

Abstract
Sarcomas represent less than 1% of all solid neoplasms in adults and over 20% in children. Their etiology is unclear, but genetic susceptibility plays an important role in this scenario. Sarcoma is central in Li-Fraumeni Syndrome (LFS), a familial predisposition cancer syndrome. In Brazil, the high prevalence of p.Arg337His mutations in the TP53 gene brings about a unique condition: a cluster of LFS. In the present work, we studied 502 sarcoma patients not selected by age or family history in an attempt to assess the impact of the so-called “Brazilian germline TP53 mutation” (p.Arg337His) on this tumor type. We found that 8% of patients are carriers, with leiomyosarcoma being the main histologic type of sarcoma, corresponding to 52.5% of the patients with the mutated TP53 gene. These findings emphasize the importance of genetic counseling and can better guide the management of sarcoma patients.
Introduction
Sarcomas are malignant neoplasms of mesenchymal origin [1]. Considered rare, they comprise less than 1% of all tumors in adults [2]. In children, sarcomas account for over 20% of solid tumors [3]. The etiology of sarcoma is not well-known, but some risk factors are well described, such as the association of HHV8 and Kaposi´s sarcoma in HIV-positive individuals, previous radiation therapy, chemical exposure (such as vinyl chloride) and lymphedema [4].
One of the most studied risk factors for sarcoma is genetic susceptibility. Several different conditions may be involved, such as neurofibromatosis (NF1), mutations in the RB gene (retinoblastoma) and Li-Fraumeni syndrome (LFS, involving mutation of the TP53 gene). Sarcoma has been known to be involved in LFS since the first description in 1969 [5] and remains in all subsequent categorizations, denoting its importance in that context [6].
In Brazil, LFS has a high prevalence, mainly due to a specific point mutation in the TP53 gene, p.Arg337His, also known as R337H [7,8]. Since the first description of this mutation, named “the Brazilian germline TP53 mutation” [9], researchers have been trying to establish its clinical behavior [10,11,12]. Families are heterogeneous, and despite having the same mutation, tumor patterns are quite different. Some families have adrenocortical tumors (ACT) and no other tumors, and some have many different tumor types. The background of this behavior has yet to be defined. In addition, regarding the clinical management of these R337H families, clinical surveillance protocols have been designed to organize patients’ follow-up in LFS. However, each tumor type has its own characteristics, hindering an application of these protocols in such heterogeneous families as those with “Brazilian germline TP53 mutation” [13].
Populational ancestry may influence epidemiology, and it is an important factor to be considered in regard to pathogenic mutations. Brazil is the largest country in South America, and its population is the product of Native Americans, Europeans and Africans that intermixed over the centuries, placing Brazil as a classical model of human admixture. Recent studies have characterized Brazilian ancestry [14]; however, to the best of our knowledge, no ancestry study on Brazilian LFS patients has been conducted so far.
To clarify this issue, we evaluated the frequency of the TP53 p.Arg337His mutation in sarcoma patients not selected by family history in a Brazilian cancer reference center. We intend to add information that contributes to the state of the art in sarcoma, a highly lethal tumor, as well as improves our understanding of frequency of the R337H mutation among sarcoma cases not selected based on clinical criteria. In addition, we also defined the ancestral substructure for all participants.
Materials and methods
Study subjects
A total of 701 patients treated at the Barretos Cancer Hospital between 2008 and 2016 were included in the study. Because a large proportion of the cases did not have blood samples available, frozen tumor tissue was analyzed for the purposes of this study. Samples were selected from the Institutional Biobank, macrodissected and revised by a board of pathologists who decided the best area to be analyzed (areas with tumor content higher than 60% and necrosis lower than 20%). A total of 199 cases were excluded due to different issues, mainly because of insufficient material, leading to a total of 502 sarcomas analyzed. In cases where the R337H mutation could be identified in the tumor, DNA from blood or normal tissue was analyzed when available.
