

Abstract
Reliable design of artificial metalloenzymes (ArMs) to access transformations not observed in nature remains a long-standing and important challenge. We report that a monomeric streptavidin (mSav) Rh(III) ArM permits asymmetric synthesis of α,β-unsaturated-δ-lactams via a tandem C-H activation and [4+2] annulation reaction. These products are readily derivatized to enantioenriched piperidines, the most common N-heterocycle found in FDA approved pharmaceuticals. Desired δ-lactams are achieved in yields as high as 99% and enantiomeric excess of 97% under aqueous conditions at room temperature. Embedding a Rh cyclopentadienyl (Cp*) catalyst in the active site of mSav results in improved stereocontrol and a seven-fold enhancement in reactivity relative to the isolated biotinylated Rh(III) cofactor. In addition, mSav-Rh outperforms its well-established tetrameric forms, displaying 11–33 times more reactivity.
Graphical Abstract
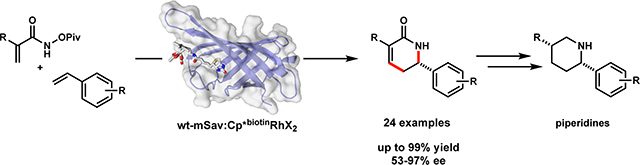
Piperidines are the most common N-heterocycle found in FDA approved drugs.1 Strategies to assemble this important structural motif are common but often involve cyclization, which necessitates the preassembly of an acyclic precursor. 2 Two-component approaches such as cycloadditions or annulations are inherently more attractive. A handful of recent [4+2] cycloaddition approaches have been devised, using N-heterocyclic carbene,3,4,5 cinchona alkaloid,6 chiral phosphoric acid7 or metal catalysis.8,9 Although each strategy is useful, they are limited to specific classes of precursors and substitution patterns. An alternative approach is to develop a methodology that enables access to α,β-unsaturated-δ-lactams from simple precursors under mild conditions.10,11 In this context, the coupling of acrylamides with abundant α-olefins via C-H activation and annulation would provide direct access to α,β-unsaturated-δ-lactams in a single step.
Rh(III) catalysis has been extensively demonstrated to mediate C-H activation reactions involving C(sp2)-H bonds proximal to a directing group.12 The subsequent functionalization with alkenes is less well-developed but has been documented for benzhydroxamate esters13 and unsaturated oxime esters14 delivering N-heterocycles in good yield. The direct union of acrylamide hydroxamate esters with alkenes has not been disclosed.15
In collaboration with the Ward group, 16 we have previously described a tetrameric streptavidin (tSav) system containing a biotinylated Rh(III) cofactor for the asymmetric synthesis of dihydroisoquinolones using benzhydroxamate esters and acrylate partners (Fig 1ii).17 In concurrent work, Cramer reported a small-molecule chiral Rh(III) catalyst for a complementary coupling reaction involving styrene partners (Figure 1iii).18
Figure 1.

a) i. tetrameric streptavidin (tSav) ArM; ii. Previously developed tSav Rh(III) benzhydroxamate coupling; iii. Cramer’s enantioselective Chiral Cp variant. b) iv. monomeric streptavidin (mSav) ArM. v. Target transformation (this work). [tSav (PDB ID: 3RY1) and mSav (PDB ID: 4JNJ) were complexed with the biotin cofactor in AutoDock and rendered in PyMOL.]
The most common ArM platform developed to date is the biotin-tetrameric (strept)avidin (biotin-tSav) system, pioneered by Whitesides19 and Ward20 (Fig 1a).These ArMs utilize high affinity (up to KD ~10−14 M) interactions between tSav and biotin-metal conjugates. tSav-based ArMs have been utilized in an increasing number of transition-metal mediated transformations.16 Herein, we introduce a new ArM platform based on monomeric streptavidin (mSav) and demonstrate its use in the direct enantioselective coupling of acrylamide hydroxamate esters and styrenes (Fig 1v). The reaction allows rapid access to piperidines – the most common N-heterocycle found in FDA-approved pharmaceuticals.1,21
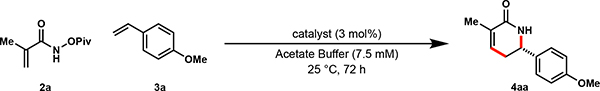
Initial investigations examined the coupling of methacrylamide 2a with styrene 3a.22 The use of Cp*Rh(III) as a catalyst under aqueous buffer conditions delivers 4aa in modest yield (entry 1, Table 1).23 The use of biotinylated Rh cofactor as catalyst provides 4aa in 15% yield. Incorporation of the cofactor into wt tSav leads to further diminished yields and very low selectivities (entry 3, Table 1).
