

Abstract
Tandem repeat diseases include the neurodegenerative disorders known as polyglutamine (polyQ) diseases, caused by CAG repeat expansions in the coding regions of the respective disease genes. The nine known polyQ disease include Huntington’s disease (HD), dentatorubral–pallidoluysian atrophy (DRPLA), spinal bulbar muscular atrophy (SBMA), and six spinocerebellar ataxias (SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17). The underlying disease mechanism in the polyQ diseases is thought principally to reflect dominant toxic properties of the disease proteins which, when harboring a polyQ expansion, differentially interact with protein partners and are prone to aggregate. Among the polyQ diseases, SCA3 is the most common SCA, and second to HD in prevalence worldwide. Here we summarize current understanding of SCA3 disease mechanisms within the broader context of the broader polyQ disease field. We emphasize properties of the disease protein, ATXN3, and new discoveries regarding three potential pathogenic mechanisms: 1) altered protein homeostasis; 2) DNA damage and dysfunctional DNA repair; and 3) nonneuronal contributions to disease. We conclude with an overview of the therapeutic implications of recent mechanistic insights.
Keywords: Ataxin-3, ATXN3, Spinocerebellar Ataxia, SCA3, Machado Joseph disease, MJD, Polyglutamine disease, Neurodegenerative disease, deubiquitinase
1. Introduction
Among the tandem repeat diseases, a subset of neurodegenerative diseases are caused by exonic CAG trinucleotide repeat expansions that are translated into abnormally long polyglutamine (polyQ) tracts in the disease proteins (Klockgether, Mariotti, and Paulson 2019). Currently, nine disorders comprise the so-called polyQ diseases, including Huntington’s disease (HD), Dentatorubral–pallidoluysian atrophy (DRPLA), spinal bulbar muscular atrophy (SBMA), and six distinct spinocerebellar ataxias (SCAs) including SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17 (Klockgether, Mariotti, and Paulson 2019). PolyQ diseases share several key features including that they: 1) are autosomal dominant disorders except SBMA, which is X-linked and sex-limited; (2) principally affect the central nervous system (CNS) although the peripheral nerves and muscle are variably affected; (3) usually present in adulthood and slowly progress over many years; (4) display neuropathological accumulation of disease protein aggregates, largely in neurons; and (5) are typically fatal disorders with no approved treatments to alter the disease course (Paulson et al. 2017).
Here we review what is known about the disease mechanisms underlying SCA3, the most common polyQ SCA, and relate this information to the broader polyQ field. We begin by introducing the disease protein, ATXN3, and summarizing the relevance of ATXN3 protein function and protein context to disease pathogenesis. We then highlight new discoveries regarding three potential pathogenic mechanisms in SCA3: 1) perturbations in cellular protein homeostasis; 2) disruption of DNA damage repair pathways; and 3) contributions of nonneuronal cells. We conclude with a discussion of how a thorough understanding of pathogenic mechanisms in SCA3 and related polyQ diseases is necessary to identify the most compelling therapeutic targets for these devastating, fatal, and currently untreatable disorders.
2. Genetic, Clinical, and Neuropathological features of SCA3
The neurodegenerative disorder now designated as SCA3 was first recognized in the 1970s in several families of Azorean descent as a heritable “ataxia-plus” disorder with overlapping clinical and pathological characteristics (Coutinho and Andrade 1978). Two of the identified families presenting with this dominantly inherited ataxia were descended from William Machado and Antone Joseph, leading to the disease designation Machado-Joseph Disease (MJD), a label still regionally used, particularly in Portuguese-speaking countries. In 1993, the genetic mutation causing SCA3 was mapped to chromosome 14q32.1 by two separate groups: one group studying the Azorean-linked MJD lineages and another European group which had designated the disease SCA3 after discovering that the genetic locus in families exhibiting similar symptoms to SCA1 and SCA2 was distinct from these two previously identified diseases (Takiyama et al. 1993; Gispert et al. 1993). In 1994, cloning of the SCA3 and MJD genetic loci identified the same polyQ-encoding CAG trinucleotide expansion, leading to convergence of these diseases into a single disorder, referred to here as SCA3 (Kawaguchi, Okamoto, Taniwaki, Aizawa, Inoue, Katayama, Kawakami, Nakamura, Nishimura, Akiguchi, and et al. 1994).
The CAG repeat expansion in SCA3 resides in exon 10 of the ATXN3 gene (Haberhausen et al. 1995; Kawaguchi, Okamoto, Taniwaki, Aizawa, Inoue, Katayama, Kawakami, Nakamura, Nishimura, Akiguchi, Kimura, et al. 1994) (Figure 1). In healthy individuals, this CAG repeat ranges from 12 to 44, whereas in affected individuals the CAG repeat ranges from around 56 to 87 (Paulson 2007; Maciel et al. 1995; Maruyama et al. 1995; Durr et al. 1996). Individuals harboring CAG repeat lengths ranging from 45 to 55 present incomplete penetrance of SCA3 symptoms (Ashizawa, Öz, and Paulson 2018). As with other polyQ diseases, the CAG repeat length inversely correlates with age of disease onset and directly correlates with severity of disease (Maciel et al. 1995; Maruyama et al. 1995; Durr et al. 1996). SCA3 also exhibits “anticipation”, a phenomenon shared across most polyQ diseases (Maciel et al. 1995; Maruyama et al. 1995). Anticipation refers to the tendency for expanded microsatellite repeats (e.g., the expanded CAG repeat in mutant ATXN3) to become increasingly long in subsequent generations (Durr et al. 1996), thereby typically causing more severe, earlier onset disease in offspring. This variability in CAG repeat length, in conjunction with (still poorly understood) genetic modifiers, contributes to the high variability in the clinical presentation of disease. Albeit rare, homozygous SCA3 patients have been reported, wherein an individual inherits two copies of the expanded mutant ATXN3 gene that leads to more severe symptoms and earlier onset (Carvalho et al. 2008; Sobue et al. 1996). To our knowledge, no humans that are haploinsufficient or null for ATXN3 have been reported.
Figure 1. Schematic of ATXN3 protein.

The N-terminal Josephin domain contains the deubiquitinase (DUB) catalytic site (C14), as well as nuclear export sites (NES) and a small ubiquitin-like modifier [SUMO]-interacting motif (SIM). The C-terminal tail contains two or three ubiquitin-interacting motifs (UIMs), a putative nuclear localization signal (NLS) and the variable polyQ repeat. Repeat lengths of normal and pathogenic polyQ are shown. ATXN3 can be post-translationally modified by ubiquitin (Ub) at K117, sumoylation (S) at K166 and K356, and phosphorylation at S12, S236, S256, S260, S261, S335, and S347.
SCA3 disease is believed to be the most common dominantly inherited form of ataxia, affecting approximately 1:50,000-100,000 people, and is second to HD in prevalence among the polyQ diseases (Durr 2010; Gardiner et al. 2019). While Portuguese SCA3 families have been shown to arise from two haplotypes, it has been established that the majority of SCA3 families worldwide result from one intragenic haplotype (Gaspar et al. 2001). Still significant regional and ethnic differences in SCA3 prevalence have been reported, with the greatest prevalence reported in East Asian countries including China, Japan and Taiwan, as well as Portugal and Brazil (Buijsen et al. 2019). Symptom onset in SCA3 typically begins in adulthood between the third to fifth decade of life and progresses slowly with age (Rub et al. 2013). SCA3 patients exhibit a wide range of progressive motor impairments, including prominent cerebellar ataxia with abnormal gait, impaired balance, limb incoordination, dystonia, spasticity, dysarthria, dysphagia, and oculomotor abnormalities (Rub et al. 2013; Paulson et al. 2017). Parkinsonism with or without tremor also occurs in a subset of SCA3 patients (Park, Kim, and Jeon 2015). Though the severity and rate of decline varies across SCA3 patients, failure in brainstem-associated functions usually leads to death within 10-15 years of symptom onset (Diallo et al. 2018).
Progressive motor impairment in SCA3 results from neuronal dysfunction and neuronal cell loss in somatosensory and motor nuclei spanning the brainstem, cerebellum, midbrain, spinal cord, striatum, and thalamus (Rub et al. 2013). A prominent gross anatomical feature in SCA3 patients is enlargement of the fourth ventricle resulting from atrophy of neurons in the basilar pons and deep cerebellar nuclei, and loss of pontocerebellar fibers and spinocerebellar tracts (Durr et al. 1996; Rub et al. 2013). Post-mortem SCA3 disease brains exhibit significant degeneration of dopaminergic neurons of the substantia nigra and vestibular nuclei (Rub et al. 2004; Rub et al. 2013; Durr et al. 1996). SCA3 also leads to degeneration of motor neurons of the cranial nerve nuclei, red nucleus, subthalamic nucleus, globus pallidus, and some thalamic nuclei (Rub et al. 2003; Rub et al. 2013; Rüb et al. 2002; Rub et al. 2002). Spinal cord atrophy can occur in anterior horn, dorsal root ganglia, and dorsal nuclei (Fahl et al. 2015). Unlike in most other SCAs, olivary nuclei and the cerebellar cortex including Purkinje cells are relatively spared in SCA3 (Rub et al. 2013).
The cerebral cortex is affected in SCA3 patients but to a lesser extent than the brainstem and cerebellar nuclei (Yamada et al. 2001). By MRI, SCA3 patients exhibit reduced gray matter volume throughout temporal, frontal, parietal and insular areas (Lopes et al. 2013). Magnetic resonance spectroscopy (MRS) studies of SCA3 patients have identified metabolic abnormalities in white matter tracts beginning in premanifest SCA3 (Joers et al. 2018; Adanyeguh et al. 2015). Symptomatic SCA3 patients display mild cognitive impairments in executive function and memory, likely reflecting cerebral involvement in disease and/or disruptions to cerebellar-cerebral circuitry (Lopes et al. 2013). Sleep disorders are also common amongst patients, including REM sleep behavior disorder and restless leg disorder (Pedroso et al. 2011; Seshagiri et al. 2018). SCA3 pathology is not limited to the CNS: patients exhibit progressive peripheral neuropathy particularly in distal limbs leading to muscle atrophy and areflexia (Suga et al. 2014; Linnemann et al. 2016).
Accumulation of ubiquitinated protein aggregates or inclusions containing the mutant polyQ-expanded ATXN3 are found throughout vulnerable brain regions in SCA3 (Schmidt et al. 1998; Sittler et al. 2018; Paulson, Perez, et al. 1997). Neuronal nuclear inclusions (NNI) make up the majority of aggregates, though smaller neuronal cytoplasmic inclusions (NCIs) and distal axonal aggregates also occur (Hayashi, Kobayashi, and Furuta 2003; Seidel et al. 2010; Seidel et al. 2017). Similarly to aggregates in other polyQ diseases, ATXN3 NNI stain positively for many other proteins including ubiquitin, heat shock factor proteins, proteasomal subunits, transcription factors, autophagy-associated chaperones such as p62, other polyQ proteins, and non-expanded wildtype ATXN3 (Chai, Koppenhafer, Bonini, et al. 1999; Uchihara et al. 2001; Chai, Koppenhafer, Shoesmith, et al. 1999; Seidel et al. 2010; Seidel et al. 2017). The exact role that ATXN3 aggregation plays in SCA3 remains unclear, although it is generally accepted that the sequestration of critical protein quality control machinery into protein aggregates may contribute to neuronal stress. The same is likely true for other polyQ diseases (Paulson et al. 2017).
Scientists still do not fundamentally understand why only selective brain regions degenerate in SCA3 or other polyQ diseases, or why the CAG repeat length threshold for disease varies among the polyQ diseases. Clearly, understanding the normal functions of each polyQ protein will be critical to elucidating the cause of these distinct disease features in the polyQ disorders.
3. The SCA3 disease protein ATXN3
In polyQ diseases, most evidence points to aberrant actions of the disease protein as the principal driver of disease (reviewed in (Paulson et al. 2017)). The clinical and neuropathological differences between diseases likely reflects the unique protein context in which the polyQ expansion is embedded in each disease. Accordingly, to understand SCA3, we need to understand the SCA3 disease protein, ATXN3. ATXN3 (Figure 1) is a small, evolutionarily conserved protein that is ubiquitously expressed in the body and localized throughout the cell (Linhartová et al. 1999; Schmitt et al. 1997; Rodrigues et al. 2007; Paulson, Das, et al. 1997; Ichikawa et al. 2001). It is a deubiquitinase (DUB) implicated in protein quality control pathways (reviewed in (Costa and Paulson 2012)). DUBs are proteases that cleave ubiquitin from proteins, removing single ubiquitin modifications or cleaving polyubiquitin chains (Reyes-Turcu, Ventii, and Wilkinson 2009). ATXN3 binds polyubiquitinated proteins through three ubiquitin interaction motifs (UIMs) that flank the polyQ track, but the protein also contains additional ubiquitin binding sites in the catalytic domain known as the Josephin domain. ATXN3 binds the proteasome and preferentially cleaves ubiquitin chains that are four or more ubiquitins in length -- remarkably, the same chain length needed to target substrates efficiently to the proteasome (Thrower et al. 2000).
