

Summary
The genomes of mammalian neurons contain uniquely high levels of non-CG DNA methylation that can be bound by the Rett syndrome protein, MeCP2, to regulate gene expression. How patterns of non-CG methylation are established in neurons and the mechanism by which this methylation works with MeCP2 to control gene expression is unclear. Here we find that genes repressed by MeCP2 are often located within megabase-scale regions of high non-CG methylation that correspond with topologically-associating domains of chromatin folding. MeCP2 represses enhancers found in these domains that are enriched for non-CG and CG methylation, with the strongest repression occurring for enhancers located within MeCP2-repressed genes. These alterations in enhancer activity provide a mechanism for how MeCP2 disruption in disease can lead to widespread changes in gene expression. Hence, we find that DNA topology can shape non-CG DNA methylation across the genome to dictate MeCP2-mediated enhancer regulation in the brain.
Keywords: MeCP2, enhancer, transcription, topologically-associating domains, DNA methylation, non-CG methylation, cerebral cortex, Rett syndrome
Graphical Abstract
eTOC Blurb
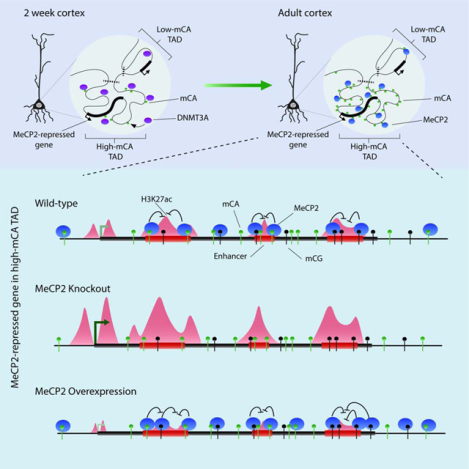
Clemens and Wu et al. demonstrate that DNMT3A establishes non-CG DNA methylation across topologically-associating domains in neurons, resulting in broad regions of high and low methylation. Within regions of enriched DNA methylation, MeCP2 represses the activity of enhancers, regulating promoter activation for genes controlled by these enhancers.
Introduction
The development and function of the mammalian brain requires precise control of gene expression (Cholewa-Waclaw et al., 2016). While DNA methylation at CG dinucleotides is used to control genes in all cells, neurons also utilize a unique form of non-CG DNA methylation for gene regulation (Lister et al., 2013; Xie et al., 2012). Non-CG DNA methylation is deposited by DNA methyltransferase 3A (DNMT3A), accumulating specifically in neurons postnatally until methylated cytosine (mC) at non-CG sites is nearly equal to mC at CG sites (mCG) (Lister et al., 2013). DNMT3A disruption in the mouse brain results in ablation of mC at non-CG sites and neurological phenotypes (Nguyen et al., 2007), supporting the importance of this methyl-mark.
Studies indicate that Methyl-CpG binding Protein 2 (MeCP2) has high affinity for mC in CA dinucleotides (mCA) similar to its classical substrate, mCG (Chen et al., 2015; Kinde et al., 2015; Guo et al., 2014). MeCP2 is expressed at near-histone levels in neurons and binds widely across the genome, with some enrichment at regions high in mCG and mCA sites (Chen et al., 2015; Gabel et al., 2015; Skene et al., 2010). The protein interacts with the NCoR-HDAC3 corepressor complex and can block reporter expression in vitro, suggesting an important function of MeCP2 is gene repression (Lyst and Bird, 2015). Loss of MeCP2 causes the neurological disorder Rett syndrome, and duplication of MeCP2 leads to autism spectrum disorder (Amir et al., 1999; Van Esch et al., 2005). Transcriptomic studies of brains from Rett syndrome patients and MeCP2 knockout (MeCP2 KO) or overexpression (MeCP2 OE) mice detect only subtle changes across many genes however, making it difficult to differentiate direct targets of MeCP2 regulation from secondary gene expression effects (Chahrour et al., 2008; Tudor et al., 2002). Defining how MeCP2 mediates its broad, subtle expression effects is thus recognized as an important challenge to address (Ip et al., 2018; Lyst and Bird, 2015).
Recent analysis has revealed that MeCP2 represses genes marked by high mCA levels (Chen et al., 2015; Gabel et al., 2015; Lagger et al., 2017). Notably, these genes are not substantially enriched for mCA at promoters, a canonical site of action for methylation, but show high mCA in their transcribed regions (“gene bodies”) and flanking sequences. In addition, MeCP2 KO causes a relative upregulation of genes genome-wide that is correlated with both the length and mC level of the gene body (Gabel et al., 2015; Sugino et al., 2014). These observations have led to the proposal that MeCP2 regulates expression by binding to mC in gene bodies to repress transcription (Kinde et al., 2016).
While these findings provide insight into mCA and MeCP2 in the brain, it is not known how high mCA levels are established at MeCP2-repressed genes. In addition, while MeCP2 represses the mRNA of long, high-mCA genes, it has not been demonstrated that this occurs through direct repression of transcription, and alternative post-transcriptional mechanisms have been proposed (Johnson et al., 2017). Finally, the mechanism by which binding of MeCP2 to mC outside of promoters can drive subtle, but critical, gene repression is unknown.
Here we examined how high mCA levels are established at MeCP2-repressed genes and explored how MeCP2 functions with mCA to control transcription. We find that mCA patterning in neurons is associated with chromatin folding, and that genes most strongly repressed by MeCP2 often land in mCA-enriched topologically-associating domains (TADs). We uncover evidence that MeCP2 binds to mCA and mCG within mCA-enriched TADs to repress activity of enhancer elements and cause downregulation of promoter activity for target genes. These findings provide insight into how disruption of MeCP2 drives neurological dysfunction in Rett syndrome and related disorders.
Results
To explore how high levels of mCA are established at MeCP2-repressed genes and to determine how MeCP2 reads out mC to control gene expression, we analyzed the cerebral cortex, a brain region where MeCP2 mutations disrupt gene expression and physiology (Pacheco et al., 2017; Shepherd and Katz, 2011). We integrated our data with epigenomic datasets for this brain region (Dixon et al., 2012; Sloan et al., 2016; Stroud et al., 2017) to search for features that can explain patterns of high mCA at genes repressed by MeCP2. We focused on mCA, the most prevalent, highest affinity non-CG site for MeCP2 (Gabel et al., 2015), but obtained similar results for minor non-CG methylation sites (mCT, mCC) in our studies.
Combined RNA-seq analysis of cortex from MeCP2 KO and MeCP2 OE mice (see methods) identified 884 “MeCP2-repressed genes” significantly upregulated in MeCP2 KO and downregulated in MeCP2 OE and 843 “MeCP2-activated genes” affected in the opposite manner in these mutants (Figure 1A; Table S1). These genes overlap with genes identified by meta-analysis of non-cortical brain regions in MeCP2 mutants (Gabel et al., 2015), indicating many MeCP2-regulated genes are shared across brain regions (Figure S1A, B). As in other studies (Gabel et al., 2015; Renthal et al., 2018; Sugino et al., 2014), cortical MeCP2-repressed genes are longer than the genome average (Figure S1D) and are enriched for mCA (mCA/CA, or “mCA level”) in and around the gene, with little mCA enrichment at their transcription start site (TSS) (Figure 1B, C; S1C). We also detected global length- and mCA-associated upregulation of genes in MeCP2 KO and downregulation in MeCP2 OE (Figure S1E, F), with dysregulation most correlated with gene body mCA levels compared to gene-flanking regions (Figure S1G, H). These results confirm that MeCP2 represses long, high-mCA genes and provide a high-confidence set of MeCP2-regulated cortical genes for our analyses.
Figure 1. Non-CG methylation in cerebral cortex is associated with domains of chromatin folding.

A. Exonic RNA changes for all genes dysregulated in MeCP2 KO and OE (FDR <0.1) (left), and an example MeCP2-repressed gene, Sdk1 (right). ***, B-H adjusted p<10−8 Wald test.
B. Aggregate mCA/CA levels for MeCP2-regulated genes. Mean mCA/CA for 1kb bins shown at kilobase (left) and megabase (right) scale. “Metagene” is 50 equally-sized bins within gene bodies.
C. Genome browser view of mC at Sdk1.
D. Hi-C interactions and mC for a genomic region including two MeCP2-repressed genes, Sdk1 and Auts2 (red). TAD-like structures visible in Hi-C interactions (blue).
E. mCA/CA for TADs containing MeCP2-regulated genes (see methods). ***, p < 10−8 Wilcoxon test.
F. Aggregate mCA/CA at boundaries of TADs containing MeCP2-repressed genes. Analysis of true TADs (red) or TADs shuffled around MeCP2-repressed genes (pink, see methods). mCA/CA drop-off at true boundaries is significantly different from shuffled boundaries. p<10−3 (see Figure S2C, methods).
G. Aggregate mCA/CA as in F, but for TADs containing MeCP2-repressed genes with the top 33% most differential mCA/CA levels compared to the neighboring TAD (see methods).
H. Cross-correlation of mCA/CA levels for regions in and around TADs (see methods) containing MeCP2-repressed genes, all genes, and shuffled control TADs.
See also Figure S1, S2 and Table S1, S2.
Data from cerebral cortex of 7–10 week-old animals. n=6 each (MeCP2 KO, wild type) and n=5 each (MeCP2 OE, wild type) for RNA-seq, n=2 wild type for DNA methylation (Stroud et al., 2017). Hi-C data (Dixon et al., 2012).
Large-scale non-CG DNA methylation profiles are linked to chromatin topology.
We next explored how high mCA levels are established at MeCP2-repressed genes. We observed that mCA enrichment can extend for megabases around these genes (Figure 1B, C) (Kinde et al., 2016), suggesting that regional mCA varies on a megabase scale. High-throughput chromatin conformation capture (Hi-C) analysis of the mouse cortex (Dixon et al., 2012, 2016) has detected TADs on a similar megabase scale. TADs are regions of cis-interactions, where enhancers interact with promoters in the same TAD more often than with promoters outside of the TAD (Spielmann et al., 2018). We noted instances where high mCA surrounding MeCP2-repressed genes dropped off at transition points in Hi-C interactions similar to those delineating TADs (Figure 1D). A correlation between mCA patterns and TADs could have important implications for regulation of gene expression within each TAD. We therefore investigated if TADs in the cerebral cortex (Dixon et al., 2012; Weinreb and Raphael, 2016, see methods) delineate regions of high mCA at MeCP2-repressed genes (Table S2).
We find that TADs containing MeCP2-repressed genes are significantly enriched for mCA compared to TADs genome-wide (Figure 1E). Further, we observe a drop-off of mCA levels at boundaries of TADs containing MeCP2-repressed genes that is not detected in control TADs generated by shuffling genomic locations (Figure 1F; S2B, C). While the shift in mCA across all boundaries returns toward the genome average (Figure 1F), a dramatic drop-off in mCA at boundaries is detected when high-mCA TADs containing MeCP2-repressed genes are adjacent to lower mCA regions (Figure 1G). Such strong mCA enrichments and drop-offs at boundaries are not observed for shuffled TADs (Figure 1F, G; S2B, see methods) or for potential sources of sequencing bias (Figure S2B), indicating that these mCA patterns are not due to technical artifacts. Cross-correlation analysis (Rao et al., 2014, see methods) shows that mCA levels are more highly correlated for regions in the same TAD than for regions in neighboring TADs (Figure 1H). This relationship is detected for all TADs but is most prominent for TADs containing MeCP2-repressed genes. Control TADs shuffled across the genome do not show similar correlations (Figure 1H, see methods). These findings suggest that TADs are units of organization for mCA levels in the genome and that MeCP2-repressed genes often occupy high-mCA TADs.
