

Abstract
The Cdc48 ATPase (p97 or VCP in mammals) and its cofactor Ufd1/Npl4 extract poly-ubiquitinated proteins from membranes or macromolecular complexes for subsequent degradation by the proteasome. How Cdc48 processes its diverse and often well-folded substrates is unclear. Here, we report cryo-EM structures of the Cdc48 ATPase in complex with Ufd1/Npl4 and poly-ubiquitinated substrate. The structures show that the Cdc48 complex initiates substrate processing by unfolding a ubiquitin molecule. The unfolded ubiquitin molecule binds to Npl4 and projects its N-terminal segment through both hexameric ATPase rings. Pore loops of the second ring form a staircase that acts as a conveyer belt to move the polypeptide through the central pore. Inducing the unfolding of ubiquitin allows the Cdc48 ATPase complex to process a broad range of substrates.
One sentence summary
The Cdc48 ATPase initiates processing of diverse poly-ubiquitinated substrates by unfolding and translocating an attached ubiquitin molecule.
The Cdc48 ATPase in Saccharomyces cerevisiae and its p97 (VCP) orthologs in metazoans play essential roles in many cellular processes (1–3). The Cdc48 ATPase generally separates individual, poly-ubiquitinated targets from membranes or binding partners. Its central role in protein quality control is highlighted by mutations in p97, which can cause several neurodegenerative proteopathies (4–6). Cdc48 (p97) often acts upstream of the 26S proteasome. For example, in endoplasmic reticulum (ER)-associated protein degradation (ERAD), Cdc48 (p97) extracts poly-ubiquitinated, misfolded proteins from the ER and transfers them to the proteasome (7). In contrast to the proteasome (8), Cdc48 (p97) does not require flexible polypeptide segments in its substrates. The proteasome uses such segments to initiate substrate processing: it inserts them into the center of the hexameric ATPase ring of the 19S regulatory particle, so that the ATPases can move the polypeptide into the proteolytic 20S particle (9, 10). In most scenarios, Cdc48 (p97) requires a poly-ubiquitin chain, but the chain can be attached even to well-folded substrates that are inappropriate for direct degradation by the proteasome (11, 12). How Cdc48 (p97) can process a broad range of folded proteins without an unstructured segment from which to initiate unfolding is unclear. One hypothesis is that a ubiquitin molecule serves as a universal initiating signal for translocation into the pore, regardless of the substrate to which it is attached (13).
Cdc48 (p97) belongs to the ATPases associated with diverse cellular activities (AAA) family of ATPases. It consists of an N-terminal (N) domain and two ATPase domains (D1 and D2) (14, 15) (Fig. 1A). Six monomers form a double-ring structure with a central pore. The N domains are located on one side of the double-ring (“cis”-side) and can be either in an “up-conformation” above the D1 ring, or in a “down-conformation” co-planar with the D1 ring (14, 15). Cdc48 binds various cofactors, which determine substrate specificity, target the ATPase to different cellular locations, or modify the ubiquitin chain attached to the substrate (16). One of the most important cofactors is the heterodimeric Ufd1/Npl4 (UN) complex, which allows Cdc48 to process substrates with Lys48-linked ubiquitin chains (17). Npl4 consists of an N-terminal Ubx-like domain (UBXL), two Zn2+-finger domains (zf-Npl4), an MPN domain with sequence similarity to isopeptidases, including the proteasomal de-ubiquitinase (DUB) Rpn11, and a C-terminal domain (CTD) (13, 18) (Fig. 1A). Mammalian (but not yeast) Npl4 contains an additional C-terminal Zn2+-finger domain that binds ubiquitin (19). Npl4 forms a tower on the cis-side of Cdc48, above the entrance into the D1 pore (13). Ufd1 consists of a ubiquitin-binding UT3 domain and a flexible UT6 domain that contains an Npl4 binding site and two short SHP motifs that interact with Cdc48 (Fig. 1A).
Fig. 1. Generation and cryo-EM analysis of a substrate-engaged Cdc48 ATPase complex in ATP.

(A) Domain structures of Cdc48, Ufd1, and Npl4. (B) Scheme showing the step-wise assembly of the ATPase complex. The poly-ubiquitinated Eos substrate was generated as in fig. S1A, bound via a SBP tag to streptavidin beads, and incubated sequentially with Ufd1/Npl4 cofactor and Cdc48. After incubation with different nucleotides, the complex was eluted with biotin. (C) Complex generated with ATP and Cdc48 carrying a Walker B mutation in D2 (E588Q). The eluted complex was analyzed by SDS-PAGE and Coomassie blue staining. (Ub)n, poly-ubiquitin. (D) Cryo-EM reconstruction of the complex in two different views. The domains of Cdc48, cofactor, and ubiquitin molecules attached to substrate are shown in different colors. The subunits in the hexameric ATPase ring are labeled A-F. The refined, unsharpened map is shown in transparent grey over the final composite map sharpened with a B-factor of −150. The densities for the N domains, Npl4’s UBXL domain, and Ufd1’s UT6 domain are from the unsharpened map.
Substrate is initially recruited to the ATPase complex by the binding of the poly-ubiquitin chain to the UN cofactor (20). The cofactor binds Lys48-linked ubiquitin chains, although more complex chains are also recognized (21). A minimum of five Lys48-linked ubiquitins is required for efficient binding (22). Biochemical experiments show that Cdc48 then uses ATP hydrolysis in the D2 domains to move the polypeptide through its central pore, thereby unfolding the substrate (22). The D2 domains contain conserved pore loops with canonical aromatic residues, which are required for polypeptide translocation by Cdc48 and related ATPases, such as the eukaryotic 19S proteasome or the bacterial Clp ATPases (23). The D1 domains of Cdc48 (p97) lack the aromatic residues in their pore loops and only need to bind, but not hydrolyze, ATP for substrate unfolding to occur (21, 22). It is unclear how a folded substrate can reach through the D1 ring and contact the D2 pore loops that power translocation.
The mechanism by which the Cdc48 (p97) ATPase complex processes its poly-ubiquitinated substrates remains poorly understood. p97 in isolation has a narrow or even occluded central pore, which led to the assumption that polypeptides cannot pass through it (24). Nevertheless, archaeal Cdc48 homologs and certain mutants of p97 seem to be able to translocate polypeptides through their pore into the interior of the proteolytic chamber of the 20S proteasome (25, 26). However, these substrates were peptides and non-ubiquitinated proteins. Furthermore, unlike the eukaryotic protein, archaeal Cdc48 has canonical pore loops in both D1 and D2. A cryo-EM structure of an archaeal Cdc48 complex showed a polypeptide chain inside the pore, which originated from another Cdc48 molecule (27).
Here, we use cryo-electron microscopy (cryo-EM) to determine structures of the Cdc48 ATPase in complex with its UN cofactor and poly-ubiquitinated substrate. The structures reveal how the ATPase initiates the processing of a broad range of folded substrates and how it translocates polypeptides through its hexameric double-ring.
Architecture of a substrate-engaged Cdc48 complex
To obtain structures of a functional Cdc48 complex, we purified proteins of S. cerevisiae, for which substrate processing has been demonstrated in vitro (22). We assembled the complex in a stepwise manner (Fig. 1B). First, we generated a poly-ubiquitinated substrate in vitro (fig. S1A) (22). The substrate contains a single chain of 6–13 Lys48-linked ubiquitin molecules. The chain is attached to a polypeptide segment preceding the fluorescent mEos3.2 (Eos) protein and a streptavidin-binding peptide (SBP). The poly-ubiquitinated substrate was bound to streptavidin beads, incubated with S. cerevisiae UN, and then with Cdc48 and nucleotides (Fig. 1B). After elution with biotin (Fig. 1C and fig. S1B), the sample was analyzed by cryo-EM.