The preferential activity of ATXN3 toward longer poly-ubiquitin chains coupled with its known interaction with several ubiquitin ligases suggest that ATXN3 (Scaglione et al. 2011; Durcan et al. 2011; Matsumoto et al. 2004) edits ubiquitin chain length and composition on substrates destined for proteasomal degradation. The function of ATXN3, however, likely extends beyond the ubiquitin proteasome system (UPS) to other processes including macroautophagy, transcriptional regulation, cytoskeletal organization, and DNA damage repair (Doss-Pepe et al. 2003; Costa and Paulson 2012; Li et al. 2002; Araujo et al. 2011; Evert et al. 2003; Rodrigues et al. 2010; Gao et al. 2019; Gao et al. 2015; Burnett, Li, and Pittman 2003).
ATXN3 possesses several domains that contribute to its function (Figure 1). The structured globular amino-terminus contains the highly conserved Josephin domain responsible for catalytic cleavage of ubiquitin chains. The flexible C-terminal tail contains three ubiquitin interacting motifs (UIMs) that facilitate ATXN3 binding to poly-ubiquitinated chains (Costa and Paulson 2012). ATXN3 is itself regulated by mono-ubiquitination on Lys117, which enhances DUB activity (Todi et al. 2009). Dozens of ATXN3 isoforms have been identified, but most studies have focused on two full-length isoforms: one expressing all three UIMs (3UIM ATXN3) and one lacking the third UIM (2UIM ATXN3) (Harris et al. 2010). 3UIM ATXN3 appears to be the major isoform expressed in brain (Harris et al. 2010). The presence or absence of the third UIM may alter the specificity of ATXN3 towards poly-ubiquitin chains and substrates (Weishaupl et al. 2019).
Knocking out ATXN3 in vitro or in mice leads to increased levels of total ubiquitinated proteins, suggesting ATXN3 is a promiscuous DUB (Schmitt et al. 2007). Indeed, studies have identified over 100 substrates or binding partners of ATXN3 (Paulson et al. 2017). Through the editing of ubiquitin chains, ATXN3 can alter a substrate’s stability or activity in various ways depending on the substrate and the length or type of ubiquitin chain (Paulson et al. 2017). In vitro studies have shown that ATXN3 preferentially edits chains of at least four ubiquitin moieties (Winborn et al. 2008). A tetraubiquitin chain is also the minimum length required for poly-ubiquitinated substrates to be recognized by the 26S proteasome (Thrower et al. 2000). By binding the minimum length poly-ubiquitin chain on substrates, ATXN3 may prevent complete removal of the ubiquitin chain by other DUBs, and thus facilitate its recognition by the 26S proteasome (Li et al. 2015). ATXN3 DUB activity can also prevent or diminish substrate delivery to the proteasome, such as with the autophagy-related substrate Beclin1 and DNA damage repair and cell cycle proteins, Chk1 and p53 (Tu et al. 2017; Liu et al. 2016; Ashkenazi et al. 2017).
The presence of a polyQ expansion does not prevent mutant ATXN3 from binding poly-ubiquitin chains or deter its enzymatic activity in vitro (Winborn et al. 2008; Burnett, Li, and Pittman 2003). However, the polyQ expansion may alter its substrate specificity or protein-protein interactions in subtle ways that are not yet fully understood. Indeed, total levels of poly-ubiquitinated proteins are increased in the SCA3 brain, which may support some loss-of-function contributions to disease (Sittler et al. 2018). Broad disruptions to UPS as a result of sequestration of UPS regulators into mutant ATXN3 aggregates also likely contribute to the accumulation of poly-ubiquitinated proteins in SCA3 (Chai, Koppenhafer, Shoesmith, et al. 1999).
ATXN3 localizes both to the cytoplasm and nucleus to a varying degree depending on the cell type, and its transport across the nuclear membrane appears to be highly dynamic. Full-length ATXN3 possesses two nuclear export sequences (NES) and a putative nuclear localization sequence (NLS) that facilitate active transport across the nuclear membrane (Antony et al. 2009; Sowa et al. 2018) (Figure 1). In many cell types ATXN3 predominantly localizes to the cytoplasm under basal conditions (Antony et al. 2009), but cellular stressors such as oxidative stress or heat shock trigger rapid nuclear localization of ATXN3 (Reina, Zhong, and Pittman 2010). As noted earlier, the abnormal concentration and aggregation of mutant ATXN3 in neuronal nuclei is a defining feature of SCA3 neuropathology (Rub et al. 2013; Paulson, Perez, et al. 1997). Interestingly, preventing nuclear localization of mutant ATXN3 mitigates many disease features, whereas forcing mutant ATXN3 into the nucleus enhances disease in SCA3 mice (Bichelmeier et al. 2007).
ATXN3 is subjected to numerous post-translational modifications (PTMs), including ubiquitination at K117, SUMOylation, and phosphorylation (Figure 1). All three of these PTMs alter ATXN3 behavior and function. Ubiquitination at K117 potentiates the DUB activity of ATXN3 (Todi et al. 2010; Todi et al. 2009), and phosphorylation at specific sites can alter nucleocytoplasmic localization of ATXN3 (Mueller et al. 2009). While ATXN3 can bind SUMO and be SUMOylated (Zhou et al. 2013; Almeida et al. 2015), it is not yet clear how SUMO binding or SUMOylation alters ATXN3 function or clearance. In one study, mutating a SUMO binding site in ATXN3 accelerated the degradation of expanded but not normal ATXN3 (Almeida et al. 2015), suggesting that SUMO effects could be particularly germane to the mutant form of the disease protein. More work is needed to assess how these various posttranslational modifications might modulate disease pathogenesis.
3.1. Role of altered protein homeostasis
Perturbations in protein homeostasis represent a recurring theme in the polyQ diseases including SCA3 (Paulson et al. 2017; Klockgether, Mariotti, and Paulson 2019). The three major arms of protein quality control (PQC) --the UPS, molecular chaperones, and autophagy -- have all been implicated in SCA3 and other polyQ diseases. In these diseases, a consequence of continually producing aggregate-prone proteins (e.g. polyglutamine expanded ATXN3) is that PQC pathways may fail to keep pace. Moreover, the aggregation of ATXN3 and other polyQ disease proteins can sequester components of PQC pathways, exacerbating the problem. Among polyQ diseases, this issue is compounded in the case of SCA3 since the protein, a DUB, itself directly participates in PQC. In this section we review the role of ATXN3 in PQC pathways and the evidence for impairment of such pathways in disease.
As mentioned earlier, ATXN3 is a quality control DUB most closely linked to the UPS. PolyQ-expanded ATXN3 continues to bind and cleaves polyQ chains in simple cellular and in vitro assays. Under more complex physiological conditions, however, relatively little is known about how polyQ expansion affects the capacity of ATXN3 to regulate ubiquitination, the type of chain linkages it can cleave, and the efficiency of proteasomal delivery of substrates. Ubiquitin and proteasomal subunits are known to be sequestered in intraneuronal aggregates in SCA3 disease brain and mouse models (Seidel et al. 2010; Chai, Koppenhafer, Shoesmith, et al. 1999; Schmidt et al. 2002). While such sequestration may constrict neuronal UPS capacity, this has not been firmly established. But robust impairment of UPS activity may not be required to have a deleterious effect in SCA3. We suggest that even rather modest effects on UPS capacity could have profound indirect effects on other aspects of ubiquitin signaling within neurons or other cell types. Figure 2 illustrates these other components of ubiquitin signaling, to which greater attention should be paid in studies of SCA3 and other polyQ disorders.
Figure 2. Components of the Ubiquitin Signaling System.
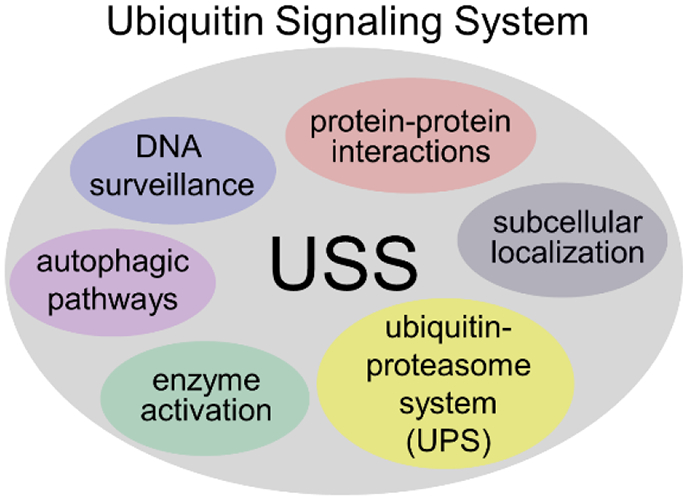
Ubiquitin conjugation to proteins serves diverse roles beyond the ubiquitin proteasome system (UPS), including roles in DNA repair, autophagy, subcellular localization and trafficking, and alterations in protein interactions. As a DUB, ATXN3 has been most closely associated with the UPS but increasingly is linked to other ubiquitin-dependent pathways shown here. Accordingly, in studies of SCA3 and other polyQ diseases, more attention should be paid to broader impairments in the ubiquitin signaling system beyond the UPS. Perturbations in ubiquitin homeostasis in one arm (e.g. the UPS) could have important, indirect effects on other arms of the ubiquitin signaling system (USS).
In various neurodegenerative diseases, an overwhelming of the UPS by accumulation of misfolded proteins can trigger aggresome formation, a cellular pathway that serves to consolidate proteotoxic aggregates (Kopito 2000). Aggresomes are dynamic perinuclear structures regulated by elements of the UPS machinery, including E3 ligases, proteasomal subunits and K63-linked poly-ubiquitin chains, and the autophagy-linked protein p62 (Olzmann, Li, and Chin 2008). Formation of aggresomes also relies on microtubule-dependent transport of aggregated proteins. Non-pathogenic ATXN3 exhibits a preference towards K63 ubiquitin linkages (Winborn et al. 2008), and directly interacts with many cytoskeletal proteins, proteasomal regulators, and autophagic proteins involved in aggresome formation, including tubulin, dynein, HDAC6, Parkin, CHIP, and p62 (Bonanomi et al. 2014; Mazzucchelli et al. 2009; Scaglione et al. 2011; Zhou et al. 2014). ATXN3 also localizes to, and regulates formation of, aggresomes containing misfolded disease proteins implicated in neurodegenerative disease, including mutant SOD1 and CFTR (Wang, Ying, and Wang 2012; Burnett and Pittman 2005). The ability of ATXN3 to mediate aggresome formation is ablated by mutating catalytic sites or removing UIMs, suggesting that ATXN3’s DUB-dependent roles are critical to aggresome formation (Burnett and Pittman 2005).
For over two decades, studies have established a link between SCA3 and a variety of molecular chaperones (Chan et al. 2000; Warrick et al. 1999; Chai, Koppenhafer, Bonini, et al. 1999; Vos et al. 2010; Ito et al. 2016; Seidel et al. 2012). Numerous chaperones, when overexpressed, can mitigate disease features in SCA3 model systems either by improving the “handling” of mutant ATXN3 or by boosting overall protein homeostasis, or both. Earlier studies established a general role for Hsp40 in Hsp70 chaperones, with more recent studies identifying specific brain-expressed chaperones as particularly relevant to SCA3 (e.g. HSPB7, HSPB8-BAG3, and DNAJC8). While many studies support a role for a classic ATP-dependent action of molecular chaperones to re-fold or disaggregate mutant ATXN3, some studies support links between the implicated chaperones and the autophagy-lysosomal system (Pavel et al. 2016). Just as various components of the ubiquitin signaling system may affect one another, the three major arms of PQC are closely interlinked and can profoundly affect one another.
Links between SCA3 and autophagy have recently emerged. In SCA3 disease brain, for example, levels of key autophagy proteins are dysregulated (Sittler et al. 2018). Boosting cholesterol pathways in the brain can improve autophagy and mitigate disease features in a mouse model of SCA3 (Nobrega et al. 2019). Similarly, the protein translation inhibitor Cordycepin and the calpain inhibitor calpeptin mitigate disease features in SCA3 disease models, presumably through a mechanism associated with enhanced autophagy (Marcelo et al. 2019; Watchon et al. 2017)
Intriguingly, ATXN3 appears to be directly tied to autophagy: Several studies have identified regulatory roles for wildtype ATXN3 in macroautophagy. In particular, ATXN3 binds to and regulates levels of Beclin1, a key protein within the autophagy pathway involved in nucleation of autophagosomes (Ashkenazi et al. 2017). The ATXN3-Beclin1 interaction stabilize Beclin1, preventing its degradation by the proteasome (Ashkenazi et al. 2017). PolyQ-expanded mutant ATXN3 similarly binds to Beclin1, but appears to facilitate its delivery and degradation by the proteasome, resulting in reduced Beclin 1 levels (Ashkenazi et al. 2017). Cellular and animal models of SCA3 exhibit altered expression of key autophagy proteins that may be caused in part by polyQ-dependent destabilization of Beclin1. SCA3 patient-derived fibroblasts exhibit reduced levels of Beclin1, decreased autophagic flux, and impaired formation of autophagosomes (Onofre et al. 2016). Beclin1 levels are significantly reduced in SCA3 patient brains, while other key-autophagy proteins, including ATG-12, LAMP-2, Rab7, Rab1A, and LC3, are significantly increased (Sittler et al. 2018). Autophagy proteins like p62 and LC3 also localize to ATXN3 aggregates in vulnerable brain regions in SCA3 patients (Sittler et al. 2018). Thus, perturbations of PQC in SCA3 may well be the culminating effect of accumulation of mutant ATXN3 aggregates, sequestration of key UPS and autophagic regulators, and destabilization of Beclin1.
