

SUMMARY
Metformin is the front-line treatment for type 2 diabetes worldwide. It acts via effects on glucose and lipid metabolism in metabolic tissues, leading to enhanced insulin sensitivity. Despite significant effort, the molecular basis for metformin response remains poorly understood, with a limited number of specific biochemical pathways studied to date. To broaden our understanding of hepatic metformin response, we combine phospho-protein enrichment in tissue from genetically engineered mice with a quantitative proteomics platform to enable the discovery and quantification of basophilic kinase substrates in vivo. We define proteins whose binding to 14-3-3 are acutely regulated by metformin treatment and/or loss of the serine/threonine kinase, LKB1. Inducible binding of 250 proteins following metformin treatment is observed, 44% of which proteins bind in a manner requiring LKB1. Beyond AMPK, metformin activates protein kinase D and MAPKAPK2 in an LKB1-independent manner, revealing additional kinases that may mediate aspects of metformin response. Deeper analysis uncovered substrates of AMPK in endocytosis and calcium homeostasis.
Graphical Abstract
In Brief
Metformin is a potential anti-aging and anti-cancer therapy and a treatment for diabetes. Stein et al. investigate metformin-induced signaling in the liver, using 14-3-3 binding to identify phosphorylation events acting as dominant regulators of target protein activity. Kinases (PKD, MK2) activated by metformin independent of LKB1/AMPK and other targets of metformin are identified.
INTRODUCTION
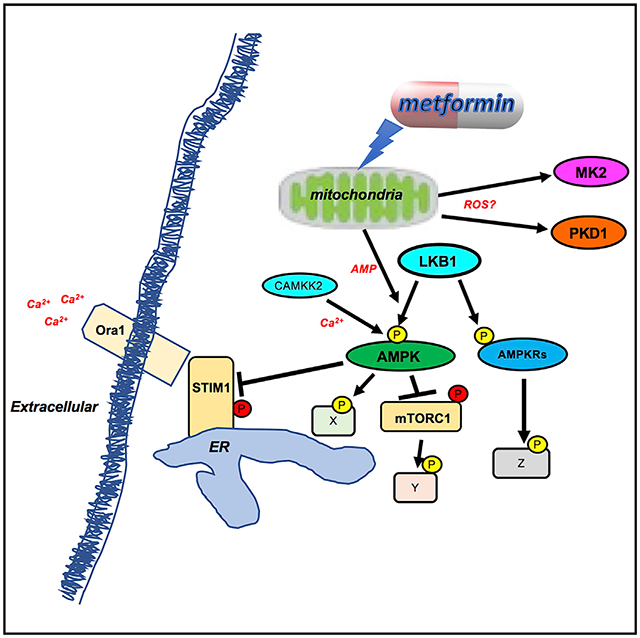
Metabolic equilibrium is essential to the survival of all organisms, both at the single and multi-cellular level (DeBerardinis and Thompson, 2012). To maintain this balance, organisms must sense and respond to decreased intracellular ATP at early stages of energy depletion, to engage mechanisms to restore ATP levels before its loss becomes catastrophic (Hardie et al., 2012). As with many cell biological processes, kinase-mediated signaling cascades have proven integral for the rapid response to metabolic changes (Hotamisligil and Davis, 2016). The hetero-trimeric energy sensing 5′-adenosine monophosphate (AMP) activated protein kinase (AMPK) complex, and the nutrient-sensing mammalian target of rapamycin complex 1 (mTORC1) represent two ancient counter-acting pathways that control anabolism and catabolism across all eukaryotic organisms (Inoki et al., 2012; Laplante and Sabatini, 2012). Genetic studies in diverse model organisms have revealed a conserved function of AMPK as a metabolic sensor that enables adaptive changes in growth, differentiation, and metabolism under conditions of low energy. AMPK has been shown to be a central regulator of cell growth and metabolism in mammals, hypothesized to play important roles in the suppression of both cancer and metabolic disease (Hardie et al., 2016; Garcia and Shaw, 2017). The kinase that phosphorylates the activation loop Threonine172 of AMPK under low ATP conditions is LKB1 (STK11), a tumor suppressor inactivated in ~20% of non-small cell lung adenocarcinomas (Hardie and Alessi, 2013). Additionally, AMPK can be directly phosphorylated at the same residue by the calcium-sensitive kinase calcium/calmodulin-dependent protein kinase kinase 2/beta (CAMKK2), which occurs transiently under conditions of calcium flux, unlike the sustained activation by LKB1 under low energy conditions. Over the past decade, there has been significant progress in the identification of downstream substrates of AMPK that act as effectors in the cellular response to low energy (reviewed in Hardie et al., 2016; Garcia and Shaw, 2017). The functional conservation of a core set of AMPK substrates mediating metabolism and growth (e.g., acetyl-CoA carboxylase, Raptor, ULK1) make it likely that developing methodologies for a more comprehensive decoding of AMPK substrates will lead to fundamental insights into the minimal number of cellular targets required to move a cell from an anabolic pro-growth state to a nutrient-limited catabolic state.
Metformin is a mild mitochondrial complex I inhibitor with modest bioavailability, known to lower hepatic lipogenesis and gluconeogenesis and promote insulin sensitivity (He and Wondisford, 2015; Tan et al., 2016). Hepatic LKB1 has been found to be required for metformin in mice on a high fat diet, and AMPK and its regulation of the downstream target ACC was also reported as critical for the ability of metformin to restore insulin sensitivity during high fat diet (Cao et al., 2014; Duca et al., 2015; Fullerton et al., 2013). However, the relative role of AMPK following metformin may be dependent on the metabolic conditions, and some therapeutic actions of metformin are likely AMPK-independent (Foretz et al., 2010; He and Wondisford, 2015). Thus, a more comprehensive understanding of metformin response is critical. In addition to directly phosphorylating AMPKα1 and AMPKα2, LKB1 is a master kinase that directly activates a family of 12 related kinases, collectively known as the AMPK-related kinase family (AMPKRs) (Alessi et al., 2006). AMPKRs control diverse cellular processes including cell polarity, cytoskeleton, transcription, and cell growth via phosphorylation of a number of downstream substrates. Notably, the SIK subfamily of AMPKRs have been shown to control hepatic gluconeogenesis independently of AMPK (Itoh et al., 2015), and thus undiscovered targets in metabolism downstream of LKB1 may exist. To date, the annotated substrates of the AMPKR family in various systems have suggested a high degree of cell-type/tissue specificity or redundancy as to which AMPKR family member is responsible for phosphorylating downstream substrates and governing diverse biological processes (Goodwin et al., 2014; Mirouse and Billaud, 2011). Therefore, to accurately annotate the correct kinase-substrate relationships that govern a physiological outcome, one must annotate these relationships in the most relevant systems possible, preferably in a quantitative manner to enable hierarchical ranking of potential substrates.
14-3-3 is a family of phospho-protein binding proteins that interact with many intracellular targets directly binding to phosphorylated serine residues that lie within a specific amino acid consensus motif, most commonly Arg/Lys-x-x-pSer-X-Pro (Yaffe, 2002). Engagement of 14-3-3 proteins with different groups of target phospho-proteins cause coordinated shifts in cellular behavior in response to growth factors and different stress stimuli (Mackintosh, 2004). Thus, following a growth factor or stress stimulus, kinase phosphorylation of select substrates induces 14-3-3 binding, often resulting in a subcellular localization change or disruption of protein-protein interactions for the target substrates.
In this study, we describe a quantitative proteomic approach to simultaneously test, in tissue, both the pharmacological and genetic determinants that dictate metformin response in liver to mediate hepatic metabolism. Through a combination of stable isotope labeling in mammals (SILAM), selective 14-3-3ζ enrichment, and conditional genetic models, we identify, quantify, and hierarchically cluster interacting phospho-proteins across multiple conditions in mouse liver (Bustos, 2012; Chen et al., 2011; McClatchy and Yates, 2008). This platform can be applied to any tissue of interest and is not limited to the pharmacological agent, genetic model, or enrichment strategy used in this study.
RESULTS
Quantitative In Vivo Enrichment Methodology
Metabolic stable isotope labeling is a powerful strategy that allows relative quantification across numerous conditions while simultaneously removing instrument bias from precursor selection, a requirement in all post-metabolic labeling strategies. Technological advances have enabled isotopic labeling of entire organisms (i.e., mice) for investigation of complex biological processes and pathologies only observed in multi-cellular models of disease (MacCoss et al., 2005; McClatchy et al., 2007; Venable et al., 2007; Wu et al., 2004). To date, most metabolic labeling technologies have been limited to studies of protein expression in disease models, although increasing efforts are aimed at quantifying posttranslational modifications, such as protein phosphorylation in signaling pathway dynamics. Common phospho-enrichment strategies for large-scale proteomic studies such as immobilized metal affinity chromatography (IMAC) are more efficient at the peptide level and using them to quantitate dynamics in a discovery-based format requires identification and quantification of individual peptides in each experimental condition, complicating the assessment of signaling dynamics (Batalha et al., 2012; Fíla and Honys, 2012; Thingholm et al., 2009). Here, we report a platform that integrates organismal metabolic labeling with selective protein level enrichment of basophilic kinase substrates in disease-relevant tissues. This platform enables the quantification of dynamic responses of signaling pathways to genetic and pharmacological perturbation in an unbiased manner (Figure 1). Applying this approach to phosphorylation events in response to metformin, we take advantage of the inherent affinity properties and target binding specificity of the phospho-scaffolding protein 14-3-3ζ, which has been previously used as an enrichment approach for phospho-proteins (Jin et al., 2004; Johnson et al., 2010; Yaffe, 2002), combined with the SILAM strategy in a ratio-of-ratio format. This enables investigation of more than two conditions and allows for a more linear quantification of larger ratios compared with direct ratio formats, as previously shown (MacCoss et al., 2003, 2005). To integrate this labeling and enrichment strategy directly in complex tissue lysate and facilitate data interpretation, we develop a computational platform to enable translation of derived data into heatmap format. Our approach allows simultaneous observation of trends within and across enriched and un-enriched analyses, correlating affinity with protein expression and enabling hierarchical clustering and ontological analysis of statistically significant proteins. Motif analysis of potential phosphorylation sites on identified proteins responsible for 14-3-3ζ interaction can reveal potential sites of regulation by the AMPKR family, the Akt-mTOR-S6K axis, and other basophilic kinases regulated by metformin independent of LKB1 status.
Figure 1. Quantitative In Vivo Proteomics of Differential 14-3-3 Interactors in Murine Liver.

Experimental design and workflow of the proteomic analysis of genetic and pharmacological sensitivity to LKB1 and Metformin in murine liver. Three biological replicates for each condition are tested.
The method begins with isotopic labeling of wild-type background strain mice with an 15N spirulina diet (McClatchy and Yates, 2014) (Figure S1). Efficient labeling of the liver is achieved, where labeling reached 97% permitting a high degree of quantification. Harvested labeled liver is used as an internal standard for all experimental conditions analyzed. Because all experimental conditions are quantitated against the same internal standard, comparison across experimental conditions is possible through a ratio-of-ratio approach. Tissue homogenate from the internal standard is mixed 1:1 with tissue homogenate from each unlabeled experimental condition, a single enrichment is done for either 14-3-3ζ-GST or GST alone, and protein expression is analyzed in a complementary un-enriched homogenate from each experimental condition. Enriched proteins are analyzed by multidimensional protein identification technology (MudPIT) fractionation and high resolution tandem mass spectrometry (MS/MS) (Washburn et al., 2001). We assess each condition of interest in biological triplicate at the enriched and protein expression level (Figure 1). Following background subtraction and statistical filtering, the remaining proteins are translated into heatmaps that plot trends across the enriched and un-enriched samples independently and correlate the trends across all biological conditions between each level of analysis depicted by a different color (red and blue, respectively). The distillation of all conditions into a single representative map enables clustering by enrichment trends through unsupervised approaches to provide robust groups of proteins with highly similar trends.
The analysis of metformin response in wild-type and liver-specific LKB1 knockout mice provides a model system to systemically define regulatory phosphorylation events acutely induced by metformin that require, or are independent of, LKB1-dependent signaling. Deletion of LKB1 in the liver of adult mice has been shown to promote dramatic hyperglycemia, which reduces the therapeutic efficacy of metformin in lowering blood glucose (Shaw et al., 2005). AMPK is critical for suppression of mTORC1 in the liver after metformin treatment (Howell et al., 2017), but several AMPK-related kinases (AMPKRs) also play key roles in hepatic metabolism (Itoh et al., 2015; Patel et al., 2014). Beyond AMPK, it remains unknown whether other basophilic kinases may be stimulated by metformin, and the use of 14-3-3 as a targeted affinity reagent helps illuminate other potential regulatory events that may be metformin-induced but LKB1-independent.
