-6-(3-pyridyl)phenyl]acetonitrile (2d) was prepared from 2-(2-bromo-6-chloro-phenyl)acetonitrile (410 mg, 1.8 mmol) using general procedure A. The crude material was purified by chromatography on silica gel, to yield the desired product (330 mg, 82%). LCMS (ESI) [M+H]+ = 229.0
2-[2-chloro-6-(6-methoxy-3-pyridyl)phenyl]acetonitrile (2e) was prepared using general procedure A. The crude material was purified by RP-HPLC to yield the desired product (86 mg, 51%). LCMS (ESI) [M+H]+ = 259.0
2-(2-chloro-6-isobutoxy-phenyl)acetonitrile (2f). 2-chloro-6-hydroxy-benzaldehyde (1.0 g, 6.4 mmol) and isobutyliodide (1.9 mL, 16 mmol) were combined in 8.0 ml anhydrous DMF, treated with potassium carbonate (1.3 g, 9.6 mmol) and heated at 90 °C in a sealed vessel for two days. The reaction was cooled to room temperature, diluted with water and extracted with ethyl acetate. The ethyl acetate extract was washed with aqueous sodium metabisulfite solution and then brine and concentrated under vacuum. The crude residue was purified on a silica gel column to give 2-chloro-6-isobutoxybenzaldehyde (930 mg, 69%). LCMS (ESI) [M+H]+ = 213.0.
A 200 ml round bottom flask fitted with addition funnel and nitrogen purged was charged with potassium tert-butoxide (20% in THF, 10 mL, 18 mmol) and cooled in a −45 °C bath. A solution of 1-Isocyanomethanesulfonyl-4-methyl-benzene (1.7 g, 8.8 mmol) in 12 ml anhydrous THF was added dropwise over 10 min. After an additional 10 min, a solution of 2-chloro-6-isobutoxy-benzaldehyde (930 mg, 4.4 mmol) in 12 ml THF was added dropwise over 10 min. After two hours the reaction was allowed to warm to room temperature and then the solvent was removed under vacuum. The residue was taken up in 30 ml water and acidified to pH 6 with 10% acetic acid. The product was extracted with ethyl acetate (3 × 30 ml) and the combined extracts were washed with saturated aqueous sodium bicarbonate and then brine, then dried over sodium sulfate, filtered and concentrated under vacuum. The crude residue was purified on a silica gel column to give the desired product (950 mg, 97%). LCMS (ESI) [M+H]+ = 223.9.
6-([1,1'-biphenyl]-2-yl)pyrido[2,3-d]pyrimidine-2,7-diamine (4b): A solution of 2-([1,1'-biphenyl]-2-yl)acetonitrile (34 mg, 0.17 mmol) in ethoxyethanol (0.5 mL) was treated with 25% sodium methoxide in methanol (31 μL, 0.14 mmol) followed by 2,4-diaminopyrimidine-5-carbaldehyde (20 mg, 0.14 mmol) and then heated at 125 °C for 30 min. The reaction was diluted with water and acidified with TFA. The solution was purified by RP-HPLC to give the desired product (30 mg, 49%). HRMS (ESI) calculated [M+H]+ 314.1405, found 314.1399; 1H NMR (DMSO-d6, 400 MHz): δ 8.78 (1H, s), 7.84 (1H, s), 7.57 (1H, t, J = 7.2 Hz), 7.52 (1H, t, J = 7.6 Hz), 7.46 (1H, d, J = 7.2 Hz), 7.39 (1H, d, J = 7.2 Hz), 7.28–7.21 (5H, m).
6-[2-(3-pyridyl)phenyl]pyrido[2,3-d]pyrimidine-2,7-diamine (4c): General procedure B: A solution of 2-[2-(3-pyridyl)phenyl]acetonitrile (97 mg, 0.50 mmol) in ethylene glycol (1.0 mL) was treated with sodium methoxide (25% in methanol) (0.11 mL, 0.50 mmol) and 2,4-diamino-pyrimidine-5-carbaldehyde (62 mg, 0.45 mmol). The reaction was heated at 140 °C for 2 h, then cooled to rt, acidified with TFA (0.08 mL, 1 mmol) and purified by RP-HPLC to give the desired product (65 mg, 30%). HRMS (ESI) calcd [M+H]+ 315.1358, found 315.1355; 1H NMR (DMSO-d6, 400 MHz): δ 8.94 (1H, s), 8.04 (2H, d, J = 8.0 Hz), 7.93 (1H, s), 7.85 (3H, br s), 7.63–7.54 (4H, m), 7.48 (1H, t, J = 6.8 Hz), 1.96 (2H, br s), 1.75 (2H, br s).
6-[2-chloro-6-(3-pyridyl)phenyl]pyrido[2,3-d]pyrimidine-2,7-diamine (4d). was prepared from 2-[2-chloro-6-(3-pyridyl)phenyl]acetonitrile (33 mg, 0.14 mmol) using general procedure B. The crude material was purified by RP-HPLC to yield the desired product (10 mg, 15%). HRMS (ESI) calcd [M+H]+ 349.0968, found 349.0959; 1H NMR (DMSO-d6, 400 MHz): δ 8.78 (1H, s), 8.43-8.41 (2H, m), 7.85 (1H, s), 7.72 (1H, dd, J = 8.0, 0.4 Hz), 7.65–7.60 (2H, m), 7.49 (1H, dd, J = 8.0 ,0.4 Hz), 7.31 (1H, dd, J = 7.6, 5.2 Hz).
6-[2-chloro-6-(6-methoxy-3-pyridyl)phenyl]pyrido[2,3-d]pyrimidine-2,7-diamine (4e). was prepared using general procedure B (34 mg, 27%). HRMS (ESI) calcd [M+H]+ 379.1074, found 379.1059; 1H NMR (DMSO-d6, 400 MHz): δ 8.80 (1H, s), 8.02 (1H, d, J = 2.4 Hz), 7.86 (1H, s), 7.67 (1H, d, J = 8.0 Hz), 7.59 (1H, t, J = 8.0 Hz) 7.48 (1H, dd, J = 8.8, 2.4 Hz), 7.44 (1H, d, J = 7.6 Hz), 6.70 (1H, d, J = 8.4 Hz), 3.77 (3H, s).
6-(2-chloro-6-isobutoxy-phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (4f): A solution of 6-(2-chloro-6-isobutoxy-phenyl)-2-methylsulfinyl-pyrido[2,3-d]pyrimidin-7-amine (44 mg, 0.11 mmol) in 1.5 mL anhydrous THF was treated with 5M ammonia in methanol (0.45 mL, 2.3 mmol) and heated to 70 °C in a sealed vial overnight. The reaction was concentrated under vacuum and the crude residue was purified by RP-HPLC to give the desired product (23 mg, 60%). HRMS (ESI) calcd [M+H]+ 344.1278, found 344.1276; 1H NMR (DMSO-d6, 400 MHz): δ 8.90 (1H, s), 7.93 (1H, s), 7.46 (1H, t J = 8.0 Hz), 7.21 (1h, d, J = 8.0 Hz), 7.14 (1H, d, J = 8.4 Hz), 3.80–3.72 (2H, m), 1.86–1.81 (1H, m), 0.79 (6H, d, J = 6.8 Hz).