3.2. DNA Damage & Repair Pathways in SCA3
DNA is continually exposed to exogenous and endogenous stressors leading to DNA breaks (Madabhushi, Pan, and Tsai 2014). Damaged or improperly repaired DNA can become oncogenic, activate pro-apoptotic pathways, or induce cellular senescence (Jackson and Bartek 2009; Iyama and Wilson 2013). Cells have developed complex DNA damage signaling and repair mechanisms, collectively called the DNA damage response (DDR), to deal with a variety of DNA insults (Jackson and Bartek 2009; Iyama and Wilson 2013). Activation of DDR pathways can trigger widespread transcriptional changes (Gregersen and Svejstrup 2018) and activate protein quality control pathways including the UPS and macroautophagy (Xie and Jarosz 2018). Conversely, inhibition of macroautophagy can directly inhibit DNA damage repair processes (Wang, Zhu, and Zhao 2017).
Increased accumulation of damaged DNA in neurons is characteristic of normal aging, but is also a key feature of many neurodegenerative diseases including in Alzheimer’s Disease, Parkinson’s Disease, and HD (Shanbhag et al. 2019; Milanese et al. 2018; Ferlazzo et al. 2014; Amirifar et al. 2019; Madabhushi, Pan, and Tsai 2014; Massey and Jones 2018). Multiple recessively inherited ataxias are directly caused by mutations in genes encoding key DNA damage repair proteins. For instance, mutations in the ataxia telangiectasia mutated (ATM) gene, a global regulator of DNA damage signaling and cell cycle arrest, causes Ataxia Telangiectasia, a recessive ataxia affecting both CNS and peripheral tissues (Rothblum-Oviatt et al. 2016; Shiloh and Ziv 2013). Several recessive ataxias exhibiting CNS-limited degeneration are caused by mutations in DNA single-strand break (SSB) repair genes, suggesting the CNS may be exceptionally vulnerable to disruptions to SSB repair (Caldecott 2003; Massey and Jones 2018).
Mounting evidence suggests that compromised DNA repair also contributes to SCA3 disease pathogenesis. Normal and polyQ-expanded ATXN3 are both rapidly recruited to DNA damage foci in cells exposed to genotoxic stress, though it remains unclear what mediates ATXN3 nuclear import and translocation to damaged chromatin (Pfeiffer et al. 2017). Enhanced accumulation of DNA damage has been observed in SCA3 patient post-mortem tissue (Chatterjee et al. 2015) and transgenic animal models of SCA3 (Kazachkova et al. 2013). Expression of polyQ-expanded mutant ATXN3 also decreases DNA repair efficiency and enhances vulnerability of cells following irradiation or exposure to genotoxins (Liu et al. 2016). And recent GWAS studies have identified DNA mismatch repair genes as modifiers of age-of-onset in polyQ diseases collectively, although the particular relevance of mismatch repair pathways to SCA3 is limited by the relatively small SCA3 population size included in this study (Bettencourt et al. 2016). Figure 3 highlights what is currently known about the normal roles for ATXN3 in mediating the DNA damage response (DDR) and how DNA repair may be impaired by polyQ-expanded mutant ATXN3.
Figure 3. Normal and pathogenic roles of ATXN3 in the DNA damage response (DDR).
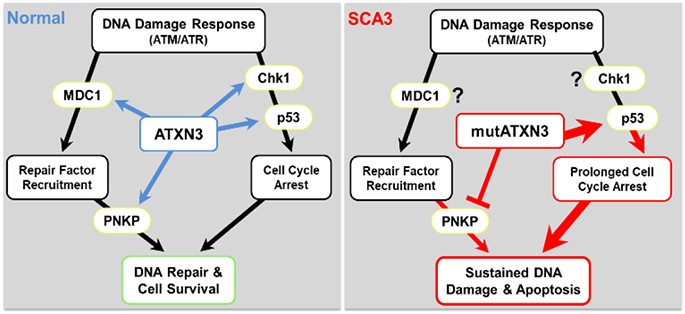
The DDR is a series of complex interconnected pathways that function to detect, signal, and repair DNA damage. DDR is initiated by phosphorylation of ATM or ATR apical kinases. Activated ATM/ATR phosphorylate mediator proteins (i.e. MDC1) that remodel damaged chromatin leading to recruitment of DNA repair factors (i.e. PNKP) that process and repair DNA. Cell cycle regulators (i.e. Chk1 and p53) are also activated by ATM/ATR to stall cell cycle progression during DNA repair. (Left) Normal ATXN3 regulates the kinetics of early and late events in the DDR process. As a deubiquitinase, ATXN3 stabilizes MDC1, Chk1, and p53 through the removal of poly -ubiquitin chains, and promotes PNKP phosphatase activity through an unclear mechanism, to promote DNA repair and cell survival. (Right) Conversely, poly glutamine-expanded mutant ATXN3 (mutATXN3) inhibits PNKP-dependent DNA repair and prolongs p53-signaling through enhanced p53-binding and stabilization. Unrepaired DNA damage and/or sustained p53-activation by mutATXN3 leads to increased activation of pro-apoptotic pathways and cell vulnerability to DNA damage. (MDC1=mediator of DNA damage checkpoint protein 1; Chk1=checkpoint protein 1; p53=tumor protein 53; PNKP=poly nucleotide kinase-phosphatase; ATM=ataxia telangiectasia mutated; ATR=ataxia telangiectasia and Rad3-related protein).
The existing literature suggests versatile DUB-dependent roles for ATXN3 in fine-tuning the temporal and spatial dynamics of key DDR proteins at DNA damage foci. DDR pathways are activated by two apical DNA kinases, ATM or ATR (ATM and rad3-related) kinase (Marechal and Zou 2013; Smith et al. 2010). Several studies demonstrate that ATXN3 stabilizes early targets of ATM- and ATR-coupled DDR pathways to promote DNA repair and checkpoint signaling. MDC1 (mediator of DNA damage checkpoint 1) is one of the earliest proteins activated by ATM (Lou et al. 2006). Activated MDC1 localizes to damaged DNA where it recruits E3 ligases that ubiquitylate surrounding chromatin, a necessary step for recruitment of scaffolding and repair factors like 53BP1 and BRCA1 (Mailand et al. 2007). MDC1 stability and dwell-time at chromatin is tightly regulated by the antagonistic enzymatic activities of ATXN3 and E3 ubiquitin ligase RNF4 (Pfeiffer et al. 2017). Interestingly, the recruitment of both RNF4 [a known SUMO-targeted ubiquitin ligase (STUbL)] and ATXN3 to sites of DNA damage can be abolished through inhibition of DDR-coupled SUMOylation, suggesting ATXN3 may act as a SUMO-targeted DUB within the DDR pathway (Pfeiffer et al. 2017). As a DUB, ATXN3 stabilizes the ATR downstream targets Chk1 (Checkpoint Kinase 1) and p53 (tumor protein 53), both of which function to delay cell cycle progression and promote DNA repair (Liu et al. 2016; Tu et al. 2017). Chk1 and p53 can also activate pro-apoptotic pathways in the presence of sustained unrepaired DNA and replicative stress (Fridman and Lowe 2003; Matsuura et al. 2008). In these studies, depleting endogenous ATXN3 led to increased levels of the poly-ubiquitinated DDR substrates (i.e. MDC1, Chk1, or p53), compromised DNA repair and checkpoint signaling, and enhanced cellular vulnerability to genotoxic stress (Chatterjee et al. 2015; Gao et al. 2015; Liu et al. 2016). Interestingly, over-expressing non-expanded ATXN3 in vitro only partially rescued or actually enhanced cellular vulnerability to genotoxins (Pfeiffer et al. 2017; Tu et al. 2017), suggesting that ATXN3 levels and activity are tightly regulated within the DDR.
Clearly, loss of ATXN3 can impair multiple DNA repair processes. But how the polyQ expansion alters ATXN3 DUB-dependent roles in DDR is less clear. Mutant ATXN3 aggregation has been shown to sequester wildtype ATXN3 (Uchihara et al. 2001), potentially depleting cellular stores of ATXN3 and leading to decreased ATXN3 interactions with and destabilization of DDR substrates. On the other hand, enhanced nuclear localization (which is characteristic of mutant ATXN3) could lead to inappropriate or prolonged mutant ATXN3 stabilization of DDR proteins. Some evidence supports the latter hypothesis. In zebrafish and mouse models of SCA3, mutant ATXN3 exhibited an increased affinity for p53, leading to prolonged activation of p53 and p53 target genes and enhanced p53-dependent neurodegeneration (Liu et al. 2016). PolyQ-expanded mutant ATXN3 appeared to have equivalent Chk1 DUB capacity in HEK293 cells (Tu et al. 2017), but whether this is true in neuronal cells or in vivo models of SCA3 remains unclear. In addition, no studies have investigated whether polyQ-expanded mutant ATXN3 exhibits altered interactions with the DUB substrate MDC1.
Normal and mutant ATXN3 also interact with PNKP (polynucleotide kinase 3’-phosphatase), an enzyme directly involved in processing and repair of DNA damage. Two studies in 2015 showed that ATXN3 binds to and enhances PNKP 3’-phosphatase activity, promoting DNA repair (Chatterjee et al. 2015; Gao et al. 2015). However, how ATXN3 enhances PNKP activity was not clear from these studies. Conversely, polyQ-expanded mutant ATXN3 inhibited PNKP activity, possibly as a result of PNKP sequestration into ATXN3 aggregates (Chatterjee et al. 2015). Knockdown of normal ATXN3 or PNKP, or expression of mutant ATXN3 increased the accumulation of DNA damage and persistent activation of the ATM/p53-dependent DNA repair pathway, suggesting ATXN3 may be a key regulator of PNKP-dependent DNA repair (Chatterjee et al. 2015; Gao et al. 2015). A more recent study demonstrated that ATXN3 and PNKP also stably associate with transcription-coupled DNA repair (TCR) complexes composed of POLR2A, CBP, and the polyQ-containing HTT protein (Gao et al. 2019). In this study, expression of polyQ-expanded HTT or depletion of endogenous ATXN3 led to impaired TCR. Knocking down ATXN3 led to increased poly-ubiquitination of CBP, suggesting CBP is a DUB-substrate of ATXN3. More investigation is required, however, to elucidate the normal and pathogenic roles of ATXN3 in TCR.
To summarize, in this section we have highlighted four recently described ATXN3 DUB substrates or interacting proteins whose functions span DNA repair factor recruitment, cell cycle arrest, and DNA repair (Figure 3). Based on recent studies, we suggest that wildtype ATXN3 stabilizes, or regulates, proteins at various step of the DDR process to promote efficient repair and cell survival. However, as evidenced by widespread changes in protein ubiquitylation in the Atxn3 knockout mouse brain (Schmitt et al. 2007), ATXN3 likely has more, as yet unidentified, DDR substrates. Future studies are also needed to discern how wildtype ATXN3 recruitment to, and removal from, DNA damage foci is regulated. Insights into normal ATXN3 kinetics within the DDR could shed light on how mutant ATXN3 directly or indirectly disrupts DNA repair. Future studies aimed at understanding the relevance of loss-of-function or gain-of-function mutant ATXN3 contributions to deficient DNA repair may also inform gene-silencing therapies for SCA3. Finally, the relevance of different DDR pathways varies across proliferating, non-proliferative dividing cells, and long-lived non-dividing cells (Massey and Jones 2018), all of which are present in the CNS. Improved understanding of cell type-specific differences in DNA repair pathways in the CNS could point to regional and cell-type specific vulnerabilities to neurodegeneration in SCA3 and related neurodegenerative diseases.
3.3. Non-neuronal contributions to SCA pathogenesis
Due to the selective degeneration of neuronal populations, most research in polyQ disorders has taken a neuron-centric point of view. It is important to note, how ever, that glial cells, once only considered secondary supporting cells, are now recognized as vital components of the CNS that contribute greatly to neuronal health. All three types of glia cells in the brain -- astrocytes, microglia and oligodendrocytes -- have recently been implicated in SCA3 disease pathology and more broadly in polyQ diseases. All three cell types may interact with neurons to abrogate disease pathogenesis.