We first determined a structure in the presence of ATP. To favor an early stage of substrate processing, we used a slowly translocating Cdc48 mutant with a Walker B mutation in D2 (E588Q) (22). Under the conditions used, the fluorescence of photo-converted Eos remained unchanged, indicating that the protein remained folded (22). Thus, the state analyzed corresponds to Eos being localized to the cis-side of the Cdc48 complex, before being pulled through the central pore. The sample was then subjected to cryo-EM single-particle analysis. 3D classification and refinement resulted in an overall density map at 3.9 Å resolution. The map was locally refined to 3.8 Å for the D1 ring and Npl4 tower, and to 4.3 Å for the D2 ring (Fig. 1D; fig. S2; table S1). Models were built for the ATPase subunits of the D1 and D2 rings and for the central tower formed by Npl4. As in the Chaetomium thermophilum Cdc48 complex lacking substrate (13), Npl4’s tower is anchored by its two Zn2+-finger domains and an N-terminal bundle (NTB) domain to the D1 ATPase ring (fig. S3). The ß-strand finger, a stalk emerging from the MPN domain, is positioned above the pore in the substrate-free structure, but is moved away from the pore axis in the substrate-bound structure (fig. S3). Density for Cdc48’s N domains and Npl4’s UBXL domain are of low local resolution (Fig. 1D). As in the C. thermophilum complex, the N domains are all in the “up-conformation,” and the UBXL domain provides an additional Cdc48 anchor point for Npl4. Ufd1 is associated with Npl4 through its UT6 region (17), for which density is visible but not of sufficient resolution for modeling. No density for the folded UT3 domain of Ufd1 was visible, as in the previous structure lacking substrate (13). This domain is likely flexible, consistent with its ability to crosslink promiscuously to different regions of the complex (13).
Interaction of the Cdc48 complex with poly-ubiquitin
Density for the substrate includes two folded ubiquitins bound at the top of the Npl4 tower (Ub1 and Ub2), and an unfolded ubiquitin molecule (unUb) extending from one of the folded ubiquitins through a groove in Npl4 all the way through both ATPase rings (Fig. 2A). The folded ubiquitin molecules fit unambiguously as rigid bodies into the map. The C-terminus of Ub2 projects towards the Lys48 of Ub1, suggesting that these ubiquitin molecules are linked (Figs. 2B, C). The arrangement of the linked ubiquitin molecules is much less compact than in a crystal structure of Lys48-linked oligo-ubiquitin (28). The solvent-exposed position of Lys48 of Ub2 suggests that it could be linked to yet another ubiquitin molecule. The juxtaposition of the C-terminus of Ub1 to Lys48 of unUb and density for the branch point (Figs. 2C, D) indicate that these ubiquitin molecules are linked through an isopeptide bond. unUb traverses the Npl4 groove with two largely linear segments joined by a prominent kink (Fig. 2A). Further identification of unUb is based on short, bulky density for Phe45 (Fig. 2D), elongated density for Arg42 (Fig. 2E), the location of Pro38 and Pro37 at the kink (Fig. 2E), and helical density corresponding to the segment of Asp32 to Ile23, which also forms a helix in folded ubiquitin (Fig. 2F). This helix interacts with Npl4 through residues that are pointing into the interior in folded ubiquitin (Fig. 2F). Residues preceding Ile23 are located in the D1 and D2 rings, with the N-terminus positioned inside D2.
Fig. 2. Ubiquitin molecules bound to the Npl4 cofactor of Cdc48.

(A) Side view of the density map of the Cdc48 complex, with subunit E removed to show the interior of the central pore. Two folded ubiquitin molecules (Ub1 and Ub2) are shown in green and an unfolded ubiquitin molecule (unUb) in yellow. (B) Top view of the map, with the Npl4 tower in the foreground. (C) Magnified view of the boxed area in (B). Density for the folded ubiquitins is shown in transparent grey. Models for folded ubiquitin models (PDB 1UBQ), fit as rigid bodies into the map, are in green ribbon presentation, and unfolded ubiquitin in yellow ribbon presentation. The C-terminal flexible tails of folded ubiquitins were fit into the map. The position of Lys48 in each ubiquitin is indicated. (D) Magnified view of the density enclosed by a broken line in (C), together with a model of unfolded ubiquitin. Prominent features used for modeling are indicated. (E) Magnified view of the kink in unfolded ubiquitin indicated in the boxed area in (A), together with a model. (F) Superposition of the helix I23-D32 in unfolded ubiquitin (yellow) with the corresponding helix in folded ubiquitin, both in ribbon presentation. The segment bound to the Npl4 groove is shown in green. Some residues in the helix face Npl4 in the complex (Ile23, Val26, Ile30), while others face away (Lys27, Asn25).
The unUb-binding Npl4 groove is lined with conserved amino acid residues (Fig. 3A). We mutated several of these residues and tested whether they affect the ability of the Cdc48 complex to unfold substrate. To this end, the fluorescent substrate was irradiated, resulting in cleavage of Eos into two associated polypeptide segments; these segments separate irreversibly upon Cdc48-mediated unfolding, resulting in the loss of fluorescence (22). Several mutations in the Npl4 groove reduced unfolding without affecting the association of Npl4 with Ufd1 (Fig. 3B–D; fig. S4A–B), and even stronger inhibition was seen with multiple mutations. The observed effects are consistent with our structural model for the interaction between Npl4 and unUb, in which Tyr461 of Npl4 interacts with Val26 and Ile23 of unUb, Phe349 intercalates between Ile30 and Gln31 of unUb, Lys165 interacts with Glu34 of unUb, Ile249 interacts with Phe45, and Arg253 interacts with the backbone of Ala46 of unUb (fig. S4C–F). Substrate binding to the Npl4 groove is also supported by hydrogen/deuterium exchange (HDX) mass spectrometry (MS) experiments: several peptides derived from segments forming the groove were protected from HDX in the presence of substrate (Fig. 3E; fig. S5). Some other regions of Npl4 were also protected in the presence of substrate, including the top of the Npl4 tower, which interacts with Ub1 (Fig. 3E; fig. S5). The protection of the Zn2+-fingers in the presence of substrate (Fig. 3E) may be caused indirectly by strengthening the interaction of Npl4 with Cdc48, as substrate is not close to these regions.
Fig. 3. Binding of unfolded ubiquitin to a conserved Npl4 groove.

(A) Surface model of the Npl4 tower with residues colored according to the degree of their conservation, as calculated by the ConSurf Server. The conserved groove to which unfolded ubiquitin binds is enclosed by a broken line. The positions of the b-strand finger and Zn2+-finger 1 (ZF-1) are indicated. (B) The highlighted conserved residues in the Npl4 groove, colored according to their position, were mutated to test their effect on substrate unfolding by the Cdc48 ATPase complex. (C) UN cofactor containing wild-type Ufd1 and either wild-type (WT) or the indicated Npl4 mutant was purified (fig. S4) and tested together with Cdc48 for unfolding of poly-ubiquitinated, photo-converted Eos, measured as loss of fluorescence. The curves show the mean and standard deviation of three replicates. (D) Experiments as in (C) were done with selected Npl4 mutants and controls. The initial unfolding rates were determined and normalized to that of WT. Shown are the mean and standard deviation of three replicates. (E) Regions of Npl4 protected by substrate against HDX in the Cdc48/UN complex (in blue). HDX MS was performed with Cdc48/UN/ADP/BeFx in the absence or presence of poly-ubiquitinated substrate at different time points (fig. S5). (F) As in (E), but in the absence of Cdc48.
The HDX MS also showed that peptides of Ufd1’s UT6 domain are protected by substrate (figs. S6; S7), likely the segments for which density is seen next to Ub1 (Fig. 1C). The invisible UT3 domain also showed substrate protection in several regions (figs. S6; S7), consistent with NMR data indicating that UT3 binds both ubiquitin and poly-ubiquitin (29). Taken together, the UN cofactor binds one unfolded ubiquitin and 3–4 folded ubiquitin molecules. If UN binds 5 ubiquitins, this would explain the minimum chain length required for stable binding (22).
The Npl4 tower contains an MPN domain homologous to that of the proteasomal DUB Rpn11 and other isopeptidases (30). However, Npl4’s MPN domain is enzymatically inactive, and the cleft that accommodates the C-terminus of a ubiquitin molecule in Rpn11 (31) is blocked by the UT6 region of Ufd1 (Fig. 1D) (13). Rpn11 lacks the groove that is seen in Npl4. Thus, despite structural similarities between Rpn11 and Npl4, they interact differently with ubiquitin. The UN complex alone can bind ubiquitin chains (32), but the presence of Cdc48 seems to be required for ubiquitin unfolding; HDX MS experiments performed in the absence of Cdc48 showed substrate protection of Npl4 at its top, where folded ubiquitin binds, but not in the groove, where unfolded ubiquitin binds (Fig. 3F; fig. S5).