Validation of Hepatic LKB1 Deletion and Metformin Pharmacological Activity
Utilizing the conditional Stk11fl/fl genetic mouse model and wild-type littermates, selective hepatic deletion of the LKB1 locus is achieved by tail vein injection of adenoviral cre-recombinase in a single dose (Shaw et al., 2004). Nine days after viral administration, animals are fasted overnight, re-fed for 4 h, then administered either saline vehicle or metformin at the therapeutic dose of 250 mg/kg for 2 h. Livers are collected, homogenized, and soluble proteome used for 14-3-3ζ-GST or GST-alone enrichments, and signaling is validated around the canonical signaling axis through western blot (Figure 2A). Following MS/MS analysis by MudPIT, ProLUCID is used for peptide identification and identified proteins quantified using the Census algorithm (Park et al., 2008, 2014; Xu et al., 2015). Comparing vehicle-treated LKB1-competent and -deleted conditions by volcano plot, prior to background subtraction and statistical filtering, identified known substrates of AMPK increased in 14-3-3ζ binding in wild-type animals, including ULK1 and LIPE (hormone sensitive lipase) (Figure 2B). Conversely, validated substrates of the mTOR-S6K pathway are observed in LKB1-deleted 14-3-3ζ enrichments including LPIN1 and PFKFB2, giving confidence that our enrichment approach yielded relevant, validated substrates of the kinase signaling pathways of interest and confirming that our model system recapitulated the genetic conditions intended for analysis of acute metformin response. When plotting the isotopic distribution of heavy and light peptides identified in 14-3-3ζ enriched samples, a normal distribution around 0 is observed, indicating approximately equivalent global enrichment of heavy and light peptides, giving confidence that the vast majority of enriched proteins should be quantifiable within each condition (Figure S2A). Following analysis comparing all three biological replicates at the protein level for the LKB1 wild-type, vehicle-treated condition to determine how stochastic our enrichment is, we find that 71.9% of the enriched proteins are recovered in 2 of 3 biological replicates, and 53.3% of enriched proteins are identified in all three biological replicates adding confidence that our enrichment is reproducible and robust, and enriched proteins should pass replicate statistical filters during further refinement and analysis (Figure S2B). Data from all biological replicates are next filtered for removal of background within each condition using a GST alone control, then further filtered by requiring an ANOVA p value of less than or equal to 0.05 for a protein quantitative measurement in 3 of the 4 biological conditions. In the un-enriched analysis across all 4 conditions, a total of 8,033 proteins are identified. 1,944 proteins are identified in 14-3-3ζ-GST-enriched samples in at least 2 of 3 replicates following background sub-traction, and following application of the statistical filter, 1,022 proteins are deemed high-confidence 14-3-3 interactors (Figure 2C). Furthermore, when comparing our 1,944 identified 14-3-3ζ binding proteins at the protein level to a previous study in liver (Huttlin et al., 2010) that employed the traditional enrichment strategy of SCX-IMAC at the peptide level, we found that the degree of overlap in identified proteins is 14.5% of the total proteins identified in both studies. This indicates that each enrichment strategy is complimentary, not redundant, and suggests that our orthogonal enrichment strategy may lead to identification and quantitation of unknown kinase substrates (Figure S2C). Additionally, when primary sequences of identified proteins in each study are analyzed for candidates containing a 14-3-3 consensus motif, we find that ~75% of proteins identified in either study containing a 14-3-3 motif (Gardino et al., 2006) are identified in the 14-3-3 enrichment, and 64% of these proteins are exclusive to our study, proving that our enrichment strategy is specific (Figure S2D). Last, we performed a similar analysis but searching for proteins containing the derived AMPK consensus motif (Dale et al., 1995; Gwinn et al., 2008), and found 82% of the proteins containing this motif in both studies are identified in the 14-3-3 enrichment approach, and 73% of these proteins are exclusive to our study, lending further confidence that our enrichment strategy may identify substrates of AMPK (Figure 2D). Following normalization and application of a hierarchical clustering algorithm described in detail below, we cluster the 1,022 high-confidence 14-3-3 interacting proteins (Figure 2E).
Figure 2. Efficacy of Proteomic Screen Compared to Traditional Phospho-Proteomic Approach.

(A) Western blot analysis in biological triplicate of extracted mouse livers for proteomic screen. Hepatic deletion and metformin induction of signaling validated by downstream substrates of AMPK and mTOR.
(B) Volcano plot for comparison of genetic sensitivity in vehicle treated 14-3-3 pull-downs from 3 biological replicates. Proteins that pass statistical and fold change criteria are highlighted in red and blue dots and found in the regions of the plot highlighted in yellow. Yellow lines represent the p value (<0.05) and fold change thresholds (>1.5).
(C) Venn diagram of total proteins identified in lysate analysis of liver homogenate across 4 biological conditions versus high confidence 14-3-3 interactors following background subtraction and statistical filtering.
(D) Venn diagram of proteins that contain the derived AMPK consensus motif to previous SCX-IMAC study.
(E) Hierarchical clustering of 1,022 high confidence 14-3-3 interactors by trend within enriched samples
Normalization and Clustering of 14-3-3 Binding Events
To enable comprehensive analysis of the acquired data, and to annotate proteins possessing distinct trends in regards to 14-3-3ζ binding, we develop an algorithm to normalize, merge, and cluster statistically significant proteins based on trends within the 14-3-3ζ enrichments. To distill the acquired data into heatmaps, the simple normalization function of Ci/(C1+C2+C3+C4) is used for recovery of a given protein within the enriched and un-enriched data. Following normalization, statistically significant proteins are clustered based on trends within the 14-3-3ζ enriched data using an unsupervised clustering approach based on Euclidean distance. Additionally, the algorithm compares the correlation of trends within 14-3-3ζ enriched and un-enriched analyses and plots the correlation in a hanging bar separated from the heatmap labeled “trend.” This trend bar allows one to infer if recovery within the enriched samples is expression based or due to specific enrichment changes, potentially caused by phosphorylation of the protein of interest in a given condition. Therefore, while analyzing the generated heatmaps of statistically significant proteins, distinct predictive patterns are expected within the 14-3-3ζ enriched data for each pathway of interest and within the AMPKR family itself. LKB1 is the master regulator of the AMPKR family and inhibits the mTORC1-signaling axis through AMPK direct phosphorylation of TSC2 and Raptor, a core subunit of the mTORC1 complex (Gwinn et al., 2008; Inoki et al., 2003). Within the AMPKR family, previous data indicate that AMPKα1/2 are the only members that are activated by metformin through interaction with AMPKγ proteins that directly bind adenine nucleotides (Figure 3A) (Lizcano et al., 2004). Therefore, in LKB1-competent conditions, 14-3- 3ζ binding of AMPK-dependent substrates would be expected to increase in response to metformin over vehicle-treated enrichments, and upon LKB1 deletion, 14-3-3 binding would be attenuated, rendering potential AMPK substrates both genetically and pharmacologically sensitive (Figure 3B). For potential substrates of other AMPKR family members whose activity should not be regulated by metformin, we expect 14-3-3 enrichment in LKB1 intact conditions with no relative change in response to metformin administration. However, upon LKB1 deletion, 14-3-3 binding would be reduced, thus rendering substrates of other AMPKR family members genetically sensitive but pharmacologically insensitive. For potential mTOR-S6K-dependent substrates, an inverse correlation would be expected to that observed for AMPK. Additionally, proteins that are solely sensitive to metformin treatment independent of LKB1 genotype, both in an inducing and suppressive subset, would be expected within the dataset (Figure 3B). After merging data into a single heatmap, all theoretical patterns described above are observed, and the appended dendrogram suggests that clustering is robust (Figure 2E).
Figure 3. Comprehensive Global Analysis of Differential 14-3-3 Interactors in Murine Liver.

(A) Schematic of pathways contributing to potential 14-3-3 interactors within the screen.
(B) Corresponding expected patterns for contributing pathways within normalized heatmaps and hierarchical clustering analysis. 14-3-3 enrichment patterns are depicted in red, lysate analysis for expression profiles are depicted in blue. Trend bar depicts correlation between 14-3-3 enrichment and lysate patterns across conditions.
(C) Western blot analysis in biological triplicate of extracted mouse liver homogenate of activation status of additional basophilic kinases.
(D) Representative cluster of potential MK2/PKD substrates.
(E) Western blot analysis of extracted mouse liver homogenate extending MK2/PKD activation to phenformin.
(F) Representative cluster of potential AMPK substrates.
(G) Representative cluster of potential AMPKR substrates.
(H) Representative cluster of potential AKT substrates.
Manual inspection of the resultant trends for known AMPK substrates indicates that observed proteins within the dataset exhibit varying patterns, but the majority of well-established substrates, including ULK1 and CRTC2, bind to 14-3-3 in an LKB1- and metformin-dependent pattern as expected (Figure S3A). The AMPK substrate BAIAP2 also emerges as both a metformin- and LKB1-dependent interactor of 14-3-3 (Banko et al., 2011). These changes do not appear to be expression-based, as evidenced by lack of a positive trend correlation between the enriched and un-enriched conditions. Moreover, for AMPK substrates like TSC2 and PFKFB3 that are not induced to bind 14-3-3 in a metformin/LKB1-dependent manner, their patterns may result from 14-3-3 binding dictated by another signaling pathway on sites other than, or in addition to, the known AMPK sites in these proteins under varying biological conditions. For example, in TSC2, the well-annotated AKT phosphorylation sites (Ser939 and Ser981) are known to dictate 14-3-3 binding to TSC2 (Cai et al., 2006), whereas AMPK phosphorylation of TSC2 on Ser1387 (Ser1345 in rat) has not been reported to induce 14-3-3 binding. Similar observations are made when manually looking at validated substrates of the AKT-mTOR-S6K axis present within the refined dataset, where the protein PFKFB2 exhibited the expected Akt-mTOR-S6K substrate pattern (Figure S3B) (Novellasdemunt et al., 2013). However, ULK1 (a shared substrate between both signaling arms) binding appears to be predominated by AMPK regulation within the liver, consistent with reports that AMPK phosphorylation induces 14-3-3 binding to ULK1 (Mack et al., 2012). Last, when examining known substrates of AMPK and AKT-mTOR-S6K pathways before and after applying the ANOVA p value filter, the majority of known substrates present within the dataset are retained following application of the filtering criteria. This analysis adds confidence that our statistical parameters are sound, and positive controls for each pathway are retained for use as guides for cluster selection moving forward (Figures S3A and S3B).
Hierarchical Clustering to Elucidate Potential Kinase Substrates
Because the intention of this study is to develop a discovery platform for basophilic kinase substrates, we use unsupervised clustering independent of a priori knowledge based solely on characteristics of protein enrichment within the derived dataset. To that end, from the global clustering previously performed, we derive 62 individual clusters grouped and merged using a greedy algorithmic function based on the trend across conditions as well as the magnitude of change (Figure 3B; Data S1). This enables us to group proteins based solely on the trends within our data and the correlation of known substrates of each pathway with potential substrates to be further validated in the context of hepatic metabolism. Furthermore, to extend the breadth of the screen to identify potential substrates of additional basophilic kinases that are also known to induce 14-3-3 binding in their substrates, the original liver homogenates are immunoblotted for activating phosphorylation events in the kinases; RSK, CHK1, PKD, AKT, and MK2 (Figure 3C). Through this analysis, we are able to identify distinct activation patterns for several of these kinases, most notably, both MK2 and PKD are activated by administration of metformin, which is independent of LKB1 status. Based on their activation pattern, we identify proteins whose 14-3-3 binding mirrored the metformin-induced but LKB1-independent activation of PKD and MK2 (Figure 3D).
The activation of PKD and MK2 in response to metformin treatment is unexpected and previously unreported. PKD is known to be activated by oxidative stress, and it is possible that increased mitochondrial reactive oxygen species (ROS) after metformin treatment is responsible for its activation (Cobbaut and Van Lint, 2018; Eisenberg-Lerner and Kimchi, 2012). MK2 is also a stress-activated serine/threonine kinase with a major role in cytokine response as well as numerous other biological processes including endocytosis, cell migration, cell-cycle progression, chromatin remodeling, and response to DNA damage (Reinhardt and Yaffe, 2009). In response to various stress agonists, MK2 is phosphorylated and activated by the MAP kinase p38-alpha/MAPK14, leading to phosphorylation and regulation of specific downstream protein substrates determined by the stress queue. To further examine the nature and extent of PKD and MK2 activation, we treated control or LKB1-liver-specific knockout mice with metformin as well as its more potent analog, phenformin, which has 100-fold higher inhibition of complex I and induces greater ROS as a result (Dykens et al., 2008; Shackelford et al., 2013). We observe that metformin and phenformin both induce MK2 and PKD in an LKB1-independent manner, although PKD activation seems proportional to the strength of complex I inhibition and predicted ROS levels, unlike MK2 and AMPK activation that each appear to be stimulated equivalently with both biguanides.
Examining derived LKB1-dependent clusters, those exhibiting predicted patterns for AMPK substrates arise (clusters 2, 9, 10, 19, 41, 48, and 54). Cluster 48 (Figure 3F) contains the previously annotated AMPK substrate cingulin, a cytosolic protein that localizes to tight junctions within epithelial cells and interacts with microtubules through the n-terminal head region, which appears to be regulated by AMPK phosphorylation (Yano et al., 2013). This cluster also contains numerous additional proteins of diverse functions never proposed to be direct substrates of AMPK, including the endoplasmic reticulum (ER) calcium sensor stromal interaction molecule 2 (STIM2), as well as the phosphoesterase PRUNE (Kobayashi et al., 2006). This cluster is one of several noted above that harbor potential substrates of AMPK and garner further investigation. Importantly, nearly all of these clusters predicted to contain potential AMPK substrates did harbor reported AMPK substrates (cluster 2: TRP53BP2 [Banko et al., 2011]; cluster 10: CRTC1 [Koo et al., 2005]; cluster 19: plectin [PLEC1] [Gregor et al., 2006]; cluster 54: BAIAP2 [Banko et al., 2011]).