In contrast to MeCP2-repressed genes, TADs containing MeCP2-activated genes display lower mCA levels (Figure 1E; S2A). Genomic mCG levels do not show prominent megabase-scale variation, but some mCG depletion occurs in TADs containing MeCP2-activated genes (Figure 1D; S2A, B). Increased correlation of mCG within TADs is observed (Figure S2D), suggesting TADs do organize modest regional mCG fluctuations.
As an added test of the association between topology and regional mCA, we analyzed Hi-C data from the embryonic cortex (Bonev et al., 2017), prior to the deposition of mCA. Topology at this time point (Table S3) is similarly associated with adult mCA (Figure S2E–H), indicating that chromatin folding early in development is predictive of adult mCA patterns. We further generalized our findings using an independent cortical mC dataset (Lister et al., 2013), as well as Hi-C and mC data from the cerebellum (Mellén et al., 2017; Yamada et al., 2019). This analysis indicated that the association between genome topology and mCA levels is robustly detected in independent datasets and brain regions (Figure S2I–K).
We next explored how mCA is established in TADs during development. DNMT3A deposits mCA in mouse cortex from birth to six weeks of age (Lister et al., 2013), and DNMT3A binding in kilobase-scale regions at two weeks predicts mCA levels at eight weeks (Figure S3A; Stroud et al., 2017). We therefore asked if megabase-scale DNMT3A binding at two weeks supports a role for TADs in influencing large-scale mCA patterning. We find that DNMT3A ChIP-seq signal at two-weeks shows higher cross-correlations within TADs than across TAD boundaries (Figure 2A) and predicts TAD mCA in the adult cortex (Figure 2B). DNMT3A ChIP also drops off at boundaries of TADs containing MeCP2-repressed genes (Figure 2C), while input controls do not (Figure S3B). Analysis of topology from embryonic neurons and DNMT3A at a later timepoint yielded similar results (Figure S3C–F, see methods). These findings indicate that DNMT3A binding during postnatal development is shaped by TAD structures, defining a long-term mCA “set-point” for each TAD into adulthood.
Figure 2. Domain-associated DNMT3A defines megabase- and kilobase-scale mCA levels.

A. Cross-correlations in and around TADs for DNMT3A ChIP at 2 weeks (see methods).
B. Comparison of DNMT3A ChiP/Input at 2 weeks and mCA/CA levels at 8 weeks for each TAD. ***, p<10−8.
C. Aggregate DNMT3A ChIP/Input at 2 weeks at boundaries of TADs containing MeCP2-repressed genes. Black line and ribbon indicate the mean and standard deviation of shuffled TAD boundaries (see methods).
D. MeCP2 ChIP/Input in TADs for quartiles of TAD mCA/CA. ***, p<10−8.
E. Comparison of TAD mCA/CA levels and mCA/CA levels at kilobase-scale genomic elements in each TAD at 8 weeks of age (see methods). ***, p<10−8.
See also Figure S3 and Table S2, S4.
Data from cerebral cortex of 2 or 8 week-old animals. Per time point: n=2–3 for DNMT3A ChIP-seq, n=2 wild type for DNA methylation (Stroud et al., 2017).
To examine how TAD mCA affects MeCP2 binding, we quantified MeCP2 ChIP-seq signal (Kinde et al., 2016) in the adult cortex. MeCP2 ChIP signal is extremely broad, showing only very modest fluctuations across the genome (Chen et al., 2015; Gabel et al., 2015). In the context of these small effects however, we find that MeCP2 levels in TADs associate with TAD mCA set-points (Figure 2D) and that MeCP2 cross-correlation is higher within TADs than across TAD boundaries (Figure S3G). Analysis of MeCP2 binding when mCA is blocked by deletion of DNMT3A (Figure S3H) (Kinde et al., 2016) revealed relative reductions in MeCP2 TAD binding that correlate with the wild-type mCA levels of each TAD (Figure S3I). Thus, high TAD mCA levels are associated with some enrichment of MeCP2 binding, and when mCA is lost, large-scale MeCP2 profiles are measurably altered.
We then considered how TAD-associated mCA levels could impact gene regulation. Methylation levels for regulatory elements and genes are thought to be determined by local sequence features and activity level at these sites (Schübeler, 2015). However, the TAD mCA set-point could act on top of these local determinants to drive consistently higher or lower mCA across all elements within each TAD. Indeed, we detect robust correlations between the TAD set-point mCA level and mCA levels of gene bodies and enhancers inside each TAD (Figure 2E). This correlation occurs only for elements found within the TAD, breaking down immediately outside of the TAD (Figure S3J). Correlations in mCA levels between elements in the same TAD are also stronger than for elements on different sides of a TAD boundary (Figure S3K). In contrast, mCA levels in TSS regions have limited correlation with TAD mCA levels, suggesting that these elements escape the TAD mCA set-point. Control analysis randomizing TAD or genomic element locations (see methods) confirmed that TAD boundaries delineate transitions in mCA set-points for enhancers and genes (Figure S3J, K) and showed that only true TSS regions escape TAD mCA levels (Figure S3L, M). mCG levels at these elements did not show strong TAD-associated signals (Figure S3L, M). These findings indicate that mCA levels at enhancers and genes in each TAD are linked to the mCA set-point for the TAD. By influencing mCA at genes and enhancers, TAD-associated mCA levels can directly impact regulation of genes within each TAD by MeCP2.
Loss of MeCP2 leads to promoter activation at MeCP2-repressed genes.
We next investigated how MeCP2 reads out mCA in TADs to affect transcription. Consistent with analysis above (Figure 2E), the high TAD mCA set-point for MeCP2-repressed genes is associated with high mCA in and around these genes but little mCA enrichment at the TSS (Figure 1b; S1C). This suggests that MeCP2 binds to mCA outside of the promoter to repress these genes. Based on the long length of MeCP2-repressed genes and the correlation between repression and gene body mCA (Figure S1D, G, H), we considered several possible regulatory mechanisms for MeCP2. For example, MeCP2 binding to gene body mCA might block transcribing RNA polymerase, resulting in premature termination and reduced mRNA. Alternatively, binding of MeCP2 to mCA at regulatory elements could act at a distance to block promoter activation. To test these and other possible mechanisms, we carried out genomic analysis of RNA intermediates and histone marks associated with transcription in MeCP2 mutant mice.
We first performed RNA-seq on nuclear RNA from MeCP2 KO and wild-type cortex to enrich for pre-mRNA (Figure 3A) and analyzed intronic reads to assess changes in transcription (Boswell et al., 2017). We reasoned that if MeCP2 normally reduces the processivity of RNA polymerase II to cause premature termination of repressed genes, increased transcript completion in the MeCP2 KO would result in more intronic reads at the 3’ end of genes but no change in reads at the 5’ end. Alternatively, if MeCP2 represses promoter activation, loss of MeCP2 would cause a consistent increase in intronic reads along the entire gene. Finally, if post-transcriptional mechanisms affect these genes, no intronic changes would be seen. Differential expression analysis of introns detected highly concordant effects with mRNA from whole cells, with introns also showing upregulation of long and high-mCA genes relative to shorter and lower-mCA genes (Figure 3B; Figure S4A–F; Table S1). Analysis of intronic RNA changes across the length of significantly upregulated genes detected a consistent increase of intronic reads across the pre-mRNA (Figure 3C; S4G, H). These results suggest that subtle changes in mRNA for MeCP2-repressed genes result from concomitant subtle changes in pre-mRNA transcription and that loss of MeCP2 leads to promoter activation, rather than changes in polymerase processivity.
Figure 3. Disruption of MeCP2 leads to promoter-associated transcriptional dysregulation.

A. Left, genome browser view of nuclear total RNA-seq and ChIP-seq from MeCP2 KO and wild type at an MeCP2-repressed gene, Zmat4. Right, overlay of MeCP2 KO and wild type signal illustrating subtle increases for blue highlights at left.
B. Changes in RNA-seq signal in the MeCP2 KO for exons in whole cortex RNA (Exonic), or introns in nuclear RNA (Intronic). Genesets defined in combined analysis of exonic RNA (see Figure 1A). ***, p<10−8 Wilcoxon test.
C. Profile (black, gray) and fold-change (orange) of intronic reads from RNA-seq for the first 100kb of upregulated genes in MeCP2 KO versus wild type (Figure S4B). Normalized median of 1kb bins is plotted for genes >100kb (see methods, and Figure S4G, H).
D. Fold-changes in ChIP signal in MeCP2 KO at MeCP2-regulated genes identified in intronic RNA analysis. Value for Zmat4 is indicated by a point on each plot. **, p<10−3; ***, p<10−8 Wilcoxon test.
E. Fold-changes in gene expression as in panel B, but for MeCP2 OE. ***, p<10−8 Wilcoxon test.
F. Profile of intron expression and fold-change as in panel C, but for MeCP2 OE. (see also Figure S4I).
G. Fold-changes in ChIP signal as in panel D but for MeCP2 OE. *, p<0.05; **, p<10−3; ***, p<10−8 Wilcoxon test.
See also Figure S4 and Table S1.
Data from cerebral cortex of 7–10 week-old animals. MeCP2 KO per genotype: n=6 for RNA-seq, n=3 for H3K36me3, n=4 for H3K4me3, n=5 for H3K27ac. MeCP2 OE per genotype: n=5 for RNA-seq, n=2 each for H3K36me3 and H3K4me3, n=3 for H3K27ac.
As an independent measure of transcriptional effects, we performed ChIP-seq of histone modifications that report on transcriptional activity. Analysis of gene body Histone 3 lysine 36 trimethylation (H3K36me3), a mark associated with transcription levels (Guenther et al., 2007), revealed a subtle but significant increase in signal for MeCP2-repressed genes in the MeCP2 KO (Figure 3A, D). Analysis of Histone 3 lysine 27 acetylation (H3K27ac) and Histone 3 lysine 4 trimethylation (H3K4me3), marks associated with promoter activation (Heintzman et al., 2007; Santos-Rosa et al., 2002), showed promoter upregulation for MeCP2-repressed genes (Figure 3A, D). While subtle, these effects are consistent with the magnitude of mRNA increases for these genes. Together with our intronic RNA analysis, these findings support a model in which MeCP2 acts at a distance to repress promoter activity.
Given the small magnitude of these effects, we sought to independently test if MeCP2 controls promoter activation. Based on the opposite effects on mRNA in the MeCP2 OE compared to the MeCP2 KO, we predicted intronic RNA and histone marks should be reciprocally affected in the MeCP2 OE. Indeed, integrated RNA-seq and ChIP-seq analyses in the MeCP2 OE revealed opposite effects of those in the MeCP2 KO (Figure 3E–G; S4I, Table S1). These findings provide further support that MeCP2 represses long, high-mCA genes through promoter downregulation.
MeCP2 represses enhancers that are enriched for mCA and mCG binding sites.