Clinical and histopathological data as well as information regarding the family history of cancer were collected from the patient’s clinical chart. This study was approved by the local institutional ethical committee (approval number 866/2014). All patients provided written informed consent.
Genotyping
Genomic DNA was extracted from peripheral blood leukocytes or tumor tissue using a DNA Blood and Tissue kit (Qiagen), following the manufacturer’s instructions. For the detection of TP53 p.Arg337His, samples were PCR-amplified and analyzed by RFLP using the HhaI enzyme as described elsewhere [15]. Samples with a PCR-RFLP profile suggestive of positivity were confirmed by bidirectional Sanger sequencing. Moreover, in cases where the mutation was identified in tumor DNA, available blood or normal tissue samples were used to confirm the mutation by bidirectional Sanger sequencing.
Ancestry analysis
A panel of 46 AIM-INDELs was performed to evaluate the population admixture proportions considering four continental origins (Africa, Europe, East Asia and Native South America) [16,17,18]. Primer sequences and PCR conditions were obtained according to Pereira et al [16]. For that purpose, a 46-multiplex PCR followed by capillary electrophoresis was performed on an ABI 3500 xL Genetic Analyzer (Applied Biosystems) according to the manufacturer’s instructions. The electropherograms were evaluated, and genotypes were automatically assigned with GeneMapper v4.1 (Applied Biosystems).
Structure v2.3.3 software [19] was used to estimate the ancestry proportions of each patient. Considering the historical formation of Brazil’s population, we assumed the four major population groups (Native Americans, Europeans, Africans and East Asians, K = 4) applied to the genetic makeup of the Brazilian population. The structure consisted of 100,000 burn-in steps followed by 100,000 Markov Chain Monte Carlo (MCMC) iterations. The option ‘Use population Information to test for migrants’ was used with the Admixture model, taking into account the allele frequencies that were correlated, and allele frequencies were updated using only individuals with POPFLAG = 1. (HGDP-CEPH samples were used as references).
Statistical analysis
The data were described by means, standard deviations, minima and maxima for the quantitative variables and by the absolute and relative frequencies for the qualitative variables.
The relationship between the clinical characteristics and the R337H status was verified by chi-square or Fisher's exact test. The combined relationship of the variables was evaluated through multiple logistic regression, considering all the variables that were significant at a level of 20% in the univariate analysis.
A significance level of 5% was utilized, and the analyses were performed with SPSS v21.
Results
Among the 701 cases included, 502 were analyzed. Exclusions were mostly due to insufficient material. All sarcomas were classified according to the latest WHO (World Health Organization) classifications [20] and were distributed among the patients as described in Table 1.
Table 1. Patients’ classification according to histologic subtypes.
| Tumor subtype | Number of patients (%) |
|---|---|
| Osteosarcoma | 98 (19.6) |
| Liposarcoma | 67 (13.4) |
| Leiomyosarcoma | 65 (12.9) |
| Synovial Sarcoma | 55 (11.0) |
| Chondrosarcoma | 32 (6.4) |
| Ewing Sarcoma | 22 (4.4) |
| Rhabdomyosarcoma | 18 (3.6) |
| Dermatofibrosarcoma | 17 (3.4) |
| Myxofibrosarcoma | 15 (3.0) |
| Undifferentiated Pleomorphic Sarcoma | 15 (3.0) |
| Spindle Cell Sarcoma, NOS | 14 (2.8) |
| Undifferentiated Pleomorphic Sarcoma | 13 (2.6) |
| Angiosarcoma | 10 (2.0) |
| Malignant Peripheral Nerve Sheath Tumor (MPNST) | 7 (1.4) |
| Other | 54 (10.5) |
| Total | 502 (100%) |
NOS: Not Otherwise Specified
In the entire group, the sex distribution was balanced: 48.5% were women and 51.5% were men. The ages ranged from 1 month to 91 years, with a mean age of 40 years (median age of 43 years). It should be emphasized that, in the hospital where the study was conducted, the majority of patients were adults; however, we analyzed 58 patients under 14 years of age.