Beyond AMPK-like protein clusters, there are several clusters whose binding to 14-3-3ζ was LKB1-dependent but not induced by metformin (clusters 1, 39, and 46), as predicted for substrates of the AMPK-related kinases including the SIK, MARK, and NUAK family kinases (Figure 3G). When assessing activating phosphorylation of AKT at Thr308 and Ser473, a distinct pattern is evident for both, and corresponding clusters are evident in the data as well, one of which contains MEK1 (MAP2K1), a previously reported substrate (Procaccia et al., 2017) (Figure 3H). Two additional clusters, 40 and 56, also contain the known AKT substrate PFKFB2 and related proteins PFKFB3/PFKFB4, respectively (Novellasdemunt et al., 2013), as well as numerous metabolic proteins including PEPCK (PCK1), SDS, GOT1, and GLS2 (Li et al., 2015) (Table S1). Notably, immunoblots for several of the identified metabolic regulators whose binding to 14-3-3ζ was controlled by LKB1 genotype revealed both positive (glucokinase: GCK) and negative (SDS and GOT1) regulation at the level of protein expression, confirming the mass spectrometry analysis and correlative trend of protein expression to 14-3-3ζ binding (Figure S4).
Clusters possessing proteins that are genetically insensitive to LKB1, but show pronounced change in response to metformin, both suppressed and induced in binding to 14-3-3ζ, are also observed. Given the correlative data between activation status of these kinases across tested conditions and accompanying clusters exhibiting similar patterns of 14-3-3 binding, these potential kinase-substrate relationships garner further validation and functional characterization in follow-up studies. These potential relationships may provide an even more comprehensive molecular characterization of metformin effects in vivo.
LKB1 Is Genetically Required for a Subset of Metformin- Induced 14-3-3 Binding Proteins
The goal of the resultant platform from this study is to address unanswered biological questions about hepatic metabolism and metformin response through identification and quantification of molecular components and potential targets of the LKB1-AMPK pathway in vivo. To achieve this goal, we focus on proteins regulated in response to pharmacological and genetic perturbation in regards to 14-3-3ζ binding. To further assess and refine the validation list, we ask the simple question: how many proteins increase in 14-3-3ζ binding in wild-type livers upon metformin administration and decrease in binding upon deletion of LKB1 similar to known substrates within the data. An examination of well-studied proteins known to bind 14-3-3ζ upon phosphorylation by AMPK such as ULK1, CRTC1, and CRTC2, reveal a quantitative induction of 14-3-3 binding of 1.25 to 1.5-fold in response to metformin treatment. High basal binding in the hepatic liver lysates may prevent measurement of higher induction for these proteins. Of the 1,022 high confidence interactors, 250 proteins are induced in 14-3-3ζ binding at least 1.25-fold. Of these 250 proteins, 110 exhibit a >1.25-fold loss of binding in LKB1-deficient livers (Figure 4A). This suggests that ~45% of proteins that inducibly bind 14-3-3 in response to metformin are LKB1-dependent.
Figure 4. Differential 14-3-3 Interactors in Response to Metformin Action and Hepatic LKB1 Loss in Murine Liver, Potential AMPK Substrates.

(A) Venn diagram depicting proteins of both metformin and LKB1 dependence. Of 1,022 high confidence 14-3-3 interactors, 250 increase in binding to 14-3-3 1.25-fold or greater in response to metformin administration (dark green). Of those 250 proteins, 110 proteins are reduced in binding by 1.25-fold or more (sum of blue and yellow) and of theses 110 proteins, 56 are reduced in 14-3-3 binding by 1.5-fold or greater in response to LKB1 loss (yellow).
(B) Heatmap depicting 56 high confidence 14-3-3 interactors with both genetic (LKB1) and pharmacological sensitivity (metformin) as defined by previous thresholds (1.25-fold metformin induction, 1.5-fold loss upon LKB1 deletion). Purple arrows indicate known AMPK substrates from previous studies that satisfy the derived criteria.
Previous immunoblot analysis suggested LKB1-loss was a stronger biological determinant than the magnitude of metformin-induced phosphorylation (Figure 2A), so we queried how many of the metformin-induced 14-3-3ζ binders were lost by more than 1.5-fold when LKB1 is deleted. Using this stringent cutoff off in LKB1-deficient livers resulted in the identification of 56 metformin-induced 14-3-3 binding proteins (Figure 4A). Because our goal was to determine downstream components that may contribute to metformin response in an LKB1-dependent manner, this subset is the most promising to pursue for functional validation. Among these 56 proteins are a number of well-studied AMPK substrates, including ULK1, CRTC1, CRTC2, BAIAP2, CGN, and CGNL1 (indicated with purple arrows in Figure 4B; highlighted in Table S1). This refined list contains proteins of diverse biological function, ranging from Rab5 signaling (RABEP1), PDZ domain containing adaptors (PDLIM1, PDLIM5, and MPP6), and calcium signaling and homeostasis components (STIM1 and STIM2).
A comparison of these 56 to proteins identified in three previously published screens for AMPK substrates, all employing orthogonal methodologies in cell lines or diverse tissues (Ducommun et al., 2015; Hoffman et al., 2015; Schaffer et al., 2015), reveals common and unique targets from each approach (those found in 2 or more screens highlighted in blue in Table S2). In addition to cross-referencing our list with previous AMPK screens, we examine the candidate AMPK substrates from each screen with proteins that immunoprecipitated endogenously with overexpressed AMPK subunits in a previous interactome study of autophagy (Behrends et al., 2010). This comparison revealed that of our 56 candidates, two had been previously found to co-immunoprecipitate with AMPK: STIM1 and STIM2 (Table S2).
To gain insight into the fraction of proteins within pathways known to be modulated by metformin that may also regulated by 14-3-3 binding, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis to examine the 14-3-3 binding patterns of each process. The key metformin-regulated processes of gluconeogenesis (Figure S4A) and insulin signaling (Figure S4B) have one or two proteins whose binding appears to be metformin-induced and LKB1-genotype-sensitive. From this analysis, it is evident that regulated proteins represent only a fraction of the relevant pathway components, similar to other known pathways controlled by AMPK where phosphorylation of one or two rate-limiting component(s) is observed. Interestingly, one of the most regulated AMPK-like proteins was Rabep1 (Rabaptin-5), a protein in the GO process of endocytosis (Figure 5A). Its direct interacting partner, Rab-GEF1 (Rabex-5), bore a signature resembling a potential AMPKR substrate, justifying further investigation of this potential biological connection (Horiuchi et al., 1997; Lippé et al., 2001).
Figure 5. AMPK Regulates Endocytosis through Phosphorylation of Rabep1.

(A) Heatmap of Gene Ontology process: endocytosis.
(B) Dose-response curve of metformin at 2 h and 5 h time points in isolated primary hepatocytes from hepatic AMPK WT and conditional AMPK DKO mouse livers.
(C) Schematic of domain architecture of Rabep1 highlighting location of S407 and clustal alignment of Rabep1 S407 across species showing conservation of AMPK consensus motif.
(D) HEK293T cells transfected with Myc-Rabep1 WT or Ser407Ala as indicated and treated for 60 min with vehicle or 50 μM 991. Myc immunoprecipitations conducted in lysates and probed with anti-AMPK pMOTIF antibody.
(E) Phosphorylation sites on Rabep1 identified by immunoprecipitation of transiently transfected Myc-Rabep1 WT in HEK293T cells after treatment with vehicle or 2 mM phenformin for 1 h and analyzed by MS/MS. Data reported in spectral counts indicated as stacked bars and graphed as a ratio of the observed modified versus unmodified spectra per site. Numbers in black overlay onto bars indicate number of observed spectral counts for each version of a given site. Blue portions of bar indicated modified, red portions indicate unmodified.
(F) HEK293T cells transfected with Myc-Rabep1 WT or Myc-Rabep1 Ser407Ala, as indicated, pre-treated for 30 min with DMSO or 1.3 mM Gö6976 for 30 min prior to co-treatment with 200 nM TPA and/or 2 mM phenformin or vehicle for an additional 60 min. Myc immunoprecipitates for Rabep1 immunoblotted with anti-PKD pMOTIF and anti-AMPK pMOTIF antibodies and re-probed with anti Myc for loading controls. Lysates probed for pPKD Ser744/748 and pPKD Ser916 as read outs of PKD activation and pJNK as readout of downstream PKD activity.
Validation of RABEP1 as a Direct AMPK Substrate in Response to Biguanide Treatment
Given this unexpected connection to Rab5-mediated endocytosis, we first explored Rabep1 as a potential AMPK substrate. To validate Rabep1 as a downstream target of AMPK signaling, hepatocytes were extracted from conditional liver-specific AMPK knockout mice following tamoxifen treatment or vehicle in vivo. Following extraction and plating, hepatocytes were starved overnight and insulin administered for 20 min prior to conducting a dose response curve of metformin at 0, 0.5, 1, and 2 mM for 2 h or 5 h (Figure 5B). Strikingly, Rabep1 exhibits clear changes in electrophoretic mobility in response to metformin in a dose-dependent manner that is ablated in AMPK-deleted hepatocytes and mirrors a dose-responsive effect observed in phosphorylation of classical AMPK substrates such as Raptor, ULK1, and ACC. Given the clear metformin-induced mobility shift in Rabep1, and ablation in the AMPK-deficient cells, we further examine whether Rabep1 may serve as a direct AMPK substrate. Sequence analysis reveals a candidate AMPK phosphorylation site at Ser407, a highly conserved residue bearing all the hallmarks of the previously described AMPK optimal motif (Gwinn et al., 2008) (Figure 5C). Rabep1 Ser407 was previously described to be phosphorylated under specific conditions by PKD (Christoforides et al., 2012), which is also true for a limited number of previously reported AMPK substrates including the class IIa family of HDACs (HDAC4, HDAC5, HDAC7, HDAC9) (Mihaylova and Shaw, 2011). Indeed, the optimal motif for PKD overlaps with that of AMPK (Döppler et al., 2005), making it intriguing that metformin can activate PKD independent of LKB1.
To further explore whether AMPK is directly regulating Rabep1, we made use of potent small molecule AMPK activators, which directly bind the AMPK hetero-trimer and activate its kinase activity without causing change to cellular ATP/AMP levels (Xiao et al., 2013). We also make use of a phospho-specific antibody generated against phospho-peptides conforming to the optimal AMPK substrate sequence. Use of such AMPK motif antibodies has been used previously to define AMPK substrates when comparing wild-type cells to AMPK genetic knockout cells (Ducommun et al., 2015; Gwinn et al., 2008). It is important to note that the antibody will also recognize the phosphorylation of proteins on sites by kinases broadly related to AMPK, because many of their substrates share common elements with the optimal AMPK motif. We find that myc-tagged Rabep1 immunoprecipitated from HEK293T cells treated with the direct AMPK activator 991 shows increased reactivity with the AMPK substrate motif antibody (Figure 5D). Mutation of Ser407 to Alanine results in loss of reactivity, suggesting antibody binding requires phosphorylation of this specific residue. The high basal phosphorylation of exogenous myc-tagged Rabep1 could be from an alternate kinase like PKD, or could even be from endogenous AMPK activation, which is quite high in HEK293T cells compared to many normal cells like MEFs (note high basal P-AMPK and P-ACC in these cells in Figure 5D). To corroborate the detection of Ser407 using this motif antibody, we perform mass spectrometric analysis to define all phosphorylation sites on Rabep1 in the basal- and phenformin-treated state in HEK293T cells. Phenformin readily causes mitochondrial complex I inhibition across cell types, unlike metformin that needs a specific cell surface transport family to enter cells (Jensen et al., 2016), and does not readily enter HEK293T cells. This unbiased analysis reveals Ser407 phosphorylation to be induced by phenformin treatment. No other phosphorylation sites detected in Rabep1 during this analysis are induced following phenformin treatment (Figures 5D and S4C).
Next, we use a direct ATP-competitive PKD/PKC inhibitor (Gö6976) (Nhek et al., 2010) to determine whether AMPK is inducing PKD activity and subsequent Rabep1 phosphorylation, rather than AMPK directly phosphorylating Rabep1. Cells are next treated with phenformin to activate AMPK or TPA to promote activation of PKD in a well-established pathway mediated by PKC isoforms (Nhek et al., 2010; Storz et al., 2004). Comparing with vehicle controls, both phenformin and TPA induce similar levels of phosphorylation of Rabep1 Ser407, as detected using the phospho-AMPK motif antibody described above (Figure 5F, compare lanes 3 and 4). The inhibitor Gö6976 greatly reduces Rabep1 phosphorylation in response to TPA but has no effect on Rabep1 phosphorylation induced by phenformin. Collectively, this data suggests that phenformin-induced phosphorylation of Rabep1 is independent of PKD in this context.
