

Abstract
Iron delivery to the developing brain is essential for energy and metabolic support needed for processes such as myelination and neuronal development. Iron deficiency, especially in the developing brain, can result in a number of long-term neurological deficits that persist into adulthood. There is considerable debate that excess access to iron during development may result in iron overload in the brain and subsequently predispose individuals to age-related neurodegenerative diseases. There is a significant gap in knowledge regarding how the brain acquires iron during development and how biological variables such as development, genetics and sex impact brain iron status. In this study, we used a mouse model expressing a mutant form of the iron homeostatic regulator protein HFE, (Hfe H63D), the most common gene variant in Caucasians, to determine impact of the mutation on brain iron uptake. Iron uptake was assessed by using 59Fe bound to either transferrin or H-ferritin as the iron carrier proteins. We demonstrate that at postnatal day 22, mutant mice brains take up greater amounts of iron compared to wildtype. Moreover, we introduce H-ferritin as a key protein in brain iron transport during development and identify a sex and genotype effect demonstrating female mutant mice take up more iron by transferrin while male mutant mice take up more iron from H-ferritin at PND22. Furthermore, we begin to elucidate the mechanism for uptake using immunohistochemistry to profile the regional distribution and temporal expression of transferrin receptor and Tim-2, the latter is the receptor for H-ferritin. These data demonstrate that sex and genotype have significant effects on iron uptake and that regional receptor expression may play a large role in the uptake patterns during development.
Graphical Abstract
Iron delivery to the developing brain is a critical process that is needed for essential metabolic support in processes such as myelination. Iron deficiency can result in a number of long-term neurological deficits that persist into adulthood. In this study we examine sex and genotype as variables that impact iron uptake in the developing brain. Our data show critical differences in iron uptake by males and females of a mutant mouse model during development. We also begin to profile the receptors of transferrin and H-ferritin during development. These data may have significant implications in the treatment of iron deficiency.
Introduction:
Iron is a crucial micronutrient to the brain and is required for such fundamental processes as DNA synthesis, myelination, and cellular metabolism (MacKenzie et al. 2008; Black 2003). The developing brain not only requires a substantial amount of iron but it also must be delivered in a timely manner to establish time sensitive connections within the brain. In two studies of adult neuronal connectivity, Algarin and colleagues demonstrated altered brain connectivity patterns in former iron deficient anemia individuals, suggesting long-lasting effects in adult brain function as a result of iron deficiency in adolescence (Algarín et al. 2003; Algarin et al. 2017). Iron deficiency during development often results in long-term measurable phenotypes such as poor cognitive function, motor deficits, increased hearing impairment, and other neurological disorders (Connor et al. 2011; Connor et al. 2003; Dunham et al. 2017). Decreased iron levels may also lead to depressed energy production and metabolism (Zimmermann and Hurrell 2007). Elevated levels of iron can cause increased oxidative stress through Fenton Chemistry. Thus, understanding the regulation of iron uptake into the brain and the dysregulation that may occur with disease is critical to healthy brain function. Studies of iron uptake into the brain have mainly focused on the adult brain; iron uptake into the developing brain is a heretofore underexplored area of study that may hold important therapeutic strategies for iron supplementation and intervention to treat brain iron deficiency.
A common genetic mutation that results in iron accumulation in numerous tissues including in the brain is the H63D mutation of the human iron homeostatic regulator protein (HFE) which is now under investigation in a number of neurodegenerative conditions as a disease modifier (Liu et al. 2011; Nandar et al. 2013; Nandar et al. 2014). The H63D variant of the HFE gene is the most common genetic variation in Caucasians and appears in 1 in 200 people (Girouard et al. 2002). Wild-type HFE functions by limiting transferrin (Tf) interaction with transferrin receptor 1 (TfR1) at the cell membrane, while the mutant H63D form fails to limit Tf binding to TfR1 (Nandar and Connor 2011). The lack of interaction between the HFE mutant protein and TfR1 results in increased uptake of transferrin-bound iron into the cell. Previous studies by our laboratory have demonstrated the presence of HFE at the blood-brain barrier (BBB) (Duck and Connor 2016), demonstrating a role for HFE in regulating iron uptake both into the brain as well as the endothelial cells that comprise the BBB. Although the existing clinical and biological paradigm is that individuals with hemochromatosis are protected from brain iron overload by the BBB, recent MRI studies have found they contain higher brain iron loads (Nandar and Connor 2011). We have recently demonstrated that adult H67D (mouse homolog to human H63D) mutant mice have increased total brain iron at three months of age, but no difference in rate of brain iron uptake (Duck et al. 2018). These key findings are suggestive evidence for a critical set point in the adult brain that limits or at least highly regulates the rate of iron uptake into the brain. To build upon this previous study, we aim to examine iron uptake in the developing brain. The H67D mouse is an exciting model to interrogate the mechanisms and regulation of iron uptake into the brain.
Classical studies of iron delivery to the brain have mainly focused on the ability of Tf to deliver iron both during development as well as in the adult brain (Fishman et al. 1987; Taylor and Morgan 1990). However, we have recently demonstrated the ability for H-ferritin (FtH1), to cross the BBB (Chiou et al. 2018c; Chiou et al. 2018b) and provide a substantial amount of iron into the brain (Fisher et al. 2007). This is a significant departure from the established literature that posited FtH1 as simply an iron storage protein (Arosio et al. 2009). Furthermore, our previous studies have demonstrated that the receptor for FtH1 is the T-cell immunoglobulin and mucin domain 1 (Tim-1) in humans (Chiou et al. 2018a) and Tim-2 in rodents (Todorich et al. 2008; Han et al. 2011; Chen et al. 2005). In this study we have chosen to focus on FtH1 rather than L-ferritin, as we previously demonstrated minimal L-ferritin transport in a model of the BBB (Fisher et al. 2007). Furthermore, the only known receptors for L-ferritin are not present on the BBB, thus L-ferritin is not considered as a likely source of iron for the developing brain (Chiou and Connor 2018).