Table 1.
Reaction Optimization and Controls.
 | |||
|---|---|---|---|
| entry | catalyst | NMR yield (%)a | enantiomeric excess (%)b |
| 1 | Cp*RhCl2 | 25 | 0 |
| 2 | Cp*biotinRhCl2 | 15 | 0 |
| 3 | wt-tSav:Cp*biotinRhCl2 | 9 | −26 |
| 4c | N118K-K121E-tSav:Cp*biotinRhCl2 | 3 | 0 |
| 5d | wt-mSav:Cp*biotinRhCl2 | 44 | 92 |
| 6 | wt-mSav:Cp*biotinRhCl2 | 99 | 91 |
| 7 | wt-mSav:Cp*biotinRhCl2 | 58 | 82 |
2a (3.0 μmol), 3a (1.5 μmol), catalyst, in 200 μL of acetate buffer (62.5 mM NaOAc, 100 mM NaCl, pH 7.4) with 3 μL MeOH.
Conversion and yield determined by 1H NMR analysis relative to a trimethyl(phenyl)silane internal standard.
Enantiomeric excess determined by HPLC analysis.
1 mol% catalyst.
200 μL of NaCl buffer (100 mM NaCl, pH 7.4) used.
Similarly, a mutant tSav (N118K/K121E) that proved to be a highly reactive ArM in our previous work17 did not provide the desired δ-lactam (4aa) in appreciable yield (Table 1, entry 4). In an effort to improve the reaction, we turned our attention to monomeric streptavidin (mSav, Fig 1–iv). We reasoned that the use of mSav, with a more exposed Rh binding site relative to tSav, could serve as a competent ArM template, and may simplify ArM tuning and analysis. Like tSav, biotin is bound tightly by mSav (KD ~2 nM 24,25,26) but has been engineered to resist tetramerization by replacing hydrophobic amino-acid residues at the barrel-barrel interface with charged ones.
In the event, 1 mol% wt-mSav: Cp*biotinRhCl2 ArM in acetate buffer, enables the coupling of acrylamide (2a) and paramethoxystyrene (3a) to provide the desired δ-lactam (4aa) in 44% yield and 92% enantiomeric excess (ee), (Table 1, entry 5).27 A modest increase in metalloenzyme catalyst loading results in substantially higher yield and virtually identical selectivity, delivering the desired δ-lactam (4aa) in 99% yield and 91% ee (Table 1, entry 6). The use of 100 mM NaCl in place of acetate buffer leads to 58% yield (Table 1, entry 7).
The wt-mSav:Cp*biotinRhCl2 catalyzed reaction proved broadly tolerant to the coupling partners employed (Scheme 1). With respect to the styrene partner (Scheme 1a), enantioselectivities are best with para-substituted styrenes, regardless of electronic character. Meta-substituted styrenes are also tolerated, affording good to high enantioselectivities, while a single ortho-substituted styrene leads to somewhat decreased selectivity. Styrene itself is a poor substrate, proceeding in modest selectivity and yield (4ab). Importantly, all substrates give the desired δ-lactam products as single regioisomers. Substitution on the acrylamide is well tolerated regardless of steric demand affording product with enantioselectivities that match the corresponding methacrylamide system (Scheme 1b). However, aryl- and alkoxy- substitution results in diminished yields (4ca, 4ea and 4ed).
Scheme 1.

a) Methyl acrylamide styrene scope. b) Extended acrylamide styrene scope.