6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-amine (6a): 4-amino-2-methylsulfanyl-pyrimidine-5-carbaldehyde (3.1 g, 18 mmol) was dissolved in 40 mL anhydrous dimethylformamide and cooled in an ice/methanol bath (−10 °C) for 15 minutes under nitrogen atmosphere. Sodium hydride (60% in mineral oil, 0.73 g, 18 mmol) divided into 5 portions was added over the course of 5 minutes keeping the reaction mixture cooled with stirring. After an additional 5 minutes a solution of 2,6-dichlorobenzylacetonitrile (4.9 g, 27 mmol) in 12 mL anhydrous dimethylformamide was added dropwise via syringe. The reaction mixture was stirred at −10 °C for 30 minutes then allowed to warm to room temperature overnight after which time the reaction mixture was cooled in an ice bath and slowly quenched with 50 mL saturated aqueous ammonium chloride. After stirring for 20 minutes the product was extracted into DCM. The combined DCM extracts were dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The residue was re-dissolved in ethyl acetate and washed with brine. The organic layer was filtered through a sintered glass funnel and the filtrate was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to dryness. The residue was purified by silica gel chromatography eluting with ethyl acetate/dichloromethane to afford 710 mg (11%) 6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-amine LCMS (ESI): [M+H]+ = 336.9.
6-[2-chloro-6-(3-pyridyl)phenyl]-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-amine (6d): 2-[2-chloro-6-(3-pyridyl)phenyl]acetonitrile (210 mg, 0.94 mmol) was dissolved in DMF (3 mL) and THF (1.5 mL) and cooled in an ice bath. Sodium hydride (60% in mineral oil,39 mg, 0.98 mmol) was added followed by 4-amino-2-methylsulfanyl-pyrimidine-5-carbaldehyde (160 mg, 0.94 mmol) and the reaction was allowed to warm to rt. After stirring overnight, the reaction was poured into ice and acidified to pH 4 with 10% aq. acetic acid. The product was extracted with ethyl acetate (2 x) and the combined organics were washed with water and then brine and dried with sodium sulfate, filtered and concentrated under reduced pressure. The oil was taken up in DCM and purified by silica gel chromatography eluting with methanol/dichloromethane to afford 110 mg (30%) 6-[2-chloro-6-(3-pyridyl)phenyl]-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-amine LCMS (ESI): [M+H]+ = 380.0.
6-(2-chloro-6-isobutoxy-phenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-amine (6f). A solution of 2-(2-chloro-6-isobutoxy-phenyl)acetonitrile (950 mg, 4.3 mmol) in 6 ml anhydrous THF was cooled to −10 °C and treated with sodium hydride (60% in mineral oil, 200 mg, 4.9 mmol) (60% oil dispersion). After 10 min a solution of 4-amino-2-methylsulfanyl-pyrimidine-5-carbaldehyde (720 mg, 4.3 mmol) in 14 mL anhydrous DMF was added dropwise over 10 min. After 2h the reaction was warmed to 60 °C and stirred overnight. After cooling to room temperature, the reaction was diluted with water (30 mL) and extracted with ethyl acetate (3 × 75 mL). The combined extracts were washed with brine (3 × 30 mL), dried with sodium sulfate and concentrated under vacuum. The crude residue was purified on a silica gel column to give the desired product (950 mg, 60%). LCMS (ESI) [M+H]+ = 375.0.
6-(2-chloro-6-isobutoxy-phenyl)-2-methylsulfinyl-pyrido[2,3-d]pyrimidin-7-amine (6g): A solution of 6-(2-chloro-6-isobutoxy-phenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-amine (43 mg, 0.11 mmol) in DCM (2 mL) was treated with 2-(benzenesulfonyl)-3-phenyl-oxaziridine (30 mg, 0.11 mmol) and stirred overnight at rt. The reaction was concentrated under vacuum and used directly in the next step. LCMS (ESI) [M+H]+ = 391.0.
6-(2,6-dichlorophenyl)-2-methylsulfanyl-8H-pyrido[2,3-d]pyrimidin-7-one (7a): 6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-amine (200 mg, 0.60 mmol) was dissolved in TFA (5 mL) and cooled in an ice bath. Sodium nitrite (110 mg, 1.6 mmol) was added and the reaction was stirred in the bath for 15 minutes, then at room temperature for 2.5 h, after which time the reaction was slowly added to a mixture of saturated aqueous sodium bicarbonate, solid sodium carbonate and ice (15-20 mL). The product was extracted with ethyl acetate (2 × 15 mL) and the combined organic layers were dried with sodium sulfate, filtered and concentrated under vacuum giving 190 mg (96%) of 6-(2,6-dichlorophenyl)-2-methylsulfanyl-8H-pyrido[2,3-d]pyrimidin-7-one LCMS (ESI): [M+H]+ = 338.0.
6-[2-chloro-6-(3-pyridyl)phenyl]-2-methylsulfanyl-8H-pyrido[2,3-d]pyrimidin-7-one (7d): 6-[2-chloro-6-(3-pyridyl)phenyl]-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-amine (110 mg, 0.28 mmol) was dissolved in TFA (2 mL) and cooled in an ice bath. Sodium nitrite (52 mg, 0.75 mmol) was added and the reaction was allowed to warm to rt. After 90 min, the reaction was slowly added to a mixture of saturated aqueous sodium bicarbonate, solid sodium carbonate and ice. The product was extracted with ethyl acetate (2x) and the combined organics were washed with brine, dried with sodium sulfate, filtered and concentrated under reduced pressure to give 93 mg (88%) 6-[2-chloro-6-(3-pyridyl)phenyl]-2-methylsulfanyl-8H-pyrido[2,3-d]pyrimidin-7-one LCMS (ESI): [M+H]+ = 381.0.
7-chloro-6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidine (8a): 6-(2,6-dichlorophenyl)-2-methylsulfanyl-8H-pyrido[2,3-d]pyrimidin-7-one (190 mg, 0.57 mmol) was combined with phosphorous oxychloride (1.1 mL, 11 mmol) and dimethylformamide (1.0 μL) at room temperature. The reaction was heated at 95 °C. An additional charge of phosphorous oxychloride (1.1 mL, 11 mmol) was added after 5 min. After 2 h, the phosphorous oxychloride was removed by concentration under reduced pressure. The residue was treated with ice and cold water in an ice bath to obtain a solid that was collected by filtration, rinsing with water. After further drying under vacuum, 200 mg (96%) of 7-chloro-6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidine was obtained LCMS (ESI): [M+H]+ = 356.0.