The goal of this study was to develop models that may impact brain iron uptake during development. The two variables under investigation were sex and genotype. Specifically, we determined if the presence of the H67D gene variant altered brain iron uptake during development. Moreover, given the evidence for FtH1 uptake in our cell culture models (Chiou et al. 2018a; Chiou et al. 2018c) and adult mice (Fisher et al. 2007), we compared FtH1 iron delivery with Tf during development. We hypothesized that the H67D genotype and sex influences the relative levels of uptake of iron during development similar to that seen in our previous study in adults (Duck et al. 2018; Wade et al. 2019). We have observed differences in adult iron uptake, and have determined in this study that these differences appear first during development. In addition to measuring uptake as a function of development, we also assessed the developmental expression of TfR, FtH1, and Tim-2 in order to begin to address and characterize the potential mechanisms for the differential uptake observed.
Materials and Methods:
H-ferritin Preparation
H-ferritin was prepared according to previously published methods (Todorich et al. 2008; Chiou et al. 2018a). Briefly, FtH1 with a poly-His tag was subcloned into a pET30a(+) plasmid, to be cloned into BL21 Escherichia coli. Isopropyl-β-D-thio-galactoside (IPTG, ThermoFisher #15529019) was used to induce expression. Subsequently, bacteria were lysed and FtH1 protein collected via nickel column according to the manufacturer’s instructions (GE Healthcare Bio-Sciences #17526801). Protein concentration was determined via bicinchoninic assay (BCA, Pierce #23225) prior to use. Custom-made materials will be shared upon reasonable request.
Protein Radiolabeling
We used our previously published methods for labeling Tf and FtH1 with 59Fe (Chiou et al. 2018c; Duck et al. 2017). Briefly, 59Fe (Perkin Elmer #NEZ037500) was complexed with 1 mM nitriloacetic acid (NTA, Sigma #N0253), 0.5 M sodium bicarbonate (NaHCO3), and 6.17 mM ferric chloride (FeCl3) at a ratio of 100 µL NTA: 6.7 µL FeCl3: 23.3 µL NaHCO3: 50 µCi 59FeCl3 to create the 59Fe-NTA complex, adjusted for cases where greater or less than 50 µCi was necessary. The 59Fe-NTA complex was incubated with iron poor transferrin (apo-Tf, Sigma #T1147) or FtH1 for 30 minutes to allow for iron loading. Free iron was subsequently separated from solution using G-50 Sephadex QuickSpin columns (Sigma #11273973001) according to the manufacturer’s instructions.
Genotyping
Wild-type or H67D knock-in mice (Jackson Laboratory, Stock #: 023025, RRID:Hfetm1.1Jrco) were genotyped prior to uptake studies as described in our previous publications (Nandar et al. 2013; Nandar et al. 2014; Duck et al. 2018). Briefly, DNA was extracted from tail clips using a DNeasy blood and tissue kit (Qiagen). PCR was performed using forward primer (5’ AGG ACT CAC TCT CTG GCA GCA GGA GGT AAC CA 3’) and reverse primer (5’ TTT CTT TTA CAA AGC TAT ATC CCC AGG GT 3’). PCR conditions used were: 94°C for 15 min followed by 39 cycles of 94°C for 45 sec, 58°C for 45 sec, 72°C for 90 sec, further followed by 72°C for 10 minutes. Amplified DNA was further digested using the BspHI restriction enzyme for 2 hours at 37°C and then separated by 2% agarose gel electrophoresis.
Uptake Studies
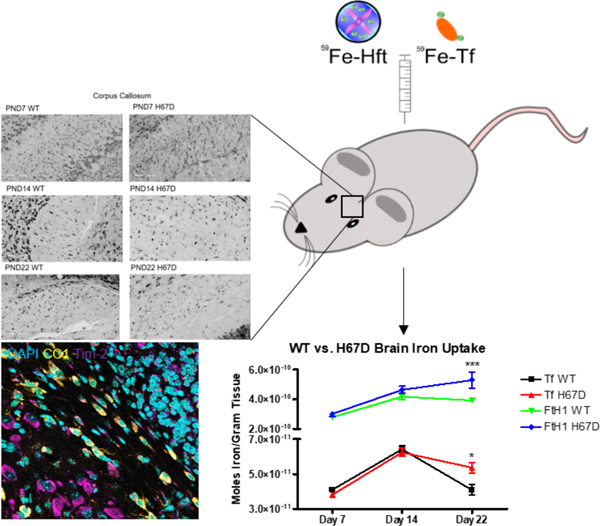
Uptake studies were performed as previously described, with mice sample size determined based on previous study results (Duck et al. 2018). Briefly, 5 wild-type males, 5 wild-type females, 5 H67D/H67D males, and 5 H67D/H67D females at postnatal day 7, 14, or 22 received a single intraperitoneal injection of 3.4 mg/kg body weight 59Fe-Tf or 59Fe-FtH1 (60 total mice per injection condition) (Fig. 1). 24 hours after injection, mice were anesthetized using a ketamine/xylazine cocktail (100 mg per kg body weight/10 mg per kg body weight), blood was drawn via cardiac puncture and mice were transcardially perfused with 0.1M phosphate-buffered saline (PBS, pH 7.4). Plasma was separated from whole blood fractions by centrifugation at 2000 x g for 15 minutes. Whole brains and liver were collected and weighed immediately, and radioactivity in liver, brain, and 50 µL plasma measured on a Beckman Gamma 4000 (Beckman Coulter). Blank tube values were subtracted from final counts to control for background counts. All procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, formally approved by the Pennsylvania State University College of Medicine International Animal Care and Use Committee under Protocol #45975, and reported in accordance with Animal Research: Reporting In Vivo Experiments. Mice are given food and water ad libitum with a maximum number of mice per cage of 5 in Cage type T II (Velaz). Mice were randomized by taking equal numbers of mice from each litter for injections, assigned by computer based randomization (Excel, RAND() function) wherein mice from each group (M vs. F, WT vs. H67D) were assigned to a list for injection of either 59Fe-Tf or 59Fe-Hft. RAND() assigns a random number between 0 and 1 to an animal and these animals were then sorted into equal groups of 5 using the =ROUNDUP(RANK(Animal, Range)/Group size, 0) function. Following this, the Sort function was used to group the mice into their respective groups prior to injection. Person assigning subjects to groups was blinded to genotype and injection protein. No exclusion criteria was pre-determined and no animals died.