A plausible catalytic cycle for this reaction28 is proposed in Scheme 2. Metalation of the amide by rhodium generates intermediate I. C-H activation occurs, presumably via a concerted-metalation deprotonation (CMD) mechanism, providing fivemembered rhodacycle II. A deuterium labeling experiment illustrates that the C-H activation step is reversible, suggesting that the concerted metalation/deprotonation (CMD) is not the turnover limiting step (Scheme 2b). Subsequent alkene coordination and migratory insertion would give seven-membered rhodacycle IV. Available evidence suggests this step is the enantiodetermining event. N-O bond cleavage and reductive elimination then occurs to form transient Rh(III) intermediate V. Protodemetallation regenerates the Rh(III) catalyst and closes the catalytic cycle.
Scheme 2.

a) Proposed mechanism. b) Labelling experiment. c) Mutational study.
While the Cp*biotinRhCl2 cofactor alone delivers the desired δ-lactam (4aa) with no appreciable selectivity (0% ee, Table 1, entry 2), its significantly reduced reactivity was a surprise (15% yield with 3 mol% catalyst loading, compared to 99% yield with an equivalent of the mSav artificial metalloenzyme). To begin to evaluate the molecular dictates of reactivity and stereocontrol, we determined that mutation of tyrosine 112 (Y112), which neighbors the putative Cp*biotinRh pocket has a signficant effect on the transformation. In comparison to the ArM featuring wt-mSav, the Y112A mutant provides the desired product in low yield and enantiomeric excess (37% and 61%, respectively). This observation is consistent with its likely role as a rigidifying element through π-stacking to the Cp framework on the catalyst.17,29 Despite significant decreases in reactivity and selectivity, Y112A maintains affinity for biotin (see Supporting Information).
In order to derivatize the resulting δ-lactam products into piperidines, the coupling of 2a and 3a to provide 4aa was performed at a 0.15 mmol scale providing identical results to the reaction performed on a 1.5 μmol scale (99% yield, 91% ee) (Scheme 3a). Hydrogenation of 4aa affords the reduced lactam 6aa in 99% yield and 10:1 dr. Subsequent reduction of 6aa with LiAlH4 furnishes the desired piperidine 5aa in 81% yield and 7:1 dr (Scheme 3).
Scheme 3.

a) Preparative scale δ-lactam synthesis and piperidine derivatization. b) Lactam scope. c) Piperidine scope.
Indeed, the derivatization can proceed under exceedingly mild reduction conditions. Treatment of a range of δ-lactams (6) formed in good diastereoselectivity following hydrogenation with BH3•SMe2 provides the corresponding piperidines in good yield and comparable diastereoselectivity to the LiAlH4 reduction (Scheme 3b and c). Notably, these reduction conditions are tolerant of ester functionalities (5fa) with a slight erosion of diastereoselectivity, likely due to competitive imine/enamine tautomerization of an intermediate.