7-chloro-6-[2-chloro-6-(3-pyridyl)phenyl]-2-methylsulfanyl-pyrido[2,3-d]pyrimidine (8d): 6-[2-chloro-6-(3-pyridyl)phenyl]-2-methylsulfanyl-8H-pyrido[2,3-d]pyrimidin-7-one (93 mg, 0.24 mmol) phosphorous oxychloride (0.89 mL, 9.8 mmol) and dimethyl formamide (1.9 μL, 0.020 mmol) were combined and stirred heated at 95 °C. After 4h, the reaction was concentrated under reduced pressure and dried further under high vacuum. The material was taken up in DCM dripped slowly into ice cold sat. sodium bicarbonate. The layers were separated, and the aqueous layer was extracted again with DCM. The combined organic layers were dried with sodium sulfate, filtered and concentrated under reduced pressure to give 68 mg (70%) 7-chloro-6-[2-chloro-6-(3-pyridyl)phenyl]-2-methylsulfanyl-pyrido[2,3-d]pyrimidine LCMS (ESI): [M+H]+ = 399.0.
tert-butyl N-[3-[6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate (10h): 7-chloro-6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidine (35 mg, 0.10 mmol) was combined with acetonitrile (0.30 mL) 1,4-dioxane (0.30 mL), tert-butyl N-(3-azabicyclo[3.1.0]hexan-6-yl)carbamate (21 mg, 0.11 mmol) and N,N''-diisopropylethylamine (0.03 mL, 0.20 mmol) and the reaction was heated at 80 °C. Additional portions of diisopropylethylamine and tert-butyl N-(3-azabicyclo[3.1.0]hexan-6-yl)carbamate were added and the temperature was increased to 90 °C. After heating overnight, the solvent was removed by concentration under reduced pressure. The residue was taken up in DCM (1 mL) and washed with 1M citric acid (2 × 1 mL), water, and sat. aqueous sodium bicarbonate, then it was dried over sodium sulfate, filtered and concentrated under reduced pressure yielding 66 mg of crude tert-butyl N-[3-[6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate, which was taken forward without further purification LCMS (ESI): [M+H]+ = 518.1.
tert-butyl N-[3-[6-(2,6-dichlorophenyl)-2-methylsulfinyl-pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate (11h): tert-butyl N-[3-[6-(2,6-dichlorophenyl)-2-methylsulfanyl-pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate (51 mg, 0.10 mmol) was dissolved in DCM (1 mL) and 3-chloroperbenzoic acid (15 mg, 0.090 mmol) was added at rt. After 2 h, the reaction was washed with sat. aqueous sodium bicarbonate, dried with sodium sulfate, filtered and concentrated under reduced pressure to yield 60 mg of crude tert-butyl N-[3-[6-(2,6-dichlorophenyl)-2-methylsulfinyl-pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate which was taken forward without further purification LCMS (ESI): [M+H]+ = 534.0.
1-[2-amino-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-7-yl]pyrrolidine-3-carboxamide (12a) was prepared by the method in Scheme 2 using pyrrolidine-3-carboxamide hydrochloride in step d and gaseous ammonia instead of 5 M ammonia in methanol in step f. HRMS (ESI) calculated [M+H]+ 403.0841, found 403.0822; 1H NMR (DMSO- d6, 400 MHz): δ 8.94 (1H, s), 7.91 (1H, s), 7.65-7.61 (2H, m), 7.51 (1H, t, J = 8.0 Hz), 7.41 (1H, s), 6.91 (1H, s), 2.90 (1H, br s), 2.02 (1H , br s), 1.91 (1H br s).
[1-[2-amino-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-7-yl]pyrrolidin-3-yl]methanol (12b) was prepared by the method in Scheme 2 using pyrrolidin-3-ylmethanol in step d, gaseous ammonia instead of 5 M ammonia in methanol in step f. HRMS (ESI) calculated [M+H]+ 390.0888, found 390.0883; 1H NMR (DMSO- d6, 400 MHz): δ 8.88 (1H, s), 7.85 (1H, s), 7.58 (1H, d, J = 4.0 Hz), 7.57 (1H, d, J = 3.6 Hz), 7.46 (1H, t, J = 8.0 Hz), 3.31–3.27 (2H, m), 3.21–3.18 (2H, m), 2.21 (1H, q, J = 6.8 Hz), 1.82–1.79 (1H, m), 1.60-1.55 (1H, m).
tert-butyl N-[3-[2-amino-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate (12h): tert-butyl N-[3-[6-(2,6-dichlorophenyl)-2-methylsulfinyl-pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate (52 mg, 0.10 mmol) was dissolved in THF (2 mL) and ammonia (5 M in methanol) (0.49 mL, 2.5 mmol) at rt. The reaction was then heated at 65 °C for 15 h, after which time the reaction was concentrated to an oil to give crude tert-butyl N-[3-[2-amino-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate which was used in the next step without further purification LCMS (ESI): [M+H]+ = 487.0.
[3-amino-1-[2-amino-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-7-yl]pyrrolidin-3-yl]methanol (13c) was prepared by the method in Scheme 2 using tert-butyl N-[3-(hydroxymethyl)pyrrolidin-3-yl]carbamate in step d and gaseous ammonia instead of 5 M ammonia in methanol in step f. HRMS (ESI) calculated [M+H]+ 405.0997, found 405.0979; 1H NMR (DMSO- d6, 400 MHz): δ 8.86 (1H, s), 8.14 (3H, br s), 7.83 (1H, s), 7.59 (2H, appt, J = 8.4 Hz), 7.47 (1H, t, J = 8.0 Hz), 5.63 (1H, s), 3.40–3.18 (8H, m), 2.07–2.00 (1H, m), 1.93-1.88 (1H, m).
7-[3-(aminomethyl)-3-fluoro-pyrrolidin-1-yl]-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-2-amine (13d) was prepared by the method in Scheme 2 using tert-butyl N-[(3-fluoropyrrolidin-3-yl)methyl]carbamate in step d and gaseous ammonia instead of 5 M ammonia in methanol in step f. HRMS (ESI) calculated [M+H]+ 407.0954, found 407.0943; 1H NMR (DMSO- d6, 400 MHz): δ 8.85 (1H, s), 8.13 (3H, br s), 7.83 (1H, s), 7.61 (1H, d, J = 8.0 Hz), 7.57 (1H, d, J = 8.0 Hz), 7.47 (1H, t, J = 8.4 Hz), 3.78–3.68 (2H, m), 3.62 (1H, br s), 3.31 (2H, br s), 3.16 (1H, br s), 3.05 (1H, br s), 2.16–2.10 (1H, m), 2.06–2.01 (1H, m).
1-[2-amino-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-7-yl]-3-(aminomethyl)pyrrolidin-3-ol (13e) was prepared by the method in Scheme 2 using tert-butyl N-[(3-hydroxypyrrolidin-3-yl)methyl]carbamate in step d and gaseous ammonia instead of 5 M ammonia in methanol in step f. HRMS (ESI) calculated [M+H]+ 405.0997, found 405.0982; 1H NMR (DMSO- d6, 400 MHz): δ 8.87 (1H, s), 7.83 (3H, s), 7.64 (1H, d, J = 8.0 Hz), 7.61 (1H, d, J = 8.4 Hz), 7.51 (1H, t, J = 8.0 Hz), 2.92 (2H, s), 1.86 (2H, t, J = 6.8 Hz).