Validation and Identification of AMPK Phosphorylation Sites on STIM1/2
Of the proteins detected in our screen, one whose 14-3-3 binding was most robustly induced was the ER calcium sensor STIM2. This protein shows clear metformin-dependent binding to 14-3-3ζ, as well as sensitivity to deletion of the catalytic subunits of AMPK as read out by electrophoretic mobility, placing it downstream of AMPK (Figure 5B). Furthermore the highly related and obligate interactor STIM1 is also present within the candidate AMPK substrate list and exhibits a similar AMPK/metformin-dependent mobility shift, although to a lesser degree than Rabep1 and STIM2 (Figure 5B). Looking at proteins within the dataset annotated as components of the GO term “calcium ion binding,” STIM1 and STIM2 emerge as the proteins whose binding to 14-3-3ζ is greatly increased by metformin in WT mice, but whose binding to 14-3-3ζ is largely reduced or not detected under LKB1 KO conditions (Figure 6A). To directly examine whether STIM1 or STIM2 binding to 14-3-3ζ is regulated both by metformin and AMPK, we employed a conditional mouse model for AMPK. This model possesses floxed alleles for both catalytic subunits of the AMPK complex, AMPKα1 and AMPKα2, crossed to a transgenic inducible cre-recombinase fused to the estrogen receptor fragment under control of the albumin promoter to selectively delete AMPK in the liver upon tamoxifen administration. Using this model and the same metformin treatment regimen in the original dataset, 14-3-3ζ-GST or GST-alone pull-downs are conducted in liver homogenate and read out by western blot using the AMPK substrate ULK1 as a control. This approach reveals a clear association of STIM1 and STIM2 with 14-3-3ζ that is enhanced by metformin and largely abolished in the AMPK DKO conditions (Figure 6B, upper panel). Additionally, a clear mobility shift in endogenous STIM2 is observed in total liver lysate following metformin treatment in wild-type, but not AMPK, DKO livers (Figure 6B, lower panel). Moreover, endogenous STIM2 co-immunoprecipitates with endogenous AMPKα1/2 in MEFs, further validating that it physically interacts with AMPK (Figure 6C), a finding previously reported in an overexpression proteomic interaction dataset conducted in HEK293T cells (Behrends et al., 2010). We next examine whether endogenous STIM1 or STIM2 is modified following a dose-response curve with the small molecule direct AMPK activator 991 in wild-type or AMPK DKO primary hepatocytes. Paralleling AMPK-dependent site-specific phosphorylation of Raptor and ACC, endogenous STIM1 and STIM2 undergo a mobility shift on SDS-PAGE in a 991 dose-dependent manner, suggestive of their phosphorylation (Figure 6D). Finally, we observe mobility shifts in endogenous STIM1 and STIM2 in vivo following treatment with the widely bioavailable 991 analog MK-8722 that has been demonstrated to yield robust direct AMPK activation in rodent and primate models across a wide variety of tissues. Treatment of wild-type mice with MK-8722 leads to robust phosphorylation of AMPK itself and its substrates ACC, Raptor, and ULK1 whose phosphorylation was ablated in AMPK DKO liver homogenates (Figure 6E). Paralleling these events is a clear mobility shift in endogenous STIM1, STIM2, and Rabep1 that are also ablated in the AMPK DKO genetic background, further validating these three proteins as hepatic AMPK substrates.
Figure 6. AMPK Phosphorylates Stim1 and Stim2 in Response to Metformin in Mouse Liver.

(A) Heatmap of Gene Ontology process: calcium ion binding, purple arrows indicate STIM1 and STIM2.
(B) Recombinant 14-3-3-GST or GST pull-downs in AMPK WT or AMPK DKO liver homogenates in biological duplicate for each biological condition.
(C) Co-immunoprecipitation in mouse embryonic fibroblasts either WT or AMPK DKO for STIM2 and AMPKα using two different polyclonal antibodies against Stim2 full-length sequence.
(D) Primary mouse hepatocytes from wild-type or AMPK DKO mice extracted and treated with a dose-response curve of 991 at 0, 10, 25, and 50 μM and compared to 2 mM phenformin for 60 min.
(E) AMPK WT and DKO mice treated with 30 mg/kg MK8722 for 4 h, and liver homogenates analyzed by western blot analysis.
Analyzing STIM1 and STIM2 for the presence of candidate-optimal AMPK phosphorylation motifs reveals both possess optimal sites that could be phosphorylated by AMPK. Ser521 in STIM1 and Ser680 in STIM2, in particular, match the AMPK consensus perfectly and are well conserved across evolution (Figure 7A). Each protein also contains additional potential regulatory sites, especially STIM2, which exhibits the more pronounced mobility shift and whose secondary sites would be predicted to be Ser346 and Ser261. Furthermore, the secondary site S261 on STIM2 is mostly conserved in STIM1 at Ser257. These conserved sites are predicted to reside proximal to the conserved hinge region of the protein (Figure 7B), which sits at the ER membrane on the cytosolic side and may affect conformational dynamics of STIM homo/heterodimers as well as oligomerization upon activation of store operated calcium entry (SOCE). Neither optimal site is conserved across paralogs, because STIM1 Ser521 is found as an alanine in STIM2, and the region in STIM2 surrounding the S680 site is completely absent in the STIM1 primary sequence (Figure 7B).
Figure 7. AMPK Regulates Calcium Homeostasis through Phosphorylation of Stim1 and Stim2 to Terminate Store Operated Calcium Entry.

(A) Clustal alignments of STIM1 S257, S512, S521, and STIM2 S261, S346, S680 across species showing conservation of AMPK consensus motif at identified potential sites of regulation.
(B) Schematic depicting location of potential AMPK sites of regulation on STIM1 and STIM2 on structure of each protein relative to orientation to cytosol and ER lumen.
(C) Representative MS/MS trace for identified STIM1 Ser521 site in STIM1 IP-MS in AMPK WT MEFs.
(D) Regulated phosphorylation sites on STIM1 identified in STIM2 endogenous IP-MS in primary hepatocytes reported by spectral count.
(E) Mouse embryonic fibroblasts stably expressing empty vector, flag-STIM1-WT, flag-STIM1-Ser521Ala in a WT or AMPK DKO background treated with vehicle, 50 μM MK-991 (9) or 2 mM phenformin (P) for 1 h. Flag immunoprecipitates blotted with antibody raised against the STIM1 Ser521 site and re-probed with antiflag antibody for loading.
(F) Reduction of SOCE in AMPK WT MEFs but not AMPK DKO MEFs in response to AMPK activation quantitated by single cell microscopy using fura2-AM. Cells treated with vehicle or 2 mM phenformin in DMEM + 10% FBS for 30 min prior to analysis. Following pre-treatment, media was removed and replaced with Ca2+-free HBSS containing 2 mM phenformin prior to analysis. ER Ca2+ stores were emptied with 2 μM thapsigargin in Ca2+-free conditions before addition of buffer containing 2 mM Ca2+.
(G) Same Fura2-AM measurements without Thapsigargin treatment to empty ER calcium stores. Induction of SOCE in response to calcium addition only seen in AMPK DKO cells in the absence of Tg treatment in phenformin-treated cells.
(H) Controls traces for (F), switching the media from calcium-free to calcium-containing media in the absence of phenformin treatment.
(I) Control traces for (F) with treatment of phenformin in the presence of calcium containing media at all times. All Fura2-AM graphs (F–I) show mean fura2 ratio of 60 (WT) and 60 (KO) cells across 2 biological replicates containing 30 single-cell readings each.
Next, employing IP-MS, we attempt to detect all phosphorylation events on STIM1 and STIM2 in primary hepatocytes. We first corroborate earlier data to demonstrate STIM1 and STIM2 mobility shifts on SDS-PAGE in response to metformin, which are abolished in AMPK DKO hepatocytes (Figure S5A), paralleling 991 treatment of these cells (Figure 6D). Utilizing these conditions, we immunoprecipitate endogenous STIM1 and STIM2 from WT and AMPK DKO hepatocytes with and without metformin treatment to determine the stoichiometry of detected phosphorylation events. This analysis reveals a number of phosphorylation sites in endogenous STIM1 and STIM2 (Figures S5 and S6). Focusing on the most-regulated phosphorylation sites in response to metformin or AMPK genotype, we observe strong induction of the S521 site in STIM1 (Figure 7C) and the S680 site in STIM2, as well as the conserved Ser257/Ser261 sites in STIM1 and STIM2, respectively, along with several additional sites in STIM2, including Ser346 (Figures 7D and S5D). To further validate whether these sites in STIM1 and STIM2 may be AMPK-dependent in multiple cell types, we additionally tested mouse embryonic fibroblasts (MEFs) and human HEK293T cells treated with phenformin or the AMP mimetic, AICAR. We again observe a clear AMPK-dependent mobility shift in both STIM1 and STIM2 following these treatments in MEFs (Figure S7A). Mass spectrometry reveals AMPK-dependent induction of Ser521 in endogenous STIM1 in all cell types as well as AMPK-dependent induction of Ser261 and Ser680 in STIM2 (Figures S7B and S7C). Furthermore, to assess whether AMPK can directly phosphorylate STIM1 and STIM2 in vitro, Flag-tagged STIM1 and STIM2 constructs were immunoprecipitated from HEK293T cells and incubated with recombinant AMPK. Using the AMPK substrate motif antibody as a readout indicates both STIM1 and STIM2 can be phosphorylated by AMPK in vitro (Figure S7D).
To further our investigation of STIM1 regulation by AMPK, an antibody raised against phospho-Ser521 STIM1 was created and demonstrates that phosphorylation of this site is robustly induced in wild-type MEFs by phenformin as well as the direct AMPK activating compound 991 (Figure 7E). The observed induction is largely abolished by a Ser521Ala mutant or by deletion of both catalytic subunits of AMPK in the AMPK DKO MEFs. This observation indicates that the antibody is selective for P-Ser521 and further supports that AMPK is responsible for phosphorylation of this residue.
STIM1 and STIM2 are well-studied components of store-operated calcium entry (SOCE) (Collins and Meyer, 2011; Lewis, 2011; Soboloff et al., 2012). STIM1/2 sense ER calcium stores via their ER luminal EF hand motif, and when ER calcium is low, STIM1/2 homo/heterodimers oligomerize and translocate along with domains of ER to fuse with the plasma membrane where they meet the cell surface transmembrane protein Orai1 to gate this calcium channel open allowing extracellular calcium to flow into the cytosol. Replenished cytosolic calcium can subsequently be passaged into the ER via the ATPase pump, SERCA, to replenish ER calcium stores (Hogan and Rao, 2015). To assess the role of AMPK in SOCE through STIM1/2 phosphorylation, we utilize the ratiometric dye, Fura2-AM, to measure cytosolic calcium levels in the AMPK MEFs. In WT cells, phenformin lowers calcium entry following thapsigargin (TG)-induced depletion of ER stores, whereas in AMPK DKO MEF phenformin treatment did not reduce calcium entry compared to vehicle-treated DKO controls (Figure 7E). Furthermore, in AMPK-deficient cells pre-treated with phenformin, the initial peak induced by TG treatment (that is representative of initial ER calcium stores) is severely blunted in comparison to the vehicle-treated control. Taken together, these data indicate that AMPK has a protective effect over ER calcium stores, maintaining homeostatic levels to avoid ER stress and in prolonged situations, apoptosis. To further extend and support this hypothesis, the same measurements were repeated in the absence of TG treatment to isolate the effects of biguanide treatment on cellular calcium levels. Following treatment with phenformin for 30 min, wild-type cells have a very low response to addition of extracellular calcium at a physiologically relevant concentration of 2 mM, while AMPKα1/2-deficient cells have significant calcium intake, further supporting a role of AMPKs protective effects over cellular calcium homeostasis (Figure 7F). Control traces demonstrate that in untreated WT or AMPK DKO cells, switching the media alone did not trigger calcium influx nor did continuous treatment with phenformin in the presence of calcium-containing media. (Figures 7G and 7H, respectively). Thus, in AMPK WT cells, phenformin treatment induces STIM1/2 phosphorylation and correlated with an inhibition of SOCE, whereas in AMPK-deficient cells, STIM1/2 are not phosphorylated after phenformin treatment, and SOCE is still observed. Collectively, our data indicate that AMPK activation suppresses or terminates store-operated calcium entry, and in cells lacking AMPK, sustained induction of calcium entry persists in an unregulated manner.