We next considered how binding of MeCP2 to high levels of mC found outside of the TSS could lead to promoter downregulation. One way MeCP2 could achieve this is through enhancers. Since our analysis shows that high-mCA set-points of TADs containing MeCP2-repressed genes would lead to high mCA at enhancers in the TAD (Figure 1E; 2E), we considered that repression of these enhancers by MeCP2 could underlie regulation of MeCP2-repressed genes. To date however, effects of MeCP2 on enhancer activity have not been examined. Enhancer activation is associated with H3K27ac signal at these sites (Creyghton et al., 2010), and visualization of our H3K27ac ChIP signal at enhancers near MeCP2-repressed genes suggests subtly increased acetylation in the MeCP2 KO (Figure 4A). Combined differential analysis of H3K27ac signal in MeCP2 KO and OE cortex genome-wide (see methods) identified significantly altered enhancers (Figure 4B; S5A; Table S4). Quantification of mCA and mCG at dysregulated enhancers detected enriched and depleted mCA levels at MeCP2-repressed and MeCP2-activated enhancers respectively, while mCG levels showed limited variation at these regions (Figure 4C; S5B).
Figure 4. MeCP2 represses enhancers enriched for mCA and mCG binding sites.

A. Left, overlaid wild type and MeCP2 KO H2K27ac ChIP-seq signal at two MeCP2-repressed genes, Zmat4 and Efna5. Right, close-up view of enhancers indicated in blue at left.
B. Fold-changes of H3K27ac ChIP in MeCP2 KO or MeCP2 OE for changed enhancers identified in combined analysis of H3K27ac ChIP-seq from MeCP2 KO and OE (FDR<0.1).
C. Aggregate mC at MeCP2-regulated enhancers, centered at the midpoint for each enhancer. Mean values plotted for 100 bp bins.
D. Enrichment significance (see methods) for mCNN trinucleotide density at MeCP2-repressed and MeCP2-activated enhancers.
E. MeCP2 ChIP/Input for MeCP2-regulated enhancers. ***, p<10−8 Wilcoxon test.
F. Fold-changes of H3K27ac in MeCP2 KO across deciles of mCA/kb (left) and mCG/kb (right) for all enhancers. Spearman rho shown for mC and change in H3K27ac at enhancers. ***, p<10−8.
G. Fold-changes of H3K27ac, as in panel F, but for MeCP2 OE. ***, p<10−8.
H. Spearman correlations between H3K27ac fold-change at enhancers in the MeCP2 KO and mC/kb for 500bp bins across these enhancers. Plots centered at the summit of H3K27ac ChIP peaks (see methods).
I. Spearman correlations, as in panel H, but for H3K27ac fold-change in the MeCP2 OE.
See also Figure S5 and Table S4.
Data from cerebral cortex of 7–10 week-old animals. MeCP2 KO per genotype: n=5 for H3K27ac. MeCP2 OE per genotype: n=3 for H3K27ac. n=2 wild type for DNA methylation (Stroud et al., 2017) and MeCP2 ChIP-seq data (Kinde et al., 2016).
Given the high affinity of MeCP2 for mCG (Meehan et al., 1989), but limited mCG/CG signal at MeCP2-regulated enhancers, we further considered if the number of mCG sites rather than the level of mCG contributes to enhancer repression by MeCP2. The number of mC binding sites for MeCP2 at enhancers is determined both by per-base methylation at CA and CG sites (e.g. mCG/CG) and dinucleotide frequencies at each enhancer (e.g. CG/kb). This is particularly relevant for CG dinucleotides, which are depleted from the genome and non-uniformly distributed (Bird, 1980). We therefore quantified mC “density” (mC/kb) at dysregulated enhancers and detected a robust signal for mCG density, which is driven by the presence or absence of CG dinucleotides (Figure 4C; S5B). These findings suggest that both mCA and mCG do contribute to MeCP2 enhancer regulation, but mCA enrichment is driven by per-base methylation, while mCG enrichment is driven by CG frequencies.
To further explore the link between high-affinity sites for MeCP2 and enhancer regulation, we examined enhancer effects at higher stringency. While mCA is the highest affinity non-CG dinucleotide for MeCP2 (Gabel et al., 2015), the third nucleotide also affects MeCP2 binding, with mCAC showing highest affinity for MeCP2 (Lagger et al., 2017). We find that mCAC is most significantly associated with MeCP2-regulated enhancers compared to lower affinity sites (Figure 4D; S5C). Consistent with the presence of high-affinity MeCP2 sites at dysregulated enhancers, MeCP2 ChIP signal is enriched at MeCP2-repressed enhancers (Figure 4E). Analysis of high-stringency MeCP2-repressed enhancers, selected based on enriched MeCP2 binding, yields similar changes in H3K27ac and mC enrichment (Figure S5A, B). This supports a direct role for MeCP2 in enhancer regulation.
While significantly dysregulated enhancers display robust signal for mCA and mCG, methylation at enhancers occurs in a continuous distribution genome-wide (Figure S5D). Thus, every enhancer may be regulated by MeCP2 to some degree, with the number of mC sites determining the repression level. Indeed, when we compared mC density and H3K27ac changes at all enhancers in the genome, we detected a positive correlation between mCA and mCG density and H3K27ac changes in the MeCP2 KO and a negative correlation in the MeCP2 OE (Figure 4F, G). While mC density is enriched in and around MeCP2-repressed enhancers (Figure 4C), we find that enhancer H3K27ac dysregulation in MeCP2 mutants best correlates with mC density at enhancer centers (Figure 4H, I). Thus, mC specifically within the enhancer is most important for regulation by MeCP2. Together, these findings support a model in which repression of enhancers by MeCP2 genome-wide is proportional to the mC density at each enhancer.
We also noted minor increases of H3K27ac at sites outside of stringently defined enhancer regions in the MeCP2 KO (Figure S5E). Examination of a set of possible regulatory elements (ATAC-seq peaks), defined across 13 mouse tissues (Cusanovich et al., 2018), showed that these regions overlap with enhancers as well as sites of H3K27ac enrichment not formally called as enhancers (Figure S5E, F, see methods). Analysis of these regions genome-wide revealed noisier, but detectable correlations between H3K27ac changes and mC density at these putative regulatory sites (Figure S5F, G, H). Thus, while a larger absolute change in H3K27ac is observed at called enhancers in MeCP2 mutants (Figure S5F), altered H3K27ac outside of enhancer regions may also contribute to effects observed in these mice.
We next tested the role of mCA and mCG in enhancer repression by selectively disrupting mCA in neurons. Conditional Dnmt3a deletion in the brain ablates mCA while preserving mCG, and leads to overlapping, but smaller effects on MeCP2-repressed genes compared to MeCP2 mutants (Gabel et al., 2015). We speculated that these partial expression effects arise due to loss of enhancer repression by mCA, but preserved repression by mCG. To test this, we deleted Dnmt3a from post-mitotic neurons in a DNMT3A Baf53b-cKO mouse (see methods) and measured effects on mC, RNA, and H3K27ac. Bisulfite-seq revealed ablation of mCA, but retention of mCG, at enhancers and other genomic elements (Figure 5A, S6A–C), and RNA-seq showed significant, but partial, dysregulation of MeCP2-regulated genes (Figure 5B). H3K27ac ChIP-seq similarly detected robust, but partial, dysregulation of MeCP2-regulated enhancers (Figure 5B). Significantly up- and downregulated enhancers defined in the DNMT3A Baf53b-cKO (see methods) displayed enrichment or depletion of mCA, but equivalent mCG (Figure 5C, S6D, E). Unthresholded analysis showed that, like MeCP2 mutants, H3K27ac changes correlate with wild-type mCA density at enhancers genome-wide (Figure 5D). In contrast, no association was detected for mCG (Figure 5D). Together, the robust but smaller effects on MeCP2-regulated enhancers and lack of mCG-associated changes in the DNMT3A mutant demonstrate the role of mCA in enhancer repression by MeCP2 and also support a function for mCG in this regulation.
Figure 5. mCA-associated enhancer de-repression in DNMT3A Baf53b-cKO.

A. mCG/CG and mCA/CA levels at all enhancers for wild type and DNMT3A Baf53b-cKO. Data shown are mean +/− SEM. **, p<10−3; ***, p<10−8two-tailed t-test.
B. Fold-changes for mRNA of MeCP2-regulated genes (left) and H3K27ac at MeCP2-regulated enhancers (right) in MeCP2 KO and DNMT3A Baf53b-cKO. ***, p<10−8 Wilcoxon test.
C. Aggregate wild-type mC profiles for enhancers significantly dysregulated in the DNMT3A Baf53b-cKO (see Figure S6D).
D. Fold-changes of H3K27ac across deciles of mCA/kb (left) and mCG/kb (right) for all enhancers in the DNMT3A Baf53b-cKO. Spearman rho shown for mC and H3K27ac change at enhancers. *, p<0.05; ***, p<10−8.
See also Figure S6 and Table S1, S4.
Data from cerebral cortex of 7–10 week-old animals. Per genotype: n=6 for DNA methylation, n=4 for RNA-seq, n=6 for H3K27ac ChIP-seq.
Enhancer repression by MeCP2 is associated with MeCP2-mediated gene regulation.
We next investigated if enhancer dysregulation is linked to altered gene expression in MeCP2 mutants. As predicted by our analysis above (Figure 2E), enhancers in high-mCA TADs containing MeCP2-repressed genes are enriched for mCA (Figure 6A), suggesting possible repression by MeCP2. Analysis of gene-enhancer links by several methods (TAD overlap, GREAT analysis (McLean et al., 2010), and Hi-C) revealed that MeCP2-repressed enhancers are significantly associated with MeCP2-repressed genes (Figure 6B). In light of the genome-wide association between mC density and repression of H3K27ac at enhancers (Figure 4F, G), we considered that many high-mCA enhancers in TADs with MeCP2-repressed genes might undergo upregulation in the MeCP2 KO that are below the statistical significance cutoff. Supporting this idea, H3K27ac changes for enhancers in TADs with prominent MeCP2-repressed genes displayed trends toward upregulation (Figure 6C). Indeed, the population of enhancers found in TADs containing MeCP2-repressed genes showed significant upregulation as a group in the MeCP2 KO and downregulation in the MeCP2 OE (Figure 6D).
Figure 6. MeCP2-repressed enhancers are linked to MeCP2-repressed genes.

A. mCA/kb of enhancers in TADs containing MeCP2-regulated genes. **, p<10−3; ***, p<10−8 Wilcoxon test.
B. Associations between MeCP2-regulated enhancers and MeCP2-regulated genes. Comparisons are: enhancers in the same TAD as dysregulated genes (“Same TAD”), assigned to dysregulated genes by GREAT analysis (McLean et al., 2010) (“GREAT”), enriched for enhancer-promoter contacts by Hi-C (“Hi-C-linked”), and found in dysregulated genes (“Intragenic”). Median significance (color) and log2 enrichment (number) are shown for true enhancers compared to resampled control enhancers (see methods).
C. Log2 fold-changes of H3K27ac in MeCP2 KO for enhancers near two MeCP2-repressed genes. Blue, TADs overlapping MeCP2-repressed genes; red, enhancers in MeCP2-repressed gene; pink, other enhancers in the same TAD; gray and black, extragenic and intragenic enhancers in other TADs. *, significantly changed enhancers.