In conclusion, we have developed an artificial metalloenzyme that efficiently catalyzes an enantioselective tandem C-H activation and [4+2] annulation reaction to afford δ-substituted lactams. This metalloenzyme accepts a diverse array of acrylamide and styrene coupling partners. The mSav metalloenzyme platform demonstrates superior reactivity relative to its tSav counterpart and the free cofactor alone. Efforts to understand the crucial factors responsible for improved reactivity are currently underway.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the Keck Foundation (T.R.). Partial support was provided by NIGMS (GM80442, T.R. and GM107520, B.R.M.). M.B. acknowledges the EU for a Marie Curie Fellowship. N.S. acknowledges the Royal Thai government for a DPST graduate scholarship. I.S.H. acknowledges the National Science Foundation for a predoctoral fellowship.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Detailed experimental procedures, characterization data, copies of 1H NMR and 13C NMR spectra for all isolated compounds (PDF)
REFERENCES
- (1).Vitaku E; Smith DT; Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency, of nitrogen heterocycles among U.S FDA approved pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (2).For a comprehensive recent review on piperidine synthesis, see: Nebe, M.M.; Opatz, T. Adv. Heterocycl. Chem. 2017, 122, 191–244. [Google Scholar]
- (3).Zhao X; Ruhl KE; Rovis T N-heterocyclic-carbene-catalyzed asymmetric oxidative hetero-Diels-Alder reactions with simple aliphatic aldehydes. Angew. Chem. Int. Ed. 2012, 51, 12330–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).White NA; DiRocco DA; Rovis T Asymmetric NHCCatalyzed Addition of Enals to Nitroalkenes: Controlling Stereochemistry Via the Homoenolate Reactivity Pathway To Access δ-Lactams. J. Am. Chem. Soc. 2013, 135, 8504–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Xu J; Jin Z; Chi YR Organocatalytic Enantioselective γ-Aminoalkylation of Unsaturated Ester: Access to Pipecolic Acid Derivatives. Org. Lett. 2013, 15, 5028–5031. [DOI] [PubMed] [Google Scholar]
- (6).Jia WQ; Chen X-Y; Sun L-H; Ye S Organocatalytic [4 + 2] cyclocondensation of α,β-unsaturated acyl chlorides with imines: highly enantioselective synthesis of dihydropyridinone and piperidine derivatives. Org. Biomol. Chem. 2014, 12, 2167–2171. [DOI] [PubMed] [Google Scholar]
- (7).Potter TJ; Kamber DN; Mercado BQ; Ellman JA Rh(III)-Catalyzed Aryl and Alkenyl C-H Bond Addition to Diverse Nitroalkenes. ACS Catal. 2017, 7, 150−153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Weilbeer C; Sickert M; Naumov S; Schneider C The Bronsted Acid-Catalyzed, Enantioselective Aza-Diels-Alder Reaction for the Direct Synthesis of Chiral Piperidones. Chem. Eur. J. 2017, 23, 513–518. [DOI] [PubMed] [Google Scholar]
- (9).Grigorjeva L; Daugulis O Cobalt-Catalyzed, Aminoquinoline-Directed Coupling of sp2 C-H Bonds with Alkenes. Org. Lett. 2014, 16, 4684–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sigman MS; Yuan Q Palladium-catalyzed enantioselective relay heck arylation of enelactams: Accessing a,b-unsaturated d-lactams. J. Am. Chem. Soc. 2018, 140, 6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Park Y; Chang S Asymmetric formation of γ-lactams via C-H amidation enabled by chiral hydrogen-bond-donor catalysts. Nat. Catal 2019, DOI: 10.1038/s41929-019-0230-x. [DOI] [Google Scholar]
- (12).(a) For selected reviews, see: Colby, D. A; Bergman RG; Ellman JA Rhodium-catalyzed C-C bond formation via heteroatom-directed C-H bond activation. Chem. Rev 2010, 110, 624–655; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Satoh T; Miura M Oxidative Coupling of Aromatic Substrates with Alkynes and Alkenes under Rhodium Catalysis. Chem. Eur. J 2010, 16, 11212–11222; [DOI] [PubMed] [Google Scholar]; (c) Patureau FW; Wencel-Delord J; Glorius F Cp*Rh-Catalyzed C-H Activations: Versatile Dehydrogenative Cross-Couplings of Csp3 C-H Positions with Olefins, Alkynes, and Arenes. Aldrichimica Acta 2012, 45, 31; [Google Scholar]; (d) Song GY; Wang F; Li XW, C-C, C-O, and C-N bond formation via rhodium(III)-catalyzed oxidative C-H