The patients’ clinical analysis revealed advanced cancer stages (stages III and IV) in the vast majority of the group: 27.2% with stage IV (n = 132) and 40.9% (n = 199) with stage III, followed by 25.7% and 6.2% with stages II and I, respectively. When we evaluated the clinical stage in relation to age, there was no statistical significance by Cox logistic regression analysis (p = 0.030).
We found that 40 cases (8%) carried the p.Arg337His mutation. In 90% of the cases (36/40), the origin of the mutation could be verified (DNA from the blood or normal tissue was available), with all of them being germline. Comparative analysis of the sequencing profile between tumor and germline DNA suggested loss of the wild-type allele in 80% of the cases (S1 Fig). Among the four cases with only tumor available for testing, loss of the wild-type allele was observed in two cases. Table 2 provides a detailed description of the 40 mutated cases.
Table 2. Characteristics of patients with p.Arg337His mutations.
| ID |
Age at diagnosis | Mutation origin | Histologic subtype | Families' tumors (gender, age at diagnosis) | Vital status |
|---|---|---|---|---|---|
| 1 | 19 | germline | leiomyosarcoma | prostate leiomyosarcoma (M, 49), non-smoking lung cancer (M, 60) | alive |
| 2 | 37 | germline | myxofibrosarcoma | breast cancer (F, 60) | alive |
| 3 | 28 | germline | liposarcoma | malignant CNS (F, 28), colorectal cancer (M, 50), prostate cancer (M, 40) | alive |
| 4 | 46 | germline | leiomyosarcoma | None | alive |
| 5 | 59 | germline | leiomyosarcoma | None | dead |
| 6 | 52 | germline | myxofibrosarcoma | stomach cancer (M, 53), breast cancer (F, 51) | alive |
| 7 | 52 | germline | leiomyosarcoma | colorectal cancer (F, 60) | dead |
| 8 | 25 | germline | myxofibrosarcoma | malignant CNS (F, 45), leiomyosarcoma (M, 64), melanoma (F, 67) | dead |
| 9 | 47 | not tested | Undifferentiated Pleomorphic Sarcoma | None | dead |
| 10 | 63 | germline | leiomyosarcoma | breast cancer (F, 29; F, 37; F, 50; F, 80, F, 63), colorectal cancer (M, 58), esophagus cancer (M, 52) | alive |
| 11 | 60 | germline | Undifferentiated Pleomorphic Sarcoma | esophagus cancer (M, 70), pediatric cancer (F, 12) | dead |
| 12 | 48 | germline | Synovial sarcoma | breast cancer (F, 35), non-smoking lung cancer (F, 20), colorectal cancer (M, 30) | alive |
| 13 | 46 | germline | fibrosarcoma | head and neck cancer (F, 50) | dead |
| 14 | 37 | germline | liposarcoma | breast cancer (F, 70; F, 58), bilateral breast cancer (F, 50), sarcoma (M, 49), colorectal cancer (M, 58), urothelial cancer (F, 66), multiple tumors in one individual (F, 60; F, 58) | dead |
| 15 | 29 | germline | leiomyosarcoma | prostate cancer (M, ?; M, ?) | dead |
| 16 | 49 | germline | leiomyosarcoma | pediatric leukemia (F, 6), breast cancer (F, 30) | alive |
| 17 | 37 | not tested | leiomyosarcoma | Sarcoma (M, 34) | dead |
| 18 | 60 | germline | leiomyosarcoma | leukemia (M, 40), non-smoking lung cancer (F, 78), head and neck cancer (M, 25) | alive |
| 19 | 68 | germline | undifferentiated pleomorphic sarcoma | no information | dead |
| 20 | 52 | germline | myxoid sarcoma | lung cancer (M, 60), stomach cancer (M, 50; M, 50) | alive |
| 21 | 35 | not tested | Osteosarcoma | stomach cancer (M, ?) | dead |