D. Fold-changes in H3K27ac ChIP in MeCP2 KO (top) and MeCP2 OE (bottom) for enhancers in TADs containing MeCP2-repressed, MeCP2-activated, or no dysregulated genes (“Same TAD”), or for enhancers in these genes (“Intragenic”). ***, p<10−8 Wilcoxon test.
E. Genic distributions of MeCP2-regulated enhancers. **, p<10−3; ***, p<10−8 chi-squared test (see methods).
F. Fold-changes of H3K27ac ChIP in MeCP2 KO (top) or MeCP2 OE (bottom) for intragenic and extragenic enhancers across deciles of enhancer mCA/kb (left) and mCG/kb (right). Spearman rho for correlation of enhancer mC and change in H3K27ac. ***, p<10−8.
G. Aggregate observed/expected Hi-C interaction frequencies for regions inside and outside of genes (see methods).
H. Observed/expected Hi-C interactions between enhancers and promoters for intragenic or distance-matched extragenic enhancers (see methods). ***, p<10−8 Wilcoxon test.
I. Running-average plot of intronic RNA fold-change in the MeCP2 KO (top) and MeCP2 OE (bottom) versus gene body mCA/kb for genes containing various numbers of enhancers. Mean changes are plotted for genes sorted by gene body mCA/kb (bins of 201 genes with a 1-gene step).
See also Figure S5 and Table S1, S2, S4.
Data from cerebral cortex of 7–10 week-old animals. MeCP2 KO per genotype: n=6 for RNA-seq, n=5 for H3K27ac. MeCP2 OE per genotype: n=3 for H3K27ac, n= 5 for RNA-seq. n=2 wild type for DNA methylation (Stroud et al., 2017). Hi-C data from E14.5 cortical neurons (Bonev et al., 2017).
While this analysis linked MeCP2 enhancer repression to gene regulation, we sought to understand our previous finding that MeCP2 mutant mRNA changes are better correlated with mC levels in long genes than with mC levels in gene-flanking regions (Figure S1F–H; S4E, F) (Kinde et al., 2016). We speculated this signal might reflect preferential repression of intragenic enhancers by MeCP2. Indeed, H3K27ac changes at prominent MeCP2-repressed genes in the MeCP2 KO showed stronger increases at intragenic enhancers than at extragenic enhancers (Figure 6C). Analysis of all MeCP2-repressed enhancers showed they tend to be intragenic (Figure 6E), and significantly overlap with MeCP2-repressed genes (Figure 6B). As a population, enhancers in MeCP2-repressed genes are more robustly upregulated in the MeCP2 KO and downregulated in the MeCP2 OE than all enhancers in the same TAD (Figure 6D). Furthermore, while H3K27ac repression by MeCP2 is correlated with mC density for both intragenic and extragenic enhancers, for a given mC density, intragenic enhancers are more repressed (Figure 6F).
Given the susceptibility of intragenic enhancers to MeCP2 repression, we investigated their interactions with promoters of target genes. Hi-C data revealed that intragenic sequences interact more with promoters than extragenic sequences do (Figure 6G), and intragenic enhancers contact promoters of their cognate gene more than equidistant extragenic enhancers do (Figure 6H). Quantitative 3C analysis of the MeCP2 KO detected no evidence of altered interaction strength between enhancers and promoters of MeCP2-repressed genes (Figure S5I). This suggests that intragenic enhancers are privileged over extragenic enhancers to control their cognate genes, but that MeCP2 does not regulate looping between enhancers and promoters. Rather, MeCP2 may affect the degree of gene activation driven by enhancers once they interact with their target promoter.
If the repression of genes containing high gene body mCA is largely due to repression of enhancers in these genes, we reasoned that gene body mCA and gene upregulation in the MeCP2 KO should only be tightly linked for genes that contain enhancers and not be linked for genes that do not contain enhancers. Because long genes contain more enhancers than shorter genes (Figure S5J), MeCP2 intragenic enhancer repression could also explain the dysregulation of longer genes observed in MeCP2 mutants (Figure S1F–H) (Gabel et al., 2015; Sugino et al., 2014). We therefore examined gene expression effects in MeCP2 mutants for genes that do or do not contain intragenic enhancers. We detect little correlation between gene dysregulation and gene body mCA density for genes that do not contain enhancers but a robust correlation for genes containing intragenic enhancers (Figure 6I, S5L). Importantly, we observe these effects when controlling for gene length (Figure S5K). Consistent with the idea that enhancers specifically contribute to this effect, we find that H3K27ac changes in intragenic enhancers are more predictive of the gene expression changes in MeCP2 mutants than H3K27ac changes in non-peak sequences (Figure S5M). These results suggest that dysregulation of intragenic enhancers contributes to the dysregulation of long, highly-methylated genes that we previously observed in MeCP2 mutants.
Discussion
Our findings suggest a model in which TADs shape DNMT3A activity across the neuronal genome during early postnatal development, establishing mCA set-points for genes and enhancers within each TAD (Figure 7A). In mature neurons, MeCP2 reads-out mC to repress enhancer elements and control gene expression. Because MeCP2 most potently represses highly-methylated, intragenic enhancers, it has the largest impact on genes in high mCA TADs that contain multiple enhancers (Figure 7B).
Figure 7. A model of TAD-associated mCA and enhancer repression by MeCP2 in neurons.

A. DNMT3A activity during early postnatal development establishes high or low mCA set-points within TADs.
B. In mature neurons, MeCP2 reads-out this mCA and mCG, repressing enhancers. MeCP2 most strongly affects intragenic enhancers, resulting in repression of genes found within high-mCA TADs that contain multiple enhancers.
The enriched DNMT3A binding and resulting high-mCA set-point we detect in TADs with MeCP2-repressed genes provides insight into the origin of high mCA previously noted at these genes (Kinde et al., 2016; Lagger et al., 2017). The large fluctuations in mCA observed between TADs can occur because of the dynamic distribution of mCA across the genome. This contrasts with consistently high mCG across large regions. While sequence and activity states of genes and enhancers are known to affect their methylation status (Schübeler, 2015), we show here that TAD mCA set-points also influence mCA levels at genes and enhancers. Enhancers in high-mCA TADs are enriched for mCA and are more repressed by MeCP2 compared to enhancers in low-mCA TADs. In this way, TAD mCA patterns set the stage for MeCP2-mediated gene regulation in the brain.
Previously, we and others have proposed that binding of MeCP2 in genes might block RNA polymerase processivity to downregulate expression (Cholewa-Waclaw et al., 2019; Kinde et al., 2016). Surprisingly, we did not detect disrupted processivity in the MeCP2 mutants, but instead observed changes in promoter activity. Consistent with this finding, a parallel study used multiple methodologies to identify regulation of transcription initiation by MeCP2 (Boxer et al., 2019). Our identification of intragenic enhancers that are dysregulated in MeCP2 mutants and analysis linking them to genes (Figure 6I, S5J–M) supports the model in which MeCP2 preferentially represses long, highly-methylated genes but points to enhancer repression as a mediator of this regulation.
MeCP2 binds nearly ubiquitously across the genome and its relevant sites of action have not been clear. Through genome-wide H3K27ac analysis, we uncovered a role for MeCP2 in enhancer repression that is driven in part by TAD-scale fluctuations in levels of mCA. While identification of enhancers significantly changed in MeCP2 mutants revealed critical determinants for MeCP2 regulation (e.g. mCA and mCG density), our unthresholded analysis suggests that these enhancers represent only extreme examples along a continuum, in which graded repression by MeCP2 that is proportional to mC density occurs at enhancers genome wide. Alterations in pervasive tuning of enhancers may explain why disruption of MeCP2 leads to profound nervous system deficits, despite the small magnitude of effects observed for individual genes.
Consistent with some role for mC sites in recruiting MeCP2 to the genome, MeCP2 ChIP-seq shows a measurable association with mCA and mCG sites (Figure 2D, S3I). However, the binding of the protein is very broad, and signal is also present at demethylated sites. Thus, mC sites may modulate MeCP2 repression of enhancers when it is bound, rather than being a strict determinant of binding. Future analyses will be necessary to dissect the role of mC sites in MeCP2 recruitment versus modulation of its repressive activity.
Our findings suggest a model in which MeCP2 at enhancers can induce histone deacetylation to block their activating effects on target promoters. This could occur through the interaction between MeCP2 and the NCoR histone deacetylase complex that is critical for MeCP2-mediated gene repression (Kokura et al., 2001; Lyst et al., 2013; Nott et al., 2016). Alternatively, MeCP2 could modulate NCoR functions on nonhistone proteins to affect enhancer activity.
A striking finding from our study is that MeCP2 represses intragenic enhancers more than extragenic enhancers, and intragenic enhancer dysregulation is closely linked to gene changes in MeCP2 mutants. We observed that intragenic enhancers more readily contact their cognate promoters than extragenic enhancers, suggesting intragenic enhancer effects in MeCP2 mutants are particularly impactful for gene expression. Notably, intragenic enhancer regulation by MeCP2 provides an explanation for the gene length and gene body mC signature that we previously observed for MeCP2-repressed genes (Kinde et al., 2016). These findings also suggest that the presence of an enhancer within a gene can fundamentally alter the nature of its regulation. Future studies can dissect how MeCP2 preferentially represses intragenic enhancers, providing insight into the unique regulatory environment of these elements.
Our study focused on mCA at MeCP2-repressed genes, but enhancer repression by MeCP2 may also explain MeCP2-activated genes. The MeCP2-activated enhancers we identify here are depleted of mC sites (Figure 4C; S5B–D). If MeCP2 primarily represses enhancers through mC, then H3K27ac changes at “activated” enhancers could reflect a relative lack of de-repression in the MeCP2 KO and escape from repression in the MeCP2 OE. MeCP2-activated enhancers are associated with MeCP2-activated genes (Figure 6B), suggesting the relative changes in gene expression may stem from enhancer effects. Future studies can test if a single model of enhancer repression by MeCP2 applies, or if MeCP2 directly activates genes by other mechanisms.
While robust changes occur at enhancers in MeCP2 mutants, the protein binds broadly across the genome and disruption of MeCP2 has effects on acetylation outside of stringently defined enhancer sequences (Figure S5E) (Boxer et al., 2019). While some of this signal appears to originate at subthreshold enhancers (Figure S5E), MeCP2 is likely to have repressive effects on chromatin outside of enhancers that can contribute to gene regulation.
Multiple datasets support a role for MeCP2 in repressing long, high-mCA genes (Gabel et al., 2015; Rube et al., 2016; Sugino et al., 2014), but a recent study proposed that long gene upregulation in MeCP2 mutants results from post-transcriptional rather than transcriptional effects (Johnson et al., 2017). Another study suggested long-gene dysregulation in individual MeCP2 datasets does not reach statistical significance (Raman et al., 2018). Here, independent RNA-seq and ChIP-seq analyses further show that MeCP2 preferentially affects long, high-mCA genes. Notably, a parallel study using multiple methodologies and large-replicate datasets has also verified these effects (Boxer et al., 2019). Our additional insights into enhancer regulation not only support these effects but also provide a mechanism for how they can occur.
Our findings have important implications for Rett syndrome and MeCP2 duplication disorder. Mutation of MeCP2 in these disorders is likely to disrupt enhancer activity in high-mCA TADs, driving altered gene expression, and contributing to disease pathology. In this way, our study suggests that Rett syndrome and MeCP2 duplication syndrome are disorders that stem in part from disruption of enhancer control.