7-[3-(aminomethyl)pyrrolidin-1-yl]-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-2-amine (13f) was prepared by the method in Scheme 2 using 3-(N-Boc-aminomethyl)pyrrolidine in step f and gaseous ammonia instead of 5 M ammonia in methanol in step f. HRMS (ESI) calculated [M+H]+ 389.1048, found 389.1035; 1H NMR (DMSO- d6, 400 MHz): δ 8.88 (1H, s), 7.85 (1H, s), 7.81 (3H, br s), 7.60-7.57 (2H, m), 7.47 (1H, t, J = 8.0 Hz), 3.57 (1H, br s), 3.07 (2H, br s), 2.82–2.78 (1H, m), 2.75–2.70 (1H, m), 2.38–2.31 (1H, m), 1.96–1.92 (1H, m), 1.65–1.60 (1H, m).
7-[4-(aminomethyl)-1-piperidyl]-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-2-amine (13g) was prepared by the method in Scheme 2 using tert-butyl N-(4-piperidinylmethyl)carbamate in step d and 2-(benzenesulfonyl)-3-phenyl-oxaziridine instead of 3-chloroperbenzoic acid in step e. HRMS (ESI) calculated [M+H]+ 403.1205, found 403.1188; 1H NMR (DMSO-d6, 400 MHz): δ 8.96 (1H, s), 7.94 (1H, s), 7.72–7.64 (5H, m), 3.92 (2H, br d, J = 12 Hz), 2.91 (2H, t, J = 12.8 Hz), 2.68 (2H, t, J = 6.4 Hz), 1.79 (1H, br s), 1.65 (2H, d, J = 11.6 Hz), 1.06 (2H, q, J = 10.0 Hz).
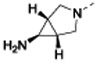
7-((1R,5S,6s)-6-amino-3-azabicyclo[3.1.0]hexan-3-yl)-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-2-amine (13h): tert-butyl N-[3-[2-amino-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate (48 mg, 0.10 mmol) was dissolved in DCM (0.50 mL) and TFA (0.50 mL) at room temperature. After 45 minutes, the reaction was concentrated under vacuum. The residue was taken up in dimethylformamide and purified by reverse phase HPLC (1” Phenomenex Luna column, 0.1% TFA in water/ACN) to afford 18 mg (35% over four steps) 7-[(1R,5S)-6-amino-3-azabicyclo[3.1.0]hexan-3-yl)-6-(2,6-dichlorophenyl)pyrido[2,3-d]pyrimidin-2-amine. HRMS (ESI) calculated [M+H]+ 387.0891, found 387.0886; 1H NMR (DMSO-d6, 400 MHz): δ 8.83 (1H, s), 8.04 (3H, br s), 7.77 (1H, s), 7.59 (2H, d, J = 8.0 Hz), 7.48 (1H, t, J = 8.4 Hz), 3.42–3.33 (4H, m), 2.15 (1H, s), 1.95 (2H, s).
7-[(1R,5S)-6-amino-3-azabicyclo[3.1.0]hexan-3-yl)-6-[2-chloro-6-(3-pyridyl)phenyl]pyrido[2,3-d]pyrimidin-2-amine (14a). The compound was prepared according to the procedure for 12a, to give 17 mg of 14a HRMS (ESI) calcd [M+H]+ 430.1547, found 430.1525; 1H NMR (DMSO- d6, 400 MHz): δ 8.88 (1H, s), 8.41 (1H, d, J = 6.4 Hz), 8.31 (1H, d, J = 2 Hz), 8.05 (3H s), 7.89 (1H, s), 7.74 (1H, d, J = 7.6 Hz), 7.63 (1H, t, J = 8.0 Hz), 7.49 (1H, d, 7.6 Hz), 7.44 (1H, d, J = 8.0 Hz), 7.25 (1H, dd, J = 7.6, 4.8 Hz), 3.34 (1H, br s), 3.21–3.16 (2H, m), 2.09 (1H, br s), 1.95 (2H, br s).
7-[(1R,5S)-6-amino-3-azabicyclo[3.1.0]hexan-3-yl)-6-[2-chloro-6-(4-pyridyl)phenyl]pyrido[2,3-d]pyrimidin-2-amine (14b) The compound was prepared according to the procedure for 14c: HRMS (ESI) calculated [M+H]+ 430.1547, found 430.1526; 1H NMR (DMSO- d6, 400 MHz): δ 8.88 (1H, s), 8.44 (2H, d, J = 5.6 Hz), 8.06 (3H, br s), 7.87 (1H, s), 7.76 (1H, d, J = 7.2 Hz), 7.65 (1H, t, J = 7.6 Hz), 7.50 (1H, d, J = 7.2 Hz), 7.09 (2H, d, J = 4.8 Hz), 2.12 (1H, s), 1.96 (2H, s).
7-[(1R,5S)-6-amino-3-azabicyclo[3.1.0]hexan-3-yl)-6-[2-chloro-6-(5-fluoro-6-methoxy-3-pyridyl)phenyl]pyrido[2,3-d]pyrimidin-2-amine (14c): tert-butyl N-[3-[2-amino-6-(2-bromo-6-chloro-phenyl)pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate (15 mg, 0.030 mmol) 1,4-dioxane (0.60 mL), 1 M sodium carbonate (0.15 mL), (5-fluoro-6-methoxy-3-pyridyl)boronic acid (5.3 mg, 0.030 mmol) and Pd(dppf)Cl2 DCM complex (2.3 mg, 0.0028 mmol) were combined in a vial under nitrogen at rt. The reaction was stirred at 100 °C for 1h. The product was extracted with ethyl acetate and the combined organic layers were dried with sodium sulfate, filtered and concentrated under reduced pressure to give 20 mg of crude tert-butyl N-[3-[2-amino-6-[2-chloro-6-(5-fluoro-6-methoxy-3-pyridyl)phenyl]pyrido[2,3-d]pyrimidin-7-yl]-3-azabicyclo[3.1.0]hexan-6-yl]carbamate that was used in the next step without further purification. LCMS (ESI): [M+H]+ = 578.1.
The compound was prepared according to the procedure for 13h. HRMS (ESI) calculated [M+H]+ 478.1558, found 478.1538; 1H NMR (DMSO- d6, 400 MHz): δ8.84 (1H, s), 8.06 (3H, br s), 7.84 (1H, s), 7.71 (2H, appd, J = 7.2 Hz), 7.60 (1H, t, J = 7.6 Hz), 7.47 (1H, d, J = 7.6 Hz), 7.39 (1H, d, J = 11.2 Hz), 3.86 (3H, s), 2.08 (1H, s), 1.94 (2H, s).
7-[(1R,5S)-6-amino-3-azabicyclo[3.1.0]hexan-3-yl)-6-[2-chloro-6-(2-methylthiazol-5-yl)phenyl]pyrido[2,3-d]pyrimidin-2-amine (14d) The compound was prepared according to the procedure for 14c HRMS (ESI) calculated [M+H]+ 450.1267, found 450.1254; 1H NMR (DMSO- d6, 400 MHz): δ 8.84 (1H, s), 8.02 (3H, br s), 7.76–7.73 (3H, m), 7.65 (1H, d, J = 8.0 Hz), 7.57 (1H, t, J = 8.0 Hz) 2.03 (1H, s), 1.95 (2H, s).