DISCUSSION
The need for technologies to annotate and discover signal transduction axes has been long recognized, although investigation of in vivo signaling networks has proven technically challenging. Our platform addresses this need by combining pharmacologic and genetic perturbations with quantitative proteomics to identify and quantify signaling pathway components in whole tissues. By incorporating protein behavior into meaningful topographical heatmaps, the simultaneous visualization of temporal relationships of a given protein across both genetic and pharmacological conditions and the contextual relationships become evident. Here, we apply this approach to identify and quantify regulatory phosphorylation events in the liver in response to metformin administration. 14-3-3 binding is one of the best established mechanisms by which a single kinase can inhibit a diverse set of downstream targets, all of different form and function (Pennington et al., 2018). We additionally can assign a rough estimate (~44%) of how much of the acute biochemical phosphorylation events following metformin relate to the LKB1/AMPK axis (Figure 4A). This finding simultaneously implies there are other stress-stimulated kinases that are activated following metformin treatment. Our discovery that PKD and MK2 are acutely activated in parallel with AMPK, and in an LKB1-independent manner in response to metformin treatment, is quite unexpected, although both MK2 and PKD are known to be activated by oxidative stress (Cobbaut et al., 2017; Storz, 2007; Tian et al., 2014). Future studies using CRISPR and conditional knockout mice for PKD isoforms and MK isoforms can be utilized to assess the role of these kinases in the response to metformin and what aspects of metformin action require each stress-responsive kinase.
The discovery that Rabep1 (Rapaptin-5) is a direct substrate of AMPK (Figure 5), and that its interactor Rabex-5/RabGef1 may also be one (Figure 5A), indicates a potential role for AMPK in Rab-5-dependent endocytosis, which is notably important in autophagy (Dou et al., 2013). Rab5 directly binds the Vps15 component of the Vps34/Beclin complex (Christoforidis et al., 1999; Salamon et al., 2015) to promote autophagosome formation (Christoforidis et al., 1999). The phosphorylation of Rabep1 Ser407 has previously been shown to stimulate its function; indeed, a Ser407Asp phospho-mimetic mutant exhibited enhanced integrin recycling, in contrast to the Ser407Ala mutant that mimicked RNAi-induced loss of Rabep1 (Christoforides et al., 2012). Assuming AMPK phosphorylation of Ser407 has a similar effect as PKD-induced phosphorylation of Ser407, this suggests AMPK may enhance Rabep1/Rapaptin-5 recruitment of the Beclin-Vps34 complex to promote autophagosome formation. Given that AMPK has been reported to directly phosphorylate Vps34 and Beclin (Kim et al., 2013), as well as their upstream regulator ULK1/Atg1 (Egan et al., 2011; Kim et al., 2011; Russell et al., 2014), the potential involvement of AMPK in controlling endosome and autophagosome recycling via Rabep1 and/or Rabex-5/RabGEF1 will be an area for further investigation. The shared regulation of the same phosphorylation site in downstream substrates by PKD and AMPK in response to unique and overlapping stress stimuli (e.g., Ser407 in Rabep1) will be an interesting area for future studies.
The suppression of store-operated calcium entry via direct phosphorylation of STIM1 and STIM2 by AMPK is a very unexpected direct connection between energy homeostasis, the response to mitochondrial insults, and the control of intracellular calcium. STIM1 and STIM2 act as direct sensors for the level of Ca2+ stored in the endoplasmic reticulum, so the suppression of STIM1/2 function by AMPK activation allows the energy state of the cell to recalibrate the sensitivity of this branch of calcium sensing machinery. Given that AMPK is rapidly activated in response to nearly all known mitochondrial inhibitors (Toyama et al., 2016)—including metformin (El-Mir et al., 2000; Owen et al., 2000; Wheaton et al., 2014)—the suppression of SOCE by AMPK indicates cross-talk between mitochondrial damage and the ability of the ER to actively restore calcium levels via SOCE. The regulation of SOCE by AMPK phosphorylation of STIM1 and STIM2 provides a molecular basis for previous observations that energy depletion following mitochondrial depolarization resulted in defective STIM1 activation and resultant inhibition of SOCE (Singaravelu et al., 2011). In addition, AICAR was previously shown to result in STIM1 phosphorylation and inhibition of SOCE, although it was hypothesized this was due to AMPK-dependent activation of the p38 MAPK. Furthermore knockdown of AMPKα1 enhanced SOCE induced by thrombin (Sundivakkam et al., 2013), consistent with our findings here.
AMPK itself is additionally activated by CAMKK2 following transient cytoplasmic calcium flux that accompanies hormonal signaling (Anderson et al., 2008; Tamás et al., 2006). CAMKK2 is the closest kinase homolog to LKB1, so it is unsurprising that CAMKK2 and LKB1 co-evolved to phosphorylate the same residue in the same substrate, however, the distinct conditions and kinetics of AMPK activation by calcium (rapid and transient) and by energetic stress (rapid and sustained) suggests the direct connection of AMPK to downstream regulators of calcium homeostasis mediates a feedback loop within the AMPK pathway. It will be important for future studies to define the extent of STIM1/2 phosphorylation by AMPK following hormones versus metabolic stresses and to determine whether STIM1 or STIM2 are spatially or temporally connected to specific pools of AMPK (i.e., AMPK isoforms or specific posttranslational modified AMPK populations). Functionally, we show that in the absence of AMPK, intracellular calcium levels are destabilized, especially in response to biguanides, and the consequence of this regulation of calcium homeostasis on hepatic metabolism, metabolic dysfunction, and diabetes merits more investigation. STIM1 deficiency also causes effects on mitochondrial morphology (Henke et al., 2012), so the nexus of AMPK, mitochondria, and ER function (Singaravelu et al., 2011) suggests an unappreciated broad overarching layer to the mechanistic details of how AMPK maintains cellular homeostasis following metabolic stress. A very recent study suggests that the STIM2 plays a specific role in recruiting CAMKK2 to AMPK (Chauhan et al., 2019), so future studies to examine the temporal regulation of STIM1/2 and CAMKK2 localization and phosphorylation following metformin treatment in hepatocytes is warranted. While this manuscript was in revision, a phospho-proteomics of exercise across species identified Ser257 and Ser521 of STIM1 as being induced by AMPK in all contexts and further detailed their roles in SOCE (Nelson et al., 2019). Future comparisons of the phospho-proteomics of response to exercise versus metformin may reveal additional interesting common nodes between these two broadly beneficial interventions.
In summary, our approach constitutes a robust and versatile discovery platform for the interrogation of in vivo signaling dynamics enabling the identification and quantification of substrates and molecular components in the most relevant physiological setting. Our findings reveal unexpected biochemical processes controlled by metformin and other biguanides, and furthers the understanding of the interplay between mitochondrial perturbations and calcium homeostasis that will be important to investigate further as metformin continues to be investigated as an anti-cancer and anti-aging therapy, in addition to its widespread use as the frontline treatment for type 2 diabetes.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Reuben Shaw (shaw@salk.edu). All plasmids generated in this study have been deposited to Addgene. All unique / stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Cell-lines
All cell lines were grown at 37°C maintained in an atmosphere of 5%CO2. Cells were grown in Dulbecco’s modified Eagles medium (DMEM) with 10% fetal bovine serum (Hyclone) and generated stable cell-lines were maintained under continuous antibiotic selection. Transgenic expressing STIM1 wt and S521A cells were generated by stable infection of lentivirus expressing cDNA of interest and puromycin resistance (pLenti-PGK-STIM1-(WT or S521A)-puro). Transient expression of Rabep1 WT and S407A in HEK293T was achieved through transfection of a 50% confluent dish of HEK293T with 1ug of plasmid of interest using lipofectamine 2000 (pCL-neo-Myc-Rabep1-(WT or S407A). Transfected cells collected 24 hr post and normalized by protein amount. All AMPK activators used in cell culture dissolved in DMEM +10% FBS except 991, which was reconstituted in DMSO. Thapsigargin, TPA and Gö6976 all dissolved in DMSO.
SILAM Labeling
FVB Mice following weaning were fed a diet designed in collaboration with Harlan Labs devoid of all protein content except from spirulina grown on a 15N source for 10 weeks as previously described. Animals were sacrificed through cervical dislocation and livers harvested, snap frozen prior in liquid nitrogen and stored at −80°C prior to homogenization.
Hepatic Deletion of LKB1 or AMPK and administration of Metformin or MK8722
Liver specific adenoviral-Cre-recombinase mediated deletion in Stk11lox/lox mice (Shaw et al., 2005) was done by tail vein injection of 1 × 109 PFUs/mouse in 8-week-old males and placed into separate cages for the remainder of the experiment. 9 days post-injection animals were fasted for 16 hours, and then reefed for 4 hours when mice were injected intraperitoneally with 250-mg/kg metformin (or 100mg/kg Phenformin when noted) in 0.9% saline for 2 hours or with equal volume of 0.9% saline vehicle. For the MK8722 experiment in Figure 6E, wild-type controls and L-AMPK-DKO mice were fasted for 16 hours, and then refed for 1 hour prior to receiving drug treatment. 30mg/kg MK8722 (Merck), a direct AMPK activator, or equal volume of vehicle were given to the mice by oral gavage for a total of 4 hours treatment before sacrificing the animals. Liver samples were collected and processed as described above. Following 2-hour treatment, mice were sacrificed by cervical dislocation and livers snap frozen in liquid nitrogen and stored at −80°C till ready for homogenization. Homozygous AMPKa1fl/fl and AMPKa2fl/fl mice previously described (Howell et al., 2017) on an FVB genetic background were bred with or without albumin-CreERT2 expression (Imai et al., 2000) were crossed to generate wild-type controls (AMPKa1fl/fl/a2fl/fl) or inducible liver-specific deletion of both catalytic subunits of AMPK (L-AMPK-DKO). In some experiments validating hits from the screen, Stk11lox/lox mice crossed to albumin-CreERT2 were used to create a second model of inducible LKB1 deletion in the adult mouse liver. The final cohorts of L-LKB1 or L-AMPK-DKO were generated by tamoxifen (1 mg) treatment every other day for a total of 3 i.p. injections. Experiments, or primary hepatocyte isolation, were carried out approximately 2 weeks post-tamoxifen injection. All animal care and treatments were in accordance with the Salk Institute guidelines for the care and use of animals (IACUC protocol 08-045). Male mice were used for all SILAM studies and mice of both genders were used for primary hepatocyte generation.
Primary hepatocytes
AMPKa1fl/fla2fl/fl mice ± Albumin-CreERT2 expression received tamoxifen injections every other day for a total of 3 injections (1mg/mouse/injection). Primary hepatocytes were isolated about 2 weeks later by first perfusing livers with Hank’s balanced salt solution (HBSS: 5.4 mM KCl, 0.44mM KH2 PO4, 138mM NaCl, 4.2 mM NaHCO3, 0.34 mM Na2HPO4, 5.6 mM glucose, 55 mM HEPES, 0.6 mM EGTA, pH 7.4), followed by collagenase buffer (HBSS without EGTA and supplemented with 5mM CaCl2 and 0.025% Collagenase (Sigma C5138)). Trypan blue exclusion test was used to assess cell viability. Cells were resuspended in DMEM (Corning 10-017), 5% FBS and penicillin/streptomycin and 1.6-1.8 million cells plated in TPP p60 plates. Media was changed after 4h to DMEM with antibiotics but lacking FBS and experiments were performed the next day. Cells were treated with 10nM insulin (Sigma I5500) for 20min before metformin (Sigma Phr1084) or thapsigargin treatments.
METHOD DETAILS
Antibodies and reagents
Cell Signaling Antibodies used at 1:1000 in 5% BSA in TBS-T: P-AMPKα Thr172 (#2535), AMPK α1/2 (#2532), P-ACC Ser79 (#3661), ACC (#3662), P-4EBP1 Ser65 (#9452), 4EBP1 (#9452), GAPDH (#5174), LKB1 (#3047), Stim1 (#5668), Stim2 (#4917), AMPK pMOTIF (5759), Myc (#2272), Rabex5 (#7622), P-PKD Ser744/748 (#2054), P-PKD Ser916 (#2051), PKD (#2052), P-JNK Thr183/Tyr185 (#4668), JNK (#9252), GST (#2622), 14-3-3ζ/δ (#7413), P-Raptor Ser792 (#2083), Raptor (#2280). Additional Cell Signaling reagents used within the study: Myc-Sepharose (#3400), Normal Rabbit IgG (#2729) and TPA (#4174). P-Ser521 Stim1 antibody was produced specially by Cell Signaling Technology and used at 1:1000 in 5% BSA in TBS-T. BD Biosciences Antibodies used at 1:1000 in 5% BSA in TBS-T: Rabaptin-5 (#610676). Proteintech Antibodies used: Stim2 for Immuno-precipitation (#21192), Stim1 for Immunoprecipitation (#11565), GOT1 for Immuno-blot (1:500 in 5% BSA in TBS-T) (#14886). Antibody for GCK from Santa Cruz Biotechnology (#7098) used at 1:200 in 5% BSA in TBS-T. Antibody for SDS from Abcam (#ab68536) used at 1:400 in 5% BSA in TBS-T. Antibody for TMEM24 from Aviva Systems Biology (#ARP47080) and used at 1:800 in 5% BSA in TBS-T. Actin (A5441), and Flag polyclonal (F4725) were purchased from Sigma and used at 1:5000 in 5% BSA in TBS-T. Metformin hydrochloride was obtained from Sigma (PHR1084). Dimethyl Sulfoxide (DMSO) was obtained from Sigma (D2660). AICAR was obtained from Toronto Research Chemicals Inc. (A611700) and dissolved in DMEM (Thermo scientific) with 10% FBS (GIBCO-Life Technologies). Gö6976 was obtained from EMD Biosciences and dissolved in DMSO. Thapsigargin was obtained from Life Technologies (T7459) and dissolved in DMSO. Phenformin hydrochloride was obtained from Sigma (P7045) and dissolved in DMEM (Thermo scientific) with 10% FBS (GIBCO-Life Technologies). MK-8722 was obtained from Glixx Labs (GLXC-11455).