Astrocytes:
Astrocytes help regulate the transmission of electrical impulses in the CNS by assisting with ion homeostasis at neuronal synapses (Liddelow and Barres 2017). They also function to protect cells (both neurons and other glia) from stress and reduce the spread of inflammatory cells to areas of disease damage (Sofroniew 2009). When CNS tissue is damaged, astrocytes are activated. Astrocyte activation can be visualized by increased glial fibrillary acid protein (GFAP) immunostaining, which occurs in regions of disease pathology in SCA3 (Scherzed et al. 2012; Rub et al. 2002; Rüb et al. 2002). Similarly, multiple SCA3 mouse models demonstrate increased GFAP-positive astrocytes in brainstem and cerebellar white matter (Silva-Fernandes et al. 2010; Switonski et al. 2015; Cemal et al. 2002). Whether astrocyte activation in SCA3 is cell autonomous or an indirect byproduct of neuronal damage/stress remains to be tested. Studies in other polyQ animal models expressing mutant proteins selectively in astrocytes recapitulate some aspects of disease (Yang et al. 2017; Bradford et al. 2009), and conditional knockdown of mutant HTT in astrocytes conferred partial therapeutic benefit in a HD mouse model (Wood et al. 2019). These results imply a possible synergistic toxicity of mutant polyQ proteins in neurons and astrocytes that remains to be tested in SCA3 models.
Microglia:
Microglia are the immune cells of the CNS that, when activated by insult or injury, transform into cells that phagocytose dead or dying cells and cellular debris and release proinflammatory signals such as cytokines, proteases and oxygen radicals to mitigate damage (Hickman et al. 2018). While reactive microgliosis is observed in SCA3 patients (Evert et al. 2001; Rub et al. 2002; Rüb et al. 2002), follow-up studies exploring the basis of microglial activation in SCA3 are essentially lacking. One study of serum cytokine levels found eotaxin to be increased in asymptomatic SCA3 patients relative to symptomatic SCA3 patients and normal controls (da Silva Carvalho et al. 2016). Eotaxin is secreted by activated astrocytes, whereas microglia predominantly express its receptor (Parajuli et al. 2015). High eotaxin expression can lead to exacerbated ROS and putative cell death (Parajuli et al. 2015). Early increased expression of eotaxin before the onset of symptoms may suggest glial involvement in early stages of disease. Understanding how eotaxin levels are regulated in SCA3 remains to be determined.
In another polyQ disease, SCA1, studies in mouse models have demonstrated that glial pathology (microglia and astrocytes) correlates with disease onset and severity and is induced by neuronal dysfunction (Cvetanovic et al. 2015). Interestingly, reducing microglia early in the SCA1 disease process improved motor deficits in SCA1 mice (Qu et al. 2017). In HD, activated microglia are associated with peripheral cytokine levels in asymptomatic HD patients (Politis et al. 2015) and mutant HTT expressed only in mouse microglia is sufficient to trigger neurodegeneration (Crotti et al. 2014). Assessment of early microglial dysfunction in SCA3 and other polyQ diseases may provide insight into the early pathogenic mechanisms of disease prior to symptom onset.
Oligodendrocytes:
As myelin-producing cells that ensheath and support neuronal axons, oligodendrocytes constitute the main component of white matter tracks in the CNS. They play integral roles in myelin-rich white matter which is vulnerable in SCA3 (Rüb, Brunt, and Deller 2008; Kang et al. 2014). Based on brain imaging, white matter changes in the brainstem appear to be an early disease feature in human SCA3 disease (Lukas et al. 2006; Rezende et al. 2018; Kang et al. 2014; D'Abreu et al. 2009; Guimaraes et al. 2013).
Data in SCA3 mouse models support transcriptional dysregulation in oligodendrocytes, with the top identified disrupted pathways being myelination, axon ensheathment and cholesterol biogenesis (Ramani et al. 2017; Toonen et al. 2018). High cholesterol levels are required for myelin membrane growth (Saher et al. 2005), as well as axon growth and synapse remodeling in the adult brain (Dietschy 2009). Because of the blood-brain barrier, most cholesterol in the CNS must be produced via de novo synthesis (Orth and Bellosta 2012). CNS neurons can produce enough cholesterol to survive, but proper synapse formation demands additional cholesterol generated by surrounding oligodendrocytes and astrocytes (Mauch et al. 2001; Martin, Pfrieger, and Dotti 2014). A recent study that restored neuronally synthesized cholesterol in the SCA3 mouse brain by overexpressing the rate-limiting enzyme for brain cholesterol efflux was able to rescue many aspects of SCA3 animal model pathology and behavior (Nobrega et al. 2019). Previous research links perturbations in cholesterol homeostasis to many neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, HD and SCA3 (Vance 2012). Analysis of HD mouse and human disease brain tissue revealed significantly decreased cholesterol biosynthesis due to the downregulation of cholesterol biosynthesis genes in cells expressing mutant HTT (Huang et al. 2015). A more recent study showed that removing mutant HTT protein from oligodendrocytes partially rescued aspects of HD pathology (Ferrari Bardile et al. 2019). Together, these noted alterations in myelination and cholesterol biosynthesis strongly support a significant role for early oligodendrocyte dysfunction in SCA3 and possibly other polyQ diseases.
Clearly, nonneuronal cells of the CNS contribute to pathogenesis of SCA3 and other polyQ diseases. Future studies should seek to tease apart the relationship of these cell types to disease progression rather than simply captures static snapshot of disease in postmortem samples. Disease-specific stem cell lines represent a powerful tool with which to assess the neuronal and nonneuronal molecular and cellular aspects of the human disease processes. Currently there are multiple human SCA3 induced pluripotent stem cells that can be used to study disease pathogenesis and test potential therapies (Hansen et al. 2016; Koch et al. 2011; Ou et al. 2016; Ouyang et al. 2018; Ritthaphai et al. 2018; Chuang et al. 2019). Our group recently reported the first NIH-approved human SCA3 embryonic stem cell line which recapitulates well-established SCA3 molecular phenotypes both at the stem cell and differentiated states (Moore et al. 2019). An important gap in SCA3 model systems is the lack of conditional mouse models in which scientists can express or silence mutant ATXN3 in neurons or various nonneuronal cell types in order to define cell-autonomous versus non-cell autonomous contributions to disease.
4. Recurring themes in polyglutamine diseases
Understandably, the above sections on perturbations in protein homeostasis, DNA damage and repair pathways, and functions of nonneuronal cells in disease pathogenesis focus on SCA3 and its disease protein. But the observations are not unique to this polyQ disorder. Rather, they represent recurring themes across essentially the entire polyQ disease spectrum. For example, the widespread presence of mislocalized and aggregated disease protein in essentially all polyQ diseases (possibly except SCA6) underscores the likely involvement of perturbed protein homeostasis in the entire disease class, although the precise details will differ based on the specific disease protein, its normal functions, and its normal interacting partners. Evidence increasingly suggests there are failures in DNA integrity and, more generally, nuclear integrity in polyQ diseases. Studies of DRPLA, for example, elegantly link failures in nuclear integrity to problems in autophagy (Baron et al. 2017), and genetic linkage and studies in model systems have linked DNA repair pathways to HD and several polyQ ataxias (reviewed in (Maiuri et al. 2019; Jones, Houlden, and Tabrizi 2017)). The role of non-neuronal cells in disease pathogenesis is well illustrated by the central importance of Bergmann glia to the process of cerebellar degeneration in SCA7 (Furrer et al. 2011), and activation of microglia may be a common feature in polyQ diseases, as shown in mouse models of SCA1 and HD (Cvetanovic et al. 2015; Crotti et al. 2014). These shared elements of pathogenesis across the spectrum of polyQ diseases raise the intriguing prospect of disease-modifying therapy that is applicable to more than one such disease. At the same time, we must acknowledge that, in each disease, the polyQ expansion occurs in vastly different proteins with entirely different functions and few shared interacting partners. This disease-specific protein context is the principal reason why each disorder is clinically distinct with its own characteristic pattern of neurodegeneration. As discussed below, it is also the reason why the search for disease-specific therapies must continue at the same time that broadly applicable therapies are sought.
5. Therapeutic implications
Although the genetic cause of SCA3 disease has been known for decades, SCA3 is still a fatal, untreatable disease. Strides are being made towards better disease understanding and potential treatment (Ashizawa, Öz, and Paulson 2018; Duarte-Silva and Maciel 2018; McLoughlin et al. 2018). Nevertheless, much remains unknown about how the CAG expansion in the ATXN3 gene causes brain dysfunction and cell death, manifesting in a characteristic clinical syndrome. As discussed here and in other reviews (Ashizawa, Öz, and Paulson 2018; Matos, de Almeida, and Nóbrega 2018), growing evidence supports therapeutic development toward a number of generalizable polyQ pathogenic pathways, including disrupted UPS, autophagic dysfunction, overextended cellular stress pathways, and impaired DNA damage repair (Wang 2018). It should be noted, that all of these pathways are not exclusive to neurons, but rather contribute to universal functions within all CNS cells. Therefore, development of any therapeutic intervention will need to consider target engagement across multiple cell types.
The most obvious target for therapeutic intervention is suppression or modification of the mutant protein in order to ameliorate disease phenotypes. The lack of overt phenotypes following germline or conditional knockout of endogenous ATXN3 in mice suggests that ATXN3 is not an essential protein in mammals (Boy et al. 2009; Schmitt et al. 2007). Thus, reducing total levels of ATXN3 protein would likely be well tolerated in SCA3 patients. To target a causative gene, two main types of oligonucleotide-based therapeutic strategies are currently used: RNA interference (RNAi) and antisense oligonucleotides (ASO). Both strategies have been used in SCA3 animals models, leading to differing levels of success depending on the delivery, target, and timing of therapeutic intervention (Moore et al. 2017; McLoughlin et al. 2018; Costa et al. 2013; Rodriguez-Lebron et al. 2013; Alves et al. 2008; Alves et al. 2010; Toonen et al. 2016; Toonen et al. 2017; Nobrega et al. 2013). ASOs in general have moved more quickly into human clinical trials for neurodegenerative diseases (Bennett, Krainer, and Cleveland 2019). The first ASO-mediated gene modifying therapy gained FDA-approval for the treatment of spinal muscular atrophy (SMA) in 2016 (Finkel et al. 2016; Finkel et al. 2017). The positive safety profile and extraordinary efficacy of ASOs in SMA has boosted interest in ASO treatment of other genetic neurodegenerative diseases. ASO targeting the HTT transcript for the treatment of HD is currently in phase 3 clinical trials, following several successful preclinical studies and a phase 1/2a clinical trial (Lane et al. 2018; Tabrizi et al. 2019; Kordasiewicz et al. 2012). The success and continued clinical development of ASOs as potential treatment for other genetic neurodegenerative diseases solidify ASOs as a feasible and potentially powerful therapy for SCA3 and other polyQ diseases.
Many therapeutic interventions downstream of the mutant protein have been tested in SCA3 disease. SCA3 therapies that target protein aggregation and stimulate protein clearance through UPS and autophagy were recently summarized in a review (Matos, de Almeida, and Nóbrega 2018). Toward the goal of impeding protein aggregation, heat shock proteins (HSP) have been extensively studied as polyQ protein chaperones that could play a protective role by refolding mutant proteins and increasing degradation; chaperones that have been studied include Hsp40, Hsp70, and Hsp90 (Warrick et al. 2005; Bilen and Bonini 2007; Silva-Fernandes et al. 2014). Pharmacological inhibitors of Hsp90 (e.g.17-AAG and 17-DMAG) that alleviate inhibition of heat shock transcription factor 1 (HSF1), have been shown to reduce aggregation and toxicity in SBMA mouse models (Rusmini et al. 2011), and in SCA3 C. elegans and mouse models, (Silva-Fernandes et al. 2014). These studies reveal the therapeutic potential for Hsp90 inhibitors via a HSF1-dependent mechanism in SCA3 and perhaps other polyQ diseases. More recently DNAJC8 (Ito et al. 2016), HSPB7 (Wu et al. 2019), and the novel Dictyostelium chaperone, SRCP1 (Santarriaga et al. 2015; Santarriaga et al. 2018) have been described as potential chaperone regulators of the early polyQ aggregation steps. While there is strong evidence for pathogenic dysregulation of HSF1 and downstream chaperones in many neurodegenerative diseases, including HD, Alzheimer’s Disease and Parkinson’s disease (Gomez-Rastor, Burchfiel, and Thiele 2018), further testing in vertebrate SCA3 animal models of HSF1 activators and novel chaperones that directly impede polyQ aggregation will be required to determine the therapeutic utility of chaperone treatments.
Protein clearance therapies have also been explored extensively in SCA3 and the broader polyQ disease field with some success. In the context of autophagy, genetic Beclin1 overexpression or pharmacological manipulation by Rapamycin to enhance autophagy has been shown to be neuroprotective in cell and animal models. Some caution is required toward dosing and target, however, as a recent combinatorial therapy with mTOR-inhibitor and Lithium Chloride, autophagy inducers, led to increased neurotoxicity in SCA3 mice (Duarte-Silva et al. 2016).
Studies of SCA3 patient serum and cellular models have defined increased levels of reactive oxygen species and decreased antioxidant capacity (de Assis et al. 2017). Increasing antioxidant levels to reduce oxidative stress and subsequent DNA damage levels could be therapeutic in polyQ diseases, as has already been shown to be the case in ATM-deficient mice (Browne et al. 2004). Oxidative stress is known to increase nuclear localization of ATXN3 (Reina, Zhong, and Pittman 2010), and therefore therapies to ameliorate stress levels and reduce ATXN3 entrance into the nucleus might be therapeutically beneficial.