The N-terminus of unUb reaches the D2 ring in the absence of ATP binding or hydrolysis
To confirm that the N-terminus of unUb is located inside the D2 ATPase ring, we performed photo-crosslinking experiments. Asp602 in the D2 domain of FLAG-tagged Cdc48 (Cdc48-FLAG) was replaced by the photo-reactive amino acid p-benzoylphenylalanine (Bpa) using amber codon suppression (33, 34). According to the structure, the Asp602 residues of several D2 subunits are close to the N-terminus of unUb (Fig. 4A).
Fig. 4. Insertion of the N-terminus of ubiquitin into the D2 ring of Cdc48.

(A) Side view of the model of the Cdc48 complex with the unfolded ubiquitin molecule in yellow. The boxed region is magnified to show the N-terminus unfolded (Ub NT; in yellow) in proximity of Asp602 residues (sticks) of several D2 subunits (in ribbon presentation). (B) Cdc48-FLAG containing Bpa at position 602 was incubated with UN and dye-labeled, poly-ubiquitinated sfGFP (Dy-Ub(n)-sfGFP) in the presence of ADP or ATP. The sample was irradiated, as indicated, Cdc48-FLAG and crosslinked products were isolated with FLAG-antibody beads, and analyzed by SDS-PAGE and fluorescence scanning. Where indicated, the samples were treated with the DUB Usp21 before analysis. The stars indicate crosslinks between Dy-Ub(n)-sfGFP and one or more Cdc48 molecules. The intensity of the crosslinks was quantitated and normalized to that of the sample in ATP (numbers underneath the lanes). The lower panel shows an immunoblot using Cdc48 antibodies. (C) As in (B), but in the presence of ADP, and analysis of the crosslinked products by nano-liquid chromatography/mass spectrometry after trypsin digestion. The indicated peptides of ubiquitin and Cdc48 were crosslinked through Bpa (B). Colored glyphs highlight fragment ions detected in the MS/MS spectrum, and indicate that the Bpa was crosslinked to either Met1 or Gln2 of ubiquitin (fig. S8).
Cdc48-FLAG containing Bpa at position 602 was incubated in the presence of ADP or ATP with poly-ubiquitinated super-folder GFP that was labeled with DyLight800 (Dy-Ub(n)-sfGFP). After UV irradiation, Cdc48-FLAG and crosslinked products were recovered with FLAG-antibody beads, and the bound material analyzed by SDS-PAGE and fluorescence scanning. Crosslinked products were only seen when the UN complex was present and the samples were irradiated (Fig. 4B; lanes 3 and 6 versus lanes 1, 2, 4, 5). The major products correspond to one Dy-Ub(n)-sfGFP molecule crosslinked to one or more Cdc48 molecules. Crosslinks were strongest in ATP (lane 6). Although we were not able to detect crosslinks in ADP in our previous experiments (22), we now observe crosslinks in ADP (lane 3), perhaps owing to improved substrate and enzyme preparations. These results are consistent with those of others (35). When the ADP sample was treated with a non-specific DUB (Usp21), most of the label was lost (lane 9 versus lane 3; 84% reduction), indicating that crosslinking occurred primarily to ubiquitin; the remaining fluorescence is likely due to incomplete DUB cleavage. The ATP sample retained a higher percentage of fluorescence after DUB treatment (lane 12), consistent with some dye-labeled sfGFP being pulled into the central pore and forming crosslinks to D2.
To directly test whether the N-terminus of unUb is located in proximity to position 602 of Cdc48, we performed photo-crosslinking experiments in the presence of ADP and subjected the samples to trypsin digestion, followed by nano LC-MS/MS analysis (fig. S8). The results show that the Bpa residue at position 602 of Cdc48 crosslinked to Met1 or Gln2 of ubiquitin (Fig. 4C). No other crosslinks between tryptic peptides of Cdc48 and ubiquitin were detected, and the crosslinked product was absent from samples generated in the absence of UN or irradiation (fig. S9). Together, these results show that the N-terminus of unUb inserts across both ATPase rings without the need of ATP binding or hydrolysis.
Mechanism of polypeptide translocation
In the D1 ring, the unUb polypeptide interacts with only two ATPase subunits. Contact is made by both subunits through Met288 in the pore loop (Fig. 5A, B). All subunits of the D1 ring seem to be in the ATP-bound state (Fig. 5A; fig. S10), as judged from the density for the bound nucleotide and Arg finger of the neighboring subunit, as well as the distance of the Arg to the nucleotide (fig. S10). The D1 ring is almost planar, allowing interaction of the Npl4 tower through its Zn2+-finger and NTB domains (fig. S3C). In the D2 ring, the extended polypeptide is contacted by the canonical aromatic pore loop residues of four ATPase subunits (labeled B-E in Fig. 5C). These subunits form a staircase and are positioned to propel the substrate to the trans-side of Cdc48 (Fig. 5C). The three highest substrate-engaged D2 domains B-D are in the ATP-bound state, and domain E seems to be in the ADP-bound state (fig. S10). The substrate-disengaged domains A and F are in the ADP-bound or nucleotide-free state, as there is no clear density for the nucleotide and their nucleotide-binding sites are too far away from the respective Arg-fingers of the neighboring domains to bind ATP (fig. S10). The D1 and D2 domains both behave as rigid bodies (fig. S11). Superposition of the Cdc48 monomers on the basis of the D1 domains shows that the staircase arrangement of the D2 domains is caused by rigid body movements relative to D1 (fig. S11B). The angle between the D1 and D2 subunits changes dramatically from subunits B to E, causing a displacement of the D2 pore loop by more than 18Å (fig. S11B).
Fig. 5. The N-terminal segment of unfolded ubiquitin in the central pore of the ATPase rings.

(A) Top view of the density map of the substrate-engaged Cdc48 complex in ATP, cut to the surface of the D1 ring. Substrate density is shown in yellow. The D1 ATPases are colored individually and labeled A-F. Density for bound nucleotides is shown in green. (B) As in (A), but cut to the surface of the D2 ring. (C) The upper panel shows a cut-away side view of the map, with unUb in yellow. The boxed areas in D1 and D2 are magnified in the lower panels. Amino acids of the pore loops contacting the polypeptide chain are indicated.
To better understand the mechanism by which the polypeptide chain is moved through the ATPase rings, we determined cryo-EM structures of the substrate-engaged Cdc48 complex in the presence of ADP and the phosphate mimic BeFX. The presence of both ADP and BeFx locks the ATPase subunits into an early stage of ATP hydrolysis, whereas ADP alone corresponds to a state after ATP hydrolysis. The sample was prepared as before (fig. S1B) and subjected to cryo-EM analysis. After image processing, two major classes were obtained – one at an overall resolution of 4.1Å with a visible Npl4 tower (ADP/BeFx-1) (figs. S12–S14; table S1), the other (ADP/BeFx-2) at 3.6Å, where the density for the cofactor was averaged out. In both structures, the density for some ATPase subunits was weak (fig. S15), indicating that they are conformationally heterogeneous in the hexameric rings.
The ADP/BeFx-2 structure shows how the D2 ATPases translocate a polypeptide chain (Fig. 6A). Five of the subunits (A-E) contact the polypeptide and form a pronounced staircase with subunit A on top and subunit E on the bottom (Fig. 6B). Subunits B-E and their pore loops are at approximately the same positions as in the ATP structure (Fig. 6C). Subunit A is added at the top of the staircase, while subunit F has weak density and is thus likely flexible and disengaged (fig. S14). D2 subunits A-D have both ADP and BeFX bound, and are thus close to the ATP-bound state, while subunit E is probably in the ADP-bound state. The flexible subunit F may not be able to bind nucleotide and is thus probably in the nucleotide-free state (fig. S16). All substrate-engaged subunits contact the polypeptide through the Trp561 and Tyr562 residues in their pore loops (Fig. 6B). These residues “pinch” every other peptide bond of the extended polypeptide substrate. Thus, each power stroke of the hexameric ATPase moves two amino acids of the substrate, and the intercalation of pore loops between side chains allows translocation of a polypeptide regardless of its specific amino acid sequence. In the ADP/ BeFx −1 structure, the D2 pore loops of subunits B-E are again at approximately the same positions as in the other two structures (Fig. 6C). However, subunit A is conformationally heterogeneous and a model cannot be built; it is disengaged from the substrate. This subunit is probably in the nucleotide-free state and is caught at a state prior to rejoining the staircase at the top (fig. S17; Fig. 6C).
Fig. 6. Structures of substrate-engaged Cdc48 complex in ADP/BeFx reveal translocation mechanism.