| 22 | 53 | germline | Undifferentiated Pleomorphic Sarcoma | non-smoking lung cancer (M, 18), breast cancer (F, 44; F, 50; F, 50), colorectal cancer (F, 84), pancreas (F, ?) | dead |
| 23 | 50 | germline | leiomyosarcoma | leukemia (F, 1), hepatocellular carcinoma (M, 37) | alive |
| 24 | 50 | germline | leiomyosarcoma | malignant CNS (F, 28), adrenal cancer (F, 31), non-smoking lung cancer (F, 45), stomach cancer (M, 60) | Alive |
| 25 | 66 | germline | spindle cell sarcoma, NOS | malignant CNS (M, 60; F, 42; M, 41), breast cancer (F, 50; F, 40), bilateral breast cancer (F, 32), leukemia (M, 12), lung cancer (M, 50), stomach cancer (F, 45; M, 45), multiple tumors in one individual (F, 32), melanoma (F, 44) | Alive |
| 26 | 50 | germline | leiomyosarcoma | colorectal cancer (W, 70) | Alive |
| 27 | 53 | germline | leiomyosarcoma | sarcoma (F, 68), adrenal tumor (M, 23), non-smoking lung cancer (M, 44; F, 73; F, 49; F, 54), stomach cancer (M, 61), leukemia (F, 40), renal cancer (F, 39), GIST (M, 59) | Alive |
| 28 | 49 | germline | Malignant Peripheral Nerve Sheath Tumor | adrenal cancer (F, 1; F, 32), colorectal cancer (F, 35), stomach cancer (F, 55; F, ?), lung cancer (M, 55) | Alive |
| 29 | 61 | germline | leiomyosarcoma | malignant CNS (F, 62), lung cancer (M, 65), sarcoma (F, 76), colorectal cancer (F, 40) | Dead |
| 30 | 75 | germline | liposarcoma | None | Alive |
| 31 | 60 | germline | leiomyosarcoma | adrenal cancer (M, 5), sarcoma (F, 60) | Alive |
| 32 | 18 | germline | osteosarcoma | no information | Dead |
| 33 | 64 | germline | leiomyosarcoma | adrenal cancer (F, 2), colorectal cancer (F, 45), leukemia (F, 62) | Dead |
| 34 | 58 | germline | leiomyosarcoma | None | Alive |
| 35 | 59 | germline | leiomyosarcoma | prostate cancer (M, 83; M, 75), colorectal cancer (F, 50) | Alive |
| 36 | 61 | germline | leiomyosarcoma | breast cancer (F, 64; F, 50; F, 40; F, 50), non-smoking lung cancer (F, 80) | Alive |
| 37 | 42 | germline | leiomyosarcoma | no information | Alive |
| 38 | 41 | germline | rhabdomyosarcoma | no information | Dead |
| 39 | 82 | germline | spindle cell sarcoma, NOS | no information | Dead |
| 40 | 21 | not tested | leiomyosarcoma | colorectal cancer (F, 60) | Alive |
CNS: central nervous system; GIST: gastrointestinal stromal tumor; NOS: not otherwise specified; F: female; M: male.
When patients were stratified according to the mutational status, it was found that adulthood is the principal age group for mutation, considering those of 40 years of age or more (Fig 1). Although pediatric patients constituted 11.6% of the cohort, we did not find any R337H-positive cases in this group. The youngest patient with the TP53 mutation was 18 years old, and the oldest was 84 years old. The age distribution according to mutational status is shown in Fig 1.
Fig 1. Age distribution according to mutational status.

Information regarding cancer history in families positive for the mutation was available for 87.5% of the cases. Among those, 85.7% reported a positive family history, as shown in Table 2. In 83% of the families, there were tumors that constituted part of the LFS spectrum, although the R337H status was not verified in the relatives.