Figure 1. Timeline of the uptake study.

At PND7, PND14, and PND21, 5 wild-type male, 5 H67D male, 5 wild-type female, and 5 H67D female mice were given intraperitoneal injections with 3.4 mg/kg of either 59Fe-Tf or 59Fe-Hft. 24 hours after injection, mice were sacrificed and brains, liver, and blood was extracted to count for radioactivity.
Specific Activity
Specific activity was calculated from counts per minute (CPM) and reported as moles of iron per gram of tissue. Calculations were as follows: 10,000 CPM at a counting efficiency of 80% results in 12,500 disintegrations per minute (DPM). As stated previously, mice were injected with 3.4 mg/kg of either 59Fe-Tf or 59Fe-FtH1. Specific activity for 59Fe-Tf injection as DPM per mol: Tf at PND7 = 7.81*1013, PND14 = 4.56*1013, and PND22 = 3.15*1013. Specific activity for 59Fe-FtH1 injection as DPM per mol: FtH1 at PND7 = 4.42*1014, PND14 = 2.58*1014, and PND22 = 1.78*1014. To obtain moles iron per gram tissue, DPM per gram tissue obtained from the radioactivity measurements was divided by DPM/mol Tf or DPM/mol FtH1. Subsequently, DPM/mol protein was multiplied by either 2 or 121 for Tf and FtH1 respectively. This correction factor was applied because each Tf molecule can hold 2 iron atoms, while we have previously determined that 30 minutes of loading FtH1 with iron results in 121 atoms of iron within the core (Chiou et al. 2018b).
Immunohistochemistry
Mice at postnatal day 7, 14, or 22 were sacrificed and transcardially perfused first with Ringer’s solution followed by 4% paraformaldehyde. Brains were isolated and moved into a 4% paraformaldehyde solution overnight. Subsequently, brains were transferred to a 0.1 M PBS solution until one day prior to paraffin embedding, when they were immersed in 70% ethanol. Brains were embedded in paraffin, sagittal sections were cut at 5 µm thickness, and sections mounted on standard microscopy slides. Immunohistochemistry was performed as previously described (Snyder et al. 2009). The slides were deparaffinized in xylene and rehydrated in an ethanol gradient followed by a dH2O rinse. Antigen retrieval was performed with 10 mM citrate buffer (pH = 6.0). The slides were blocked in a 3% H2O2/methanol solution for 20 minutes, rinsed with 1X PBS, and blocked in a 2% milk/PBS solution for 1 hour. The slides were then incubated in primary antibody overnight at 4°C using anti-Tim-2 (10 µg/mL, R&D Systems, MAB1885, RRID:AB_2201833), anti-TfR (2 µg/mL, Thermo Fisher Scientific, 13–6800, RRID:AB_2533029), anti-CC1 (1:100, Calbiochem, OP80, RRID:AB_2057371), anti-Iba1 (1:200, Wako, 019–19741, RRID:AB_839504), anti-GFAP (1:1000, Agilent, Z0334, RRID:AB_10013382) or anti-FtH1 (1 µg/mL, Cell Signaling, 4393) primary antibodies. The following day, the slides were washed with PBS and secondary antibodies (1:200) were applied using an ABC Vectastain kit (Vector Labs, RRID:AB_2336827) in accordance with manufacturer’s protocol or using species-specific AlexaFluor488 and AlexaFluor555 secondary antibodies (Life Technologies). The slides treated with ABC Vectastain secondary antibodies were visualized with diaminobenzidine (DAB) application using standard procedure. DAB staining was intensified via the addition of nickel chloride to the DAB solution. Following DAB staining or fluorescent secondary antibody incubation, slides were mounted, coverslipped, and allowed to dry overnight. Fluorescent slides were mounted with Prolong Diamond Antifade Mountant with DAPI (ThermoFisher #P36962). All slides were imaged using Aperio AT2 Leica slide scanner with a 40X objective. Images were viewed using Aperio ImageScope software.