Acknowledgements/Funding
This work was funded by grants from the NIH (R01-NS038712, U01-NS106670, R21-NS111154) and HSM was supported by the National Ataxia Foundation SCA3 Special Projects Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests None to report.
References:
- Adanyeguh IM, Henry PG, Nguyen TM, Rinaldi D, Jauffret C, Valabregue R, Emir UE, Deelchand DK, Brice A, Eberly LE, Oz G, Durr A, and Mochel F. 2015. 'In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3, and 7', Mov Disord, 30: 662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida B, Abreu IA, Matos CA, Fraga JS, Fernandes S, Macedo MG, Gutierrez-Gallego R, Pereira PJ, Carvalho AL, and Macedo-Ribeiro S. 2015. 'SUMOylation of the brain-predominant Ataxin-3 isoform modulates its interaction with p97', Biochim Biophys Acta, 1852: 1950–9. [DOI] [PubMed] [Google Scholar]
- Alves S, Nascimento-Ferreira I, Auregan G, Hassig R, Dufour N, Brouillet E, Pedroso de Lima MC, Hantraye P, Pereira de Almeida L, and Deglon N. 2008. 'Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease', PloS one, 3: e3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves S, Nascimento-Ferreira I, Dufour N, Hassig R, Auregan G, Nobrega C, Brouillet E, Hantraye P, Pedroso de Lima MC, Deglon N, and de Almeida LP. 2010. 'Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: no role for wild-type ataxin-3?', Hum Mol Genet, 19: 2380–94. [DOI] [PubMed] [Google Scholar]
- Amirifar P, Ranjouri MR, Yazdani R, Abolhassani H, and Aghamohammadi A. 2019. 'Ataxia-telangiectasia: A review of clinical features and molecular pathology', Pediatr Allergy Immunol, 30: 277–88. [DOI] [PubMed] [Google Scholar]
- Antony PM, Mantele S, Mollenkopf P, Boy J, Kehlenbach RH, Riess O, and Schmidt T. 2009. 'Identification and functional dissection of localization signals within ataxin-3', Neurobiol Dis, 36: 280–92. [DOI] [PubMed] [Google Scholar]
- Araujo J, Breuer P, Dieringer S, Krauss S, Dorn S, Zimmermann K, Pfeifer A, Klockgether T, Wuellner U, and Evert BO. 2011. 'FOXO4-dependent upregulation of superoxide dismutase-2 in response to oxidative stress is impaired in spinocerebellar ataxia type 3', Hum Mol Genet, 20: 2928–41. [DOI] [PubMed] [Google Scholar]
- Ashizawa T, Öz G, and Paulson HL. 2018. 'Spinocerebellar ataxias: prospects and challenges for therapy development', Nature Reviews Neurology, 14: 590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, Squitieri F, Hardenberg MC, Imarisio S, Menzies FM, and Rubinsztein DC. 2017. 'Polyglutamine tracts regulate beclin 1-dependent autophagy', Nature, 545: 108–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron O, Boudi A, Dias C, Schilling M, Nolle A, Vizcay-Barrena G, Rattray I, Jungbluth H, Scheper W, Fleck RA, Bates GP, and Fanto M. 2017. 'Stall in Canonical Autophagy-Lysosome Pathways Prompts Nucleophagy-Based Nuclear Breakdown in Neurodegeneration', Curr Biol, 27: 3626–42 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CF, Krainer AR, and Cleveland DW. 2019. 'Antisense Oligonucleotide Therapies for Neurodegenerative Diseases', Annu Rev Neurosci, 42: 385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt C, Hensman-Moss D, Flower M, Wiethoff S, Brice A, Goizet C, Stevanin G, Koutsis G, Karadima G, Panas M, Yescas-Gomez P, Garcia-Velazquez LE, Alonso-Vilatela ME, Lima M, Raposo M, Traynor B, Sweeney M, Wood N, Giunti P, Spatax Network, Durr A, Holmans P, Houlden H, Tabrizi SJ, and Jones L. 2016. 'DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases', Ann Neurol, 79: 983–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichelmeier U, Schmidt T, Hubener J, Boy J, Ruttiger L, Habig K, Poths S, Bonin M, Knipper M, Schmidt WJ, Wilbertz J, Wolburg H, Laccone F, and Riess O. 2007. 'Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence', J Neurosci, 27: 7418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J, and Bonini NM. 2007. 'Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila', PLoS Genet, 3: 1950–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi M, Mazzucchelli S, D'Urzo A, Nardini M, Konarev PV, Invernizzi G, Svergun DI, Vanoni M, Regonesi ME, and Tortora P. 2014. 'Interactions of ataxin-3 with its molecular partners in the protein machinery that sorts protein aggregates to the aggresome', Int J Biochem Cell Biol, 51: 58–64. [DOI] [PubMed] [Google Scholar]
- Boy J, Schmidt T, Wolburg H, Mack A, Nuber S, Bottcher M, Schmitt I, Holzmann C, Zimmermann F, Servadio A, and Riess O. 2009. 'Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3', Hum Mol Genet, 18: 4282–95. [DOI] [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, and Li S. 2009. 'Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms', Proc Natl Acad Sci U S A, 106: 22480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SE, Roberts LJ 2nd, Dennery PA, Doctrow SR, Beal MF, Barlow C, and Levine RL. 2004. 'Treatment with a catalytic antioxidant corrects the neurobehavioral defect in ataxia-telangiectasia mice', Free Radic Biol Med, 36: 938–42. [DOI] [PubMed] [Google Scholar]
- Buijsen RAM, Toonen LJA, Gardiner SL, and van Roon-Mom WMC. 2019. 'Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias', Neurotherapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett BG, and Pittman RN. 2005. 'The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation', Proc Natl Acad Sci U S A, 102: 4330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett B, Li F, and Pittman RN. 2003. 'The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity', Human Molecular Genetics, 12: 3195–205. [DOI] [PubMed] [Google Scholar]
- Caldecott KW 2003. 'DNA single-strand break repair and spinocerebellar ataxia', Cell, 112: 7–10. [DOI] [PubMed] [Google Scholar]
- Carvalho DR, La Rocque-Ferreira A, Rizzo IM, Imamura EU, and Speck-Martins CE. 2008. 'Homozygosity Enhances Severity in Spinocerebellar Ataxia Type 3', Pediatric Neurology, 38: 296–99. [DOI] [PubMed] [Google Scholar]
- Cemal CK, Carroll CJ, Lawrence L, Lowrie MB, Ruddle P, Al-Mahdawi S, King RH, Pook MA, Huxley C, and Chamberlain S. 2002. 'YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit', Hum Mol Genet, 11: 1075–94. [DOI] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Bonini NM, and Paulson HL. 1999. 'Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease', J Neurosci, 19: 10338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, and Paulson HL. 1999. 'Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro', Hum Mol Genet, 8: 673–82. [DOI] [PubMed] [Google Scholar]
- Chan HY, Warrick JM, Gray-Board GL, Paulson HL, and Bonini NM. 2000. 'Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila', Hum Mol Genet, 9: 2811–20. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Saha S, Chakraborty A, Silva-Fernandes A, Mandal SM, Neves-Carvalho A, Liu Y, Pandita RK, Hegde ML, Hegde PM, Boldogh I, Ashizawa T, Koeppen AH, Pandita TK, Maciel P, Sarkar PS, and Hazra TK. 2015. 'The role of the mammalian DNA end-processing enzyme polynucleotide kinase 3'-phosphatase in spinocerebellar ataxia type 3 pathogenesis', PLoS Genet, 11: e1004749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CY, Yang CC, Soong BW, Yu CY, Chen SH, Huang HP, and Kuo HC. 2019. 'Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology', Sci Rep, 9: 1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Mdo C., and Paulson HL. 2012. 'Toward understanding Machado-Joseph Disease', Progress in Neurobiology, 97: 239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Mdo. C., Luna-Cancalon K, Fischer S, Ashraf NS, Ouyang M, Dharia RM, Martin-Fishman L, Yang Y, Shakkottai VG, Davidson BL, Rodríguez-Lebrón E, and Paulson HL. 2013. 'Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease', Molecular Therapy, 21: 1898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P, and Andrade C. 1978. 'Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions', Neurology, 28: 703–9. [DOI] [PubMed] [Google Scholar]
- Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C, Cattaneo E, Gage FH, Cleveland DW, and Glass CK. 2014. 'Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors', Nat Neurosci, 17: 513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetanovic M, Ingram M, Orr H, and Opal P. 2015. 'Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1', Neuroscience, 289: 289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Abreu A, Franca M Jr., Appenzeller S, Lopes-Cendes I, and Cendes F. 2009. 'Axonal dysfunction in the deep white matter in Machado-Joseph disease', J Neuroimaging, 19: 9–12. [DOI] [PubMed] [Google Scholar]
- da Silva Carvalho G, Saute JA, Haas CB, Torrez VR, Brochier AW, Souza GN, Furtado GV, Gheno T, Russo A, Monte TL, Schumacher-Schuh A, D'Avila R, Donis KC, Castilhos RM, Souza DO, Saraiva-Pereira ML, Torman VL, Camey S, Portela LV, and Jardim LB. 2016. 'Cytokines in Machado Joseph Disease/Spinocerebellar Ataxia 3', Cerebellum, 15: 518–25. [DOI] [PubMed] [Google Scholar]
- de Assis AM, Saute JAM, Longoni A, Haas CB, Torrez VR, Brochier AW, Souza GN, Furtado GV, Gheno TC, Russo A, Monte TL, Castilhos RM, Schumacher-Schuh A, D'Avila R, Donis KC, de Mello Rieder CR, Souza DO, Camey S, Leotti VB, Jardim LB, and Portela LV. 2017. 'Peripheral Oxidative Stress Biomarkers in Spinocerebellar Ataxia Type 3/Machado-Joseph Disease', Front Neurol, 8: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo A, Jacobi H, Cook A, Labrum R, Durr A, Brice A, Charles P, Marelli C, Mariotti C, Nanetti L, Panzeri M, Rakowicz M, Sobanska A, Sulek A, Schmitz-Hubsch T, Schols L, Hengel H, Melegh B, Filla A, Antenora A, Infante J, Berciano J, van de Warrenburg BP, Timmann D, Boesch S, Pandolfo M, Schulz JB, Bauer P, Giunti P, Kang JS, Klockgether T, and Tezenas du Montcel S. 2018. 'Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study', Lancet Neurol, 17: 327–34. [DOI] [PubMed] [Google Scholar]
- Dietschy JM 2009. 'Central nervous system: cholesterol turnover, brain development and neurodegeneration', Biological chemistry, 390: 287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doss-Pepe EW, Stenroos ES, Johnson WG, and Madura K. 2003. 'Ataxin-3 interactions with rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis', Mol Cell Biol, 23: 6469–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte-Silva S, and Maciel P. 2018. 'Pharmacological Therapies for Machado-Joseph Disease', Adv Exp Med Biol, 1049: 369–94. [DOI] [PubMed] [Google Scholar]
- Duarte-Silva S, Silva-Fernandes A, Neves-Carvalho A, Soares-Cunha C, Teixeira-Castro A, and Maciel P. 2016. 'Combined therapy with m-TOR-dependent and -independent autophagy inducers causes neurotoxicity in a mouse model of Machado-Joseph disease', Neuroscience, 313: 162–73. [DOI] [PubMed] [Google Scholar]
- Durcan TM, Kontogiannea M, Thorarinsdottir T, Fallon L, Williams AJ, Djarmati A, Fantaneanu T, Paulson HL, and Fon EA. 2011. 'The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability', Hum Mol Genet, 20: 141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr A 2010. 'Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond', The Lancet Neurology, 9: 885–94. [DOI] [PubMed] [Google Scholar]
- Durr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, Chneiweiss H, Benomar A, Lyon-Caen O, Julien J, Serdaru M, Penet C, Agid Y, and Brice A. 1996. 'Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features', Ann Neurol, 39: 490–9. [DOI] [PubMed] [Google Scholar]
- Evert BO, Vogt IR, Kindermann C, Ozimek L, de Vos RA, Brunt ER, Schmitt I, Klockgether T, and Wullner U. 2001. 'Inflammatory genes are upregulated in expanded ataxin-3-expressing cell lines and spinocerebellar ataxia type 3 brains', J Neurosci, 21: 5389–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evert BO, Vogt IR, Vieira-Saecker AM, Ozimek L, de Vos RA, Brunt ER, Klockgether T, and Wullner U. 2003. 'Gene expression profiling in ataxin-3 expressing cell lines reveals distinct effects of normal and mutant ataxin-3', J Neuropathol Exp Neurol, 62: 1006–18. [DOI] [PubMed] [Google Scholar]
- Fahl CN, Branco LM, Bergo FP, D'Abreu A, Lopes-Cendes I, and Franca MC Jr. 2015. 'Spinal cord damage in Machado-Joseph disease', Cerebellum, 14: 128–32. [DOI] [PubMed] [Google Scholar]