(A) Structures of the substrate-engaged Cdc48 complex were determined in the presence of ADP and BeFx as in Fig. 1. Shown is a side view of a ribbon-diagram model built into the higher resolution map (ADP/BeFx-2), together with density for the substrate shown in blue mesh. (B) Magnified, 90°-rotated view of the boxed region ii in (A), showing the D2 pore loop residues that contact the polypeptide. The pore loops are labeled by the ATPase subunits to which they belong. (C) Comparison of the positions of the substrate-engaged D2 pore loops in the three structures, aligned on the basis of subunit B. Note that subunit A is only engaged in ADP/BeFx-2. (D) Model for polypeptide translocation. Subunit A binds ATP and moves to the top of the staircase. The previously engaged subunits move downwards, dragging the polypeptide with them by interacting with every other peptide bond of the substrate. Subunit E hydrolyzes ATP before its disengagement. The disengaged subunit, F, then moves back to the top and starts a new cycle. (E) Magnified view of the boxed region i in (A), with Met288 in the D1 pore loops highlighted. (F) Comparison of the position of the D1 pore loops in the three structures. Note that the pore loops are almost planar in ATP (blue) and arranged as a staircase in ADP/BeFx (green and red).
Together, our structures reveal that the D2 ATPases use a “conveyor belt” mechanism to translocate a polypeptide chain through the central pore (Fig. 6D). In this model, subunit A binds to the top of the polypeptide chain when it converts from the ADP-bound or nucleotide-free state to the ATP-bound state. The subunits then move downwards, dragging the polypeptide chain with them. At the lowest substrate-engaged position, subunit E hydrolyzes ATP. Subsequently, in the ADP-bound or nucleotide-free state, subunit F disengages from the substrate, so that it can move to the top position and start a new cycle. A similar “conveyor belt” mechanism is used by other single- or double-ring ATPases (9, 10, 36–38). However, Cdc48 (p97) is unique in that each substrate-gripping pore loop contains two bulky aromatic residues (Trp-Tyr or Trp-Phe), whereas the others contain only one. The added contact may allow Cdc48 (p97) to exert more force to unfold its substrates.
In the ADP/BeFx structures, four D1 domains contact the polypeptide through their Met288 residues (Figs. 6E, fig. S15). All visible D1 domains have bound ADP/BeFx or ADP, but the flexible ones probably cannot bind nucleotide and may thus be in the nucleotide-free state (fig. S16; S17). The unfolded ubiquitin segment inside the D1 pore is located at a defined position, suggesting binding to the D1 pore residues. An interaction of the polypeptide with the D1 ring is also supported by HDX MS experiments. In the presence of ADP, essentially all regions of the D1 subunits were protected, whereas the N and D2 domains remained unaffected (fig. S18). In the presence of ADP/BeFx, substrate protected both ATPase rings (fig. S18), likely because now the substrate engaged the D2 pore loops as well.
In both ADP/BeFx structures, the D1 ATPase pore loops have a pronounced staircase arrangement (Fig. 6E, F), more pronounced than in our ATP structure and previously solved structures (13–15). The staircase arrangement in the D1 ring may explain why in the ADP/BeFx-2 structure one of the two Zn2+-finger domains of the Npl4 tower is not visible in the density map (fig. S14C). The staircase arrangement of the D1 domains seems to be caused by that of the D2 domains, as a superposition of the six subunits on the basis of D1 shows that there is no relative movement between the D1 and D2 domains (fig. S11C, D). Thus, ADP/BeFx fixes the interaction between D1 and D2.
Discussion
Our structures reveal how the Cdc48 ATPase can process a broad range of poly-ubiquitinated substrates that are well-folded, associated with binding partners, or integrated into membranes. These substrates are first recruited by binding of the poly-ubiquitin chain to Cdc48’s cofactor UN. The UN complex has several binding sites for folded ubiquitin molecules. Our structure shows two sites at the top of the central Npl4 tower, and previous studies (29) and our HDX experiments indicate one or two additional binding sites in the UT3 domain of Ufd1. However, surprisingly, one additional ubiquitin molecule is unfolded, bound to a conserved groove of Npl4 and inserted into the Cdc48 pore. Unfolding of ubiquitin is triggered simply by its binding to the Cdc48 complex and does not require ATP binding or hydrolysis by the ATPase. Binding to the Npl4 groove might provide the major driving force for unfolding, but the presence of Cdc48 is required and there may also be contributions from the interactions of the neighboring folded ubiquitins with Npl4 or Ufd1. The UN cofactor alone can bind folded ubiquitin molecules (32), but does not unfold ubiquitin. Unfolding of ubiquitin is remarkable, given that ubiquitin is an extremely stable protein that can survive boiling. Among hundreds of ubiquitin binding partners, the Cdc48 complex is the only one known to unfold ubiquitin.
Npl4 binds a segment of folded ubiquitin, which precedes Lys48 and includes a b-strand and an irregular loop. Atomic force microscopy experiments have shown that pulling on Lys48 readily unfolds ubiquitin (39), and unfolding might follow a similar trajectory when Npl4 locks unUb’s Lys48 residue in place by binding to Ub1. Furthermore, molecular dynamics simulations indicate that separation of the N-terminus represents the first unfolding intermediate of ubiquitin (40), and Npl4-mediated unfolding might occur by a similar mechanism. Indeed, insertion of the N-terminus into the central pore and engagement of the D1 domains seems to be required for the unfolding of ubiquitin. Once the N-terminal ubiquitin segment is bound to the Npl4 groove and inserted into the pore, most of the core b-sheet structure of the ubiquitin molecule would be disrupted (40), consistent with the lack of density for the C-terminal part of unUb in our structures. Notably, the a-helix of ubiquitin persists both in our structure and in simulated late unfolding intermediates (41).
After insertion, the N-terminal ubiquitin segment is placed in a position to engage the D2 pore loop residues. The pore loops can now use ATP hydrolysis to pull on the N-terminal segment. Engagement of the D2 domains may explain why their ATPase activity is stimulated by substrate addition (22). Inducing the unfolding of a ubiquitin molecule, inserting its N-terminal segment across both ATPase rings, and pulling on the N-terminal segment provides a universal initiation mechanism for substrate processing by the Cdc48/UN complex. Cdc48 and its metazoan homolog p97 or VCP also cooperate with other cofactors, and it remains to be seen whether they function similarly to the UN complex. At least in one case, a different mechanism must be in place, because the substrate is unmodified and yet pulled through the central pore (35).
The substrate-moving D2 pore loops are arranged in a staircase and act as a “conveyor belt” to pull on every other peptide bond of the polypeptide. Pulling is powered by a rotary mechanism of ATP binding, hydrolysis, and probably nucleotide release in the six D2 ATPase domains (Fig. 6D). Each D2 pore loop of Cdc48 (p97) contains two bulky aromatic residues, both of which interact with the peptide bond of the substrate. This arrangement is unique, as other studied hexameric ATPases contain only one substrate-interacting aromatic residue. The extra contact could provide added grip strength relative to other AAA proteins, as suggested by experiments in which an aromatic residue was added to the pore loop of the ClpX ATPase (42). The increased grip strength might help Cdc48 (p97) deal with difficult substrates. Interestingly, adding aromatic pore residues to the D1 domain causes cell lethality (43), suggesting that too strong a grip may also be deleterious.
Our structures lead to a model with important implications for the steps following initiation (Fig. 7). The linkages of the Npl4-bound ubiquitin molecules in our structures indicate that they are distal to the substrate attachment site. Because 3–4 folded ubiquitins are required for substrate recognition, the unfolded ubiquitin must be at least 3–4 subunits away from the distal end of the chain. However, unfolding and thus initiation of translocation can occur with any of the more proximal ubiquitin molecules, and not necessarily with the one directly attached to the substrate (the scheme in Fig. 7 indicates a situation in which there is one ubiquitin in between). When Cdc48 pulls on the N-terminus of the unfolded ubiquitin, the segment bound to the Npl4 groove must be dislodged, which in turn must cause the two ubiquitin molecules bound to the top of the Npl4 tower to be released. Eventually, the branch point of Lys48 will enter the pore, which requires the neighboring ubiquitin molecule to be unfolded (labeled Ub1 in our ATP structure). The pore then contains two polypeptide strands, the segment of the unfolded ubiquitin and the C-terminal segment of this neighboring ubiquitin molecule. It seems likely that the D2 pore loops pull on only one of these strands at any given time. How the translocating strand would be chosen remains unclear, but ultimately the substrate at the proximal end of the ubiquitin chain needs to be translocated. Perhaps, the distal end of the ubiquitin chain provides resistance to translocation because it remains bound to the UT3 domain of Ufd1 and, in mammals, to the C-terminal Zn2+-finger domain of Npl4. If substrate directly precedes the unfolded ubiquitin, it will next translocate through the central pore and be unfolded. If not, Cdc48 has to successively unfold the intervening ubiquitin molecules until it reaches and unfolds the substrate. The proposed model is consistent with biochemical data that show that at least one ubiquitin molecule of a free poly-ubiquitin chain can translocate through the Cdc48 rings (22). Our model also implies that at some point there will be three polypeptide chains in the central pore, which is conceivable given the large empty space seen in both rings in our structures containing only one strand (Fig. 5), and results obtained with other AAA ATPases (44–46).