Western Blot
Western blots were performed as previously described (Chiou et al. 2018a). Briefly, 20 µg protein solubilized in RIPA buffer (Sigma #89900) was quantified by bicinchoninic assay prior to boiling for 10 minutes, loading onto a denaturing 4–20% gradient SDS-PAGE gel (BioRad #4561094), and subsequent transfer to nitrocellulose membrane. Membranes were blocked in a 5% milk solution made in Tris-buffered saline and Tween-20 (TBS-T, pH 7.6). Blots were probed for Tim-2 (2 µg/mL, R&D Systems, RRID:AB_2201833), TfR (1 µg/mL, Thermo Fisher Scientific, RRID:AB_2533029), FtH1 (1:500, Cell Signaling, 4393), or β-actin (1:3000, Sigma-Aldrich, A178, RRID:AB_476692) overnight at 4°C in a 5% milk/TBS-T solution. Corresponding species-specific secondary antibody conjugated to HRP was used (1:5000, GE Amersham), using ECL reagents (Perkin Elmer) to visualize bands on an Amersham Imager 600 (GE Amersham) with densitometry analysis using ImageJ. The sum total of all bands was used in the quantification, because the additional bands are thought to be dimers of the proteins (near exact molecular weight matches). Shown are representative western blots. Western blots were repeated 3 times for each timepoint and each band represents homogenates pooled from 3 male and 3 female mice.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 4 Software (Graphpad Software Inc.). Data from biological replicates were averaged and are expressed as the mean ± standard deviation (SD). Specific numbers of replicates are found in each figure legend. Data were tested for normality using the Shapiro-Wilk test; data were normally distributed thus parametric statistics were used. Two-way ANOVA with Bonferroni post-hoc analysis or unpaired t-tests were used to evaluate for statistical significance where appropriate. A p-value <0.05 was considered significant. ROUT test with a Q=1% was performed in Prism for all data, but no animals or data were excluded.
Results:
Iron Uptake into the Developing Brain
To measure the uptake of iron into the brain, we loaded either Tf or FtH1 with 59Fe and injected the proteins intraperitoneally. In our first study, we examined the levels of iron uptake into the developing brain by both 59Fe-Tf and 59Fe-FtH1 (Fig. 2). Uptake of Tf peaked at PND14 and decreased at PND22. Overall, 59Fe-FtH1 uptake was significantly higher, with a peak uptake for wild-type mice at PND14 and a peak uptake for H67D mice at PND22 (Fig. 2A). There was a genotype and sex effect at PND22 for both Tf and FtH1: H67D mice had a significantly higher amount of uptake than wild-type mice at PND22, however, the increased iron uptake was from Tf in the female H67D mice (Fig. 2B) whereas in the male H67D mice there was significantly increased uptake of 59Fe-FtH1 (Fig. 2C).
Figure 2. Sex and genotype effects on brain iron uptake.

A) H67D mice take up significantly more 59Fe-Tf and 59Fe-FtH1 into the brain on PND22 (n=10 mice). B) Female H67D mice take up significantly more 59Fe-Tf into the brain on PND22 than male H67D mice. C) Male H67D mice take up significantly more 59Fe-FtH1 into the brain on PND22 than female H67D mice. For B) and C), n=5 mice for males and n=5 mice for females; means from all biological replicates ± SD were evaluated for statistical significance using two-way ANOVA with Bonferroni’s posttest for significance. *=p<0.05, ***=p<0.001.
We also measured the amount of 59Fe retained by the liver. At PND14, wild-type mice take up a significantly greater amount of 59Fe-FtH1 into their livers compared to the H67D mice (Fig. 3A). Furthermore, at PND22, H67D mice took up a greater amount of 59Fe-Tf into their livers compared to wild-type mice. At PND7, we demonstrate that both male and female wild-type mice livers take up significantly more 59Fe-Tf than their H67D counterparts (Fig. 3B). There was also a sex and genotype effect of FtH1 delivery of iron; male wild-type take up more 59Fe-FtH1 than male H67D mice into their livers on PND7, whereas female H67D mice take up more iron into their livers than wild-type mice on PND22 (Fig. 3C). While there were no significant differences in plasma levels of 59Fe-Tf, 59Fe-FtH1 levels in plasma at PND7 was significantly higher in H67D mice whereas at PND22, wild-type mice had more 59Fe-FtH1 in plasma than the H67D mice (Fig. 4A). There were no sex differences in the plasma accumulation levels of iron for either FtH1 or Tf (Fig. 4B, C).
Figure 3. Sex and genotype effects on liver iron uptake.

A) H67D mice take up significantly more 59Fe-Tf into the liver on PND22. Wild-type mice take up significantly more 59Fe-FtH1 into the liver uptake on PND14 (n=10 mice). B) Female wild-type mice take up significantly more 59Fe-Tf into their livers on PND7 than their respective H67D counterparts, but on PND22 H67D mice livers take up significantly more 59Fe-Tf. C) Male wild-type mice take up significantly more 59Fe-FtH1 at PND7 than H67D mice, while female H67D mice take up significantly more 59Fe-FtH1 on PND22 than wild-type female mice. For B) and C), n=5 mice for males and n=5 mice for females; means from all biological replicates ± SD were evaluated for statistical significance using two-way ANOVA with Bonferroni’s posttest for significance. *=p<0.05.
Figure 4. Sex and genotype effects on plasma iron content.

A) Genotype does not affect plasma iron content after injection of 59Fe-Tf. H67D mice have higher circulating levels of 59Fe-FtH1 on PND7 than wild-type mice. On PND22, wild-type mice have higher circulating levels of 59Fe-FtH1 than H67D mice (n=10 mice). B) Sex does not affect circulating levels of 59Fe after 59Fe-Tf injection. C) Sex does not affect circulating levels of 59Fe after 59Fe-FtH1 injection. For B) and C), n=5 mice for males and n=5 mice for females; means from all biological replicates ± SD were evaluated for statistical significance using two-way ANOVA with Bonferroni’s posttest for significance. *=p<0.05.