- Ferlazzo ML, Sonzogni L, Granzotto A, Bodgi L, Lartin O, Devic C, Vogin G, Pereira S, and Foray N. 2014. 'Mutations of the Huntington's disease protein impact on the ATM-dependent signaling and repair pathways of the radiation-induced DNA double-strand breaks: corrective effect of statins and bisphosphonates', Mol Neurobiol, 49: 1200–11. [DOI] [PubMed] [Google Scholar]
- Ferrari Bardile C, Garcia-Miralles M, Caron NS, Rayan NA, Langley SR, Harmston N, Rondelli AM, Teo RTY, Waltl S, Anderson LM, Bae HG, Jung S, Williams A, Prabhakar S, Petretto E, Hayden MR, and Pouladi MA. 2019. 'Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease', Proc Natl Acad Sci U S A, 116: 9622–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E, Norris DA, Xia S, Bennett CF, and Bishop KM. 2016. 'Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study', Lancet, 388: 3017–26. [DOI] [PubMed] [Google Scholar]
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC, and Endear Study Group. 2017. 'Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy', N Engl J Med, 377: 1723–32. [DOI] [PubMed] [Google Scholar]
- Fridman JS, and Lowe SW. 2003. 'Control of apoptosis by p53', Oncogene, 22: 9030–40. [DOI] [PubMed] [Google Scholar]
- Furrer SA, Mohanachandran MS, Waldherr SM, Chang C, Damian VA, Sopher BL, Garden GA, and La Spada AR. 2011. 'Spinocerebellar ataxia type 7 cerebellar disease requires the coordinated action of mutant ataxin-7 in neurons and glia, and displays non-cell-autonomous bergmann glia degeneration', J Neurosci, 31: 16269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Chakraborty A, Geater C, Pradhan S, Gordon KL, Snowden J, Yuan S, Dickey AS, Choudhary S, Ashizawa T, Ellerby LM, La Spada AR, Thompson LM, Hazra TK, and Sarkar PS. 2019. 'Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription', Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Liu Y, Silva-Fernandes A, Fang X, Paulucci-Holthauzen A, Chatterjee A, Zhang HL, Matsuura T, Choudhary S, Ashizawa T, Koeppen AH, Maciel P, Hazra TK, and Sarkar PS. 2015. 'Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3', PLoS Genet, 11: e1004834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner SL, Boogaard MW, Trompet S, de Mutsert R, Rosendaal FR, Gussekloo J, Jukema JW, Roos RAC, and Aziz NA. 2019. 'Prevalence of Carriers of Intermediate and Pathological Polyglutamine Disease-Associated Alleles Among Large Population-Based Cohorts', JAMA Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar C, Lopes-Cendes I, Hayes S, Goto J, Arvidsson K, Dias A, Silveira I, Maciel P, Coutinho P, Lima M, Zhou YX, Soong BW, Watanabe M, Giunti P, Stevanin G, Riess O, Sasaki H, Hsieh M, Nicholson GA, Brunt E, Higgins JJ, Lauritzen M, Tranebjaerg L, Volpini V, Wood N, Ranum L, Tsuji S, Brice A, Sequeiros J, and Rouleau GA. 2001. 'Ancestral origins of the Machado-Joseph disease mutation: a worldwide haplotype study', Am J Hum Genet, 68: 523–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gispert S, Twells R, Orozco G, Brice A, Weber J, Heredero L, Scheufler K, Riley B, Allotey R, Nothers C, and et al. 1993. 'Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1', Nat Genet, 4: 295–9. [DOI] [PubMed] [Google Scholar]
- Gomez-Pastor R, Burchfiel ET, and Thiele DJ. 2018. 'Regulation of heat shock transcription factors and their roles in physiology and disease', Nat Rev Mol Cell Biol, 19: 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen LH, and Svejstrup JQ. 2018. 'The Cellular Response to Transcription-Blocking DNA Damage', Trends Biochem Sci, 43: 327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes RP, D'Abreu A, Yasuda CL, Franca MC Jr., Silva BH, Cappabianco FA, Bergo FP, Lopes-Cendes IT, and Cendes F. 2013. 'A multimodal evaluation of microstructural white matter damage in spinocerebellar ataxia type 3', Mov Disord, 28: 1125–32. [DOI] [PubMed] [Google Scholar]
- Haberhausen G, Damian MS, Leweke F, and Muller U. 1995. 'Spinocerebellar ataxia, type 3 (SCA3) is genetically identical to Machado-Joseph disease (MJD)', J Neurol Sci, 132: 71–5. [DOI] [PubMed] [Google Scholar]
- Hansen SK, Stummann TC, Borland H, Hasholt LF, Tumer Z, Nielsen JE, Rasmussen MA, Nielsen TT, Daechsel JC, Fog K, and Hyttel P. 2016. 'Induced pluripotent stem cell - derived neurons for the study of spinocerebellar ataxia type 3', Stem Cell Res, 17: 306–17. [DOI] [PubMed] [Google Scholar]
- Harris GM, Dodelzon K, Gong L, Gonzalez-Alegre P, and Paulson HL. 2010. 'Splice isoforms of the polyglutamine disease protein ataxin-3 exhibit similar enzymatic yet different aggregation properties', PloS one, 5: e13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Kobayashi K, and Furuta H. 2003. 'Immunohistochemical study of neuronal intranuclear and cytoplasmic inclusions in Machado-Joseph disease', Psychiatry Clin Neurosci, 57: 205–13. [DOI] [PubMed] [Google Scholar]
- Hickman S, Izzy S, Sen P, Morsett L, and El Khoury J. 2018. 'Microglia in neurodegeneration', Nat Neurosci, 21: 1359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wei W, Wang G, Gaertig MA, Feng Y, Wang W, Li XJ, and Li S. 2015. 'Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes', Neuron, 85: 1212–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa Y, Goto J, Hattori M, Toyoda A, Ishii K, Jeong SY, Hashida H, Masuda N, Ogata K, Kasai F, Hirai M, Maciel P, Rouleau GA, Sakaki Y, and Kanazawa I. 2001. 'The genomic structure and expression of MJD, the Machado-Joseph disease gene', J Hum Genet, 46: 413–22. [DOI] [PubMed] [Google Scholar]
- Ito N, Kamiguchi K, Nakanishi K, Sokolovskya A, Hirohashi Y, Tamura Y, Murai A, Yamamoto E, Kanaseki T, Tsukahara T, Kochin V, Chiba S, Shimohama S, Sato N, and Torigoe T. 2016. 'A novel nuclear DnaJ protein, DNAJC8, can suppress the formation of spinocerebellar ataxia 3 polyglutamine aggregation in a J-domain independent manner', Biochem Biophys Res Commun, 474: 626–33. [DOI] [PubMed] [Google Scholar]
- Iyama T, and Wilson DM 3rd. 2013. 'DNA repair mechanisms in dividing and non-dividing cells', DNA Repair (Amst), 12: 620–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J. 2009. 'The DNA-damage response in human biology and disease', Nature, 461: 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joers JM, Deelchand DK, Lyu T, Emir UE, Hutter D, Gomez CM, Bushara KO, Eberly LE, and Oz G. 2018. 'Neurochemical abnormalities in premanifest and early spinocerebellar ataxias', Ann Neurol, 83: 816–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L, Houlden H, and Tabrizi SJ. 2017. 'DNA repair in the trinucleotide repeat disorders', Lancet Neurol, 16: 88–96. [DOI] [PubMed] [Google Scholar]
- Kang JS, Klein JC, Baudrexel S, Deichmann R, Nolte D, and Hilker R. 2014. 'White matter damage is related to ataxia severity in SCA3', J Neurol, 261: 291–9. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, and et al. 1994. 'CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1', Nat Genet, 8: 221–8. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, Kimura J, Narumiya S, and Kakizuka A. 1994. 'CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1', Nature Genetics, 8: 221–28. [DOI] [PubMed] [Google Scholar]
- Kazachkova N, Raposo M, Montiel R, Cymbron T, Bettencourt C, Silva-Fernandes A, Silva S, Maciel P, and Lima M. 2013. 'Patterns of mitochondrial DNA damage in blood and brain tissues of a transgenic mouse model of Machado-Joseph disease', Neurodegener Dis, 11: 206–14. [DOI] [PubMed] [Google Scholar]
- Klockgether T, Mariotti C, and Paulson HL. 2019. 'Spinocerebellar ataxia', Nat Rev Dis Primers, 5: 24. [DOI] [PubMed] [Google Scholar]
- Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, Doerr J, Ladewig J, Mertens J, Tuting T, Hoffmann P, Klockgether T, Evert BO, Wullner U, and Brustle O. 2011. 'Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease', Nature, 480: 543–6. [DOI] [PubMed] [Google Scholar]
- Kopito RR 2000. 'Aggresomes, inclusion bodies and protein aggregation', Trends Cell Biol, 10: 524–30. [DOI] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, and Cleveland DW. 2012. 'Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis', Neuron, 74: 1031–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RM, Smith A, Baumann T, Gleichmann M, Norris D, Bennett CF, and Kordasiewicz H. 2018. 'Translating Antisense Technology into a Treatment for Huntington's Disease', Methods Mol Biol, 1780: 497–523. [DOI] [PubMed] [Google Scholar]
- Li F, Macfarlan T, Pittman RN, and Chakravarti D. 2002. 'Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities', J Biol Chem, 277: 45004–12. [DOI] [PubMed] [Google Scholar]
- Li X, Liu H, Fischhaber PL, and Tang TS. 2015. 'Torward therapeutic targets for SCA3: Insight into the role of Machado-Joseph disease protein ataxin-3 in misfolded proteins clearance', Prog Neurobiol, 132: 34–58. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, and Barres BA. 2017. 'Reactive Astrocytes: Production, Function, and Therapeutic Potential', Immunity, 46: 957–67. [DOI] [PubMed] [Google Scholar]
- Linhartová I, Repitz M, Dráber P, Nemec M, Wiche G, and Propst F. 1999. 'Conserved domains and lack of evidence for polyglutamine length polymorphism in the chicken homolog of the Machado-Joseph disease gene product ataxin-3', Biochimica et Biophysica Acta - Gene Structure and Expression, 1444: 299–305. [DOI] [PubMed] [Google Scholar]
- Linnemann C, Tezenas du Montcel S, Rakowicz M, Schmitz-Hubsch T, Szymanski S, Berciano J, van de Warrenburg BP, Pedersen K, Depondt C, Rola R, Klockgether T, Garcia A, Mutlu G, and Schols L. 2016. 'Peripheral Neuropathy in Spinocerebellar Ataxia Type 1, 2, 3, and 6', Cerebellum, 15: 165–73. [DOI] [PubMed] [Google Scholar]
- Liu H, Li X, Ning G, Zhu S, Ma X, Liu X, Liu C, Huang M, Schmitt I, Wullner U, Niu Y, Guo C, Wang Q, and Tang TS. 2016. 'The Machado-Joseph Disease Deubiquitinase Ataxin-3 Regulates the Stability and Apoptotic Function of p53', PLoS Biol, 14: e2000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes TM, D'Abreu A, Franca MC Jr., Yasuda CL, Betting LE, Samara AB, Castellano G, Somazz JC, Balthazar ML, Lopes-Cendes I, and Cendes F. 2013. 'Widespread neuronal damage and cognitive dysfunction in spinocerebellar ataxia type 3', J Neurol, 260: 2370–9. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, and Chen J. 2006. 'MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals', Mol Cell, 21: 187–200. [DOI] [PubMed] [Google Scholar]
- Lukas C, Schols L, Bellenberg B, Rub U, Przuntek H, Schmid G, Koster O, and Suchan B. 2006. 'Dissociation of grey and white matter reduction in spinocerebellar ataxia type 3 and 6: a voxel-based morphometry study', Neurosci Lett, 408: 230–5. [DOI] [PubMed] [Google Scholar]
- Maciel P, Gaspar C, DeStefano AL, Silveira I, Coutinho P, Radvany J, Dawson DM, Sudarsky L, Guimaraes J, Loureiro JE, and et al. 1995. 'Correlation between CAG repeat length and clinical features in Machado-Joseph disease', Am J Hum Genet, 57: 54–61. [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, and Tsai LH. 2014. 'DNA damage and its links to neurodegeneration', Neuron, 83: 266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, and Lukas J. 2007. 'RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins', Cell, 131: 887–900. [DOI] [PubMed] [Google Scholar]
- Maiuri T, Suart CE, Hung CLK, Graham KJ, Barba Bazan CA, and Truant R. 2019. 'DNA Damage Repair in Huntington's Disease and Other Neurodegenerative Diseases', Neurotherapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelo A, Brito F, Carmo-Silva S, Matos CA, Alves-Cruzeiro J, Vasconcelos-Ferreira A, Koppenol R, Mendonca L, de Almeida LP, and Nobrega C. 2019. 'Cordycepin activates autophagy through AMPK phosphorylation to reduce abnormalities in Machado-Joseph disease models', Hum Mol Genet, 28: 51–63. [DOI] [PubMed] [Google Scholar]