STAR Methods
Contact for Reagent and Resource Sharing
Requests for reagents and resources should be directed towards the Lead Contact, Harrison Gabel (gabelh@wustl.edu). This study did not generate new unique reagents.
Experimental Model and Subject Details
Mice
MeCP2 knockout mice (B6.129P2(C)-MeCP2tm1.Bird/J) were obtained from The Jackson Laboratory. Female heterozygous mice (MeCP2−/+) were crossed with C57BL/6J male mice to generate hemizygous male knockout mice (MeCP2−/y) and wild-type male litter mates (MeCP2+/y). MeCP2 overexpression mice (FVB-Tg(MECP2)3Hzo/J) were cryo-recovered from The Jackson Laboratory. Female heterozygous mice (MeCP2Tg3/+) were crossed with FVB/NJ male mice to generate hemizygous male transgenic mice (MeCP2Tg3/y) and wild-type male litter mates (MeCP2+/y). Female Dnmt3afl/fl were provided by M. Goodell and crossed to male B6.Cg-Tg(Nes-cre)1Kln/J (Nestin-Cre+/−) to generate Dnmt3afl/+; Nestin-Cre+/−. Male Dnmt3afl/+; Nestin-Cre+/− were then crossed to female Dnmt3afl/fl to generate Dnmt3afl/fl Tg(Nes-cre)1Kln/J conditional knockout mice (DNMT3A Nestin-cKO) (Gabel et al., 2015). To generate Dnmt3afl/fl; Tg(Actl6b-cre)4092Jiwu/J conditional knockout mice (DNMT3A Baf53b-cKO), Dnmt3afW were crossed to Tg(Actl6b-cre)4092Jiwu/J (Baf53b-Cre+/+) to generate Dnmt3afl/+; Baf53b-Cre+/− Dnmt3afl/+; Baf53b-Cre+/− were then crossed to Dnmt3afl/fl producing experimental and control animals for analysis.
Method Details
Topologically associated domain analysis
Topologically associating domains were called using the TADtree algorithm (Weinreb and Raphael, 2016) on interaction matrices (Dixon et al., 2012), with the following parameters: gamma (sensitivity vs specificity tradeoff) = 200, M (number of hierarchical layers) = 1, p and q (minimum scale of interaction shift) = 3 and 12 respectively, and N (maximum number of TADs detected) = 500. To allow for manual model selection, TADtree also computes TADs for the range of 1:500, in this case. We leveraged this to apply an additional filter for consistency, wherein we selected for TADs that are called in at least 30% of all runs. When we applied this filter, we found that the effects of changing gamma were moderated, and the program performed consistently at many ranges of sensitivity vs specificity. TADs defined in the cerebral cortex by this method are ~400kb on average and range in size from ~160kb to ~2mb, a scale that is similar to regions that we observed with enriched mCA levels (Table S2). We defined TAD-related “contact domains” of interaction across the genome using the Arrowhead algorithm as previously described (Rao et al., 2014) (Table S3) at 5kb resolution from embryonic neocortex. These contact domains had a similar length distribution to TADs, with a median length of half that of TADs. These domains are established and detected before DNMT3A increases in expression and establishes mCA, and the close correlation between early TAD structure, early DNMT3A binding, and mCA patterns suggests (Figure S3C, D) that the activity of DNMT3A in a TAD during early postnatal development defines a long-term “set-point” for mCA across this region into adulthood. In addition, the fact that DNMT3A expression is low at 8-weeks and that 8-week DNMT3A binding, concurrent with adult TAD structure, is less correlated with mCA compared to 2-week DNMT3A binding (Figure S3E, F), lends more support to a stable “set-point” of mCA, rather than levels that fluctuate through adulthood.
8-week cortex Hi-C data was obtained as pre-processed observed/expected contact matrices, from http://chromosome.sdsc.edu/mouse/hi-c/download.html (Dixon et al., 2012). For analysis of neocortex Hi-C data from Bonev and colleagues (Bonev et al., 2017), raw FASTQ files were downloaded from GEO. HiC-Pro (Servant et al., 2015) was used to generate contact matrices using the mm10 mouse genome as reference. Juicer (Durand et al., 2016) was then run on the contact matrices to generate Hi-C contact matrices at 1.5kb, 5kb, and 40kb resolutions using KR normalization. Arrowhead domains were then called on the data within Juicer. For cerebellum analysis, contact domains called from 10kb-resolution Hi-C data were used (Yamada et al., 2019).
Total and nuclear RNA isolation
Cerebral cortex was dissected on ice in phosphate buffered saline from 1) MeCP2 KO and wild-type male litter mates at 7–8 weeks old, 2) MeCP2 OE and wild-type male litter mates at 7–10 weeks old, and 3) DNMT3A Baf53b-cKO and control mice at 7–8 weeks old. Total RNA was extracted from 1/16th of a whole cortex using RLT buffer following RNeasy Micro Kit (Qiagen). Nuclear RNA was isolated following a modified version of the protocol described (Mo et al., 2015). Briefly, half of a cortex was homogenized in 0.25M sucrose, 25mM KCI, 5mM MgCl2, 20mM Tricine-KOH using a glass dounce homogenizer. Nuclei were isolated via centrifugation at 10, 000g for 18 minutes at 4°C (Sorvall HB-4) by pelleting through a 30% iodixanol density gradient (Sigma D1556). RNA was isolated from nuclei by resuspending pellet in RLT buffer following the RNeasy Micro Kit (Qiagen).
RNA sequencing
RNA libraries were generated from 250ng total and nuclear RNA with NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) using a modified amplification protocol (37°C, 15 minutes; 98°C, 30 seconds; (98°C, 10 seconds; 65°C, 30 seconds; 72°C, 30 seconds)x13; 72°C, 5 minutes; 4°C hold. RNA libraries were pooled at a final concentration of 8–10nM and sequenced using Illumina HiSeq 2500 or 3000 with the Genome Technology Access Center (GTAC) at Washington University in St. Louis, typically yielding 20–30 million single-end reads per sample.
RNA sequencing analysis
Raw FASTQ files were trimmed with Trim Galore, using a quality filter of 20, then rRNA sequences were filtered out using Bowtie, using rRNA sequences from Mus Musculus obtained from the NCBI sequence database (see Key Resources Table). The unaligned reads from this step were then aligned to mm9 using STAR (Dobin et al., 2013) with the default parameters. Reads mapping to multiple regions in the genome were then filtered out, and uniquely mapping reads were converted to BED files. Intronic and exonic reads were then separated. To do this, splice-site reads were first filtered out of the BED read files, then reads that mapped entirely within exons were added to the splice-site reads to make the exonic read file. All remaining reads that overlapped introns were considered intronic reads. Finally, reads were assigned to genes using bedtools coverage -counts.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID:AB_2118291 |
| Mouse monoclonal anti-Histone H3 (tri methyl K4) | Abcam | Cat# ab1012; RRID:AB_442796 |
| Rabbit polyclonal anti-Histone (tri methyl K36) | Active Motif | Cat# 61101; RRID:AB_2615073 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| OptiPrep Density Gradient Medium | Sigma-Aldrich | Cat#: D1556 |
| Critical Commercial Assays | ||
| RNeasy Micro Kit | Qiagen | Cat#: 74004 |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | NEB | Cat#: E7420S |
| NEBNext Mulitplex Oligos for Illumina (Index Primers Set 1) | NEB | Cat#: E7335S |
| NEBNext rRNA Depletion Kit (Human/Mouse/Rat) | NEB | Cat#: E6310L |
| Ovation Ultralow Library System V2 | NuGEN | Cat#: 0344–32 |
| Ovation Ultralow Methyl-Seq Library System | NuGEN | Cat# 0535–32 |
| Deposited Data | ||
| RNA-sequencing data | This paper | GEO: GSE123373 |
| ChIP-sequencing data (H3K27ac, H3K4me3, H3K36me3) | This paper | GEO: GSE123373 |
| ENCODE H3K4me1 ChIP-seq | ENCODE | GEO: GSM769022 |
| Bisulfite-sequencing data | This paper | GEO: GSE123373 |
| MeCP2 ChIP-seq | (Kinde et al., 2016) | GEO: GSE90704 |
| DNMT3A ChIP-seq, Bisulfite-seq | (Stroud et al., 2017) | GEO: GSE104298 |
| Dixon Hi-C contact matrices | (Dixon et al., 2012) |
http://chromosome.sdsc.edu/mouse/hi-c/cortex.norm.tar.gz |
| Bonev Hi-C data | (Bonev et al., 2017) | GEO: GSE96107 |
| Bisulfite-seq | (Lister et al., 2013) | GEO: GSE47966 |
| Bisulfite-seq | (Mellén et al., 2017) | GEO: GSE95628 |
| Mus musculus mm9 genome assembly | UCSC |
http://hgdownload.soe.ucsc.edu/goldenPath/mm9/ |
| Mus musculus mm10 genome assembly | UCSC |
http://hgdownload.soe.ucsc.edu/goldenPath/mm10/ |
| Ensembl gene models | UCSC |
https://genome.ucsc.edu/cgi-bin/hgTables |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: MeCP2 KO: B6.129P2(C)-MeCP2tm1.Bird/J | The Jackson Laboratory | JAX: 003890; RRID:IMSR_JAX:003890 |
| Mouse: MeCP2 OE: FVB-Tg(MECP2)3Hzo/J | The Jackson Laboratory | JAX: 008680; RRID:IMSR_JAX:008680 |
| Mouse: DNMT3A Baf53b-cKO: Dnmt3afl/fl Tg(Actl6b-cre)4092Jiwu/J | Derived from DNMT3A fl/fl strain (described in (Kaneda et al., 2004), mice provided by M. Goodell) and Baf53b-Cre (Zhan et al., 2015), mice obtained from The Jackson Laboratory (IMSR Cat# JAX:027826, RRID:IMSR_JAX:027826). | This study |
| Mouse: DNMT3A Nestin-cKO: Dnmt3afl/fl Tg(Nes-cre)1Kln/J | Derived from DNMT3A fl/fl strain (described in (Kaneda et al., 2004), mice provided by M. Goodell) and Nes-Cre (Tronche et al., 1999), mice obtained from The Jackson Laboratory (IMSR Cat# JAX:003771, RRID:IMSR_JAX:003771). | (Gabel et al., 2015) |
| Oligonucleotides | ||
| See Table S5 for 3C primers | IDT | Custom |
| Recombinant DNA | ||
| Zmat4 BAC: RP23–21B20 | Life Technologies | Cat#: Clone Id 21B20 RPCI-23 MM BAC CLONE |
| Zmat4 BAC: RP23–320D14 | Life Technologies | Cat#: Clone Id 320D14 RPCI-23 MM BAC CLONE |
| Efna5 BAC: RP23–51F6 | Life Technologies | Cat#: Clone Id 51F6 RPCI-23 MM BAC CLONE |
| Efna5 BAC: RP23–309F7 | BACPAC Resources | Cat#: RP23–309F7 |
| Snx24 BAC: RP23–207I5 | BACPAC Resources | Cat#: RP23–207I5 |
| Tshz1 BAC: RP23–153P8 | BACPAC Resources | Cat#: RP23–153P8 |
| Actb BAC: RP23–35M10 | BACPAC Resources | Cat#: RP23–35M10 |
| Ank2 BAC: RP23–399L5 | BACPAC Resources | Cat#: RP23–399L5 |
| Zfp60 BAC: RP23–181E8 | BACPAC Resources | Cat#: RP23–181E8 |
| Software and Algorithms | ||
| DESeq2 (v1.14.1) | (Love et al., 2014) |
http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR (v3.16.5) | (Robinson et al., 2010) |
https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| SAMtools (v1.3) | (Li and Durbin, 2009) |
https://sourceforge.net/projects/samtools/files/ |
| BEDtools2 (v2.25.0) | (Quinlan and Hall, 2010) |
https://github.com/arq5x/bedtools2 |
| Bowtie2 (v2.2.5) | (Langmead and Salzberg, 2012) |
http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR | (Dobin et al., 2013) |
https://github.com/alexdobin/STAR |
| fastQC |
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
|
| MACS2 (v2.1.0) | (Zhang et al., 2008) |
https://github.com/taoliu/MACS |
| Trim galore |
https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
|
| Tadtree | (Weinreb and Raphael, 2016) |
http://compbio.cs.brown.edu/projects/tadtree/ |
| Juicer | (Durand et al., 2016) |
https://github.com/theaidenlab/juicer |
| BS-seeker2 | (Guo et al., 2013) |
https://github.com/BSSeeker/BSseeker2 |
| GREAT | (McLean et al., 2010) |
http://great.stanford.edu |
| Other | ||
For gene annotation we defined a “flattened” list of longest transcript forms for each gene, generated on Ensgene annotations, obtained from the UCSC table browser. For each gene, Ensembl IDs were matched to MGI gene names. Then, for each unique MGI gene name, the most upstream Ensgene TSS and the most downstream TES were taken as that gene’s start and stop. Based on these Ensembl gene models, we defined TSS regions and gene bodies.