Cross-linking recombinant 14-3-3ζ-GST and GST to Glutathione Sepharose
Recombinant GST or GST-14-3-3ζ were produced in E. coli as previously described (Yaffe et al., 1997) then purified on glutathione Sepharose and eluted with free reduced glutathione. Following purification, recombinant protein was cross-linked to glutathione Sepharose using Dimethyl pimelimidate-2HCl, DMP, at a theoretical titer of 1mg/ml adhering to a protocol as previously described. Following cross-linking, resin was washed twice 5 minutes each with 100 mM Glycine, pH 3.0 to remove all non-covalently linked protein, then washed 5x in phosphate buffered saline and stored and 4°C till ready for use.
Lysis of Liver Tissue
Frozen tissue was homogenized to a fine powder in an ice cold mortar packed in dry ice and incrementally equilibrated with liquid nitrogen to ensure sub-freezing temperatures. Ground tissue powder was transferred to a 5 mL culture tube equilibrated to just above freezing temperatures and ice-cold lysis buffer added (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM pyrophosphate, 50 mM NaF, 5 mM β-glycero-phosphate, 50 nM calyculin A, 1 mM Na3VO4, Roche complete protease inhibitors) on ice for 30 s using a tissue homogenizer. Homogenized lysates were then placed on a rocker at 4°C for 15 minutes then spun in a bench-top centrifuge for 15 minutes at 18,000 x g to clear homogenate of insoluble fraction and lipids. Following centrifugation, soluble proteome was removed avoiding lipid layer and transferred to a clean micro-centrifuge tube and lysates quantified by BCA assay (Pierce-Thermo-Scientific).
14-3-3ζ-GST and GST enrichments
Following homogenization and quantification soluble proteomes of experimental and 15N labeled liver lysates were mixed 1:1 (1.5mg each, 3mg total) and pre-cleared with glutathione Sepharose (500μl bed-volume) for 2 hours at 4°C. After pre-clear, resin was separated from supernatant by centrifugation for 5 minutes at 500 × g at 4°C and supernatant moved to a clean tube without disturbing bed volume of resin, resin discarded. Next, cross-linked 14-3-3ζ-GST or GST Sepharose was equilibrated 3 times in ice cold lysis buffer, then added to the pre-cleared supernatant at a theoretical ratio of 1:150 (40ml at a titer of 1mg/ml) and incubated on a rocker at 4°C overnight. Following incubation, beads were separated from the supernatant by centrifugation for 5 minutes at 500 × g at 4°C and supernatant transferred to a clean tube and stored at −80°C for later use. Resulting resin was then washed 3 times in 1ml ice-cold lysis buffer with centrifugation as above between each step. Following third wash and centrifugation, enriched proteins were eluted off resin by addition of 300 μL SDS containing boiling buffer (50 mM Tris, pH 7.5, 5% SDS, 50mM DTT) and placed on a heating block set to 90°C for 5 minutes. Supernatant containing eluted proteins were then separated from resin by centrifugation for 10 minutes at 500 × g at room temperature and supernatant transferred to a clean micro-centrifuge tube. Supernatant containing eluted proteins were next precipitated out of solution by addition of Trichloroacetic Acid (Sigma) to a final concentration of 25% and placed on ice overnight to precipitate enriched, eluted proteins out of solution. After overnight incubation, precipitated proteins were separated from supernatant by centrifugation for 30 min at 18,000 × g at 4°C. The supernatant was removed by aspiration and the pellet washed in 500 μL ice-cold acetoe twice with centrifugation for 10 minutes at 18,000 × g at 4°C following each wash. Following the second wash the resulting protein pellet was air-dried and stored at −80°C till all samples ready for preparation and digestion for proteomic analysis. For un-enriched samples 100 μg total protein (50 μg 15N labeled Standard and 50 μg unlabeled 14N experimental) was submitted to the same precipitation protocol as above.
Preparation and Digestion of Enriched, Eluted Proteins
Dried pellets were re-suspended in 60 μl of 100 mM Tris, pH 8.5, 8 M urea and reduced with 5 mM Tris-carboxyethyl phosphine (TCEP) for 20 minutes at room temperature. Following reduction of disulfide bonds with TCEP the denatured protein mix was alkylated with 10 mM Iodoacetamide (IAA) for 15 minutes in the dark at room temperature. After reduction and alkylation of disulfide bonds the denatured protein mix was diluted to a final urea concentration of 2 M and supplemented with a final concentration of Calcium Chloride of 1 mM and 5 μg of sequencing grade Trypsin (Promega) and placed in the dark on a thermal mixer (Eppendorf) set to 37°C and shaking at 800 RPM for 16 hours. Following digestion samples were stored at −80°C till ready for analysis.
Multidimensional Protein Identification Technology
When ready for analysis, corresponding samples were thawed at room temperature and acidified with formic acid to a final concentration of 5% and centrifuged at 13,200 RPM at 4°C for 15 minutes. Supernatant was then moved to a clean micro-centrifuge tube and ready for analysis. As described previously, peptide mixtures were then loaded onto a 250μm inner diameter fused-silica undeactivated microcapillary column (Agilent Technologies) fritted on one end with polymerized kasil packed first with 2.5cm of 5mm strong cation exchange material (Partisphere SCX, Whatman) followed by 2.5cm of 5μm C18 reverse phase (Aqua, Phenomenex). Following loading on a high-pressure bomb using helium as previously described, the loaded microcapillary columns were joined to 100 μm inner diameter fused-silica microcapillary columns with 5μm tips (Sutter Instruments P-2000 laser puller) packed with 15cm of 3 μm C18 reverse phase through a zero volume union with frit sleeves (Upchurch, western analytical) and installed in-line with a quaternary Agilent 1100 series HPLC pump. Fully automated 12-step chromatography runs were carried out. The flow rate was set to 250 nl·min−1. Three different elution buffers were utilized: Buffer A (5%ACN, 0.1% Formic Acid), Buffer B (80%ACN, 0.1% Formic Acid), and Buffer C (500 mM Ammonium Acetate, 5% CAN, 0.1% Formic Acid). Application of 2.5 kV distal voltage enabled electrospray of the eluted peptides directly into a LTQ Velos Pro Orbitrap Elite ion trap mass spectrometer equipped with a nano-LC based electrospray ionization source (Thermo Fisher Scientific). The mass spectrometer was operated in data-dependent mode using a TOP-20 strategy. Survey MS scans were acquired in the Orbitrap (R = 250,000 FWHM) in a mass range of 300 to 1600 m/z range, and up to 20 of the most abundant ions per full MS scan were selected and fragmented by collision-induced dissociation (normalized collision energy set equal to 35, activation Q equal to 0.25) and analyzed in the linear ion trap. Ion target values used were 1,000,000 (or 250 ms maximum fill time) for full MS survey scans and 10,000 (or 50 ms maximum fill time) for subsequent MS/MS scans. The instrument for MS/MS analysis rejected ions possessing a charge state of 1 or unknown. Dynamic exclusion was enabled with a repeat count = 1, exclusion duration of 30 s, list size of 500, and mass window of −0.51 m/z, +1.51 m/z with an expiration count set to 3 and signal-to-noise threshold of 3. Mass spectrometer scans functions and HPLC solvent gradients were centrally controlled through the Xcalibur data platform provided by the manufacturer (Thermo Fisher Scientific).
Protein Identification and Analysis
Acquired MS/MS datasets were processed to extract MS1 and MS2 scans using the software package RawXtract version 1.9.9.2 as previously described. Next, extracted files were searched against a mouse database from IPI/EPI containing isoform information. The reversed protein sequences of the target database were used as a decoy database. The ProLUCID algorithm was used to match MS/MS spectra to peptides. The search criteria were set as follows: 2 peptides required; unlimited missed cleavages were allowed; carbamidomethylation (C) was set as a fixed modification per protein, including half tryptic peptides. We only require half tryptic peptides as literature precedent has widely demonstrated that tryptic proteolytic digestion frequently generates non-tryptic peptides due to chymotrypsin impurity, self cleavage of trypsin to generate pseudotrypsin which generates chymotrypsin activity, or potential insource fragmentation frequently observed during mass spectrometric analysis and potentially due to endogenous proteases contained within the liver lysates to be interrogated that may retain activity even in the presence of protease inhibitor cocktails. Following peptide identification the output matches were parsed and filtered using DTASelect version 2, targeting a protein false discovery rate of 1% and a delta-mass requirement of less than or equal to 5 parts per million. Following filtering peptides were quantified at the MS1 precursor level using the Census algorithm.
Ca2+ measurement methods
Fura loading
MEF cells were plated onto 8-well glass chamber slides (Lab-Tek II, 155449) one day prior to imaging. On the day of experiments, cell media (DMEM + 10% FBS) were quickly mixed with freshly thawed Fura, 2AM (ThermoFisher, F1201, 1mM in 100% DMSO) to a final concentration of 5 μM, and HEPES buffer (pH 7.4) to a final concentration of 20 mM. Cells were kept in Fura-containing media in the dark at room temperature for approximately 50 min. Cells were subsequently washed once in Ca2+ free imaging buffer (HBSS) containing 0.2 mM EGTA and stored in the same buffer for an additional 10 min in the dark. Imaging system: Ratiometric imaging of Fura-loaded cells was carried out by using D340Xv2 and D380Xv2 excitation filters (Chroma), a T400lp dichroic mirror (Chroma), and an ET530/30x emission filter (Chroma). Imaging was performed on a Nikon Ti Eclipse inverted microscope equipped with a CFI S Fluor 40x/1.3 oil immersion objective, QIClick CCD camera, and Tokai Hit stage-top incubator. Dual-channel 340/380 imaging was performed using identical camera settings (typically electrical gain at 30 and integration time at 100 ms) at 37°C with a 10 s interval for 20 min. Acquired data were analyzed using Fiji Cookbook. Background subtraction was performed prior to evoking Ratio Profiler from Image Intensity Processing in Fiji. Ratio profiles were obtained from individually outlined cells and combined for statistical analysis. Ca2+ imaging of thapsigargin (Tg)-triggered Ca2+release and SOCE: Fura-loaded cells were imaged in 250 ul of Ca2+ free imaging buffer for 1 min. Cells were promptly treated with 2 mM Tg (addition of 50 ul 6 × Tg solution). Tg-triggered ER Ca2+ release was monitored for 5 min. At 6 min, cells were spiked with 2 mM Ca2+ (addition of 50 ul 7 × Ca2+ solution), and SOCE was subsequently monitored for 14 min.
Ca2+ imaging of phenformin-treated cells
2 mM phenformin was added to cell media at 37°C for 30 min. Fura loading and Ca2+ imaging were proceeded as previously described, except for the addition of 2 mM phenformin to all media, imaging buffer, and Tg/ Ca2+ solutions. For imaging SOCE in response to the phenformin treatment, Tg treatment at 1 min was skipped for both WT and DKO cells.
Control experiments
To monitor cell response to Ca2+ addition without drug treatment, Fura-loaded WT and DKO cells were imaged in 300 ul Ca2+ free imaging buffer for 6 min. At 6 min, cells were spiked with 2 mM Ca2+ (addition of 50 ul 7 × Ca2+ solution) and continuously monitored for a total of 20 min. To monitor the phenformin effect on intracellular Ca2+ levels of cells incubated in Ca2+ containing buffer, Fura-loaded WT and DKO cells were imaged in 2 mM Ca2+ imaging buffer containing 2 mM phenformin at a 1-min interval for 60 min.
QUANTIFICATION AND STATISTICAL ANALYSIS
Post-processing of Acquired Data prior to Clustering
Following peptide quantification using the Census algorithm, background was filtered from each biological condition by utilizing the quant compare algorithm embedded in the IP2 pipeline in a ratio-of-ratio approach and statistical analysis based on all peptide ratios for a given protein identified. Following creation of exclusion lists using the GST controls within each biological condition tested, quant compare files across conditions were parsed using the derived exclusion lists to generate 6 processed files for further analysis. Following background subtraction using the exclusion lists derived from the GST alone controls, pairwise comparison files were further filtered by requiring an anova pValue of less than or equal to 0.05 for proteins which contained quantitative values in at least 3 of the 4 biological conditions tested. For proteins that only possessed quantitative values for 2 of the 4 conditions or less, the proteins were retained within the list as long as the conditions that did contain quantitative values adhere to the same statistical measure. Following processing this filtering scheme produced 1022 proteins across the four biological conditions tested with high statistical confidence.
Normalization and Clustering of Statistically Significant Proteins in Heatmaps
Following filtering of background contaminants and statistical means, resulting proteins were normalized at the quantification values and then clustered using a hierarchical clustering algorithm.