Fig. 7. Model for substrate processing by the Cdc48 ATPase complex.
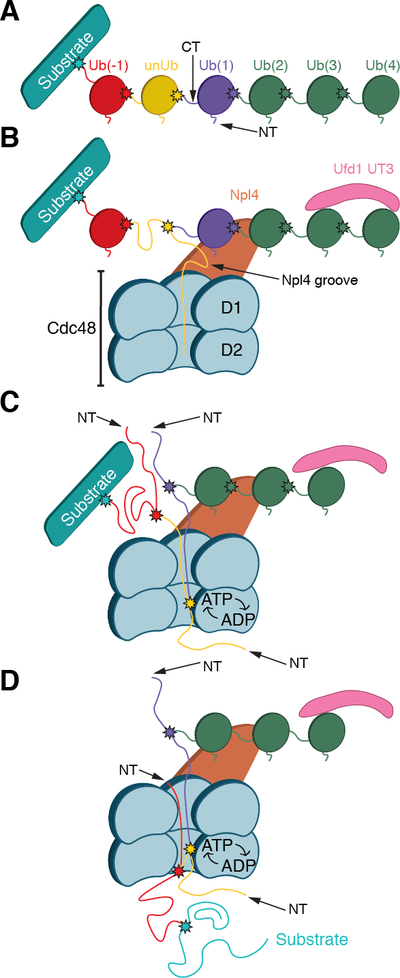
(A) Scheme of a substrate with an attached Lys48-linked poly-ubiquitin chain. In the example chosen, the ubiquitin molecule shown in yellow will be unfolded; it is separated from the substrate by one ubiquitin molecule (Ub(−1)). Ubiquitins distal to the one being unfolded are numbered (Ub(1)…Ub(4)). Lys48 branch points are indicated by stars. (B) Binding of the substrate to the Cdc48 complex. The distal folded ubiquitins Ub(1) and Ub(2) bind to the top of Npl4, and Ub(3) and possibly Ub(4) bind to the UT3 domain of Ufd1. The unfolded ubiquitin (unUb) binds to the groove in Npl4 and projects its N-terminus across both ATPase rings. (C) ATP hydrolysis in the D2 ring pulls the N-terminus of the unfolded ubiquitin through the central pore, moving the branch point into the ATPase rings and causing unfolding of attached Ub(1). Pulling on the proximal side of the ubiquitin chain results in unfolding of Ub(−1). (D) Ultimately the substrate is moved through the central pore and unfolded. Substrate release from the ATPase complex requires the removal of the distal ubiquitins Ub(2)-Ub(4) by a DUB.
The model can also explain why substrate release from the Cdc48 complex requires deubiquitination by a DUB (22), as the binding of the distal ubiquitin molecules to the UN cofactor would prevent their efficient release. The DUB Otu1 binds to the N domains of Cdc48, and cleaves the ubiquitin chain following ATP hydrolysis by the D1 domains, which triggers the movement of the N domains to their “down-conformation”. The substrate released by Otu1 contains chains with up to ten ubiquitin molecules (22). Assuming that these ubiquitins can rapidly refold on the trans-side of Cdc48, many of the released molecules could be degraded directly by the 26S proteasome, which requires a minimum of four ubiquitins for recognition (47). However, some ubiquitin molecules might be degraded concomitantly with the substrate itself (48). The proteasome might start degrading the substrate emerging from the trans-side of Cdc48 even before it is released from the cis side, which may explain its cooperation with Cdc48 in the absence of a DUB (12).
The role of the D1 ATPase ring in substrate processing by Cdc48 requires further investigation. The D1 ATPases control the position of the N domains, and our results now suggest that the D1 ring also makes initial contact with the N-terminal segment of unfolded ubiquitin. During substrate processing, the D1 subunits probably maintain an almost planar arrangement, as seen in our ATP structure. The structure indicates that subunit A of the D2 ring could re-join the top of the staircase with relatively minor changes of the D1 ring. The pronounced staircase arrangement of the D1 subunits seen in the ADP/BeFx structures may not be physiological or may be an intermediate/transient state that can be adopted only when one or more D1 subunits hydrolyze their ATP.
In summary, our results provide a mechanistic explanation for why the Cdc48 ATPase often acts upstream of the 26S proteasome. Proteasomal substrates generally need both an attached poly-ubiquitin chain and a flexible polypeptide segment (8). In contrast, the Cdc48/UN complex needs only a poly-ubiquitin chain, and can thus act on a broad range of substrates by using ubiquitin binding and unfolding to initiate their processing. Cdc48 ultimately releases unfolded, oligo-ubiquitinated polypeptides, which can then be degraded by the 26S proteasome.
Materials and methods
Purification of proteins
Wild-type Cdc48, Cdc48 (E588Q), Ufd1/Npl4, Eos3.2, Ubc2, Uba1, and Ubr1 were purified as described (22). The Ufd1/Npl4 complex used for cryo-EM and H/D exchange experiments contained untagged proteins (20). The wild-type and mutant Ufd1/Npl4 complexes used in unfolding assays contained Npl4 with a C-terminal Flag tag. S. cerevisiae ubiquitin was purchased from Boston Biochem. Bpa-incorporated Cdc48 was expressed and purified as described (22), except that a Flag-tag instead of SBP-tag was placed at the C-terminus of Cdc48. The sfGFP used in the photo-crosslinking experiment has a re-designed surface: all lysine residues at the surface were mutated to arginine (this construct was originally termed sffrGFP5 (49)). The gene was re-cloned with His-SUMO tag at N-terminus and SBP tag at C-terminus, and purified as described for mEos3.2, except that an anion-exchange column was used instead of a cation-exchange column. The catalytic domain of yeast Usp21 (Usp21cat, residue 196–565) was purified as described (50). All purified proteins were buffer-exchanged to SEC buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5 mM tris(2-carboxyethyl)phosphine) before snap-freezing.
Photoconversion of Eos3.2-containing substrate
The purified substrate protein (6 mg/mL) was placed in a 200-μl PCR tube on an ice bath. A long-wavelength UV flashlight (395–410 nm, DULEX) was positioned 5 cm from the tube, and the sample was irradiated for 1 h, with occasional mixing. Precipitated protein was removed by filtration.
Poly-ubiquitination and fluorescence-labeling of substrate
For unfolding assays, ubiquitination of photo-converted substrate was carried out as previously described (22), with some modifications. Substrate (8 μM) was incubated with S. cerevisiae ubiquitin (250 μM), Uba1 (800 nM), Ubc2 (4.63 μM), Ubr1 (800 nM), and ATP (5 mM) for 45 to 60 min at 30°C in ubiquitination buffer (50 mM Tris pH 8.0, 150 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT)). The sample was applied to Ultra HBC streptavidin agarose beads (Goldbio) for 30 min at 4°C. The resin was washed 3 times with 5 column volumes of ubiquitination buffer, and then eluted with 5 column volumes of ubiquitination buffer containing 2 mM biotin. Eluates were incubated with 5 mM HRV-3C protease for 30 min at 4°C, before gel filtration on a Superdex 200 column. After analysis by SDS-PAGE, relevant fractions were pooled and snap frozen. The concentration of the pooled substrate was determined using a FlexStation 3 Microplate Reader (Molecular Devices), with unmodified, photo-converted Eos3.2 protein as a standard. The majority of the final product contained a poly-ubiquitin chain of more than 13 ubiquitin molecules. For photo-crosslinking experiments, sfGFP-containing substrate was labeled with DyLight 800 maleimide (ThermoFisher) before subjecting it to poly-ubiquitination.
To generate substrate with shorter poly-ubiquitin chains for cryo-EM analysis, non-converted mEos3.2 was ubiquitinated as described above, except that the concentration of S. cerevisiae ubiquitin was reduced to 80 μM. In addition, the cleavage by HRV-3C protease was omitted to retain the SBP tag. The final product contained poly-ubiquitin chains of 6 to 13 ubiquitin molecules.