- Marechal A, and Zou L. 2013. 'DNA damage sensing by the ATM and ATR kinases', Cold Spring Harb Perspect Biol, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MG, Pfrieger F, and Dotti CG. 2014. 'Cholesterol in brain disease: sometimes determinant and frequently implicated', EMBO Rep, 15: 1036–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Nakamura S, Matsuyama Z, Sakai T, Doyu M, Sobue G, Seto M, Tsujihata M, Oh-i T, Nishio T, and et al. 1995. 'Molecular features of the CAG repeats and clinical manifestation of Machado-Joseph disease', Hum Mol Genet, 4: 807–12. [DOI] [PubMed] [Google Scholar]
- Massey TH, and Jones L. 2018. 'The central role of DNA damage and repair in CAG repeat diseases', Dis Model Mech, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos CA, de Almeida LP, and Nóbrega C. 2018. 'Machado-Joseph disease/spinocerebellar ataxia type 3: lessons from disease pathogenesis and clues into therapy', Journal of Neurochemistry, 148: 8–28. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Yada M, Hatakeyama S, Ishimoto H, Tanimura T, Tsuji S, Kakizuka A, Kitagawa M, and Nakayama KI. 2004. 'Molecular clearance of ataxin-3 is regulated by a mammalian E4', EMBO J, 23: 659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, Wakasugi M, Yamashita K, and Matsunaga T. 2008. 'Cleavage-mediated activation of Chk1 during apoptosis', J Biol Chem, 283: 25485–91. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, and Pfrieger FW. 2001. 'CNS synaptogenesis promoted by glia-derived cholesterol', Science, 294: 1354–7. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli S, De Palma A, Riva M, D'Urzo A, Pozzi C, Pastori V, Comelli F, Fusi P, Vanoni M, Tortora P, Mauri P, and Regonesi ME. 2009. 'Proteomic and biochemical analyses unveil tight interaction of ataxin-3 with tubulin', Int J Biochem Cell Biol, 41: 2485–92. [DOI] [PubMed] [Google Scholar]
- McLoughlin HS, Moore LR, Chopra R, Komlo R, McKenzie M, Blumenstein KG, Zhao H, Kordasiewicz HB, Shakkottai VG, and Paulson HL. 2018. 'Oligonucleotide therapy mitigates disease in Spinocerebellar Ataxia Type 3 mice', Annals of Neurology, 81: 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanese C, Cerri S, Ulusoy A, Gornati SV, Plat A, Gabriels S, Blandini F, Di Monte DA, Hoeijmakers JH, and Mastroberardino PG. 2018. 'Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson's disease', Cell Death Dis, 9: 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LR, Keller L, Bushart DD, Delatorre R, Li D, McLoughlin HS, Costa Mdo C., Shakkottai VG, Smith GD, and Paulson HL. 2019. 'Antisense oligonucleotide therapy rescues aggresome formation in a novel Spinocerebellar Ataxia type 3 human embryonic stem cell line', Stem Cell Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LR, Rajpal G, Dillingham IT, Qutob M, Blumenstein KG, Gattis D, Hung G, Kordasiewicz HB, Paulson HL, and McLoughlin HS. 2017. 'Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models', Molecular Therapy Nucleic Acids, 7: 200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller T, Breuer P, Schmitt I, Walter J, Evert BO, and Wullner U. 2009. 'CK2-dependent phosphorylation determines cellular localization and stability of ataxin-3', Hum Mol Genet, 18: 3334–43. [DOI] [PubMed] [Google Scholar]
- Nobrega C, Mendonca L, Marcelo A, Lamaziere A, Tome S, Despres G, Matos CA, Mechmet F, Langui D, den Dunnen W, de Almeida LP, Cartier N, and Alves S. 2019. 'Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia', Acta Neuropathol. [DOI] [PubMed] [Google Scholar]
- Nobrega C, Nascimento-Ferreira I, Onofre I, Albuquerque D, Hirai H, Deglon N, and de Almeida LP. 2013. 'Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice', PloS one, 8: e52396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Li L, and Chin LS. 2008. 'Aggresome formation and neurodegenerative diseases: therapeutic implications', Curr Med Chem, 15: 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onofre I, Mendonca N, Lopes S, Nobre R, de Melo JB, Carreira IM, Januario C, Goncalves AF, and de Almeida LP. 2016. 'Fibroblasts of Machado Joseph Disease patients reveal autophagy impairment', Sci Rep, 6: 28220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth M, and Bellosta S. 2012. 'Cholesterol: Its Regulation and Role in Central Nervous System Disorders', Cholesterol, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Z, Luo M, Niu X, Chen Y, Xie Y, He W, Song B, Xian Y, Fan D, OuYang S, and Sun X. 2016. 'Autophagy Promoted the Degradation of Mutant ATXN3 in Neurally Differentiated Spinocerebellar Ataxia-3 Human Induced Pluripotent Stem Cells', Biomed Res Int, 2016: 6701793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang S, Xie Y, Xiong Z, Yang Y, Xian Y, Ou Z, Song B, Chen Y, Xie Y, Li H, and Sun X. 2018. 'CRISPR/Cas9-Targeted Deletion of Polyglutamine in Spinocerebellar Ataxia Type 3-Derived Induced Pluripotent Stem Cells', Stem Cells Dev, 27: 756–70. [DOI] [PubMed] [Google Scholar]
- Parajuli B, Horiuchi H, Mizuno T, Takeuchi H, and Suzumura A. 2015. 'CCL11 enhances excitotoxic neuronal death by producing reactive oxygen species in microglia', Glia, 63: 2274–84. [DOI] [PubMed] [Google Scholar]
- Park H, Kim HJ, and Jeon BS. 2015. 'Parkinsonism in spinocerebellar ataxia', Biomed Res Int, 2015: 125273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL 2007. 'Dominantly inherited ataxias: lessons learned from Machado-Joseph disease/spinocerebellar ataxia type 3', Semin Neurol, 27: 133–42. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Das SS, Crino PB, Perez MK, Patel SC, Gotsdiner D, Fischbeck KH, and Pittman RN. 1997. 'Machado-Joseph disease gene product is a cytoplasmic protein widely expressed in brain', Ann Neurol, 41: 453–62. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, and Pittman RN. 1997. 'Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3', Neuron, 19: 333–44. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Shakkottai VG, Clark HB, and Orr HT. 2017. 'Pblyglutamine spinocerebellar ataxias - from genes to potential treatments', Nat Rev Neurosci, 18: 613–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavel M, Imarisio S, Menzies FM, Jimenez-Sanchez M, Siddiqi FH, Wu X, Renna M, O'Kane CJ, Crowther DC, and Rubinsztein DC. 2016. 'CCT complex restricts neuropathogenic protein aggregation via autophagy', Nat Commun, 7: 13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedroso JL, Braga-Neto P, Felicio AC, Dutra LA, Santos WA, do Prado GF, and Barsottini OG. 2011. 'Sleep disorders in machado-joseph disease: frequency, discriminative thresholds, predictive values, and correlation with ataxia-related motor and non-motor features', Cerebellum, 10: 291–5. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Luijsterburg MS, Acs K, Wiegant WW, Helfricht A, Herzog LK, Minoia M, Bottcher C, Salomons FA, van Attikum H, and Dantuma NP. 2017. 'Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4', EMBO J, 36: 1066–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis M, Lahiri N, Niccolini F, Su P, Wu K, Giannetti P, Scahill RI, Turkheimer FE, Tabrizi SJ, and Piccini P. 2015. 'Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington's disease gene carriers', Neurobiol Dis, 83: 115–21. [DOI] [PubMed] [Google Scholar]
- Qu W, Johnson A, Kim JH, Lukowicz A, Svedberg D, and Cvetanovic M. 2017. 'Inhibition of colony-stimulating factor 1 receptor early in disease ameliorates motor deficits in SCA1 mice', J Neuroinflammation, 14: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani B, Panwar B, Moore LR, Wang B, Huang R, Guan Y, and Paulson HL. 2017. 'Comparison of spinocerebellar ataxia type 3 mouse models identifies early gain-of-function, cell-autonomous transcriptional changes in oligodendrocytes', Human Molecular Genetics, 26: 3362–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina CP, Zhong X, and Pittman RN. 2010. 'Proteotoxic stress increases nuclear localization of ataxin-3', Hum Mol Genet, 19: 235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, and Wilkinson KD. 2009. 'Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes', Annual review of biochemistry, 78: 363–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezende TJR, de Paiva JLR, Martinez ARM, Lopes-Cendes I, Pedroso JL, Barsottini OGP, Cendes F, and Franca MC Jr. 2018. 'Structural signature of SCA3: From presymptomatic to late disease stages', Ann Neurol, 84: 401–08. [DOI] [PubMed] [Google Scholar]
- Ritthaphai A, Wattanapanitch M, Pithukpakorn M, Heepchantree W, Soi-Ampornkul R, Mahaisavariya P, Triwongwaranat D, Pattanapanyasat K, and Vatanashevanopakorn C. 2018. 'Derivation of an induced pluripotent stem cell line (MUSIi004-A) from dermal fibroblasts of a 48-year-old spinocerebellar ataxia type 3 patient', Stem Cell Res, 30: 113–16. [DOI] [PubMed] [Google Scholar]
- Rodrigues AJ, Coppola G, Santos C, Costa Mdo C., Ailion M, Sequeiros J, Geschwind DH, and Maciel P. 2007. 'Functional genomics and biochemical characterization of the C. elegans orthologue of the Machado-Joseph disease protein ataxin-3', The FASEB Journal, 21: 1126–36. [DOI] [PubMed] [Google Scholar]
- Rodrigues AJ, do Carmo Costa M, Silva TL, Ferreira D, Bajanca F, Logarinho E, and Maciel P. 2010. 'Absence of ataxin-3 leads to cytoskeletal disorganization and increased cell death', Biochim Biophys Acta, 1803: 1154–63. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lebron E, Costa Mdo C., Luna-Cancalon K, Peron TM, Fischer S, Boudreau RL, Davidson BL, and Paulson HL. 2013. 'Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice', Mol Ther, 21: 1909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, and Lederman HM. 2016. 'Ataxia telangiectasia: a review', Orphanet J Rare Dis, 11: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rub U, Brunt ER, de Vos RA, Del Turco D, Del Tredici K, Gierga K, Schultz C, Ghebremedhin E, Burk K, Auburger G, and Braak H. 2004. 'Degeneration of the central vestibular system in spinocerebellar ataxia type 3 (SCA3) patients and its possible clinical significance', Neuropathol Appl Neurobiol, 30: 402–14. [DOI] [PubMed] [Google Scholar]
- Rüb U, Brunt ER, and Deller T. 2008. 'New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado-Joseph disease)', Current Opinion in Neurology, 21: 111–16. [DOI] [PubMed] [Google Scholar]
- Rub U, de Vos RA, Brunt ER, Schultz C, Paulson H, Del Tredici K, and Braak H. 2002. 'Degeneration of the external cuneate nucleus in spinocerebellar ataxia type 3 (Machado-Joseph disease)', Brain Res, 953: 126–34. [DOI] [PubMed] [Google Scholar]
- Rüb U, de Vos RA, Schultz C, Brunt ER, Paulson HL, and Braak H. 2002. 'Spinocerebellar ataxia type 3 (Machado–Joseph disease): severe destruction of the lateral reticular nucleus', Brain, 125: 2115–24. [DOI] [PubMed] [Google Scholar]
- Rub U, Del Turco D, Del Tredici K, de Vos RA, Brunt ER, Reifenberger G, Seifried C, Schultz C, Auburger G, and Braak H. 2003. 'Thalamic involvement in a spinocerebellar ataxia type 2 (SCA2) and a spinocerebellar ataxia type 3 (SCA3) patient, and its clinical relevance', Brain, 126: 2257–72. [DOI] [PubMed] [Google Scholar]
- Rub U, Schols L, Paulson H, Auburger G, Kermer P, Jen JC, Seidel K, Korf HW, and Deller T. 2013. 'Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7', Prog Neurobiol, 104: 38–66. [DOI] [PubMed] [Google Scholar]
- Rusmini P, Simonini F, Crippa V, Bolzoni E, Onesto E, Cagnin M, Sau D, Ferri N, and Poletti A. 2011. '17-AAG increases autophagic removal of mutant androgen receptor in spinal and bulbar muscular atrophy', Neurobiol Dis, 41: 83–95. [DOI] [PubMed] [Google Scholar]
- Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, and Nave KA. 2005. 'High cholesterol level is essential for myelin membrane growth', Nat Neurosci, 8: 468–75. [DOI] [PubMed] [Google Scholar]
- Santarriaga S, Haver HN, Kanack AJ, Fikejs AS, Sison SL, Egner JM, Bostrom JR, Seminary ER, Hill RB, Link BA, Ebert AD, and Scaglione KM. 2018. 'SRCP1 Conveys Resistance to Polyglutamine Aggregation', Mol Cell, 71: 216–28 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarriaga S, Petersen A, Ndukwe K, Brandt A, Gerges N, Bruns Scaglione J, and Scaglione KM. 2015. 'The Social Amoeba Dictyostelium discoideum Is Highly Resistant to Polyglutamine Aggregation', J Biol Chem, 290: 25571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione KM, Zavodszky E, Todi SV, Patury S, Xu P, Rodriguez-Lebron E, Fischer S, Konen J, Djarmati A, Peng J, Gestwicki JE, and Paulson HL. 2011. 'Ube2w and ataxin-3 coordinated regulate the ubiquitin ligase CHIP, Mol Cell, 43: 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzed W, Brunt ER, Heinsen H, de Vos RA, Seidel K, Burk K, Schols L, Auburger G, Del Turco D, Deller T, Korf HW, den Dunnen WF, and Rub U. 2012. 'Pathoanatomy of cerebellar degeneration in spinocerebellar ataxia type 2 (SCA2) and type 3 (SCA3)', Cerebellum, 11: 749–60. [DOI] [PubMed] [Google Scholar]