Developmental Expression of Iron Related Proteins
To examine the mechanism of uptake of Tf and FtH1, we performed a characterization of the expression profiles of the receptors. For this, we used a separate group of mice that did not have any injections and collected whole brains at PND7, 14, and 22. To begin, we used western blot analyses on whole brain lysates to probe for levels of Tim-2, the receptor for FtH1 in mice (Todorich et al. 2008; Chen et al. 2005) (Fig. 5A), and TfR (Fig. 5B). We also probed for FtH1 as a surrogate for the amount of iron stored (Fig. 5C), with β-Actin used as a loading control (Fig. 5D). At PND7, there was significantly more TfR and FtH1 in the H67D mouse brains compared to wild-type mice but no difference in Tim-2 expression (Fig. 5E). At PND14 there were no significant differences between wild-type and mutant mice in any of the proteins measured. At PND22, Tim-2 was significantly increased in the H67D mouse brains compared to wild-type (Fig. 5E). Throughout development in both genotypes, Tim-2 expression was highest at PND7 and 14 and decreased at PND22. Wild-type TfR expression peaked at PND14 whereas the inverse was true in H67D mice – PND14 had the lowest amount of observed TfR. FtH1 expression remained consistent throughout development.
Figure 5. Tim-2, Transferrin Receptor, and H-ferritin expression during development.

Shown are representative western blots of expression for Tim-2 (A), TfR (B), FtH1 (C), and β-actin (D) on whole mouse brain homogenates. Western blots were repeated 3 times for each timepoint; each band represents homogenates pooled from 3 male and 3 female mice. E) Densitometric analyses of western blots reveal significantly elevated Tim-2 expression in H67D mice at day 22 and elevated TfR expression in H67D mice at day 7. For quantification we used the sum total of all of the bands because additional bands are thought to be dimers (near exact molecular weight matches). FtH1 expression was increased in H67D mice at day 7. Wild-type versus H67D means ± SD were evaluated for statistical significance by unpaired t-tests, n=3 biological replicates. *=p<0.05, ***=p<0.001.
Next, we examined the cellular distribution of Tim-2 in vivo. Though we have previously published evidence for the presence of Tim-2 on mouse oligodendrocytes which is able to mediate FtH1 uptake (Todorich et al. 2008), we wanted to determine if other cell types may use Tim-2 as a receptor for FtH1 during development. Consistent with our previous results (Todorich et al. 2008), we found robust Tim-2 staining on CC1-positive cells, denoting mature oligodendrocytes (Fig. 6A). We did not observe any Iba1-positive cells that were positive for Tim-2 (Fig. 6B, white arrows). We did observe that astrocytes robustly stain for Tim-2 (Fig. 6C). Throughout all the stains, we observed strong staining for Tim-2 in cells that were negative for CC1, Iba1, and GFAP, suggesting neuronal staining.
Figure 6. Tim-2 expression in oligodendrocytes, microglia, and astrocytes.

Slides were stained for Tim-2 (magenta) or CC1/Iba1/GFAP (yellow); nuclei were counterstained with DAPI (cyan). A) Mouse corpus callosum stained for CC1 (yellow), a marker for myelinating oligodendrocytes, and Tim-2 (magenta) reveal expression of Tim-2 in oligodendrocytes. There is similarly robust Tim-2 staining in CC1-negative cells, likely to be neurons. B) Dual-staining in the neocortex for Iba1 and Tim-2 reveals microglia (Iba1+, white arrows) do not express Tim-2. C) Hippocampus staining for GFAP (yellow), an astrocytic marker, demonstrates colocalization with Tim-2 (magenta), marked by white arrowheads. The white arrow marks cells surrounding a blood vessel positive for Tim-2 staining, likely to be endothelial cells of the BBB. There can again be seen robust staining for Tim-2 in non-GFAP+ cells, most likely to be neurons. All brain slices came from wild-type PND7 female mice. Scale bars indicate 10 µm.
Distribution of Iron Related Proteins During Development
In the following study, we examined the regional distribution and expression of Tim-2 (Fig. 7), TfR (Fig. 8), and FtH1 (Fig. 9) in the brain during development.
Figure 7. Tim-2 expression throughout the brain during development.

Tim-2 staining in key regions throughout the brain in wild-type and H67D mice at day 7, day 14, and day 22. Areas focused upon in this figure are the somatosensory cortex layer 4, corpus callosum, and microvasculature. For a full description of staining patterns and expression, refer to the ‘Results’ section. Scale bars indicate 100 µm.
Figure 8. TfR expression throughout the brain during development.

TfR staining in the corpus callosum and microvasculature in the brain of wild-type and H67D mice at day 7, day 14, and day 22. For a full description of staining patterns and expression, refer to the ‘Results’ section. Scale bars indicate 100 µm.
Figure 9. FtH1 expression throughout the brain during development.

FtH1 staining in the corpus callosum and microvasculature in the brain of wild-type and H67D mice at day 7, day 14, and day 22. For a full description of staining patterns and expression, refer to the ‘Results’ section. Scale bars indicate 100 µm.