Assembly of substrate-engaged Cdc48 complex
Non-converted Eos substrate with short poly-ubiquitin chains was bound to streptavidin agarose resin (Thermo Fisher Scientific), and incubated with Ufd1/Npl4 at 1:1 molar ratio in assembly buffer (50 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT). The resin was washed to remove unbound UN complex and the beads were incubated with Cdc48 (E588Q) in assembly buffer supplemented with 1 mM ATP, or wild-type Cdc48 supplemented with 1 mM ADP, 1 mM BeSO4, and 4 mM NaF, for 60 min at 4°C. The molar ratio of Cdc48 to Ufd1/Npl4 was about 1:2.5. The beads were then washed three times with two volumes of the assembly buffer containing 1 mM of the appropriate nucleotide. Bound protein was eluted with buffer containing 2 mM biotin and nucleotide and concentrated to 4~5 mg/ml for cryo-EM analysis.
Cryo-EM sample preparation and data collection
All samples for cryo-EM were prepared with a Gatan Cryoplunge 3 instrument, maintained at 90% humidity at room temperature. Blot times of 2.5 – 3.0 s were used 20s after 3 μl sample application (2–3 mg/ml complex, 1 mM nucleotide, 0.005% tween 20) to Quantifoil Cu 1.2/1.3 200 mesh grids that were previously plasma treated in a Pelco easiGlow at 0.39 mBar, 15 mA for 60 s. To limit sample exposure to Tween 20, 3 μl of the sample mixture was quickly pipetted up and down in 0.5 μl of 0.035% tween (final concentration of 0.005% tween) immediately prior to application to the grid. Tween was used to diversify the particle orientations in ice.
For the ATP dataset, 3037 micrographs were collected on a Cs-corrected Thermo Fisher Titan Krios operating at 300 kV, with post-GIF energy filter (20 eV slit) and a Gatan K2 camera in super resolution mode (super resolution pixel size of 0.675 Å/pixel). A dose rate of 10.0 e− pixel−1 s−1 was used. The total exposure time of 10 s was divided into 50 frames (total dose of approximately 54.9 (e−/Å2), with a defocus range of −1 μm to −2.5 μm. The objective and C2 apertures were set to 70 μm.
For ADP-BeFx, 2523 micrographs were collected on a Thermo Fisher Talos Arctica operating at 200 kV (Cs 2.7 mm), with Gatan K2 camera in counting mode (1.15 Å/pixel). A dose rate of 7.4 e− pixel−1 s−1 was used. The total exposure time of 8 s was divided into 40 frames (total dose of approximately 44.7 e−/Å2), with a defocus range of −1 μm to −2.5 μm. The objective aperture was set to 70 μm and the C2 aperture was set to 50 μm.
Image processing
Frame alignment and dose-weighting was carried out using MotionCor2 (51). CTF correction was performed with CTFFind4.1 (52). All subsequent image processing steps were carried out using Cryosparc2 (53). Density visualization was performed using UCSF Chimera (54).
For the ATP dataset, 3037 micrographs were collected and 265 were discarded after CTF correction and micrograph curation. 1,588,559 particles were picked from the remaining 2,772 micrographs. After an initial round of 2D classification, 1,446,651 particles were subjected to 3D classification. Two primary classes were combined (527,552 particles). These particles were sub-classified, resulting in a class of 291,301 particles. This class was then sub-classified into three classes, two of which were chosen for further processing based on the density for Npl4. These 223,901 particles resulted in a 4.0 Å map after refinement. To better separate the particles contributing to this class, signal subtraction was used. The signal from the D2 ATPase ring was removed from the particle images in Fourier space, and classification was performed. This resulted in a class of 127,261 particles. The signal of the D2 ring was restored to these particles, and subsequent refinement resulted in a 3.9 Å map, where the D2 ring density was significantly weaker in local resolution compared to the rest of the density. To improve the features of interest, local refinement was performed. One refinement was performed locally for D1 and Npl4, which resulted in a 3.8 Å map, and a second for D2, which resulted in a 4.3 Å map.
For the ADP-BeFx dataset, 751 micrographs were discarded after CTF correction and micrograph curation. 1,017,931 particles were picked from the remaining 1772 micrographs. After 2D classification, 844,376 particles were further processed. These were then subjected to iterative rounds of 3D classification with the map obtained for the ATP sample as a 3D reference. From the 844,736 particles, 355,106 were isolated after the first round of 3D classification. A second round of 3D classification resulted in a class comprising 184,176 particles that had strong features in the density representing the Cdc48 complex. These classes were refined together, resulting in a 3.7 Å map where the density for Npl4 was very weak. This group of particles was sub-classified into three classes, resulting in one weak, featureless class, one class of 54,098 particles that had a Npl4 tower, and a third class of 101,527 particles that was lacking the tower. The tower-containing class was refined to 4.4 Å, and the tower-less class was refined to 3.8 Å. The tower-containing class was sub-classified into three classes, where the primary class, 30,118 particles, refined to 4.1 Å (ADP/BeFX-1). The tower-less class was also sub-classified into three classes, two of which were indistinguishable. Combined, the 93,395 particles refined to 3.6 Å (ADP/BeFX-2).
Model Building
Initial models were built into the map of the ATP-containing sample by using homology models for S. cerevisiae Cdc48 and Npl4, generated with the SWISS-MODEL server and the C. thermophilum complex (PDB code 6CHS) as a template. Building was then carried out in Coot. The chains were individually fit as rigid bodies into the density map sharpened with B-factors of −100 and −150. Amino acid side chain positions were modified based on the density. The crystal structure of ubiquitin (PDB code 1UBQ) was rigid-body fit into the density, and compared for fit between the unsharpened map and maps sharped with B-factors of −100 and −150. Unfolded ubiquitin was built as described in the text. The resulting model was refined against a half map with real space refinement in Phenix.
The model obtained for the ATP-containing sample was used to build those obtained with ADP/BeFx. First, ADP/BeFX-2 was built, as it had higher resolution. Chains were initially fit as rigid bodies, then the main chains and side chains were built into the map based on the density. Protomer F was not built because of low local resolution. A poly-alanine chain was built into the substrate density. The resulting model was refined against a half map with real space refinement in Phenix. Cdc48 from ADP/BeFX-2 was rigid body fit into the ADP/BeFX-1 map and chain A was removed due to low local resolution. Npl4 from the ATP-containing model was also fit as a rigid body into the map, and the first Zn2+-finger and NTB were removed from the model, as density for these regions was missing. Unfolded ubiquitin was also built based on the helical density in the Npl4 binding groove. The resulting model was refined against a half map with real space refinement in Phenix (55).
Substrate unfolding assays
Unfolding experiments were performed as previously described (13). Briefly, the poly-ubiquitinated, photo-converted mEos3.2 proteins (200 nM) were mixed with 300 nM UN variants and 400 nM Cdc48 in 50 mM HEPES pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.5 mg/ml protease-free bovine serum albumin. After a 10-min pre-incubation at 30°C, ATP regeneration mixture was added (2 mM ATP, 20 mM phosphocreatine, 100 μg/ml creatine kinase), and fluorescence (excitation, 540 nm; emission, 580 nm) was measured at 30-s intervals for 30 min, using a FlexStation 3 Microplate Reader (Molecular Devices). For each experiment, a sample with the same concentration of substrates denatured in 6 M guanidine-HCl was included as a control for background fluorescence. The relative fluorescence at time t was calculated as [(fluorescence at t)–(background fluorescence at t)] / [(fluorescence at t0)–(fluorescence at t0)]. A linear fit was performed with data points within the first 2 min to calculate the initial velocities. These rates were normalized to that of the wild type sample in the same experiment. The graphs were generated by GraphPad Prism software.