Chromatin immunoprecipitation protocol
Cerebral cortex was dissected on ice in phosphate buffered saline from 1) MeCP2 KO and wild-type male litter mates at 7–8 weeks old, 2) MeCP2 OE and wild-type male litter mates at 7–10 weeks old, 3) DNMT3A Baf53b-cKO and control mice at 7–8 weeks old. The tissue was flash-frozen in liquid nitrogen and stored at −80°C. ChIP experiments were performed on half a cortex as previously described (Cohen et al., 2011), using an alternative chromatin fragmentation method. Chromatin were fragmented with Covaris E220 sonicator (5% Duty Factory, 140 Peak Incidence Power, 200 cycles per burst, milliTUBE 1mL AFA Fiber). ChIP was performed with H3K27ac (0.025–0.1μg; Abcam ab4729), H3K4me3 (2μg; Abcam ab1012), and H3K36me3 (0.2μg; Active Motif 61101). ChIP libraries for H3K27ac, H3K4me3, and H3K36me3 were generated using Ovation Ultralow Library System V2 (NuGEN). Libraries were pooled to a final concentration of 8–10nM and sequenced using Illumina HiSeq 3000 with GTAC, yielding 15–30 million single-end reads per sample.
Chromatin immunoprecipitation analysis
Sequenced reads were mapped to the mm9 genome using bowtie2 alignment, and reads were extended based on library sizes and deduplicated to consolidate PCR duplicate reads. Deduplicated reads were used to quantify read density normalized by the number of reads per sample and by read length in basepairs. Bedtools coverage -counts was used to quantify ChIP signal at the transcriptional start site (TSS), gene body (GB), and transcriptional end site (TES). For consistency with methylation analysis, the TSS was defined as a 1kb region surrounding the TSS (+/−500bp), the GB was defined as 3kb downstream of the TSS to the end of the transcript, and the TES was defined as 2kb upstream through 3kb downstream of the end of the transcript, based on our Ensembl gene models. edgeR was then used to determine differential ChIP-signal across genotypes.
Whole-genome bisulfite sequencing
Cerebral cortex was dissected from DNMT3A Nestin-cKO or DNMT3A Baf53b-cKO and controls at 7−8 weeks, flash-frozen in liquid nitrogen, and stored at −80°C. DNA extraction and bisulfite conversion were performed as before (Gabel et al., 2015). Briefly, genomic DNA was extracted and bisulfite libraries were generated using the Ovation Ultralow Methyl-Seq Library System (NuGEN). Libraries were pooled and sequenced using Illumina MiSeq 2×150 with the Spike-In Cooperative at Washington University in St. Louis.
Chromatin conformation capture
3C assays were adapted from previously described procedures (Kim and Dekker, 2018; Lieberman-Aiden et al., 2009; Yamada et al., 2019). Half of a cerebral cortex was dounced 10x with a loose pestle in cross-linking buffer (50mM Hepes-KOH, pH 7.9; 100mM NaCl; 1mM EDTA; 0.5mM EGTA) with 1% formaldehyde at room temperature for 15 minutes. Formaldehyde was quenched with 125mM glycine and incubated for 5 minutes at room temperature. The suspension was pelleted at 1150xg for 5 minutes at 4°C and the pellet was subsequently washed with PBS and frozen at −80°C. The frozen pellet was thawed and lysed (10mM Tris-HCl, pH 8; 10mM NaCl, 0.2% IGEPAL-630, Protease Inhibitor (Complete Protease Inhibitor Cocktail, Roche)) for 15 minutes followed by douncing 25x with a loose pestle on ice. The lysed cells were strained through a 70um cell strainer (Falcon) and pelleted at 200×g for 5 minutes at 4°C. Nuclei were resuspended in 1mL of 0.5% SDS and incubated at 62°C for 10 minutes to permeabilize the nuclei. 25ul (approx. 90–100k nuclei) were used for the 3C library construction, and SDS was quenched with Triton X-100 (1% Triton, 1x NEB 3.1 Buffer). Nuclei were digested overnight at 37°C with 200U of BglII. The digested nuclei were incubated at 65°C for 15 minutes to inactivate the enzyme. Samples were then ligated for 6 hours at 16°C with 4000U of T4 DNA ligase (NEB Ligase Buffer, 0.1mg/ml BSA, Triton X-100, NEB T4 ligase). Digested and ligated 3C samples were pelleted at 3500rpm for 5 minutes at 4°C and resuspended in 200ul NEB 3.1 Buffer and brought to 1% SDS and 250mM NaCl. 3C libraries were de-crosslinked overnight at 65°C. They were then incubated with 40ug RNase A at 37°C for 1 hour, followed by 80ug of Proteinase K at 55°C for 1 hour. The samples were purified with Phenol/Chloroform/lsoamyl Alcohol (25:24:1) and subsequently ethanol precipitated (100mM Sodium Acetate) overnight at −20°C. Control libraries were generated from Bacterial Artificial Chromosomes (BACs) as previously published (Kim and Dekker, 2018). BACs (see STAR methods) were isolated using PureLink HiPure Plasmid Maxiprep Kit (Invitrogen), following the modified protocol for BAC isolation.
Quantification and Statistical Analysis
Whole-genome bisulfite analysis
Bisulfite-sequencing cannot distinguish between hydroxymethylation and methylation at cytosines, detecting both only as modified sites during sequencing. Thus, measures of methylation included in this study represent the aggregate of both forms of methylation at sites across the genome. Bisulfite data for the DNMT3A Nestin-cKO and DNMT3A Baf53b-cKO, and data obtained from GEO as FASTQ files, were adapter-trimmed, mapped to mm9, then deduplicated and called for methylation using BS-seeker2 (with bowtie2). Nonconversion rate was set to be (--XS=.3, 2), and default settings were used otherwise. The methylation levels for genes and regions were assessed by summing the number of reads mapping to Cs that supported mC and dividing that by the number of reads mapping to Cs that supported non-mC, using bedtools map −o sum. This allows sites with more read information to contribute more in determining the methylation level of the surrounding region. In order to avoid confounding effects of promoter-associated depletion of methylation, genic methylation was assessed 3kb downstream of promoters to the TES. In order to assess any potential for C site coverage bias influencing TAD-associated Bisulfite-seq results, a bed file of every C/G in the genome was generated, and bedtools coverage -counts was run on it. Bisulfite coverage bias was assessed on informative sites for methylation with bedtools map −o mean on the number of reads mapping to Cs in the region.
RNA-sequencing quantification
MeCP2-repressed and activated genes were identified by quantitative analysis of exonic reads from total RNA from the MeCP2 KO and OE. For this analysis we applied an approach similar to previous studies of other brain regions that combined results of gene expression in these two strains in order to identify the most-robustly MeCP2-regulated genes (Ben-Shachar et al., 2009; Chahrour et al., 2008; Chen et al., 2015). DESeq2 was run using default parameters on exonic reads from MeCP2 KO and their littermate control animals (n=6 per genotype). Separately, we ran DESeq2 analysis on exonic reads from MeCP2 OE and their littermate control animals (n=5). The nominal p-values output by DESeq2 for each gene in each mutant-control comparison were then combined using the Fisher method (log-sum). The resulting combined p-values were then Benjamini-Hochberg corrected, and genes with a q-value < .1 and a log2 fold-change > 0 in the KO and a log2 fold-change < 0 in the OE were labeled as MeCP2-repressed, while genes with a q-value < .1 and a log2 fold-change < 0 in the KO and a log2 fold-change > 0 in the OE were labeled as MeCP2-activated. Notably, similar results for enrichment of mCA, and gene length were observed when examining lists of genes called as significantly dysregulated in the MeCP2 KO and OE on their own. However, a smaller gene list was identified in each independent analysis, likely due to the reduced statistical power. For comparison between changes in intronic RNA and changes in exonic RNA (Figure 3; S1) a list of significantly changed genes in the MeCP2 KO compared to littermate controls based on intronic fold-changes was generated by applying the same DESeq2 analysis to intronic reads derived from total RNA-seq of nuclear RNA.
RNA-seq aggregate plots examining changes in expression over lengths of genes (Figure S4G–I) were performed by binning genes into 1kb windows, then calculating nuclear intronic coverage over each bin using bedtools coverage -hist. Each gene was then normalized by the median amount of combined coverage from the MeCP2 KO and wild-type. Finally, genes were aligned by their TSSs, and median expression levels were plotted for each bin. For any given graph, the genes are filtered such that the lengths of genes plotted is equal to or greater than the aggregate length being plotted (Boswell et al., 2017; Gray et al., 2014). Termination ratios (adapted from (Boswell et al., 2017)) were calculated in a manner similar to the aggregate plots of expression. Windows were made for every gene longer than 100kb, from the TSS to 25kb downstream of the TSS (window A), and from the TES to 25kb upstream of the TES (window B). Coverage over these windows was calculated with bedtools coverage -hist, then coverage of window A was divided by the coverage of window B, within each sample. Then, wild type/MeCP2 KO samples were divided against each other, and a normalized metric for each wild type/MeCP2 KO replicate was generated. Finally, median metrics were plotted for each wild type/MeCP2 KO replicate. Simulated metrics were generated by progressively applying exonic fold-changes to the wild-type expression throughout the gene. For example, a gene 50kb long, with an observed exonic log2 fold-change (MeCP2 KO/wild type) of −1 would be divided into 50 1kb bins. A simulated KO expression pattern would then be generated by progressing down each of the 50 bins, applying a fold-change of 1/50 * −1, 2/50 * −1, etc, to the expression of the corresponding bin in the WT.