- Schmidt T, Landwehrmeyer GB, Schmitt I, Trottier Y, Auburger G, Laccone F, Klockgether T, Volpel M, Epplen JT, Schols L, and Riess O. 1998. 'An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients', Brain Pathol, 8: 669–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Lindenberg KS, Krebs A, Schols L, Laccone F, Herms J, Rechsteiner M, Riess O, and Landwehrmeyer GB. 2002. 'Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions', Ann Neurol, 51: 302–10. [DOI] [PubMed] [Google Scholar]
- Schmitt I, Brattig T, Gossen M, and Riess O. 1997. 'Characterization of the rat spinocerebellar ataxia type 3 gene', Neurogenetics, 1: 103–12. [DOI] [PubMed] [Google Scholar]
- Schmitt I, Linden M, Khazneh H, Evert BO, Breuer P, Klockgether T, and Wuellner U. 2007. 'Inactivation of the mouse Atxn3 (ataxin-3) gene increases protein ubiquitination', Biochem Biophys Res Commun, 362: 734–9. [DOI] [PubMed] [Google Scholar]
- Seidel K, den Dunnen WF, Schultz C, Paulson H, Frank S, de Vos RA, Brunt ER, Deller T, Kampinga HH, and Rub U. 2010. 'Axonal inclusions in spinocerebellar ataxia type 3', Acta Neuropathol, 120: 449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Siswanto S, Fredrich M, Bouzrou M, den Dunnen WFA, Ozerden I, Korf HW, Melegh B, de Vries JJ, Brunt ER, Auburger G, and Rub U. 2017. 'On the distribution of intranuclear and cytoplasmic aggregates in the brainstem of patients with spinocerebellar ataxia type 2 and 3', Brain Pathol, 27: 345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Vinet J, Dunnen WF, Brunt ER, Meister M, Boncoraglio A, Zijlstra MP, Boddeke HW, Rub U, Kampinga HH, and Carra S. 2012. 'The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases', Neuropathol Appl Neurobiol, 38: 39–53. [DOI] [PubMed] [Google Scholar]
- Seshagiri DV, Botta R, Sasidharan A, Kumar Pal P, Jain S, Yadav R, and Kutty BM. 2018. 'Assessment of Sleep Spindle Density among Genetically Positive Spinocerebellar Ataxias Types 1, 2, and 3 Patients', Ann Neurosci, 25: 106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag NM, Evans MD, Mao W, Nana AL, Seeley WW, Adame A, Rissman RA, Masliah E, and Mucke L. 2019. 'Early neuronal accumulation of DNA double strand breaks in Alzheimer's disease', Acta Neuropathol Commun, 7: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, and Ziv Y. 2013. 'The ATM protein kinase: regulating the cellular response to genotoxic stress, and more', Nat Rev Mol Cell Biol, 14: 197–210. [PubMed] [Google Scholar]
- Silva-Fernandes A, Costa Mdo C, Duarte-Silva S, Oliveira P, Botelho CM, Martins L, Mariz JA, Ferreira T, Ribeiro F, Correia-Neves M, Costa C, and Maciel P. 2010. 'Motor uncoordination and neuropathology in a transgenic mouse model of Machado-Joseph disease lacking intranuclear inclusions and ataxin-3 cleavage products', Neurobiol Dis, 40: 163–76. [DOI] [PubMed] [Google Scholar]
- Silva-Fernandes A, Duarte-Silva S, Neves-Carvalho A, Amorim M, Soares-Cunha C, Oliveira P, Thirstrup K, Teixeira-Castro A, and Maciel P. 2014. 'Chronic treatment with 17-DMAG improves balance and coordination in a new mouse model of Machado-Joseph disease', Neurotherapeutics, 11: 433–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittler A, Muriel MP, Marinello M, Brice A, den Dunnen W, and Alves S. 2018. 'Deregulation of autophagy in postmortem brains of Machado-Joseph disease patients', Neuropathology, 38: 113–24. [DOI] [PubMed] [Google Scholar]
- Smith J, Tho LM, Xu N, and Gillespie DA. 2010. 'The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer', Adv Cancer Res, 108: 73–112. [DOI] [PubMed] [Google Scholar]
- Sobue G, Doyu M, Nakao N, Shimada N, Mitsuma T, Maruyama H, Kawakami H, and Nakamura S. 1996. 'Homozygosity for Machado-Joseph disease gene enhances phenotypic severity', Journal of Neurology, Neurosurgery, and Psychiatry, 60: 354–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV 2009. 'Molecular dissection of reactive astrogliosis and glial scar formation', Trends Neurosci, 32: 638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa AS, Martin E, Martins IM, Schmidt J, Depping R, Weber JJ, Rother F, Hartmann E, Bader M, Riess O, Tricoire H, and Schmidt T. 2018. 'Karyopherin alpha-3 is a key protein in the pathogenesis of spinocerebellar ataxia type 3 controlling the nuclear localization of ataxin-3', Proc Natl Acad Sci U S A, 115: E2624–E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga N, Katsuno M, Koike H, Banno H, Suzuki K, Hashizume A, Mano T, Iijima M, Kawagashira Y, Hirayama M, Nakamura T, Watanabe H, Tanaka F, and Sobue G. 2014. 'Schwann cell involvement in the peripheral neuropathy of spinocerebellar ataxia type 3', Neuropathol Appl Neurobiol, 40: 628–39. [DOI] [PubMed] [Google Scholar]
- Switonski PM, Szlachcic WJ, Krzyzosiak WJ, and Figiel M. 2015. 'A new humanized ataxin-3 knock-in mouse model combines the genetic features, pathogenesis of neurons and glia and late disease onset of SCA3/MJD', Neurobiol Dis, 73: 174–88. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, Blair NF, Craufurd D, Priller J, Rickards H, Rosser A, Kordasiewicz HB, Czech C, Swayze EE, Norris DA, Baumann T, Gerlach I, Schobel SA, Paz E, Smith AV, Bennett CF, and Lane RM. 2019. 'Targeting Huntingtin Expression in Patients with Huntington's Disease', N Engl J Med, 380: 2307–16. [DOI] [PubMed] [Google Scholar]
- Takiyama Y, Nishizawa M, Tanaka H, Kawashima S, Sakamoto H, Karube Y, Shimazaki H, Soutome M, Endo K, Ohta S, Kagawa Y, Kanazawa I, Mizuno Y, Yoshida M, Yuasa T, Horikawa Y, Oyanagi K, Nagai H, Kondo T, Inuzuka T, Onodera O, and Tsuji S. 1993. 'The gene for Machado-Joseph disease maps to human chromosome 14q', Nature Genetics, 4: 300–04. [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, and Pickart CM. 2000. 'Recognition of the polyubiquitin proteolytic signal', EMBO J, 19: 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todi SV, Scaglione KM, Blount JR, Basrur V, Conlon KP, Pastore A, Elenitoba-Johnson K, and Paulson HL. 2010. 'Activity and cellular functions of the deubiquitinating enzyme and polyglutamine disease protein ataxin-3 are regulated by ubiquitination at lysine 117', J Biol Chem, 285: 39303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todi SV, Winborn BJ, Scaglione KM, Blount JR, Travis SM, and Paulson HL. 2009. 'Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3', EMBO J, 28: 372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen LJA, Overzier M, Evers MM, Leon LG, van der Zeeuw SAJ, Mei H, Kielbasa SM, Goeman JJ, Hettne KM, Magnusson OT, Poirel M, Seyer A, ‘t Hoen PAC, and van Roon-Mom WMC. 2018. 'Transcriptional profiling and biomarker identification reveal tissue specific effects of expanded ataxin-3 in a spinocerebellar ataxia type 3 mouse model', Molecular Neurodegeneration, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen LJA, Rigo F, van Attikum H, and van Roon-Mom WMC. 2017. 'Antisense Oligonucleotide-Mediated Removal of the Polyglutamine Repeat in Spinocerebellar Ataxia Type 3 Mice', Mol Ther Nucleic Acids, 8: 232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen LJ, Schmidt I, Luijsterburg MS, van Attikum H, and van Roon-Mom WM. 2016. 'Antisense oligonucleotide-mediated exon skipping as a strategy to reduce proteolytic cleavage of ataxin-3', Sci Rep, 6: 35200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y, Liu H, Zhu X, Shen H, Ma X, Wang F, Huang M, Gong J, Li X, Wang Y, Guo C, and Tang TS. 2017. 'Ataxin-3 promotes genome integrity by stabilizing Chk1', Nucleic Acids Res, 45: 4532–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchihara T, Fujigasaki H, Koyano S, Nakamura A, Yagishita S, and Iwabuchi K. 2001. 'Non-expanded polyglutamine proteins in intranuclear inclusions of hereditary ataxias--triple-labeling immunofluorescence study', Acta Neuropathol, 102: 149–52. [DOI] [PubMed] [Google Scholar]
- Vance JE 2012. 'Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases', Dis Model Mech, 5: 746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos MJ, Zijlstra MP, Kanon B, van Waarde-Verhagen MA, Brunt ER, Oosterveld-Hut HM, Carra S, Sibon OC, and Kampinga HH. 2010. 'HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones', Hum Mol Genet, 19: 4677–93. [DOI] [PubMed] [Google Scholar]
- Wang H, Ying Z, and Wang G. 2012. 'Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains', J Biol Chem, 287: 28576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu WG, and Zhao Y. 2017. 'Autophagy substrate SQSTM1/p62 regulates chromatin ubiquitination during the DNA damage response', Autophagy, 13: 212–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z 2018. 'Experimental and Clinical Strategies for Treating Spinocerebellar Ataxia Type 3', Neuroscience, 371: 138–54. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, and Bonini NM. 1999. 'Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70', Nat Genet, 23: 425–8. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Morabito LM, Bilen J, Gordesky-Gold B, Faust LZ, Paulson HL, and Bonini NM. 2005. 'Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism', Mol Cell, 18: 37–48. [DOI] [PubMed] [Google Scholar]
- Watchon M, Yuan KC, Mackovski N, Svahn AJ, Cole NJ, Goldsbury C, Rinkwitz S, Becker TS, Nicholson GA, and Laird AS. 2017. 'Calpain Inhibition Is Protective in Machado-Joseph Disease Zebrafish Due to Induction of Autophagy', J Neurosci, 37: 7782–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weishaupl D, Schneider J, Peixoto Pinheiro B, Ruess C, Dold SM, von Zweydorf F, Gloeckner CJ, Schmidt J, Riess O, and Schmidt T. 2019. 'Physiological and pathophysiological characteristics of ataxin-3 isoforms', J Biol Chem, 294: 644–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winborn BJ, Travis SM, Todi SV, Scaglione KM, Xu P, Williams AJ, Cohen RE, Peng J, and Paulson HL. 2008. 'The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains', J Biol Chem, 283: 26436–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood TE, Barry J, Yang Z, Cepeda C, Levine MS, and Gray M. 2019. 'Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington's disease mouse model', Hum Mol Genet, 28: 487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Vonk JJ, Salles F, Vonk D, Haslbeck M, Melki R, Bergink S, and Kampinga HH. 2019. 'The N terminus of the small heat shock protein HSPB7 drives its polyQ aggregation-suppressing activity', J Biol Chem, 294: 9985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JL, and Jarosz DF. 2018. 'Mutations, protein homeostasis, and epigenetic control of genome integrity', DNA Repair (Amst), 71: 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Hayashi S, Tsuji S, and Takahashi H. 2001. 'Involvement of the cerebral cortex and autonomic ganglia in Machado-Joseph disease', Acta Neuropathol, 101: 140–4. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yang S, Guo J, Cui Y, Tang B, Li XJ, and Li S. 2017. 'Synergistic Toxicity of Polyglutamine-Expanded TATA-Binding Protein in Glia and Neuronal Cells: Therapeutic Implications for Spinocerebellar Ataxia 17', J Neurosci, 37: 9101–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Wang H, Chen D, Gao F, Ying Z, and Wang G. 2014. 'p62/sequestosome 1 regulates aggresome formation of pathogenic ataxin-3 with expanded polyglutamine', Int J Mol Sci, 15: 14997–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YF, Liao SS, Luo YY, Tang JG, Wang JL, Lei LF, Chi JW, Du J, Jiang H, Xia K, Tang BS, and Shen L. 2013. 'SUMO-1 modification on K166 of polyQ-expanded ataxin-3 strengthens its stability and increases its cytotoxicity', PloS one, 8: e54214. [DOI] [PMC free article] [PubMed] [Google Scholar]