When analyzing tumor types in mutated cases, leiomyosarcoma was the predominant subtype (Fig 2).
Fig 2. Mutational status according to sarcoma subtype.

Among the 40 patients positive for the TP53 mutation, 8 presented with more than one tumor. In almost all patients, sarcoma was the second tumor diagnosed (Table 3).
Table 3. Patients with multiple tumors.
| Patient | Sex | First tumor (age at diagnosis) | Second tumor (age at diagnosis) |
|---|---|---|---|
| 2 | F | Phyllodes breast Tumor (36) | Myxofibrosarcoma (37) |
| 4 | F | Renal cancer (38) | Leiomyosarcoma (46) |
| 10 | M | Thyroid tumor (58) | Leiomyosarcoma (63) |
| 18 | F | Breast cancer (49) | Leiomyosarcoma (60) |
| 20 | M | Prostate cancer (42) | Myxoid sarcoma (52) |
| 26 | F | Breast cancer (42) | Leiomyosarcoma (50) |
| 35 | F | Thyroid cancer (61) | Leiomyosarcoma (59) |
| 36 | F | Leiomyosarcoma (61) | Breast cancer (68) |
F = female; M = male
The vast majority of our cohort (68%) had advanced clinical stages (III and IV) of cancer. There was no statistically significant difference when comparing the clinical stages between mutated and wild-type cases (p = 0.30) (Fig 3).
Fig 3. Clinical stages among wild-type and mutated cases.

Considering the declared ethnicity, 60% of the patients from our cohort were white. Ancestry analysis was performed for 411 patients, and the results are compatible with the declared data, reporting that the median value for European ancestry was 67.1%, followed by African (15.9%), East Asian (7.2%) and Native American (9.8%) (Fig 4). Among the mutated cases, a total of 39 patients with the Brazilian founder pathogenic variant in TP53 (c.1010G>A; p.Arg337His) were analyzed (Fig 4). For these 39 individuals, the average ancestry proportions were 66.3% for European, followed by African (15.6%), East Asian (10.7%) and Native American (7.4%). Using the Mann-Whitney test, we did not find any correlation between the ancestral profile and the presence/absence of the pathogenic variant in TP53 (p.Arg337His).
Fig 4. Ancestral profile of all patients analyzed.

Left: Patients with the R337H mutation. Right: Patients with the WT phenotype.
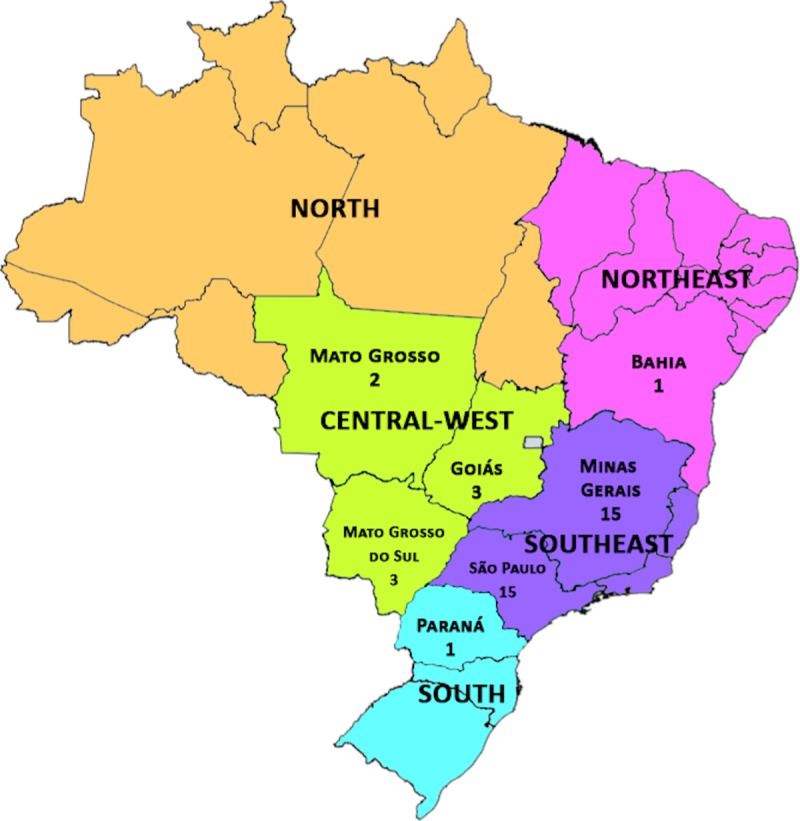
It is known that the “Brazilian germline TP53 mutation” occurs predominantly in southern Brazil [7,8,10,15,21]. However, the institution where this study took place receives patients from outside of southern Brazil. Therefore, our study could identify mutations in the northern and central-western regions, as depicted in Fig 5.