Tim-2
At PND7, 14, and 22, Tim-2 staining was present throughout the cortex in both wild-type and H67D mice (Fig. 7A). Though we observed very sparse staining in cortical layer 1, immunostaining in cortical layers 2, 3, 4, 5 and 6 was robust (data not shown). Of note is the localization of Tim-2 to the borders of the cell body, especially in the large pyramidal neurons of layer 4 and 5. Consistent with our previous studies (Todorich et al. 2008), there is robust Tim-2 staining in oligodendrocytes of the corpus callosum arranged in a tram-track orientation at PND7 (Fig. 7B). By PND14 and PND22, we observed similar clear staining of the corpus callosum. Lastly, we observed Tim-2 staining at the microvasculature throughout the brain at PND7 (Fig. 7C), which persisted throughout development (data not shown). We also observed robust staining in the Purkinje cells of the cerebellum, hippocampus, thalamus, and substantia nigra (data not shown). At all timepoints we measured, we observed no regional differences in Tim-2 staining between wild-type and H67D mice.
Transferrin Receptor
TfR staining at PND7 in the corpus callosum was very light in both wild-type and H67D mice (Fig. 8A). Similarly, staining for TfR at PND14 was also very lightl, and this staining was decreased at PND22. At PND7, we observed staining for TfR in the microvasculture (Fig. 8B) which persisted at PND14 and PND22 (data not shown), consistent with previous literature demonstrating TfR at the level of the BBB (Simpson et al. 2015). We also observed extremely dense staining in all cortical layers, the thalamus, hippocampus, substantia nigra, and cerebellum throughout development (data not shown). At all timepoints we measured, we observed no regional differences in TfR staining between wild-type and H67D mice.
H-ferritin
In stark contrast to Tim-2 and TfR expression, FtH1 staining, used here as a surrogate marker for intracellular iron, was relatively sparse except in specific regions. Throughout the developmental timepoints we examined, FtH1 was consistently highest in the corpus callosum (Fig. 9A) and was present in the microvasculature (Fig. 9B). At PND22 in the corpus callosum, staining is very heavy for FtH1, especially in what appear to be mature oligodendrocytes. Similar to Tim-2 and TfR, FtH1 staining was observed heavily at PND7 which persisted throughout development (data not shown). We also examined the cortex, thalamus, hippocampus, substantia nigra, and cerebellum for FtH1 immunostaining, finding that FtH1 was heavily localized to cortex layer 4 and the white matter throughout the brain (data not shown). However no regional differences between wild-type and H67D mice were observed.
Discussion:
The aim of this study was to examine the uptake of FtH1 bound iron as a novel iron delivery system to the developing brain. Furthermore, we determined if sex or genotype could alter the uptake of Tf or the newly discovered FtH1 delivery system. The results of this study have demonstrated that FtH1 can deliver a significantly higher amount of labelled iron during development than transferrin. These data further demonstrate that brain iron acquisition from both transport proteins are modified by age, sex, and genotype. While it has previously been shown that in general, liver and plasma iron levels tend to correspond in normal humans and HFE hemochromatosis patients (Olynyk et al. 2008; Graham et al. 2010), our single time point data demonstrate that after injection of extra iron via different carriers, we find a significant difference in the handling of this iron by H67D and wild-type mice.
For Tf uptake, the transport of iron appears highly regulated (Simpson et al. 2015), increasing at PND14 and decreasing thereafter. The decrease in brain iron uptake occurs even in the presence of a gene variant that is associated with increased iron uptake by all organs in the body including the brain (Nandar et al. 2013). For FtH1 uptake, the transport of iron into the brain does not decrease after PND14 and in the H67D mouse, FtH1 uptake continues to increase with age. There is also a sex difference as males with the H67D variant show an increase, rather than a decrease, in FtH1 uptake between PND7 and PND22. Similarly, female H67D mice take up more Tf at PND22 than male H67D mice. These results clearly demonstrate the regulation of uptake of iron from FtH1 is distinct from Tf and furthermore that regulatory mechanisms between males and females may have significant differences. Our data demonstrate that gross levels of TfR in the brain do not necessarily follow the same temporal pattern of brain iron uptake, pointing towards a delay in and possibly more regional regulation of uptake at the BBB as we have proposed (Chiou et al. 2018c). We have previously published that FtH1 and Tf transport across the BBB share a common regulatory mechanism mediated by apo-Tf and holo-Tf (Chiou et al. 2018c), suggesting that this axis may be of critical significance during development. We have also begun to examine the mechanism of uptake into the brain by profiling the different cell types of the brain and various brain regions for the receptors for Tf and FtH1 during development. By tracking these receptors during development, we have demonstrated that the uptake mechanism for iron delivered via Tf and FtH1 is present. Our data showing both Tf and FtH1 delivery to the brain during development are consistent with the demonstration that both of these proteins are involved in brain iron delivery in the adult (Duck et al. 2018).
This study shows that the temporal pattern of delivery of Tf and FtH1 to the developing brain is similar. There is peak uptake of Tf at PND14, with a decrease at PND22. Our data for Tf-mediated uptake of iron are consistent with previous studies that demonstrate a similar uptake pattern during development (Moos and Morgan 2002). Though the general decline in uptake occurs in both wild-type and H67D mice, female H67D mice take up significantly higher amounts of 59Fe-Tf than the other groups. In contrast to Tf, FtH1 uptake does not decline after PND14 in either the wild-type or H67D mouse. Furthermore, male H67D mice significantly increase the amount of 59Fe-FtH1 that they take up at PND22 compared to wild-type. The different patterns of uptake for Tf and FtH1 delivered iron suggest that Tf could be providing iron more acutely to aid in the a growth spurt of the brain that accompanies peak myelination (Hulet et al. 2002) whereas FtH1 provides iron at a steadier pace for maintence of required levels for normal brain function. For example, oligodendrocytes are the highest iron staining cells in the brain (Benkovic and Connor 1993; Connor et al. 1995) and the ability of these cells to take up a substantial amount of iron during development is necessary for proper myelination; inadequate iron delivery to oligodendrocytes frequently results in hypomyelination (Todorich et al. 2009). We have reported that FtH1 is the major source of iron for mature oligodendrocytes (Todorich et al. 2011). This study provides further evidence that FtH1 plays a significant role in delivery of iron to various organs throughout the body (Blight and Morgan 1983; Kim et al. 2013; Fisher et al. 2007; Sibille et al. 1989). While we have previously demonstrated the ability of FtH1 to deliver iron to the adult rat brain (Fisher et al. 2007), this study is the first to demonstrate FtH1 delivery during development in a mouse. We have previously shown that the FtH1 in our preparation results in at least 10 times as much iron loaded as transferrin/mole (Chiou et al. 2018b). Conceptually, FtH1 could contain 4500 atoms of iron per mole or 2000 times more iron than transferrin (Arosio et al. 2017). The source of FtH1 that may be taken up in the brain has yet to be elucidated but studies have shown FtH1 is released by multiple cell types (Chiou and Connor 2018).