-
Normalization
For each protein, the quantification values were normalized according to their summation:
where NQ is the normalized quantification value and Q is the original quantification value. Note that during the normalization procedure only the quantification values that are not quantified are considered. Clustering
A two-stage hierarchical clustering algorithm was used to cluster proteins according to quantification values:
Clustering in three groups - Proteins were categorized into three groups according to the quantification values (with nulls, with zeros, and with full values) and then clustered separately using Euclidean distance as the distance metric. We used 0.1, 0.1, and 0.07 as the distance thresholds when clustering these three groups, respectively. In each round during the clustering, a pair of quantification value vectors with the minimal Euclidian distance were selected and merged as a new average vector if their distance is below the given threshold.
Merging clusters - Step (a) generated a number of clusters, which would be merged as larger clusters till only one cluster remained in this merging step. Similarly to the clustering rules used in Step (a), two clusters with the minimal Euclidian distance were merged as a new cluster with a vector of average quantification values.
The normalization and clustering rules described above were applied on 14-3-3 data. The resultant heatmap and dendrogram were generated in parallel with corresponding normalized lysate quantification values to illustrate the protein abundance variance.
DATA AND CODE AVAILABILITY
All derived MS/MS data is deposited on MASSive (MSV000084429) and ProteomeXchange (PXD015733). The unpublished code generated during this study will be made freely available and accessible on: http://fields.scripps.edu/downloads.php
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| P-AMPKa Thr172 | Cell Signaling Technology | 2535; RRID:AB_331250 |
| AMPK α1/2 | Cell Signaling Technology | 2532: RRID:AB_330331 |
| P-ACC Ser79 | Cell Signaling Technology | 3661: RRID:AB_330337 |
| P-4EBP1 Ser65 | Cell Signaling Technology | 9451; RRID:AB_330947 |
| 4EBP1 | Cell Signaling Technology | 9452; RRID:AB_331692 |
| ACC | Cell Signaling Technology | 3662; RRID:AB_2219400 |
| GAPDH | Cell Signaling Technology | 5174; RRID:AB_10622025 |
| P-Raptor Ser792 | Cell Signaling Technology | 2083; RRID:AB_2249475 |
| Raptor | Cell Signaling Technology | 2280; RRID:AB_561245 |
| P-RSK Thr359/Ser363 | Cell Signaling Technology | 9344; RRID:AB_331650 |
| P-Chk1 Ser345 | Cell Signaling Technology | 2348; RRID:AB_331212 |
| P-PKD Ser744/748 | Cell Signaling Technology | 2054; RRID:AB_2172539 |
| P-PKD Ser916 | Cell Signaling Technology | 2051; RRID:AB_330841 |
| PKD | Cell Signaling Technology | 2052; RRID:AB_2268946 |
| P-AKT Ser473 | Cell Signaling Technology | 4060; RRID:AB_2315049 |
| P-AKT Thr308 | Cell Signaling Technology | 4056; RRID:AB_331163 |
| AKT (pan) | Cell Signaling Technology | 4691; RRID:AB_915783 |
| P-MAPKAPK-2 Thr334 | Cell Signaling Technology | 3007; RRID:AB_490936 |
| MAPKAPK2 | Cell Signaling Technology | 12155; RRID:AB_2797831 |
| Beta-Actin | Sigma Aldrich | A5441; RRID:AB_476744 |
| LKB1 | Cell Signaling Technology | 3047; RRID:AB_2198327 |
| P-ULK1 Ser555 | Cell Signaling Technology | 5869; RRID:AB_10707365 |
| Rabep1 (Rabaptin-5) | BD Biosciences | 610676; RRID:AB_398003 |
| STIM1 | Cell Signaling Technology | 5668; RRID:AB_10828699 |
| STIM2 | Cell Signaling Technology | 4917; RRID:AB_2198021 |
| AMPK pMOTIF | Cell Signaling Technology | 5759; RRID:AB_10949320 |
| Myc tag | Cell Signaling Technology | 2272; RRID:AB_10692100 |
| Rabex5 | Cell Signaling Technology | 7622; RRID:AB_10828937 |
| P-JNK Thr183/Tyr185 | Cell Signaling Technology | 4668; RRID:AB_823588 |
| JNK | Cell Signaling Technology | 9252; RRID:AB_2250373 |
| GST | Cell Signaling Technology | 2622; RRID:AB_331670 |
| 14-3-3z/d | Cell Signaling Technology | 7413; RRID:AB_10950820 |
| STIM2 (IP) | Droteintech | 21192; RRID:AB_10734322 |
| Flag | Sigma Aldrich | F7425; RRID:AB_439687 |
| STIM1 (IP) | Proteintech | 11565; RRID:AB_2302808 |
| P-STIM1 Ser521 | Cell Signaling Technology | This Study |
| GCK | Santa Cruz Biotechnology | 7908; RRID:AB_2107620 |
| TMEM24 | Aviva Systems Biology | ARP47080; RRID:AB_1294955 |
| GOT1 | Proteintech | 14886; RRID:AB_2113630 |
| SDS | Abcam | ab68536; RRID:AB_1310659 |
| Normal Rabbit IgG | Cell Signaling Technology | 2729; RRID:AB_1031062 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Metformin Hydrochloride | Sigma Aldrich | PHR1084 |
| Dimethyl Sulfoxide (DMSO) | Sigma Aldrich | D2660 |
| AICAR | Toronto Research Chemicals Inc. | A611700 |
| Phenformin Hydrochloride | Sigma Aldrich | P7045 |
| 991 (Ex229) | Selleck chemicals Inc. | S8654 |
| MK-8722 | Glixx Labs | GLXC-11455 |
| Gö6976 | EMD Millipore | 365250 |
| TPA | Cell Signaling Technology | 4174 |
| Myc-Sepharose | Cell Signaling Technology | 3400 |
| Thapsigargin (TG) | Life Technologies | T7459 |
| Fura2-AM | ThermoFisher | F1201 |
| insulin | Sigma Aldrich | I5500 |
| Collagenase | Sigma Aldrich | C5138 |
| Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) | Sigma Aldrich | C4706 |
| lodoacetamide | Sigma Aldrich | I1149 |
| Trichloroacetic acid solution (TCA) | Sigma Aldrich | T0699 |
| Sequencing Grade Modified Trypsin | Promega Corporation | V5111 |
| ANTI-FLAG M2 Affinity Gel | Sigma Aldrich | A2220 |
| Glutathione Sepharose 4B | GE life sciences | 17075601 |
| DMP (dimethyl pimelimidate) | Thermo Scientific | 21667 |
| Ethanolamine | Sigma Aldrich | 411000 |
| Sodium tetraborate decahydrate | Sigma Aldrich | S9640 |
| calyculin A | EMD Millipore | 208851 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich (Roche) | 04693132001 |
| PARTISPHERE Analytical HPLC Columns | Whatman | 4621-0507 |
| Aqua® 3 μm C18 125 Å | Phenomenex | 04A-4311 |
| Aqua® 5 μm C18 125 Å | Phenomenex | 04A-4299 |
| Undeactivated Fused Silica 0.25 mm ID, 0.35 mm OD | Agilent Technologies | 160-2250-10 |
| Protein A - Sepharose 4B | Life Technologies | 101042 |
| CAPILLARY 100UMX363UM | Polymicro Technologies (Molex) | 1068150023 |
| Deposited Data | ||
| MS/MS Data | This Paper | MassIVE: MSV000084429 |
| MS/MS Data | This Paper | ProteomeXchange: PXD015733 |
| Experimental Models: Cell Lines | ||
| AMPKa1fl/fl/a2fl/fl MEFs | Toyama et al., 2016 | N/A |
| HEK293T | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mice: LKB1fl/fl on FVB | Shaw et al., 2005 | N/A |
| Mice: LKB1fl/fl; Alb-CreERT2 on FVB | Mihaylova and Shaw, 2011 | N/A |
| Mice: AMPKa1fl/fl/a2fl/fl;Alb-CreERT2 on FVB | Howell et al., 2017 | N/A |
| Mice: FVB/NJ | Jackson Laboratories | 001800 |
| Mice: AMPKa1fl/fl/a2fl/fl on FVB | Howell et al., 2017 | N/A |
| Recombinant DNA | ||
| pGEX-4T1-14-3-3 zeta GST | Yaffe et al., 1997 | Addgene 13278 |
| pGEX-4T-1 | GE Healthcare (Sigma Aldrich) | GE28-9545-49 |
| pLenti PGK Puro DEST | Campeau et al., 2009 | Addgene 19068 |
| pLenti PGK-C-FLAG-STIM1 WT Puro | This Paper | N/A |
| pLenti PGK-C-FLAG-STIM1 S521A Puro | This Paper | N/A |
| pCL-neo-Myc-Rabaptin-5 WT | Christoforides et al., 2012 | N/A |
| pCL-neo-Myc-Rabaptin-5 S407A | Christoforides et al., 2012 | N/A |
| Software and Algorithms | ||
| RAW Xtractor (v. 1.9.9.2) | http://fields.scripps.edu/downloads.php | |
| ProLuCID algorithm | N/A | http://fields.scripps.edu/downloads.php |
| Census | The Scripps Research Institute | http://fields.scripps.edu/census |
| Unpublished code | The Scripps Research Institute | http://fields.scripps.edu/downloads.php |
| Integrated Proteomics Pipeline - IP2 | Park et al., 2008 | http://www.integratedproteomics.com/ |
| European Bioinformatic Institute (IPI) mouse protein database | N/A | ftp://ftp.ebi.ac.uk/pub/databases/IPI |
| Other | ||
| Mouse Express (15N, 98%) MOUSE FEED KIT IRRADIATED PREPARED WITH SPIRULINA | Cambridge Isotope Laboratories, Inc. | MLK-SPIRULINA-N-IR |
Highlights.