7-[(1R,5S)-6-amino-3-azabicyclo[3.1.0]hexan-3-yl)-6-(2-chloro-6-isobutoxy-phenyl)pyrido[2,3-d]pyrimidin-2-amine (14e). The compound was prepared according to the procedure for 12a: HRMS (ESI) calculated [M+H]+ 425.1856, found 425.1843, 1H NMR (DMSO- d6, 400 MHz): δ 8.89 (1H, s), 8.07 (3H, s), 7.75 (1H, s), 7.43 (1H, t, J = 8.4 Hz), 7.18 (1H, d, J = 8.0 Hz), 7.08 (1H, d, J = 8.4 Hz), 3.77-3.69 (3H, m), 3.35 (4H, br s), 2.11 (1H, s), 1.98 (2H, s), 1.79–1.76 (1H, m), 0.76–0.70 (6H, m).
Bacterial Strains.
Wild type strains of A. baumannii (ATCC 19606) and K. pneumoniae (ATCC 43816) were obtained from the American Type Culture Collection. The E. coli ΔtolC knockout was constructed at Achaogen in the DM4100 background (Coli Genetic Stock Center Database #6333). The P. aeruginosa efflux-compromised strain ΔmexAB-oprM::Cm ΔmexEF-oprN::ΩHg ΔmexCD-oprJ::Gm, also known as PAM1262, was constructed in a PAO1 background as described in Ref. 47 and kindly provided by the authors.
Minimum Inhibitory Concentrations.
MICs were determined by broth microdilution according to guidelines established by the Clinical and Laboratory Standards Institute.51 Compounds were serially diluted in two-fold increments in 100% DMSO, then diluted 10-fold in water. Inocula were prepared from cells streaked onto Mueller Hinton agar (MHA) and grown at 35 °C overnight. 10 μL of antibiotic solution was mixed with 90 μL of inoculum in cation-adjusted Mueller Hinton broth in 96-well assay plates, with a final inoculum of approximately 5 × 104 cells/well. When included, PMBN was diluted in broth prior to addition of cells, to a final assay concentration of 8 μg/mL. After incubation of assay plates at 35 °C for 18-20 hours, the lowest concentration of antibiotic that prevented visible growth was recorded as the MIC. In general, MIC values reported are geometric means of at least two independent measurements rounded to the nearest standard dilution increment.
Purification of P. aeruginosa BC.
The acetyl-CoA carboxylase (accC) gene was cloned from P. aeruginosa (PAO1) genomic DNA using forward primer with underlined EcoR1 restriction site 5’- GACTGAATTCTATGTTGGAAAAAGTGCTGATCG-3’ and reverse primer with SacI restriction site 5’-GACTGAGCTCTCAGTGCTTGTCCATACCC-3’ and inserted into the pET47b(+) expression vector (Novagen). This construct has the following N-terminal sequence preceding the WT BC sequence: MAHHHHHHSAALEVLFQGPGYQDPNS. Recombinant N-terminal His-Tagged BC was overexpressed in Rosetta™(DE3)pLysS E. coli cells (Novagen). A starter culture of 25 mL of LB broth containing kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL) was inoculated with a single colony of the Rosetta™(DE3)pLysS E. coli cells containing the His-Tagged BC construct and grown overnight at 37 °C. 20 mL of starter culture was used to inoculate 2 L of LB broth containing kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL). The culture was grown to an OD600 of 0.5 at 37 °C at which point 0.5 nM IPTG was added to induce overexpression for 3 hours. Cells were then pelleted at 10,200 g for 15 minutes and resuspended in 10 mL of Buffer A (20 mM Tris•HCl, pH 7.9, 500 mM NaCl, and 5 mM imidazole). The resulting cell slurry was lysed either by sonication (duty cycle 30; power 6, pulse for 15 sec, rest for 45 seconds for 20 rounds on ice) or using an Avestin Emulsiflex. The lysed solution was then centrifuged at 75,000 g for at least 1 hour. The supernatant was gravity flowed over a 3mL Co2+ His-binding column (Pierce) and incubated with the resin for 30 minutes with rotation at 4 °C. The column was washed with 30 mL of Buffer A and then 10 mL of Buffer B (20 mM Tris•HCl, pH 7.9, 500 mM NaCl, and 170 mM imidazole) was added to the column to elute the protein. The eluent was exchanged into Buffer C (10 mM HEPES, pH 8.0, 25 mM KCl, and 1 mM TCEP) using 10,000 MWCO filters (Amicon®) or dialysis. The buffer exchanged sample (5 mL) was applied to a 5 mL Q-Sepharose column (GE Healthcare) at 2 mL/min. After a 50 mL wash with Buffer C, protein was eluted with a linear gradient over 50 mL of Buffer D (10 mM HEPES, pH 8.0, 250 mM KCl, and 1 mM TCEP). Fractions were collected and assessed using SDS-PAGE (12 % gel; Bio-Rad). Fractions containing the desired BC at >95% purity were pooled and exchanged into 10 mM HEPES, pH 7.0, 500 mM KCl using an Amicon® 10,000 MWCO filtration unit. A bicinchoninic acid assay (Pierce™) was used to determine the final protein concentration.
Previously reported purification procedures were used to purify material for the X-ray crystallography studies18,37
P. aeruginosa BC enzyme assay.
The IC50 assay used to measure a compound’s relative inhibtion of P. aeruginosa BC was adapted from the coupled assay reported by Ref. 5. The previous study used the full ACCase multi-enzyme complex consisting of BC, carboxytransferase, and biotin carboxyl carrier protein, while the assay herein uses only the isolated BC enzyme. Both BC and ACCase catalytic activities produce inorganic phosphate (Figure S1), which in the IC50 assay is coupled to the generation of a spectroscopically active product. However, because the kcat and KM values for inorganic phosphate generation by ACCase likely differs from BC in isolation, the apparent IC50 values measured for a given inhibitor of the two different enzyme preparations are not directly comparable.
Initial rates of BC activity were measured at various concentrations of test inhibitor in 96-well plates with a final reaction volume of 300 μL. An initial solution was made such that the final concentration of each component would be 50 mM HEPES, pH 8.0, 100 mM KCl, 1 mM TCEP, 5 mM MgCl2, 0.1 mg/mL BSA, 0.005% (vol/vol) Tween 20, 150 μM 7-methyl-6-thioguanosine (Barry and Associates), 0.25 U/mL purine nucleoside phosphorylase (Sigma), 2 mM ATP, 10 mM sodium carbonate, and 50 mM biotin. This solution was incubated at room temperature for 30 minutes. Afterwards, purified P. aeruginosa BC was added to achieve a final apparent concentration of 15 nM and 270 μL of the resulting solution was distributed into the wells of a 96-well plate containing 30 μL of a 10X concentration of test inhibitor (previously diluted in <1% DMSO) in 3-fold serial dilutions across the plate row. Reaction progress was monitored by the increase in absorbance at 360 nm. Initial rates at each inhibitor compound were plotted and fitted to a standard IC50 equation.