In this study, we performed immunostaining for intracellular FtH1 to understand the regional expression of FtH1 in the brain during development as a surrogate marker for cellular iron status (Fig. 9). It was noted that FtH1 staining becomes more punctate and region-specific during development, suggesting that there is a higher utilization of iron in specific regions of the brain during development. This suggests future studies on regional brain delivery via FtH1 are warranted. The data supported previous reports that oligodendrocytes and glial cells of the white matter tracts are the cells that accumulate the most iron (Connor et al. 1992; Connor and Menzies 1996) and FtH1 (Schonberg et al. 2012; Hulet et al. 1999a; Hulet et al. 2002). Here, we expand those findings and demonstrate that specific neurons in the hippocampus and pyramidal neurons in the cortex stain for FtH1 as well. That FtH1 staining in oligodendrocytes is highest during peak myelination strongly coincides with the necessity of iron for the myelination process and supports the reports that chronic iron deficiency frequently leads to hypomyelination in both rodent (Ortiz et al. 2004) and human models (Roncagliolo et al. 1998; Algarín et al. 2003). Consistent with these results, we confirmed the presence of Tim-2 for iron acquisition via extracellular FtH1 on oligodendrocytes (Fig. 6A).
We also demonstrate that there is a sex effect on iron uptake, a phenomena we have described previously in the adult H67D mouse (Duck et al. 2018); female H67D mice accumulate more iron in their brain than male H67D mice (Duck et al. 2018). At the earlier time points the pattern of iron uptake from either FtH1 or Tf was similar between the different sexes and genotypes. However, at PND22, there is increased Tf-iron uptake in the H67D female mice compared to the other groups, whereas FtH1-mediated iron uptake is greater in the H67D males compared to the other groups. These uptake data may account for reports of a higher volume of total brain iron in males (Hahn et al. 2009) compared to females, with the caveat that our differences were only seen in the mutant mice. We have also previously published that the adult H67D mouse brain has a higher overall iron load (Nandar et al. 2013; Duck et al. 2018), suggesting that this imbalance in iron uptake is established at PND22 and persists into adulthood. Overall, this is the first study to reveal a sex and genotype effect on brain iron acquisition during development. The mechanism regulating these uptake differences, which is occurring prior to hormonal influences associated with puberty will be explored in future studies.
Our data further support the concept that iron uptake is tightly regulated and suggests the potential for a set-point for iron delivery that is established during development and maintained throughout adulthood. We propose there is a critical window during which the brain is growing that is amenable to relatively high levels of iron uptake. This concept is evident in studies on nutritionally iron deficient animals where iron supplementation started at PND4 can correct brain iron deficiency yet supplementation at PND21 was not able to correct early iron losses (Felt et al. 2006; Beard et al. 2006; Unger et al. 2012). Our data suggest that once significant brain growth has plateaued, there are regulatory molecules such as apo-Tf and holo-Tf (Chiou et al. 2018c) from the brain signaling to the endothelial cells of the BBB regarding brain iron status. The observation that the H67D mutation and sex both affect iron uptake by Tf and FtH1 demonstrates that multiple factors exist in tandem to influence the regulation of iron uptake. Importantly, we demonstrate that the H67D mutation may result in an aberrant set-point, as there is higher uptake of 59Fe-Tf and 59Fe-FtH1 at PND22 than wild-type. This point is further illustrated in the adult brain, as brain iron in the H67D adult mouse brain is significantly higher than wild-type yet iron accumulation in the brain 24 hours after injection with 59Fe-Tf does not differ from wild-type (Duck et al. 2018). This implies that the H67D mouse brain has a higher set-point for iron uptake than the wild-type, and this persists into adulthood. Furthermore, it has been shown that H67D mutant mice have altered brain iron handling proteins such as TfR, H- and L-ferritin, and Tim-2 (Nandar et al. 2013), suggesting that in these mice the altered brain iron profile is a result of altered regulation.
The data from this study could have significant potential implications in the clinical treatment of brain iron deficiency during development. The data herein demonstrates the concept that in the mouse there may be a specific time during normal development that iron supplementation may be most effective, as Tf uptake decreases after PND14 while FtH1 uptake levels off at PND14. This is significant, as it is still controversial regarding at what age iron supplementation should be started in the treatment of iron deficiency, or if iron supplementation in an early age would lead to adult disease phenotypes. For example, there are studies demonstrating that in mice, early exposure to iron overload during a critical period of development (PND10–17) may result in higher rates of a Parkinsonian disease phenotype (Billings et al. 2016). Similarly, Kaur et al. demonstrate that increased neonatal iron intake results in adult Parkinsonian phenotypes (Kaur et al. 2007). Furthermore, in β-thalassemia patients, iron supplementation in children leads to impaired neurocognitive function (Elalfy et al. 2017). The ability to override the set point and normal regulatory mechanisms for brain iron uptake in these models could be due to significant inflammation and subsequent compromise in BBB integrity that would be associated with infusion of intravenous iron or gavage of carbonyl iron molecules (Varatharaj and Galea 2017; Chiou and Connor 2018). While needing further studies to fully explore this concept, these results may impart specific directions for clinicians to follow in the treatment of developmental iron deficiency.