Fig 5. Brazilian map depicting the birthplace of patients with the R337H mutation (numbers in the map describe the number of patients with the R337H mutation who came from that specific state).
Discussion
Since its first description, the “Brazilian germline TP53 mutation” has been an intriguing point of discussion in oncogenetics. Garritano and cols. proved a founder effect for this particular mutation [22]. It has a preferential geographical localization and a peculiar clinical behavior. This mutation was first described in southern Brazil [9]. Our data demonstrate that the mutation is no longer geographically restricted, as some of our cases are from the central-west and northern regions, emphasizing the national importance of this condition. This spread reflects the internal migration within Brazil and has a direct impact on public health care since the mutation predisposes carriers to multiple cancer types.
We found that 8% of a nonselected, random sarcoma population are carriers of the R337H –TP53 pathogenic variant. Sarcoma is the lead tumor type in the original description of LFS and appeared in our study as an important issue. Previous Brazilian studies have found an unexpectedly high prevalence of this mutation in other types of LFS tumors, for instance, adrenocortical and plexus choroid carcinoma [10].
A review of the IARC TP53 database analyzed the types of mutations in and ages of sarcoma patients and demonstrated an age-dependent variation in these patients. They concluded that for TP53 mutation carriers, 67% of sarcomas appeared before 20 years of age [23]. However, our data demonstrate quite different behavior, with 95% of the sarcomas being diagnosed after the age of 20 and 77.5% after 40 years of age. A possible explanation for these differences comes from our institution´s characteristics, where the Clinical Oncology service is bigger and older than the Pediatric Oncology service. However, we analyzed 58 sarcoma patients under 14 years of age, which is a robust number compared to those of previous Brazilian studies [10].
Regarding histopathology, following the latest WHO sarcoma classification, our data show a regular distribution pattern [1]. When only patients with the R337H mutation were analyzed, a particular subtype, leiomyosarcoma, was revealed, accounting for 52.5% of the sarcomas. According to the IARC TP53 database, leiomyosarcoma corresponds to 9.1% of the sarcomas in TP53 mutation carriers, showing once again the particular behavior of the p.Arg337His pathogenic variant [23].
It is important to highlight that leiomyosarcomas have a poor prognosis. They do not respond well to chemotherapy and radiotherapy [24]. If resected, cure might be possible, but if this approach is not possible, mortality could be high. Therefore, early diagnosis is crucial for a better outcome.
Family history is an important indicator of hereditary cancer identification. In our registry, 75% of the patients with the TP53 mutation had family members with cancer (85.7% of those whose information regarding cancer history in families was available). Among the cancer subtypes in relatives, some were more frequent, such as lung cancer, breast cancer, malignant central nervous system tumor, colorectal and stomach cancer, leukemia and adrenal tumor. However, in approximately 15% of the mutated cases, there was no cancer family history. This emphasizes that, in Brazil, leiomyosarcoma should be considered a core LFS tumor and that the mutation should be accessed even in the absence of cancer in family history. Such a diagnosis might benefit these patients, offering specific preventive protocols to avoid second tumors.