Crystallization and Data Collection.
Crystallization conditions are summarized in Table S1. Diffraction data were collected at 100 K at the 24-ID-E beamline equipped with a CCD-based ADSC Quantum 315 detector. The images were processed using the XDS program suit52 and scaled using the Scala program.53 Data collection and data processing statistics are given in Table S1.
Crystal Structure Determination.
The molecular replacement procedure was applied to locate a solution for the 13f data set using the program Phaser.54 A monomer of E. coli BC (PDB ID: 2V58) was used as a search model. The positioned MR model was refined using the maximum likelihood refinement in REFMAC55 with the TLS parameters generated by the TLSMD server.56 Program Coot57 was used for model building throughout the refinement. The final model consists of protein residues 1-447 (molecule A), 1-447 (molecule B), two 13f molecules, 3 ethylene glycol molecules, and 432 water molecules. Alternate conformations have been built for protein residues S68, E71, N90, R111, K116, S118, R314, R423 (mol A) and I5, R37, R314 (mol B).
A monomer of H. influenzae BC (PDB: 4RZQ) was used as a search model to locate a solution for the 14a data set. The positioned MR solution was refined as described above. The final model consists of protein residues 1-445, one 14a molecule, one ethylene glycol molecule, and 30 water molecules.
Bacterial Cytological Profiling.
Cultures of E. coli (ATCC 25922) were grown in LB at 30 °C to an OD600 ~0.15. Cultures were then split and each sample treated with the test compound for 120 minutes. The cells were stained with FM 4-64 (membranes), DAPI (DNA), and Sytox Green (cell integrity). Cells were then imaged at 100X magnification using an Applied Precision DeltaVision Deconvolution microscope.
Resistance Frequency.
Spontaneous resistance frequency was measured by plating approximately 109 colony-forming units from 5 independent cultures of the efflux-compromised strain of P. aeruginosa onto MHA containing 1 at 4-, 8-, and 16-fold above the MIC. The inoculum density of each culture was determined by plating serial dilutions onto drug-free MHA. Plates were incubated at 35 °C in ambient air. After 48 hours, visible colonies on antibiotic-containing plates were counted, and several colonies from each plate were archived for MIC analysis and sequencing. The frequency for each culture and antibiotic level was calculated as the number of colonies on the antibiotic-containing plate divided by the inoculum density for that culture. The reported frequency is the mean of the frequencies from the five independent cultures.
Genome Sequencing.
Whole-genome sequences of the P. aeruginosa efflux-compromised parent and the wild type ancestral strain were determined on a PacBio RSII system at the Arizona Genomics Institute and assembled into single contigs. Both genomes were re-sequenced by Illumina HiSeq (read length 2 × 100 base pairs) to resolve minor homopolymer discrepancies, resulting in a high-quality reference genome for the parent strain. Spontaneous mutants were sequenced by Illumina HiSeq. Raw reads were filtered for quality, trimmed, and assembled against the reference genome by Maverix Biomics. Single-nucleotide variants were reported by Maverix Biomics and verified at Achaogen by inspection of the assembled reads for representative mutants of each class. The inversion junction sites in muxA and mexZ were determined by analysis of the assembled reads.
Supplementary Material
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R21AI113572. We acknowledge Andrew McCandlish for help preparing and managing grant applications. This report also includes research conducted at the Northeastern Collaborative Access Team beam lines (24ID-C and 24ID-E at the Advanced Photon Source), which are funded by the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health. The Pilatus 6M detector on 24ID-C is funded by an NIH-ORIP HEI grant (S10 RR029205) and use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy by Argonne National Laboratory, is under Contract No. DE-AC02-06CH11357. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We acknowledge the computational modeling and scientific advice of Erin Bradley and the microbiological assay contributions of Hoan Lee.
ABBREVIATIONS
- ICU
intensive care unit
- INV
inversion
- MIC
minimum inhibitory concentration
- K
Kelvin
- BC
biotin carboxylase
- IC
inhibitory concentration
- ATCC
American Type Culture Collection
- CLSI
Clinical and Laboratory Standards Institute
- MHA
Mueller-Hinton agar
- PMBN
polymyxin B nonapeptide
- DMSO
dimethyl sulfoxide
- ATP
adenosine triphosphate
- HTS
high-throughput screen
- MR
molecular replacement
- US
United States
- LB
lysogeny broth
- AMPCP
adenosine 5’-(alpha,beta-methylene)diphosphate
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- TCEP
tris(2-carboxyethyl)phosphine
- MWCO
molecular weight cut off
- LPS
lipopolysaccharide
Footnotes
ASSOCIATED CONTENT
Supporting Information: Figure S1 showing the reaction catalyzed by BC and the actyl-CoA carboxylase multienzyme complex; Text S1 describing the tests of BC essentiality in P. aeruginosa; Table S1 of X-ray crystallographic conditions and statistics; molecular formula strings
PDB ID Codes: The coordinates for X-ray co-crystal structures of 13f and 14a have been deposited in the PDB with codes 6OI9 and 6OI8, respectively.
Notes: The authors declare no competing financial interest.