Using 14-3-3 as an affinity tool reveals acute metformin-induced signaling in liver
Roughly 40% of proteins induced to bind 14-3-3 by metformin require LKB1
Metformin activates kinases PKD and MK2, independent of LKB1/AMPK
AMPK regulates store-operated calcium entry via STIM1/STIM2 phosphorylation
ACKNOWLEDGMENTS
We thank Liliana Vera and Amanda Hutchins for technical assistance with maintaining the Shaw lab mouse colony. We thank Pablo Hollstein and Claire Delahunty for critical review of the manuscript. This work was supported, in part, by NIH (R01-DK080425 and R35-CA220538 to R.J.S., P01-CA120964 to R.J.S. and L.C.C., R35 CA197588 to L.C.C., P41-GM103533 and R01-MH068770 to J.R.Y., and 1DP2GM105455-01 to B.F.L.). B.F.L. additionally thanks the Nomis Foundation, the Waitt Foundation, and the James B. Pendleton Charitable Trust for support. Further support was provided by a fellowship from the George E. Hewitt Foundation for Medical Research (K.H.) and the American Cancer Society (122123-PF-12-029-01-TBE to E.Q.T.). We thank L. Hui and G. Kasof at Cell Signaling Technology for developing the antibody to STIM1 P-Ser521 in conjunction with B.D.S. and R.J.S.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.117.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alessi DR, Sakamoto K, and Bayascas JR (2006). LKB1-dependent signaling pathways. Annu. Rev. Biochem 75, 137–163. [DOI] [PubMed] [Google Scholar]
- Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, Witters LA, Kemp BE, and Means AR (2008). Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 7, 377–388. [DOI] [PubMed] [Google Scholar]
- Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villén J, Wang B, Kim SR, Sakamoto K, et al. (2011). Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol. Cell 44, 878–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batalha IL, Lowe CR, and Roque ACA (2012). Platforms for enrichment of phosphorylated proteins and peptides in proteomics. Trends Biotechnol. 30, 100–110. [DOI] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, and Harper JW (2010). Network organization of the human autophagy system. Nature 466, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos DM (2012). The role of protein disorder in the 14-3-3 interaction network. Mol. Biosyst 8, 178–184. [DOI] [PubMed] [Google Scholar]
- Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, andWalker CL (2006). Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol 173, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Meng S, Chang E, Beckwith-Fickas K, Xiong L, Cole RN, Radovick S, Wondisford FE, and He L (2014). Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem 289, 20435–20446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan AS, Liu X, Jing J, Lee H, Yadav RK, Liu J, Zhou Y, and Gan B (2019). STIM2 interacts with AMPK and regulates calcium-induced AMPK activation. FASEB J. 33, 2957–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Synowsky S, Tinti M, and MacKintosh C (2011). The capture of phosphoproteins by 14-3-3 proteins mediates actions of insulin. Trends Endocrinol. Metab. 22, 429–436. [DOI] [PubMed] [Google Scholar]
- Christoforides C, Rainero E, Brown KK, Norman JC, and Toker A (2012). PKD controls αvβ3 integrin recycling and tumor cell invasive migration through its substrate Rabaptin-5. Dev. Cell 23, 560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis S, Miaczynska M, Ashman K, Wilm M, Zhao L, Yip SC, Waterfield MD, Backer JM, and Zerial M (1999). Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol 1, 249–252. [DOI] [PubMed] [Google Scholar]
- Cobbaut M, and Van Lint J (2018). Function and Regulation of Protein Kinase D in Oxidative Stress: A Tale of Isoforms. Oxid. Med. Cell. Longev 2018, 2138502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbaut M, Derua R, Döppler H, Lou HJ, Vandoninck S, Storz P, Turk BE, Seufferlein T, Waelkens E, Janssens V, and Van Lint J (2017). Differential regulation of PKD isoforms in oxidative stress conditions through phosphorylation of a conserved Tyr in the P+1 loop. Sci. Rep 7, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, and Meyer T (2011). Evolutionary origins of STIM1 and STIM2 within ancient Ca2+ signaling systems. Trends Cell Biol. 21, 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale S, Wilson WA, Edelman AM, and Hardie DG (1995). Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett. 361, 191–195. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Thompson CB (2012). Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döppler H, Storz P, Li J, Comb MJ, and Toker A (2005). A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J. Biol. Chem 280, 15013–15019. [DOI] [PubMed] [Google Scholar]
- Dou Z, Pan JA, Dbouk HA, Ballou LM, DeLeon JL, Fan Y, Chen JS, Liang Z, Li G, Backer JM, et al. (2013). Class IA PI3K p110β subunit promotes autophagy through Rab5 small GTPase in response to growth factor limitation. Mol. Cell 50, 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duca FA, Côté CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, and Lam TK (2015). Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med 21, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducommun S, Deak M, Sumpton D, Ford RJ, Núñez Galindo A, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, et al. (2015). Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell. Signal 27, 978–988. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Jamieson J, Marroquin L, Nadanaciva S, Billis PA, and Will Y(2008). Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol 233, 203–210. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, and Kimchi A (2012). PKD at the crossroads of necrosis and autophagy. Autophagy 8, 433–434. [DOI] [PubMed] [Google Scholar]
- El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, and Leverve X(2000). Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem 275, 223–228. [DOI] [PubMed] [Google Scholar]
- Fíla J, and Honys D (2012). Enrichment techniques employed in phospho-proteomics. Amino Acids 43, 1025–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, and Viollet B (2010). Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest 120, 2355–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. (2013). Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med 19, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, and Shaw RJ (2017). AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 66, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardino AK, Smerdon SJ, and Yaffe MB (2006). Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 143-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin. Cancer Biol 16, 173–182. [DOI] [PubMed] [Google Scholar]
- Goodwin JM, Svensson RU, Lou HJ, Winslow MM, Turk BE, and Shaw RJ (2014). An AMPK-independent signaling pathway downstream of the LKB1 tumor suppressor controls Snail1 and metastatic potential. Mol. Cell 55, 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor M, Zeöld A, Oehler S, Marobela KA, Fuchs P, Weigel G, Hardie DG, and Wiche G (2006). Plectin scaffolds recruit energy-controlling AMP-activated protein kinase (AMPK) in differentiated myofibres. J. Cell Sci 119,1864–1875. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, and Shaw RJ (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, and Alessi DR (2013). LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol. 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, and Hawley SA (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol 13, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Schaffer BE, and Brunet A (2016). AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 26, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, and Wondisford FE (2015). Metformin action: concentrations matter. Cell Metab. 21, 159–162. [DOI] [PubMed] [Google Scholar]
- Henke N, Albrecht P, Pfeiffer A, Toutzaris D, Zanger K, and Methner A (2012). Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J. Biol. Chem 287, 42042–42052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ, et al. (2015). Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 22,922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, and Rao A (2015). Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 460, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi H, Lippé R, McBride HM, Rubino M, Woodman P, Stenmark H, Rybin V, Wilm M, Ashman K, Mann M, and Zerial M (1997). A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell 90, 1149–1159. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, and Davis RJ (2016). Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol 8, a006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ, and Manning BD (2017). Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 25, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143,1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Chambon P, and Metzger D (2000). Inducible site-specific somatic mutagenesis in mouse hepatocytes. Genesis 26, 147–148. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, and Guan K-L (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Inoki K, Kim J, and Guan KL (2012). AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol 52, 381–400. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Sanosaka M, Fuchino H, Yahara Y, Kumagai A, Takemoto D, Kagawa M, Doi J, Ohta M, Tsumaki N, et al. (2015). Salt-inducible Kinase 3 Signaling Is Important for the Gluconeogenic Programs in Mouse Hepatocytes. J. Biol. Chem 290, 17879–17893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JB, Sundelin EI, Jakobsen S, Gormsen LC, Munk OL, Frøkiær J, and Jessen N (2016). [11C]-Labeled Metformin Distribution in the Liver and Small Intestine Using Dynamic Positron Emission Tomography in Mice Demonstrates Tissue-Specific Transporter Dependency. Diabetes 65,1724–1730. [DOI] [PubMed] [Google Scholar]
- Jin J, Smith FD, Stark C, Wells CD, Fawcett JP, Kulkarni S, Metalnikov P, O’Donnell P, Taylor P, Taylor L, et al. (2004). Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cyto-skeletal regulation and cellular organization. Curr. Biol 14, 1436–1450. [DOI] [PubMed] [Google Scholar]
- Johnson C, Crowther S, Stafford MJ, Campbell DG, Toth R, and MacKintosh C (2010). Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem. J 427, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, and Guan KL (2013). Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Hino S, Oue N, Asahara T, Zollo M, Yasui W, and Kikuchi A (2006). Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol. Cell. Biol 26, 898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. (2005). The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111. [DOI] [PubMed] [Google Scholar]
- Laplante M, and Sabatini DM (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS (2011). Store-operated calcium channels: new perspectives on mechanism and function. Cold Spring Harb. Perspect. Biol 3, a003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Csibi A, Yang S, Hoffman GR, Li C, Zhang E, Yu JJ, and Blenis J (2015). Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1-driven tumor cells. Proc. Natl. Acad. Sci. USA 112, E21–E29. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lippé R, Miaczynska M, Rybin V, Runge A, and Zerial M (2001). Functional synergy between Rab5 effector Rabaptin-5 and exchange factor Rabex-5 when physically associated in a complex. Mol. Biol. Cell 12,2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, and Alessi DR (2004). LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCoss MJ, Wu CC, Liu H, Sadygov R, and Yates JR 3rd. (2003). A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal. Chem 75, 6912–6921. [DOI] [PubMed] [Google Scholar]
- MacCoss MJ, Wu CC, Matthews DE, and Yates JR 3rd. (2005). Measurement of the isotope enrichment of stable isotope-labeled proteins using high-resolution mass spectra of peptides. Anal. Chem 77, 7646–7653. [DOI] [PubMed] [Google Scholar]
- Mack HI, Zheng B, Asara JM, and Thomas SM (2012). AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 8, 1197–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackintosh C (2004). Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem. J 381, 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchy DB, and Yates JR (2008). Stable Isotope Labeling of Mammals (SILAM). CSH Protoc. 2008, pdb.prot4940. [DOI] [PubMed] [Google Scholar]
- McClatchy D, and Yates J III. (2014). Stable Isotope Labeling in Mammals (SILAM) In Shotgun Proteomics, Martins-de-Souza D, ed. (Springer, New York: ), pp. 133–146. [Google Scholar]
- McClatchy DB, Dong M-Q, Wu CC, Venable JD, and Yates JR 3rd. (2007). 15N metabolic labeling of mammalian tissue with slow protein turnover. J. Proteome Res 6, 2005–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, and Shaw RJ (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol 13,1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirouse V, and Billaud M (2011). The LKB1/AMPK polarity pathway. FEBS Lett. 585, 981–985. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Parker BL, Burchfield JG, Hoffman NJ, Needham EJ, Cooke KC, Naim T, Sylow L, Ling NX, Francis D, et al. (2019). Phospho-proteomics reveals conserved exercise-stimulated signaling and AMPK regulation of store-operated calcium entry. EMBO J August 5, e102578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nhek S, Ngo M, Yang X, Ng MM, Field SJ, Asara JM, Ridgway ND, and Toker A (2010). Regulation of oxysterol-binding protein Golgi localization through protein kinase D-mediated phosphorylation. Mol. Biol. Cell 21, 2327–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novellasdemunt L, Tato I, Navarro-Sabate A, Ruiz-Meana M, Méndez-Lucas A, Perales JC, Garcia-Dorado D, Ventura F, Bartrons R, and Rosa JL (2013). Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) isoenzyme by amino acids. J. Biol. Chem 288, 10640–10651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MR, Doran E, and Halestrap AP (2000). Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J 348, 607–614. [PMC free article] [PubMed] [Google Scholar]
- Park SK, Venable JD, Xu T, and Yates JR 3rd. (2008). A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 5, 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SKR, Aslanian A, McClatchy DB, Han X, Shah H, Singh M, Rauniyar N, Moresco JJ, Pinto AFM, Diedrich JK, et al. (2014). Census 2: isobaric labeling data analysis. Bioinformatics 30, 2208–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K, Foretz M, Marion A, Campbell DG, Gourlay R, Boudaba N, Tournier E, Titchenell P, Peggie M, Deak M, et al. (2014). The LKB1-salt-inducible kinase pathway functions as a key gluconeogenic suppressor in the liver. Nat. Commun 5, 4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington KL, Chan TY, Torres MP, and Andersen JL (2018). The dynamic and stress-adaptive signaling hub of 14-3-3: emerging mechanisms of regulation and context-dependent protein-protein interactions. Oncogene 37, 5587–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccia S, Ordan M, Cohen I, Bendetz-Nezer S, and Seger R (2017). Direct binding of MEK1 and MEK2 to AKT induces Foxo1 phosphorylation, cellular migration and metastasis. Sci. Rep 7, 43078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, and Yaffe MB (2009). Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol 21, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Yuan HX, and Guan KL (2014). Autophagy regulation by nutrient signaling. Cell Res. 24, 42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamon RS, Dbouk HA, Collado D, Lopiccolo J, Bresnick AR, and Backer JM (2015). Identification of the Rab5 binding site in p110β: assays for PI3Kβ binding to Rab5. Methods Mol. Biol 1298, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer BE, Levin RS, Hertz NT, Maures TJ, Schoof ML, Hollstein PE, Benayoun BA, Banko MR, Shaw RJ, Shokat KM, and Brunet A (2015). Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large-Scale Substrate Prediction. Cell Metab. 22, 907–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, and Shaw RJ (2013). LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23, 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, and Cantley LC (2004). The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6, 91–99. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, and Cantley LC (2005). The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaravelu K, Nelson C, Bakowski D, de Brito OM, Ng SW, Di Capite J, Powell T, Scorrano L, and Parekh AB (2011). Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. J. Biol. Chem 286, 12189–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, and Gill DL (2012). STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol 13, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P (2007). Mitochondrial ROS-radical detoxification, mediated by protein kinase D. Trends Cell Biol. 17, 13–18. [DOI] [PubMed] [Google Scholar]
- Storz P, Döppler H, and Toker A (2004). Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol. Cell. Biol 24, 2614–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundivakkam PC, Natarajan V, Malik AB, andTiruppathi C (2013). Store-operated Ca2+ entry (SOCE) induced by protease-activated receptor-1 mediates STIM1 protein phosphorylation to inhibit SOCE in endothelial cells through AMP-activated protein kinase and p38β mitogen-activated protein kinase. J. Biol. Chem 288, 17030–17041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamás P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, and Cantrell DA (2006). Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J. Exp. Med 203, 1665–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Alquraini H, Mizokami-Stout K, and MacEachern M (2016). Metformin: From Research to Clinical Practice. Endocrinol. Metab. Clin. North Am 45, 819–843. [DOI] [PubMed] [Google Scholar]
- Thingholm T, Jensen O, and Larsen M (2009). Enrichment and separation of mono- and multiply phosphorylated peptides using sequential elution from IMAC prior to mass spectrometric analysis. Methods Mol. Biol. 527, 67–78. [DOI] [PubMed] [Google Scholar]
- Tian L, Chen J, Chen M, Gui C, Zhong CQ, Hong L, Xie C, Wu X, Yang L, Ahmad V, and Han J (2014). The p38 pathway regulates oxidative stress tolerance by phosphorylation of mitochondrial protein IscU. J. Biol. Chem 289, 31856–31865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Losön OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, and Shaw RJ (2016). Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable JD, Wohlschlegel J, McClatchy DB, Park SK, and Yates JR 3rd. (2007). Relative quantification of stable isotope labeled peptides using a linear ion trap-Orbitrap hybrid mass spectrometer. Anal. Chem 79, 3056–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, and Yates JR 3rd. (2001). Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol 19, 242–247. [DOI] [PubMed] [Google Scholar]
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, and Chan- del NS (2014). Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 3, e02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, MacCoss MJ, Howell KE, Matthews DE, and Yates JR 3rd. (2004). Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal. Chem 76, 4951–4959. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, et al. (2013). Structural basis of AMPK regulation by small molecule activators. Nat. Commun 4, 3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015). ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB (2002). How do 14-3-3 proteins work?- Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 513, 53–57. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, and Cantley LC (1997). The structural basisfor 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971. [DOI] [PubMed] [Google Scholar]
- Yano T, Matsui T, Tamura A, Uji M, and Tsukita S (2013). The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J. Cell Biol 203, 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All derived MS/MS data is deposited on MASSive (MSV000084429) and ProteomeXchange (PXD015733). The unpublished code generated during this study will be made freely available and accessible on: http://fields.scripps.edu/downloads.php