activation. Chem. Soc. Rev 2012, 41, 3651–3678; [DOI] [PubMed] [Google Scholar]; (e) Song GY; Li XW Substrate activation strategies in rhodium (III)-catalyzed selective functionalization of arenes. Acc. Chem. Res 2015, 48, 1007–1020; [DOI] [PubMed] [Google Scholar]; (f) Zhu C; Wang R; Falck JR Amide-Directed Tandem C-C/C-N Bond Formation though C-H activation. Chem. Asian. J 2012, 7, 1502–1514; [DOI] [PubMed] [Google Scholar]; (g) Gensch T; Hopkinson MN; Glorius F; Wencel-Delord J Mild metal-catalyzed C-H activation; examples and concepts. Chem. Soc. Rev, 2016, 45, 2900; [DOI] [PubMed] [Google Scholar]; (h) Gulías, M.; Mascareñas, J. L. Metal-Catalyzed Annulations though Activation and Cleavage of C-H bonds. Angew. Chem. Int. Ed. 2016, 55, 11000–11019. [DOI] [PubMed] [Google Scholar]
- (13).Guimond N; Gorelsky SI; Fagnou K Rhodium(III)-catalyzed heterocycle synthesis using an internal oxidant: improved reactivity and mechanistic studies. J. Am. Chem. Soc. 2011, 133, 6449–6457. [DOI] [PubMed] [Google Scholar]
- (14).Romanov-Michailidis F; Sedillo KF; Neely JM, Rovis T Expedient Access to 2,3-Dihydropyridines from Unsaturated Oximes by Rh(III) Catalyzed C-H Activation. J. Am. Chem. Soc. 2015, 137, 8892–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Grigorjeva L; Daugulis O Cobalt-Promoted Dimerization of Aminoquinoline Benzamides. Org. Lett. 2015, 17, 1204–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Heinisch T; Ward TR;. Artificial Metalloenzymes Based on the Biotin-Streptavdidin Technology: Challenges and Opportunities. Acc. Chem. Res. 2016, 49, 1711–1721. [DOI] [PubMed] [Google Scholar]
- (17).Hyster TK; Knorr L; Ward TR; Rovis T Biotinylated Rh(III) complexes in engineered streptavidin for accelerated asymmetric C-H activation. Science 2012, 338, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ye B; Cramer N Chiral Cyclopentadienyl Ligands as Stereocontrolling Element in Asymmetric C–H Functionalization. Science 2012, 338, 504–506. [DOI] [PubMed] [Google Scholar]
- (19).Wilson ME; Whitesides GM Conversion of a Protein to a Homogeneous Asymmetric Hydrogenation Catalyst by Site-Specific Modification with a Diphosphinerhodium(I) Moiety. J. Am. Chem. Soc. 1978, 100, 306–307. [Google Scholar]
- (20).Schwizer F et al. , Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (21).Rakshit S; Grohmann C; Besset T; Glorius F Rh(III)-catalyzed directed C-H olefination using an oxidizing directing group: mild, efficient, and versatile. J. Am. Chem. Soc. 2011, 133, 2350–2353. [DOI] [PubMed] [Google Scholar]
- (22).The solid acrylamide needs to be administered as a solution in methanol. Both the acrylamide and styrene are only slightly soluble in water. The metalloenzyme reaction starts out clear in appearance and gradually turns cloudy as the reaction progresses. [Google Scholar]
- (23).For optimization of the racemic reaction in organic solvent, see SI.
- (24).Demonte D; Dundas CM; Park S Expression and purification of soluble monomeric streptavidin in Escherichia coli. Appl. Microbiol. Biotechnol. 2014, 98, 6285–6295. [DOI] [PubMed] [Google Scholar]
- (25).Wu SC; Wong SL Engineering soluble monomeric streptavidin with reversible biotin binding capability. J. Biol. Chem. 2005, 280, 23225–23231. [DOI] [PubMed] [Google Scholar]
- (26).Qureshi MH; Wong SL Design, production, and characterization of a monomeric streptavidin and its application for affinity purification of biotinylated proteins. Protein Expr. Purif. 2002, 25, 409–415. [DOI] [PubMed] [Google Scholar]
- (27).Conversion of acrylamide substrate is typically >90%. In parallel studies, we have observed products derived from Lossen type rearrangement of the hydroxamate leading to low molecular weight and water soluble by-products. Excess styrene is left unreacted. [Google Scholar]
- (28).Piou T; Rovis R Electronic and Steric Tuning of a Prototypical Piano Stool Complex: Rh(III) Catalysis for C-H Functionalization. Acc. Che. Res 2018, 51, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hunter CA; Sanders JKM The nature of π-π interactions. J. Am. Chem. Soc, 1990, 112, 5525–5534. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.