REFERENCES
- (1).Antibiotic Resistance Threats in the United States, 2013. U.S. Centers for Disease Control and Prevention; [Online] http://www.cdc.gov/drugresistance/threat-report-2013. (accessed Feb 27, 2019). [Google Scholar]
- (2).Burnham J; Olsen M; Kollef M Re-Estimating Annual Deaths Due to Multidrug-Resistant Organism Infections. Infection Control and Hostpital Epidemiology. 2019, 40, 112–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cassini A; Hogberg LD; Plachouras D; Quattrocchi A; Hoxha A; Simonsen GS; Colomb-Cotinat M; Kretzschmar ME; Devleesschauwer B; Cecchini M; Ouakrim DA; Oliveira TC; Struelens MJ; Suetens C; Monnet DL; Burden of AMR Collaborative Group. Attributable Deaths and Disability-Adjusted Life-Years Caused by Infections with Antibiotic-Resistant Bacteria in the EU and the European Economic Area in 2015: a Population-Level Modelling Analysis. Lancet Infect. Dis 2019, 19, 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chen L; Todd R; Kiehlbauch J; Walters M; Kallen A Notes From the Field: Pan-Resistant New Delhi Metallo-Beta-Lactamase-Producing Klebsiella pneumoniae - Washoe County, Nevada, 2016. MMWR Morb. Mortal Wkly. Rep 2017, 66, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Miller JR; Dunham S; Mochalkin I; Banotai C; Bowman M; Buist S; Dunkle B; Hanna D; Harwood HJ; Huband MD; Karnovsky A; Kuhn M; Limberakis C; Liu JY; Mehrens S; Mueller WT; Narasimhan L; Ogden A; Ohren J; Prasad JVNV; Shelly JA; Skerlos L; Sulavik M; Thomas VH; VanderRoest S; Wang L; Wang Z; Whitton A; Zhu T; Stover CK A Class of Selective Antibacterials Derived From a Protein Kinase Inhibitor Pharmacophore. Proc. Natl. Acad. Sci. USA 2009, 106, 1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Payne DJ; Warren PV; Holmes DJ; Ji Y; Lonsdale JT 2001. “Bacterial Fatty-Acid Biosynthesis: A Genomics-Driven Target for Antibacterial Drug Discovery.” Drug Discov. Today 2001, 6, 537–544. [DOI] [PubMed] [Google Scholar]
- (7).Campbell JW; Cronan JE Jr. 2001. Bacterial Fatty Acid Biosynthesis: Targets for Antibacterial Drug Discovery. Annu. Rev. Micobiol 2001, 55, 305–332. [DOI] [PubMed] [Google Scholar]
- (8).Heath RJ; White SW; Rock CO Lipid Biosynthesis as a Target for Antibacterial Agents. Prog. Lipid Res 2001, 40, 467–497. [DOI] [PubMed] [Google Scholar]
- (9).Zhang YM; White SW; Rock CO Inhibiting Bacterial Fatty Acid Synthesis. J. Biol. Chem 2006, 281, 17541–17544. [DOI] [PubMed] [Google Scholar]
- (10).Wright HT; Reynolds KA Antibacterial Targets in Fatty Acid Biosynthesis. Curr. Opin. Microbiol 2007, 10, 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Parsons JB; Rock CO Is Bacterial Fatty Acid Synthesis a Valid Target for Antibacterial Drug Discovery? Curr. Opin. Microbiol 2011, 14, 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cronan JE Jr.; Waldrop GL Multi-Subunit Acetyl-CoA Carboxylases. Prog. Lipid Res 2002, 41, 407–435. [DOI] [PubMed] [Google Scholar]
- (13).Mochalkin I; Miller JR; Narasimhan L; Thanabal V; Erdman P; Cox PB; Prasad JV; Lightle S; Huband MD; Stover CK Discovery of Antibacterial Biotin Carboxylase Inhibitors by Virtual Screening and Fragment-Based Approaches. ACS Chem. Biol 2009, 4, 473–483. [DOI] [PubMed] [Google Scholar]
- (14).Cheng CC; Shipps GW Jr.; Yang Z; Sun B; Kawahata N; Soucy KA; Soriano A; Orth P; Xiao L; Mann P; Black T Discovery and Optimization of Antibacterial AccC Inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 6507–6514. [DOI] [PubMed] [Google Scholar]
- (15).Ogden A; Kuhn M; Dority M; Buist S; Mehrens S; Zhu T; Xiao D; Miller JR; Hanna D Evaluation of Pharmacokinetic/Pharmacodynamic Relationships of PD-0162819, a Biotin Carboxylase Inhibitor Representing a New Class of Antibacterial Compounds, Using In Vitro Infection Models. Antimicrob. Agents Chemother 2012, 56, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Silvers MA; Robertson GT; Taylor CM; Waldrop GL. Design, Synthesis, and Antibacterial Properties of Dual-Ligand Inhibitors of Acetyl-CoA Carboxylase. J. Med. Chem 2014, 57, 8947–8959. [DOI] [PubMed] [Google Scholar]
- (17).Wallace J; Bowlin NO; Mills DM; Saenkham P; Kwasny SM; Opperman TJ; Williams JD; Rock CO; Bowlin TL; Moir DT Discovery of Bacterial Fatty Acid Synthase Type II Inhibitors Usng a Novel Cellular Bioluminescent Reporter Assay. Antimicrob. Agents Chemother 2015, 59, 5775–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mochalkin I; Miller JR; Evdokimov A; Lightle S; Yan C; Stover CK; Waldrop GL Structural Evidence for Substrate-Induced Synergism and Half-Sites Reactivity in Biotin Carboxylase. Protein Sci. 2008, 17, 1706–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nikaido H; Vaara M 1985. Molecular Basis of Bacterial Outer Membrane Permeability. Microbiol. Rev 1985, 49, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Nikaido H Prevention of Drug Access to Bacterial Targets: Permeability Barriers and Active Efflux. Science. 1994, 264, 382–388. [DOI] [PubMed] [Google Scholar]
- (21).Kumar A; Schweizer HP Bacterial Resistance to Antibiotic: Active Efflux and Reduced Uptake. Adv. Drug Deliv. Rev 2005, 57, 1486–1513. [DOI] [PubMed] [Google Scholar]
- (22).Brown DG; May-Dracka TL; Gagnon MM; Tommasi R Trends and Exceptions of Physical Properties on Antibacterial Activity for Gram-Positive and Gram-Negative Pathogens. J. Med. Chem 2014, 57, 10144–10161. [DOI] [PubMed] [Google Scholar]
- (23).Tommasi R; Brown DG; Walkup GK; Manchester JI; Miller AA ESKAPEing the Labyrinth of Antibacterial Discovery. Nat. Rev. Drug Discov 2015, 14, 529–542. [DOI] [PubMed] [Google Scholar]
- (24).Mugumbate G; Overington JP The Relationship Between Target-Class and the Physicochemical Properties of Antibacterial Drugs. Bioorg. Med. Chem 2015, 23, 5218–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).O’Shea R; Moser HE Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem 2008, 51, 2871–2878. [DOI] [PubMed] [Google Scholar]
- (26).Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ Predictive Rules for Compound Accumulation Yield a Broad-Spectrum Antibiotic. Nature, 2017, 545, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Serio AW; Kubo A; Lopez S; Gomez M; Corey VC; Andrews LD; Schwatz MA; Kasar R; McEnRoe G; Aggen J; Miller G; Armstrong ES Structure, Potency and Bactericidal Activity of ACHN-975, a First-In-Class LpxC Inhibitor. 53rd Interscience Conference on Antimicrobial Agents and Chemotherapy, 2013. [poster #F-1226]. [Google Scholar]
- (28).Tari LW; Trzoss M; Bensen DC; Li X; Chen Z; Lam T; Zhang J; Creighton CJ; Cunningham ML; Kwan B; Stidham M; Shaw KJ; Lightstone FC; Wong SE; Nguyen TB; Nix J; Finn J Pyrrolopyrimidine Inhibitors of DNA Gyrase B (GyrB) and Topoisomerase IV (ParE). Part I: Structure Guided Discovery and Optimization of Dual Targeting Agents with Potent, Broad-Spectrum Enzymatic Activity. Bioorganic and Medicinal Chemistry Letters, 2013, 23, 1529–1536. [DOI] [PubMed] [Google Scholar]