Photo-crosslinking experiments
Crosslinking experiments were performed as described (22), with some modifications. Briefly, the reaction components included Cdc48-Flag D602Bpa (200 nM), Ufd1/Npl4 (500 nM), and dye-labeled polyubiquitinated sfGFP (1 μM) in reaction buffer (50 mM HEPES pH 7.5, 130 mM KCl, 10 mM MgCl2, 0.5 mM TCEP, 0.5 mg/ml protease-free BSA) supplemented with 2 mM of either ADP or ATP. The reactions were assembled on ice, incubated at 30°C for 10 min, and transferred to individual wells of a black polystyrene plate. A long-wave UV lamp (Blak-Ray) was positioned 5 cm from the plate, and the samples were irradiated for 30 min. To prevent overheating, an ice-cold metal block was placed in contact with the bottom of the plate. After irradiation, the samples were diluted 10-fold in dissociation buffer (50 mM Tris pH 8, 800 mM KCl, 1% (v/v) Triton X-100, 1 mM EDTA, 0.5 mM DTT) and incubated at room temperature for 5 min. Samples were then applied to 5 μl anti-Flag M2 magnetic beads (Sigma) equilibrated in dissociation buffer for 1 h at room temperature. Beads were washed three times, and bound protein was eluted in 50 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM TCEP, and 0.2 mg/ml 3xFlag peptide (Bimake). The eluted material was split into two halves. One half remained untreated, the other was treated with 5 μM of the DUB Usp21cat at room temperature for 30 min. All samples were subjected to SDS-PAGE and the gel scanned on an Odyssey imager (LI-COR).
Mass spectrometry analysis after photo-crosslinking
The crosslinking experiment was performed as described above, except that material bound to the anti-Flag beads was eluted with 0.2 mg/ml single Flag peptide (Sigma-Aldrich) in 50 mM Tris pH 8, 150 mM NaCl, 0.05% NP-40, 1 mM EDTA, 10% glycerol, and protease inhibitors. The treatment with Usp21cat was omitted.
Flag peptide eluates were diluted 1:1 with 100 mM ammonium bicarbonate, denatured with 0.1% Rapigest (Waters Corporation, Milford, MA), reduced with 10 mM DTT for 30 min at 56 °C, cooled for 5 min at room temperature, alkylated with 22.5 mM iodoacetamide for 30 min at room temperature protected from light, and then digested with trypsin overnight at 37 °C. Rapigest was cleaved by adding trifluoroacetic acid to a final concentration of 1% and incubating for an additional 30 min at 37 °C. After centrifugation to remove Rapigest by-products, peptides in the supernatant were desalted using C18 (SOLA-RP, ThermoFisher Scientific, Madison, WI). C18 eluates were dried by vacuum centrifugation, and residual detergent was removed using magnetic beads (56).
Peptides were analyzed by nanoflow LC-MS/MS as described (57) using a NanoAcquity UPLC system (Waters Corporation) interfaced to a QExactive HF mass spectrometer (ThermoFisher Scientific). Peptides were injected onto a self-packed pre-column (100 μm I.D. packed with 4 cm POROS 10R2, Applied Biosystems, Framingham, MA), resolved on an analytical column (30 μm I.D. packed with 50 cm 5 μm Monitor C18, Orochem, Naperville, IL), and introduced to the mass spectrometer via ESI (spray voltage = 4 kV). In initial experiments, the mass spectrometer was programmed to operate in data dependent mode, such that the 10 most abundant precursor ions in each MS scan (m/z 300–2000, 120K resolution, target=3E6) were subjected to MS/MS (target value=5E4, max fill time=50 ms, minimum threshold = 50000, isolation width=2 Da, resolution=15K, collision energy=27%). An independent replicate of each analysis was performed as described, except that an additional targeted MS/MS scan for the +4 charge state of the Cdc48-ubiquitin crosslinked peptide was executed after each dd-MS/MS cycle (target value=1E5, max fill time=100 ms, resolution=15K, collision energy=30%).
MS/MS spectra were deisotoped, charge reduced, and converted to mgf using multiplierz scripts (58). Peptide sequences were assigned using Mascot 2.6.2 to search against a custom fasta database consisting of Cdc48, Ubi4, Ufd1, Npl4, and NeD-sffrGFP5. Search parameters specified precursor and product ion mass tolerances of 10 ppm and 25 mmu, respectively, as well as variable oxidation of methionine, variable benzoylation of phenylalanine, and fixed carbamidomethylation of cysteine. Candidate cross-linked peptides were identified using CrossFinder version 1.4 (59). Additional multiplierz scripts were used to interrogate peptide-spectral-matches (PSMs) for candidate cross-linked peptides to corroborate fragment ions and identify internal fragments (60). Finally, the cross-linked Cdc48-ubiquitin peptide MS/MS spectrum was manually verified using mzStudio software (61). Only one high-confidence crosslinked product between the Cdc48 peptide containing Bpa at residue 602 and ubiquitin was detected (shown in Fig. 4C and fig. S8). In addition, crosslinks between Cdc48 peptides were detected.
HDX MS experiments
Substrate-engaged Cdc48–UN complex was prepared as described above, except that the final sample was buffer-exchanged into equilibration buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5 mM TCEP) supplemented with 1 mM of the appropriate nucleotides. The HDX experiments were performed essentially as previously described (13). Stock solutions of proteins (Cdc48-UN-substrate complex, 7.27 μM, with 1 mM ADP; 9.09 μM of this complex with 1 mM ADP plus 1 mM BeSO4 and 4 mM NaF (ADP-BeFx); Cdc48-UN complex, 10 μM, with 1 mM ADP; 10 μM of this complex with ADP-BeFx) were prepared in equilibration buffer. To initiate HDX, 1.0 μL of each protein was diluted directly from stock solutions with 18 μL labeling buffer (25 mM Tris/HCl, 150 mM NaCl, 5 mM MgCl2 and 0.5 mM TCEP, 99.9% D2O pD 7.4). The final conditions were: D2O concentration 94.6%, pHread=7.00, 21 °C. At each labeling time (0.167, 1, 10, 60, and 311 min), 19 μL of quench buffer was added (150 mM potassium phosphate pH 2.4, H2O). All subsequent steps were performed at 0 °C; the average back exchange was 30–35%. Four undeuterated controls were perfomed, one for each state. Quenched samples were digested online with immobilized pepsin using a Waters UPLC instrument with HDX technology (62). Peptides were trapped on a Waters UPLC BEH C18 1.7 μm VanGuard BEH column, desalted with 0.1% formic acid in water for 3 min at 100 μL/min, and eluted over 10 min using a 5–35% gradient of water:acetonitrile with 0.1% formic acid at a flow rate of 100 μL/min using a Waters HSS T3 1.8 μm C18, 1.0 mm x 50 mm analytical column. Deuterium incorporation was measured using a Waters Synapt G2-Si system equipped with a standard ESI source and operated in ion mobility mode. Mass spectra were acquired over a m/z range of 50–2000, and mass accuracy was confirmed by calibration with 500 fmol/μL of human glu-fibrinopeptide. Peptic peptides were identified using ProteinLynx Global Server (PLGS) 3.0 (Waters) and deuterium incorporation measured using DynamX 3.0 (Waters). The parameters used to filter peptides for identification from the four undeuterated controls were three consecutive products with 0.3 products per amino acid. In the case of Npl4, 117 peptides were followed and 139 identified. In the case of Ufd1, 79 peptides were followed and 93 identified. For Cdc48, 363 peptides were followed and 440 identified. The deuterium levels were not corrected for back exchange and are reported as relative (63). The repeatability of the analysis was +/− 0.15 relative Da and differences were considered meaningful if they were larger than 0.5 Da. In Figs. 3E, 3F, and fig. S7A, a peptide is colored as protected if it has less deuterium exchange for at-least two consecutive time points. Overlapping peptides were used to localize exchange whenever possible.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Z. Yu and R. Huang at the HHMI Janelia Research Campus cryo-EM facility for assistance with microscope operation and data collection, Z. Li, R. Walsh, S. Sterling, and S. Rawson at the Harvard cryo-EM Center for Structural Biology for assistance with microscope operation, data collection, and image processing, the SBGrid team for software and workstation support, and the ICCB-Longwood Screening Facility for use of equipment. We are grateful to W. Alexander for developing software tools for the identification and confirmation of the cross-linked peptides. We thank Marco Catipovic for critical reading of the manuscript.
Funding: This work was supported by a NIGMS grant (R01GM052586) to T.A.R. NCI grant (R01CA219850) to J.A.M., and a research collaboration between J.R.E. and the Waters Corporation. N.O.B. was supported by a fellowship from the NIH (T32GM007753). Z.J. is a Howard Hughes Medical Institute Fellow of the Damon Runyon Cancer Research Foundation, DRG-2315–18. T.A.R. is a Howard Hughes Medical Institute investigator.
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and materials availability: Cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-0665 (ATP), EMD-0666 (ADP/BeFx-1), and EMD-20000 (ADP/BeFx-2). Model coordinates have been deposited in the Protein Data Bank (PDB) under accession numbers 6OA9 (ATP), 6OAA (ADP/BeFx-1), and 6OAB (ADP/BeFx-2).