Controlled resampling
A resampling approach that controls for a desired variable was used throughout the paper. A sample set (e.g. MeCP2-repressed genes) and a control set (e.g. all other genes) are assessed for a certain characteristic (e.g. expression, length, etc). The control set is then sorted on this characteristic, and each entry in the sample set is assessed for where it would be placed in the sorted control set. Then, for each sample entry, a control entry is selected that is within 10 places of the sample entry, generating a control set the same size and variable distribution as the sample set.
TAD boundary analysis
For Figures 1F, 2C, S2B, and S2F, all TADs that intersected MeCP2-repressed, MeCP2-activated, and unchanged genes were selected, boundaries phased, and values for mC/C, coverage, GC percent, or ChIP/Input plotted in aggregate using R, python, and bedtools. Shuffling analysis was performed in these cases by moving each TAD randomly around their target genes (TADs containing MeCP2-repressed genes were shuffled around MeCP2-repressed genes, etc.). This was done once to generate a single example (e.g. Figure 1F), or up to 20 times to generate a resampling ribbon (e.g. Figure S2B). Figures 1G and S2K were generated by selecting boundaries through the following method: TADs were assigned a score, based on the difference between its mCA level and the mCA level of the next downstream TAD. Then, the 33% top scoring TADs were taken and plotted in aggregate analysis. For shuffled TAD control plots, TAD locations were randomized and the selection process for these shuffled TADs was repeated. We note that because both true TADs and shuffled TADs in these plots were selected based on high differences in average methylation between them, a reduction of signal going from left to right is predicted. However, true TADs display two aspects of mCA/CA signal that are not present in shuffled TADs, which demonstrate the organizing effects associated with TAD boundaries: 1) True TADs are more highly enriched for mCA, showing that resampling TADs eliminates an organized enrichment of mCA/CA in specific regions of the genome. 2) A steep step-down in mCA/CA occurs at true TAD boundaries, while a more gradual fall off in signal occurs at reshuffled boundaries. These two differences illustrate that ~30% shifts in mCA/CA levels are common between adjacent TADs, and they demonstrate that TAD boundaries delineate a sharp transition between mCA/CA levels at these regions. Plotted in all figures is the average of the upstream and downstream TAD boundaries, taking into account TAD orientation. Upstream boundaries (where the TAD would be on the right, instead of the left), have their orientation flipped horizontally along the boundary. The value plotted is the mean of the flipped upstream boundaries and the unflipped downstream boundaries.
TAD methylation cross-correlation
Cross-correlation matrices (e.g. Figure 1H) were generated by dividing each domain into 10 equally-sized bins, then prepending and appending 10 identically sized bins up and down-stream of the domain, making a number of domains x 30 matrix. Each column of this matrix was then correlated against each other, making a 30 × 30 correlation matrix, which was plotted in heatmap form. Shuffled TADs were generated as a negative control by randomly placing TAD-sized regions around the genome, separated from each other by similar distances as actual TADs to retain TAD structure.
In order to calculate heatmaps of correlations between mCA/CA levels of genomic elements in and outside of TADs and mean TAD mCA/CA levels (Figure S3J), TADs were again divided into 10 equally sized bins, with equal-sized regions placed upstream and downstream the domain. Enhancers, genes, and TSSs in the genome were then intersected with these regions, and Spearman correlations between TAD methylation (subtracting out the methylation of the element if necessary) and element methylation within each region were calculated.
To assess the similarity of mCA levels between individual elements inside and outside of the same TAD (i.e. TSS regions, enhancers, gene bodies; Figure S3K), each element was paired to each other element on the same chromosome. Each pair was then assessed if they paired within or between TADs. Because mCA varies with genomic distance, each intra-TAD pair was matched to the extra-TAD pair with the most similar distance between elements, and Spearman correlations were calculated on the two distance-matched sets.
Identification of enhancers
Enhancers in this study were defined by stringent criteria requiring the presence of overlapping H3K27ac and H3K4me1 peaks that occur outside of a known TSS region, or a peak of the promoter-associated histone mark H3K4me3. As noted, this led to the exclusion of some subthreshold regions of H3K27ac enrichment that may represent true regulatory elements, but ensured that we analyzed robust regulatory elements in our studies. For this analysis, bed files of H3K27ac and H3K4me3 ChIP-seq were pooled by replicate. Peaks of H3K27ac and H3K4me3 ChIP-seq were identified using the MACS2 peak calling algorithm on the pooled bed files using the pooled ChIP Input as background signal (macs2 callpeak --nomodel −q 0.05). MeCP2 KO and wild type peak files were then combined using bedtools unionbedg, and overlapping peaks were merged into single peaks using bedtools merge. Bedtools intersect was used to identify H3K27ac peaks that did not overlap with gene promoter regions (1kb around annotated TSS) or with H3K4me3 peaks from MACS2. These non-overlapping H3K27ac peaks were then further filtered for landing within an H3K4me1 peak, as called in the ENCODE-generated broadpeak file for H3K4me1 ChIP from 8-week old cortex. All H3K27ac peaks that remained after these rounds of filtering were defined as enhancers.
To identify the enhancers most robustly regulated by MeCP2, we used combined analysis of the MeCP2 KO and OE, similar to the approach used for determining gene-expression changes. We ran differential ChIP-seq analysis on H3K27ac from the MeCP2 KO and their littermate control animals (n=5), and from the MeCP2 OE and their littermate controls (n=3). Reads were quantified in all merged acetyl peaks, and edgeR was used to calculate nominal p-values and fold-changes for these peaks. These p-values were then combined using the Fisher method (log-sum) and were Benjamini-Hochberg corrected. Acetyl peaks with a combined q-value < 0.1, and a log2 fold-change > 0 in the KO and a log2 fold-change < 0 in the OE were called as MeCP2-repressed peaks, while peaks with a combined q-value < 0.1, and a log2 fold-change < 0 in the KO and a log2 fold-change > 0 in the OE were called as MeCP2-activated peaks.
Enhancers were also called as misregulated in the DNMT3A Baf53b-cKO, using edgeR. H3K27ac ChIP-seq reads from the DNMT3A Baf53b-cKO and control (n=6) were quantified in the merged acetyl peaks called from the MeCP2 KO/control. edgeR was then run on these regions, and peaks with a q-value < 0.1 were called as misregulated in the DNMT3A Baf53b-cKO.
ATAC peak analysis
To sensitively detect sub-peak-threshold histone acetylation signal that could correspond to putative regulatory elements, a compendium of all ATAC peaks detected in the genome was obtained from http://atlas.gs.washington.edu/mouse-atac/data/ (atac_matrix.binary.qc_filtered.peaks.txt) and acetylation was quantified in these regions. ATAC peaks that landed within 1kb of a TSS were filtered out, and methylation and acetylation analysis was performed on them as described for enhancers. Equal-sized control regions for enhancers and ATAC peaks were generated through a structured resampling approach. Enhancers and ATAC peaks, if located within a gene, are shuffled within that gene. If the peak is extragenic, it is shuffled between the nearest upstream and downstream enhancer/gene/ATAC peak. In each case, enhancers and ATAC peaks are restricted from landing within existing enhancers and ATAC peaks. Normalized pseudocounts of acetylation for enhancers, ATAC peaks, and resampled ATAC peaks were generated from edgeR common dispersions, running on each dataset and mouse strain separately.
Associations between enhancers and genes using Hi-C and GREAT
GREAT 3.0 (McLean et al., 2010) was used to determine potential enhancer-promoter interactions. NCBI build 37 of Mus Musculus was used as the species assembly, and bed files of enhancer regions were uploaded into the web tool. Enhancer-promoter interactions were identified for MeCP2-repressed, MeCP2-activated, and all other enhancers. All identified genes linked to these enhancers were used for further analysis. To link enhancers to promoters by Hi-C, intrachromosomal Hi-C matrices (KR-normalized) were extracted at 1.5kb resolution using Straw (Durand et al., 2016), and mean average interactions for all distances were calculated per-chromosome. Each matrix was then filtered down to only interactions between promoters and enhancers within 3mb of each other, and enhancers-promoter pairs with greater than 3 observed interactions and an observed/expected ratio over 1.5 were linked, making Hi-C-linked enhancers. Enhancers that were linked to promoters of MeCP2-repressed or MeCP2-activated genes were then used in analysis in Figure 6B.
For analysis of intragenic versus extragenic contact frequencies (Figure 6G), the same normalized contact matrices were analyzed. Genes greater than 50kb were extended on either side by their respective gene lengths, and contacts were mapped to them using bedtools intersect -wao -F 1. Each region was then split into 60 equally-sized bins (20 upstream, 20 intragenic, 20 downstream), making 3600 possible regions of interactions. Average interaction frequencies within these regions were calculated from the interaction matrix, and each gene’s intragenic and extragenic interactions were aggregated by calculating the mean of each bin of interaction.
For Figure 6H, intragenic and extragenic enhancer-promoter interactions were distance matched to control for the greater number and variability of extragenic interactions. To do this, a similar resampling approach to Figure S3K was used: for each intragenic interaction, an extragenic interaction with a similar distance was selected for comparison.
To assess the relationship between enhancer acetylation fold-change and gene fold-change (Figure S5M), partial correlations between enhancer/control region acetyl fold-change and gene fold-change were calculated by averaging the acetyl fold-changes of elements within the gene and correlating this aggregate value to that gene’s fold-change.
Quantitative 3C analysis
Quantitative PCR was employed, as previously described (Joo et al., 2016; Schaukowitch et al., 2014; Tolhuis et al., 2002), to determine enhancer-promoter interactions of enhancers using the primers listed (Table S5). Negative regions were selected from genomic restriction fragments that did not contain detectable enhancer sequences and were located nearby to the enhancer being tested but in closer proximity to the anchor TSS. Relative concentrations of enhancers and corresponding negative regions were calculated from a standard curve of BAC 3C libraries containing the targeted loci (see STAR methods). Enrichments of interactions were then calculated as the relative concentration of targeted enhancers divided by the relative concentration of the nearby negative region.
mC context enrichment analysis
Trinucleotide contexts and methylation status (a rational number determined through bisulfite coverage - # of Cs mapping to site / # of Ts mapping to site) were determined for each cytosine and guanine in the genome, and then sites were assigned to enhancers based on proximity (a site was assigned to an enhancer if it was within 1 kb of the enhancer’s center). T-tests were then run for each context, summing methyl-weighted or unweighted (for total mC per kb / C sites per kb respectively), or averaging methylation statuses (for percent mC), comparing test sets (up/downregulated enhancers, or enhancers within certain genes) to a set of resampled control enhancers with similar acetylation levels 5 times the size of the test set.
mC vs acetylation change local correlation analysis at enhancers
For both enhancers and gene local correlation analysis, 1 kb-sized bins were assessed for average methylation around and within enhancers/genes, making an N x M sized matrix of methylation, where N = # of enhancers/genes, and M = # of bins. Each column of this matrix was then correlated against the matching enhancer/gene’s fold-change in the MeCP2 KO/MeCP2 OE. For enhancers, correlation analysis was centered on summits of wild-type acetylation and MeCP2 KO acetylation and analyzed separately through MACS2.
Running-average plots
Running-average plots of mCA/CA and genic fold-change as well as total mC sites and genic fold-change were generated from means of 201 gene bins, with a 1 gene step, using the rollMean command in the zoo package of R. Length-controlled resampling was performed by selecting a gene within .75 − 1.25 times the length of each gene in the test set.