Some positive patients had multiple malignant tumors, which could suggest details to improve surveillance protocols. Since sarcomas appeared as a second tumor in 7 of 8 patients with multiple tumors in our data, cancer patients who carried the p.Arg337H mutation should continue to be monitored with clinical cancer surveillance protocols.
Ancestry was also analyzed, and no correlation with the mutation could be demonstrated. The declared ethnicity and the molecular ancestry profile analysis were similar, and no significant differences were found. Brazil was colonized by different ethnicities, resulting in a highly mixed population, and we found this phenomenon in our study population as well.
In terms of the limitations of this study, it is important to note that, although our goal was to analyze a germline mutation, due to lack of availability of normal samples (blood/normal tissue), tumor samples were evaluated instead. However, all mutated cases with germline material available had the alteration confirmed (36/40 cases), and in all cases, the germline origin of the alteration was confirmed. In addition, somatic R337H mutations in the IARC TP53 database (R19) occurred in 4 tumors among 29895 investigated (0.013%). These findings allow us to infer that the mutation was also germline in the remaining cases where germline DNA was unavailable for analysis.
In conclusion, we highlight that our data contribute to a better understanding of the “Brazilian germline TP53 mutation” and its clinical behavior, which can help to improve surveillance protocols.
Supporting information
(TIFF)
Acknowledgments
The authors would like to thank Barretos Cancer Hospital Research Support Department (NAP) for sample collection and Barretos Cancer Hospital Biobank for sample processing.
Data Availability
All relevant data are within the manuscript.
Funding Statement
This work was supported by the Barretos Cancer Hospital. EIP and RMR are recipients of CNPq Productivity Grants. There was no additional external funding received for this study.
References
- 1.Fletcher CDM (2013) World Health Organization., International Agency for Research on Cancer WHO classification of tumours of soft tissue and bone. (4th) IARC Press, Lyon [Google Scholar]
- 2.Casali PG, Abecassis N, Bauer S, Biagini R, Bielack S, et al. (2018) Soft tissue and visceral sarcomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 29: iv51–iv67. 10.1093/annonc/mdy096 [DOI] [PubMed] [Google Scholar]
- 3.Alaggio R, Coffin CM (2015) The Evolution of Pediatric Soft Tissue Sarcoma Classification in the Last 50 Years. Pediatr Dev Pathol 18: 481–494. 10.2350/15-07-1666-MISC.1 [DOI] [PubMed] [Google Scholar]
- 4.Hui JY (2016) Epidemiology and Etiology of Sarcomas. Surg Clin North Am 96: 901–914. 10.1016/j.suc.2016.05.005 [DOI] [PubMed] [Google Scholar]
- 5.Li FP, Fraumeni JF Jr. (1969) Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst 43: 1365–1373. [PubMed] [Google Scholar]
- 6.Schiavi A, Lavigne J, Turcotte R, Kasprzak L, Dumas N, et al. (2015) Using a family history questionnaire to identify adult patients with increased genetic risk for sarcoma. Curr Oncol 22: 317–325. 10.3747/co.22.2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Achatz MI, Olivier M, Le Calvez F, Martel-Planche G, Lopes A, et al. (2007) The TP53 mutation, R337H, is associated with Li-Fraumeni and Li-Fraumeni-like syndromes in Brazilian families. Cancer Lett 245: 96–102. 10.1016/j.canlet.2005.12.039 [DOI] [PubMed] [Google Scholar]