- (29).Richter MF; Hergenrother PJ The Challenge of Converting Gram-Positive-Only Compounds Into Broad-Spectrum Antibiotics. Ann. N. Y. Acad. Sci 2019, 1435, 18–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vaara M Agents That Increase the Permeability of the Outer Membrane. Microbiol. Rev 1992, 56, 395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Angus BL; Carey AM; Caron DA; Kropinski AM; Hancock RE Outer Membrane Permeability in Pseudomonas aeruginosa: Comparison of a Wild-Type with an Antibiotic-Supersusceptible Mutant. Antimicrob. Agents Chemother 1982, 21, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yoshirmura F; Nikaido H Permeability of Pseudomonas aeruginosa Outer Membrane to Hydrophilic Solutes. J. Bacteriol 1982, 152, 636–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Delcour AH Outer Membrane Permeability and Antibiotic Resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Page MG The Role of the Outer Membrane of Gram-Negative Bacteria in Antibiotic Resistance: Ajax’s Shield or Achilles’ Heel? Handb. Exp. Pharmacol 2012, 211, 67–86. [DOI] [PubMed] [Google Scholar]
- (35).Breidenstein EB; de la Fuente-Nunez C; Hancock RE Pseudomonas aeruginosa: All Roads Lead to Resistance. Trends Microbiol. 2011, 19, 419–426. [DOI] [PubMed] [Google Scholar]
- (36).Waldrop GL; Holden HM; St. Maurice M The Enzymes of Biotin Dependent CO2 Metabolism: What Structures Reveal About Their Reaction Mechanisms. Protein Sci. 2012, 21, 1597–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Broussard TC; Pakhomova S; Neau DB; Bonnot R; Waldrop GL Structural Analysis of Substrate, Reaction Intermediate, and Product Binding in Haemophilus influenzae Biotin Carboxylase. Biochemistry, 2015, 54, 3860–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cunningham ML; Kwan BP; Nelson KJ; Bensen DC; Shaw KJ. Distinguishing On-Target Versus Off-Target Activity in Early Antibacterial Drug Discovery Using a Macromolecular Synthesis Assay. Journal of Biomolecular Screening, 2013, 18, 1018–1026. [DOI] [PubMed] [Google Scholar]
- (39).Nonejuie P; Burkart M; Pogliano K; Pogliano J Bacterial Cytological Profiling Rapidly Identifies the Cellular Pathways Targeted by Antibacterial Molecules. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 16169–16174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lamsa A; Liu WT; Dorrestein PC; Pogliano K The Bacillus subtilis Cannibalism Toxin SDP Collapses the Proton Motive Force and Induces Autolysis. Mol. Microbiol 2012, 84, 486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lamsa A; Lopez-Garrido J; Quach D; Riley EP; Pogliano J; Pogliano K. 2016. Rapid Inhibition Profiling in Bacillus subtillis to Identify the Mechanism of Action of New Antimicrobials. ACS Chem. Biol 2016, 11, 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Mohammad H; Younis W; Chen L; Peters CE; Pogliano J; Pogliano K; Cooper B; Zhang J; Mayhoub A; Oldfield E; Cushman M; Seleem MN Phenylthiazole Antibacterial Agents Targeting Cell Wall Synthesis Exhibit Potent Activity In Vitro and In Vivo Against Vancomycin-Resistant Enterococci. J. Med. Chem 2017, 60, 2425–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lin L; Nonejuie P; Munguia J; Hollands A; Olson J; Dam Q; Kumaraswamy M; Rivera H Jr.; Corriden R; Rohde M; Hensler ME; Burkart MD; Pogliano J; Sakoulas G; Nizet V Azithromycin Synergizes with Cationic Antimicrobial Peptides to Exert Bactericidal and Therapeutic Activity Against Highly Multidrug-Resistant Gram-Negative Bacterial Pathogens. EBioMedicine, 2015, 2, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Nonejuie P; Trial RM; Newton GL; Lamsa A; Ranmali Perera V; Aguilar J; Liu WT; Dorrestein PC; Pogliano J; Pogliano K Application of Bacterial Cytological Profiling to Crude Natural Product Extracts Reveals the Antibacterial Arsenal of Bacillus subtilis. J. Antibiot. (Tokyo), 2016, 69, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hamby JM; Connolly JC; Schroeder MC; Winters RT; Showalter HDH; Panek RL; Major TC; Olsewski B; Ryan MJ; Dahring T; Lu GH; Keiser J; Amar A; Shen C; Kraker AJ; Slintak V; Nelson JM; Fry DW; Bradford L; Hallak H; Doherty AM Structure-Activity Relationships for a Novel Series of Pyrido[2,3-d]pyrimidine Tyrosine Kinase Inhibitors. J. Med. Chem 1997, 40, 2296–2303. [DOI] [PubMed] [Google Scholar]
- (46).Hernandez V; Crepin T; Palencia A; Cusack S; Akama T; Baker SJ; Bu W; Feng L; Freund YR; Liu L; Meewan M; Mohan M; Mao W; Rock FL; Sexton H; Sheoran A; Zhang Y; Zhang YK; Zhou Y; Nieman JA; Anugula MR; Keramane el M.; Savariraj K; Reddy DS; Sharma R; Subedi R; Singh R; O’Leary A; Simon NL; De Marsh PL; Mushtaq S; Warner M; Livermore DM; Alley MR; Plattner JJ Discovery of Novel Class of Boron-Based Antibacterials with Activity Against Gram-Negative Bacteria. Antimicrob. Agents Chemother 2013, 57, 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lomovskaya O; Lee A; Hoshino K; Ishida H; Mistry A; Warren MS; Boyer E; Chamberland S; Lee VJ Use of a Genetic Approach to Evaluate the Consequences of Inhibition of Efflux Pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother 1999, 43, 1349–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tian ZX; Mac Aogain M; O’Connor HF; Fargier E; Mooji MJ; Adams C; Wang YP; O’Gara F MexT Modulates Virulence Determinants in Pseudomonas aeruginosa Independent of the MexEF-OprN Efflux Pump. Microb. Pathog 2009, 47, 237–241. [DOI] [PubMed] [Google Scholar]
- (49).Tian ZX; Fargier E; Mac Aogain M; Adams C; Wang YP; O’Gara F Transcriptome Profiling Defines a Novel Regulon Modulated by the LysR-Type Transcriptional Regulator MexT in Pseudomonas aeruginosa. Nucleic Acids Res. 2009, 37, 7546–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Luong PM; Shogan BD; Zaborin A; Belogortseva N; Shrout JD; Zaborina O; Alverdy JC Emergence of the P2 Phenotype in Pseudomonas aeruginosa PAO1 Strains Involves Various Mutations in MexT or MexF. J. Bacteriol 2014, 196, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).CSLI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Tenth Edition. CSLI document M07-A10. Wayne, PA: Clinical and Laboratory Standards Institute; 2015. [Google Scholar]
- (52).Kabsch W XDS. Acta. Crystallogr. D Biol. Crystallogr 2010, 66(Pt 2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Evans P Scaling and Assessment of Data Quality. Acta Crystallogr. D Biol. Crystallogr 2006, 62(Pt 1), 72–82. [DOI] [PubMed] [Google Scholar]
- (54).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX:A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66(Pt 2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Collaborative Computation Project, Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. D Biol. Crystallogr 1994, 50(Pt 5), 760–763. [DOI] [PubMed] [Google Scholar]
- (56).Painter J; Merritt EA TLSMD Web Server for the Generation of Multi-Group TLS Models. J. Appl. Cryst 2006, 39, 109–11. [Google Scholar]
- (57).Emsley P; Cowtan K Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr 2004, 60(Pt 12 Pt 1), 2126–2132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.