TAD/element methylation correlation
Enhancers, transcriptional start sites, and gene bodies were intersected with TADs and contact domains, and each unique intersection was plotted (Figure 2E, S2L, M). Shuffling analysis was generated by moving elements randomly within its containing TAD. To avoid spuriously detecting a correlation due to the contribution of the elements themselves to the calculated average methylation of the TAD, the mC signal from the elements within the TAD (e.g. gene) were excluded from the calculation of the mC levels for that TAD.
Misregulated gene-enhancer linkage enrichment
To calculate the significance and magnitude of the linkage between misregulated genes and enhancers (Figure 6B), misregulated enhancers were resampled based on acetylation, and a fisher’s exact test was performed comparing the association of enhancers with misregulated genes between the misregulated and control set. This process was repeated 1000 times, and the median p-value and log2 fold-enrichment was plotted.
Data and Software Availability
All genomic data generated in this study have been uploaded to the NCBI GEO archive GSE123373
Supplementary Material
Table S1. Gene expression Table, Related to Figure 1, Figure 3, Figure 5, Figure 6, Figure S1, Figure S2, Figure S4, and Figure S6.
Supplementary Figures
Table S2. TADtree-defined TADs, Related to Figure 1, Figure 2, Figure 6, Figure S2, Figure S3, and Figure S6.
Table S4. Enhancer ChIP-seq Table, Related to Figure 2, Figure 4, Figure 5, Figure 6, Figure S3, Figure S5, and Figure S6.
Table S5. List of primers for 3C, related to STAR Methods.
Highlights.
Chromatin folding domains shape regions of enriched non-CG DNA methylation in neurons
MeCP2 represses transcription of genes in domains of enriched non-CG DNA methylation
MeCP2 represses enhancers containing a high density of methylated CA and CG sites
Intragenic enhancer dysregulation contributes to MeCP2 mutant gene expression changes
Acknowledgements
We thank Y. Liu and M. Nemera and the Gabel lab, as well as J. Dougherty, K. Kroll, J. Edwards, A. Bonni, and J. Yi for support and feedback on the manuscript. We thank L. Boxer, W. Renthal, and M. Greenberg for discussions. The following grants supported this work: NIH 5T32GM007067 and F31NS108574 to A.W.C; The Klingenstein-Simons Fellowship Fund, the G. Harold and Leila Y. Mathers Foundation, the Brain and Behavior Research Foundation, and NIMH R01MH117405 to H.W.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References Cited
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, and Zoghbi HY (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar S, Chahrour M, Thaller C, Shaw CA, and Zoghbi HY (2009). Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet 18, 2431–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP (1980). DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J-P, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell SA, Snavely A, Landry HM, Churchman LS, Gray JM, and Springer M (2017). Total RNA-seq to identify pharmacological effects on specific stages of mRNA synthesis. Nat. Chem. Biol 13, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer LD, Renthal W, Greben AW, Whitwam T, Silberfeld A, Stroud H, Li E, Yang MG, Kinde B, Griffith EC, et al. (2019). MeCP2 represses the rate of transcriptional initiation of highly methylated long genes. Mol. Cell 77 Published online November 26, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J, and Zoghbi HY (2008). MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science 320, 1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen K, Lavery LA, Baker SA, Shaw CA, Li W, and Zoghbi HY (2015). MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. U. S. A 112, E2982–E2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewa-Waclaw J, Bird A, von Schimmelmann M, Schaefer A, Yu H, Song H, Madabhushi R, and Tsai L-H (2016). The Role of Epigenetic Mechanisms in the Regulation of Gene Expression in the Nervous System. J. Neurosci 36, 11427 LP–11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewa-Waclaw J, Shah R, Webb S, Chhatbar K, Ramsahoye B, Pusch O, Yu M, Greulich P, Waclaw B, and Bird AP (2019). Quantitative modelling predicts the impact of DNA methylation on RNA polymerase II traffic. Proc. Natl. Acad. Sci 116, 14995 LP–15000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, et al. (2011). Genome-Wide Activity-Dependent MeCP2 Phosphorylation Regulates Nervous System Development and Function. Neuron 72, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, et al. (2018). A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Gorkin DU, and Ren B (2016). Chromatin Domains: the Unit of Chromosome Organization. Mol. Cell 62, 668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gécz J, et al. (2005). Duplication of the MECP2 Region Is a Frequent Cause of Severe Mental Retardation and Progressive Neurological Symptoms in Males. Am. J. Hum. Genet 77, 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, and Greenberg ME (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JM, Harmin DA, Boswell SA, Cloonan N, Mullen TE, Ling JJ, Miller N, Kuersten S, Ma Y-C, McCarroll SA, et al. (2014). SnapShot-Seq: A Method for Extracting Genome-Wide, In Vivo mRNA Dynamics from a Single Total RNA Sample. PLoS One 9, e89673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, and Young RA (2007). A Chromatin Landmark and Transcription Initiation at Most Promoters in Human Cells. Cell 130, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci 17, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen P-Y, and Pellegrini M (2013). BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics 14, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39, 311–318. [DOI] [PubMed] [Google Scholar]
- Ip JPK, Mellios N, and Sur M (2018). Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci 19, 368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, Zhao Y-T, Fasolino M, Lamonica JM, Kim YJ, Georgakilas G, Wood KH, Bu D, Cui Y, Goffin D, et al. (2017). Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med 23, 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo J-Y, Schaukowitch K, Farbiak L, Kilaru G, and Kim T-K (2016). Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat. Neurosci 19, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, and Sasaki H (2004). Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429, 900–903. [DOI] [PubMed] [Google Scholar]
- Kim TH, and Dekker J (2018). Generation of Control Ligation Product Libraries for 3C Analyses. Cold Spring Harb. Protoc 2018, pdb.prot097865. [DOI] [PubMed] [Google Scholar]
- Kinde B, Gabel HW, Gilbert CS, Griffith EC, and Greenberg ME (2015). Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. U. S. A 112, 6800–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde B, Wu DY, Greenberg ME, and Gabel HW (2016). DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc. Natl. Acad. Sci 113, 15114–15119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokura K, Kaul SC, Wadhwa R, Nomura T, Khan MM, Shinagawa T, Yasukawa T, Colmenares C, and Ishii S (2001). The Ski Protein Family Is Required for MeCP2-mediated Transcriptional Repression. J. Biol. Chem 276, 34115–34121. [DOI] [PubMed] [Google Scholar]
- Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH, Yu M, He C, Sanguinetti G, Sowers LC, et al. (2017). MeCP2 recognizes cytosine methylated tri-nucleotide and dinucleotide sequences to tune transcription in the mammalian brain. PLOS Genet. 13, e1006793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, and Bird A (2015). Rett syndrome: a complex disorder with simple roots. Nat. Rev. Genet 16, 261–275. [DOI] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, et al. (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci 16, 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan RR, Lewis JD, McKay S, Kleiner EL, and Bird AP (1989). Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 58, 499–507. [DOI] [PubMed] [Google Scholar]
- Mellén M, Ayata P, and Heintz N (2017). 5-hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes. Proc. Natl. Acad. Sci. U. S. A 114, E7812–E7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. (2015). Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S, Meletis K, Fu D, Jhaveri S, and Jaenisch R (2007). Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn 236, 1663–1676. [DOI] [PubMed] [Google Scholar]
- Nott A, Cheng J, Gao F, Lin Y-T, Gjoneska E, Ko T, Minhas P, Zamudio AV, Meng J, Zhang F, et al. (2016). Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat Neurosci 19, 1497–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco NL, Heaven MR, Holt LM, Crossman DK, Boggio KJ, Shaffer SA, Flint DL, and Olsen ML (2017). RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome. Mol. Autism 8, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman AT, Pohodich AE, Wan Y-W, Yalamanchili HK, Lowry WE, Zoghbi HY, and Liu Z (2018). Apparent bias toward long gene misregulation in MeCP2 syndromes disappears after controlling for baseline variations. Nat. Commun 9, 3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Boxer LD, Hrvatin S, Li E, Silberfeld A, Nagy MA, Griffith EC, Vierbuchen T, and Greenberg ME (2018). Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci 21, 1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rube HT, Lee W, Hejna M, Chen H, Yasui DH, Hess JF, LaSalle JM, Song JS, and Gong Q (2016). Sequence features accurately predict genome-wide MeCP2 binding in vivo. Nat. Commun 7, 11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NCT, Schreiber SL, Mellor J, and Kouzarides T (2002). Active genes are tri-methylated at K4 of histone H3. Nature 419, 407. [DOI] [PubMed] [Google Scholar]
- Schaukowitch K, Joo J-Y, Liu X, Watts JK, Martinez C, and Kim T-K (2014). Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schübeler D (2015). Function and information content of DNA methylation. Nature 517, 321. [DOI] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GMG, and Katz DM (2011). Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: Focus on Mecp2 and Met. Curr. Opin. Neurobiol 21, 827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr ARW, James KD, Turner DJ, Andrews R, and Bird AP (2010). Neuronal MeCP2 Is Expressed at Near Histone-Octamer Levels and Globally Alters the Chromatin State. Mol. Cell 37, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, et al. (2016). ENCODE data at the ENCODE portal. Nucleic Acids Res. 44, D726–D732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmann M, Lupiáñez DG, and Mundlos S (2018). Structural variation in the 3D genome. Nat. Rev. Genet 19, 453–467. [DOI] [PubMed] [Google Scholar]
- Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, et al. (2017). Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1164.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Okaty BW, Arnson HA, Kato S, Dani VS, and Nelson SB (2014). Cell-Type-Specific Repression by Methyl-CpG-Binding Protein 2 Is Biased toward Long Genes. J. Neurosci 34, 12877–12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B, Palstra R-J, Splinter E, Grosveld F, and de Laat W (2002). Looping and Interaction between Hypersensitive Sites in the Active ß-globin Locus. Mol. Cell 10, 1453–1465. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, and Schutz G (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23, 99–103. [DOI] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, and Jaenisch R (2002). Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc. Natl. Acad. Sci. U. S. A 99, 15536–15541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C, and Raphael BJ (2016). Identification of hierarchical chromatin domains. Bioinformatics 32, 1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, and Ren B (2012). Base-Resolution Analyses of Sequence and Parent-of-Origin Dependent DNA Methylation in the Mouse Genome. Cell 148, 816–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Valnegri P, Juric I, Abnousi A, Markwalter KH, Guthrie AN, Godec A, Oldenborg A, Hu M, et al. (2019). Sensory experience remodels genome architecture in neural circuit to drive motor learning. Nature 569, 708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Cao M, Yoo AS, Zhang Z, Chen L, Crabtree GR, and Wu JI (2015). Generation of BAF53b-Cre transgenic mice with pan-neuronal Cre ac- tivities. Genesis 53, 440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene expression Table, Related to Figure 1, Figure 3, Figure 5, Figure 6, Figure S1, Figure S2, Figure S4, and Figure S6.
Supplementary Figures
Table S2. TADtree-defined TADs, Related to Figure 1, Figure 2, Figure 6, Figure S2, Figure S3, and Figure S6.
Table S4. Enhancer ChIP-seq Table, Related to Figure 2, Figure 4, Figure 5, Figure 6, Figure S3, Figure S5, and Figure S6.
Table S5. List of primers for 3C, related to STAR Methods.
Data Availability Statement
All genomic data generated in this study have been uploaded to the NCBI GEO archive GSE123373