REFERENCES AND NOTES
- 1.Xia D, Tang WK, Ye Y, Structure and function of the AAA+ ATPase p97/Cdc48p. Gene. 583, 64–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Boom J, Meyer H, VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 69, 182–194 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Bodnar N, Rapoport T, Toward an understanding of the Cdc48/p97 ATPase. F1000Research. 6, 1318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watts GDJ et al. , Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet 36, 377–381 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Kimonis VE, Fulchiero E, Vesa J, Watts G, VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: Review of a unique disorder. Biochim. Biophys. Acta - Mol. Basis Dis 1782, 744–748 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Johnson JO et al. , Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron. 68, 857–864 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu X, Rapoport TA, Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol 53, 22–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A, An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat. Struct. Mol. Biol 11, 830–837 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Dong Y et al. , Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature. 565, 49–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de la Peña AH et al. , Structures of the substrate-engaged 26S proteasome reveal the mechanisms for ATP hydrolysis-driven translocation. Science (80-. ). 2, 393223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beskow A et al. , A Conserved Unfoldase Activity for the p97 AAA-ATPase in Proteasomal Degradation. J. Mol. Biol 394, 732–746 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Olszewski MM, Williams C, Dong KC, Martin A, The Cdc48 unfoldase prepares well-folded protein substrates for degradation by the 26S proteasome. Commun. Biol 2 (2019), doi: 10.1038/s42003-019-0283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bodnar NO et al. , Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1–Npl4. Nat. Struct. Mol. Biol 25, 616–622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies JM, Brunger AT, Weis WI, Improved Structures of Full-Length p97, an AAA ATPase: Implications for Mechanisms of Nucleotide-Dependent Conformational Change. Structure. 16, 715–726 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Banerjee S et al. , 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science (80-. ). 351, 871–875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hänzelmann P, Schindelin H, The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97. Front. Mol. Biosci 4, 1–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruderer RM, Brasseur C, Meyer HH, The AAA ATPase p97/VCP interacts with its alternative co-factors, Ufd1-Npl4 and p47, through a common bipartite binding mechanism. J. Biol. Chem 279, 49609–16 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Isaacson RL et al. , Detailed structural insights into the p97-Npl4-Ufd1 interface. J. Biol. Chem 282, 21361–21369 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Alam SL et al. , Ubiquitin interactions of NZF zinc fingers. EMBO J. 23, 1411–1421 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein A, Ruggiano A, Carvalho P, Rapoport TA, Key Steps in ERAD of Luminal ER Proteins Reconstituted with Purified Components. Cell. 158, 1375–1388 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blythe EE, Olson KC, Chau V, Deshaies RJ, Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci 114, E4380–E4388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodnar NO, Rapoport TA, Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell. 169, 722–735.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White SR, Lauring B, AAA+ ATPases: Achieving diversity of function with conserved machinery. Traffic. 8, 1657–1667 (2007). [DOI] [PubMed] [Google Scholar]
- 24.DeLaBarre B, Brunger AT, Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat. Struct. Biol 10, 856–63 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Barthelme D, Sauer RT, Identification of the Cdc48???20S proteasome as an ancient AAA+ proteolytic machine. Science (80-. ). 337, 843–846 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barthelme D, Sauer RT, Bipartite determinants mediate an evolutionarily conserved interaction between Cdc48 and the 20 S peptidase. Proc. Natl. Acad. Sci 110, 3327–3332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ripstein ZA, Huang R, Augustyniak R, Kay LE, Rubinstein JL, Structure of a AAA+ unfoldase in the process of unfolding substrate. Elife. 6 (2017), doi: 10.7554/eLife.25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cook WJ, Jeffrey LC, Kasperek E, Pickart CM, Structure of tetraubiquitin shows how multiubiquitin chains can be formed (1994). [DOI] [PubMed]
- 29.Park S, Isaacson R, Kim HT, Silver PA, Wagner G, Ufd1 exhibits the AAA-ATPase fold with two distinct ubiquitin interaction sites. Structure. 13, 995–1005 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Lander GC et al. , Complete subunit architecture of the proteasome regulatory particle. Nature. 482, 186–191 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Worden EJ, Dong KC, Martin A, An AAA Motor-Driven Mechanical Switch in Rpn11 Controls Deubiquitination at the 26S Proteasome. Mol. Cell 67, 799–811.e8 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Ye Y, Meyer HH, Rapoport TA, Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol 162, 71–84 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chin JW et al. , Genetic Code. 301, 964–968 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Dormán G, Prestwich GD, Benzophenone Photophores in Biochemistry. Biochemistry. 33, 5661–5673 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Weith M et al. , Ubiquitin-Independent Disassembly by a p97 AAA-ATPase Complex Drives PP1 Holoenzyme Formation. Mol. Cell (2018), doi: 10.1016/J.MOLCEL.2018.09.020. [DOI] [PubMed] [Google Scholar]
- 36.Puchades C et al. , Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science. 358, eaao0464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han H, Monroe N, Sundquist WI, Shen PS, Hill CP, The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. Elife. 6 (2017), doi: 10.7554/eLife.31324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo Y-H et al. , Cryo-EM structure of the essential ribosome assembly AAA-ATPase Rix7. Nat. Commun 10, 513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carrion-Vazquez M et al. , The mechanical stability of ubiquitin is linkage dependent. Nat. Struct. Biol 10, 738–743 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Irback A, Mitternacht S, Mohanty S, Dissecting the mechanical unfolding of ubiquitin. Proc. Natl. Acad. Sci 102, 13427–13432 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kleiner A, Shakhnovich E, The mechanical unfolding of ubiquitin through all-atom Monte Carlo simulation with a Gō-type potential. Biophys. J 92, 2054–2061 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez-Aliaga P, Ramirez L, Kim F, Bustamante C, Martin A, Substrate-translocating loops regulate mechanochemical coupling and power production in AAA+ protease ClpXP. Nat. Struct. Mol. Biol 23, 974–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esaki M, Islam MT, Tani N, Ogura T, Deviation of the typical AAA substrate-threading pore prevents fatal protein degradation in yeast Cdc48. Sci. Rep 7, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burton RE, Siddiqui SM, Kim YI, Baker TA, Sauer RT, Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J. 20, 3092–3100 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu CW, Corboy MJ, DeMartino GN, Thomas PJ, Endoproteolytic activity of the proteasome. Science (80-. ). 299, 408–411 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee C, Prakash S, Matouschek A, Concurrent translocation of multiple polypeptide chains through the proteasomal degradation channel. J. Biol. Chem 277, 34760–34765 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM, Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanna J, Leggett DS, Finley D, Ubiquitin Depletion as a Key Mediator of Toxicity by Translational Inhibitors. Mol. Cell. Biol 23, 9251–9261 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frey S et al. , Surface Properties Determining Passage Rates of Proteins through Nuclear Pores. Cell. 174, 202–217.e9 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Ye Y et al. , Polyubiquitin binding and cross-reactivity in the USP domain deubiquitinase USP21. EMBO Rep. 12, 350–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng SQ et al. , MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohou A, Grigorieff N, CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Pettersen EF et al. , UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–12 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Adams PD et al. , PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hughes CS et al. , Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol 10, 757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ficarro SB et al. , Improved Electrospray Ionization Efficiency Compensates for Diminished Chromatographic Resolution and Enables Proteomics Analysis of Tyrosine Signaling in Embryonic Stem Cells. Mosaic A J. Interdiscip. Study Lit 81, 3440–3447 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Alexander WM, Ficarro SB, Adelmant G, Marto JA, multiplierz v2.0: A Python-based ecosystem for shared access and analysis of native mass spectrometry data. Proteomics. 17, 15–16 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Mueller-Planitz F, Crossfinder-assisted mapping of protein crosslinks formed by site-specifically incorporated crosslinkers. Bioinformatics. 31, 2043–2045 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Chu N et al. , Akt Kinase Activation Mechanisms Revealed Using Protein Semisynthesis. Cell. 174, 897–907.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ficarro S, Alexander W, Marto J, mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data. Proteomes. 5, 20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wales TE, Fadgen KE, Gerhardt GC, Engen JR, High-speed and high-resolution UPLC separation at zero degrees celsius. Anal. Chem 80, 6815–6820 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wales TE, Engen JR, Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev 25, 158–170 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.