- 8.Achatz MI, Zambetti GP (2016) The Inherited p53 Mutation in the Brazilian Population. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, et al. (2001) An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A 98: 9330–9335. 10.1073/pnas.161479898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seidinger AL, Mastellaro MJ, Paschoal Fortes F, Godoy Assumpcao J, Aparecida Cardinalli I, et al. (2011) Association of the highly prevalent TP53 R337H mutation with pediatric choroid plexus carcinoma and osteosarcoma in southeast Brazil. Cancer 117: 2228–2235. 10.1002/cncr.25826 [DOI] [PubMed] [Google Scholar]
- 11.Gomes MC, Kotsopoulos J, de Almeida GL, Costa MM, Vieira R, et al. (2012) The R337H mutation in TP53 and breast cancer in Brazil. Hered Cancer Clin Pract 10: 3 10.1186/1897-4287-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giacomazzi J, Graudenz MS, Osorio CA, Koehler-Santos P, Palmero EI, et al. (2014) Prevalence of the TP53 p.R337H mutation in breast cancer patients in Brazil. PLoS One 9: e99893 10.1371/journal.pone.0099893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, et al. (2016) Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 17: 1295–1305. 10.1016/S1470-2045(16)30249-2 [DOI] [PubMed] [Google Scholar]
- 14.Kehdy FS, Gouveia MH, Machado M, Magalhaes WC, Horimoto AR, et al. (2015) Origin and dynamics of admixture in Brazilians and its effect on the pattern of deleterious mutations. Proc Natl Acad Sci U S A 112: 8696–8701. 10.1073/pnas.1504447112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Custodio G, Taques GR, Figueiredo BC, Gugelmin ES, Oliveira Figueiredo MM, et al. (2011) Increased incidence of choroid plexus carcinoma due to the germline TP53 R337H mutation in southern Brazil. PLoS One 6: e18015 10.1371/journal.pone.0018015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pereira R, Phillips C, Pinto N, Santos C, dos Santos SE, et al. (2012) Straightforward inference of ancestry and admixture proportions through ancestry-informative insertion deletion multiplexing. PLoS One 7: e29684 10.1371/journal.pone.0029684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saloum de Neves Manta F, Pereira R, Vianna R, Rodolfo Beuttenmuller de Araujo A, Leite Goes Gitai D, et al. (2013) Revisiting the genetic ancestry of Brazilians using autosomal AIM-Indels. PLoS One 8: e75145 10.1371/journal.pone.0075145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campanella NC, Berardinelli GN, Scapulatempo-Neto C, Viana D, Palmero EI, et al. (2014) Optimization of a pentaplex panel for MSI analysis without control DNA in a Brazilian population: correlation with ancestry markers. Eur J Hum Genet 22: 875–880. 10.1038/ejhg.2013.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578. 10.1111/j.1471-8286.2007.01758.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karanian M, Coindre JM (2015) [Fourth edition of WHO classification tumours of soft tissue]. Ann Pathol 35: 71–85. 10.1016/j.annpat.2014.11.003 [DOI] [PubMed] [Google Scholar]
- 21.Palmero EI, Schuler-Faccini L, Caleffi M, Achatz MI, Olivier M, et al. (2008) Detection of R337H, a germline TP53 mutation predisposing to multiple cancers, in asymptomatic women participating in a breast cancer screening program in Southern Brazil. Cancer Lett 261: 21–25. 10.1016/j.canlet.2007.10.044 [DOI] [PubMed] [Google Scholar]
- 22.Garritano S, Gemignani F, Palmero EI, Olivier M, Martel-Planche G, et al. (2010) Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: evidence for a founder effect. Hum Mutat 31: 143–150. 10.1002/humu.21151 [DOI] [PubMed] [Google Scholar]
- 23.Ognjanovic S, Olivier M, Bergemann TL, Hainaut P (2012) Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 118: 1387–1396. 10.1002/cncr.26390 [DOI] [PubMed] [Google Scholar]
- 24.Serrano C, George S (2013) Leiomyosarcoma. Hematol Oncol Clin North Am 27: 957–974. 10.1016/j.hoc.2013.07.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIFF)
Data Availability Statement
All relevant data are within the manuscript.