An unexpected finding in this study was the identification of Tim-2 on neuronal cells. We have previously identified Tim-2 as the receptor for FtH1 on oligodendrocytes (Todorich et al. 2008); indeed, FtH1 can replace Tf as the obligate source of iron for oligodendrocytes (Todorich et al. 2011). The presence of Tim-2 on neurons may then represent a novel iron uptake mechanism in neurons. Previous literature has identified neuronal acquisition of iron through a Tf-mediated pathway (Leitner and Connor 2012) or through voltage-gated calcium channels (Lopin et al. 2012), but the presence of Tim-2 may suggest FtH1 uptake in neuronal cells as well. An alternative explanation for the high abundance of Tim-2 on neurons may lie in the other function of Tim-2 as a receptor for Sema4A (Kumanogoh et al. 2002). During neural development, the semaphorin family acts as negative regulators of neural migration, causing growth cone collapse (Yukawa et al. 2005). It has also been shown that Sema4A, a semaphorin family member, can also bind to Tim-2 (Kumanogoh et al. 2002). Other binding partners to Sema4A, namely Plexin D1 (Toyofuku et al. 2007), Plexin B2 (Ito et al. 2015), and Neuropilin-1 (Delgoffe et al. 2013), have been demonstrated to facilitate axon guidance during neural development. While we have previously shown Sema4A to be cytotoxic to oligodendrocytes (Chiou et al. 2018a; Chiou et al. 2019), it is possible that the presence of Tim-2 on neurons may lead to axonal guidance either through a pro-apoptotic mechanism or through the classical Rho/vascular endothelial growth factor (VEGF) signaling pathway (Zhou et al. 2008). A second explanation may lie in the possibility for Tim-2 to non-covalently dimerize and form a separate binding site that has potentially different effects on the cell (Santiago et al. 2007). This dimerization effect may be specific to oligodendrocytes or neurons. A different function for Tim-2, and perhaps altered ability to bind to FtH1 would explain the differences between the immunostaining data for Tim-2 and a ferritin binding study performed by Hulet et al. that demonstrated ferritin binding localizes specifically to the white matter tracts and not the gray matter regions (Hulet et al. 2002; Hulet et al. 1999b).
In addition to neurons, robust Tim-2 staining was found in astrocytes and brain microvasculature throughout the development time periods interrogated (Fig. 6C). For the microvasculature, the presence of Tim-2 in endothelial cells of the BBB demonstrates a mechanism for uptake of FtH1 into the brain parenchyma. We have previously demonstrated that FtH1 can be taken up and directly cross the endothelial cells and that the human receptor for FtH1 on the BBB is Tim-1 (Chiou et al. 2018c). The abundance of Tim-2 in the BBB is further consistent with previous literature documenting FtH1 transport in vivo (Fisher et al. 2007). For astrocytes, like neurons, the presence of Tim-2 may indicate a novel iron uptake mechanism. It has been documented that astrocytes can obtain iron through a Tf-mediated mechanism in vitro (Qian et al. 1999), but the presence of Tim-2 on astrocytes may represent an alternative method for iron uptake through FtH1. Interestingly, we found that microglia, the resident immune cells of the brain, do not express Tim-2 (Fig. 6B). Previous studies have determined that CXCR4, a microglial chemokine receptor, can bind and interact with FtH1 (Li et al. 2006) suggesting that microglia may have an alternative mechanism to take up` FtH1.
In this study, while we have focused primarily on iron uptake into the brain and transport across the BBB, it is important to also consider the effects of efflux from the BBB and the brain itself. Iron efflux is typically handled by ferroportin, the only known free iron exporter, and hepcidin, a hormone that causes the degradation of ferroportin. We have previously demonstrated that hepcidin significantly impacts iron transport across the BBB in a cell culture model (Chiou et al. 2018c) and it is likely there is a similar mechanism in vivo as well. It has previously been shown that HFE plays a role as an upstream regulator of hepcidin (Pantopoulos 2008), while studies have also shown that hepcidin expression decreases in iron loaded Hfe-knockout mice (Bridle et al. 2003). In the developing brain, there is little currently known about the interplay between hepcidin levels and how a mutation in HFE would impact its’ expression and subsequently, iron uptake. Previous studies have shown that hepcidin is indeed present throughout the brain in the adult rodent (Zechel et al. 2006; Raha-Chowdhury et al. 2015), but a comprehensive analysis of hepcidin expression during development has yet to be performed. This is clearly an opportunity for additional study to interrogate another mechanism revolving around the ferroportin-hepcidin axis that may contribute to the observed increased brain iron uptake in the H67D mice.
Overall, these data provide strong evidence for a sex and genotype effect of iron uptake into the brain during development and begin to elucidate the mechanism for differential iron uptake. Importantly, this study provides developmental snapshots of Tim-2, TfR, and FtH1 expression in critical regions of the brain, demonstrating a temporal aspect for receptor expression. Moreover, the identification of Tim-2 on neurons and astrocytes represents a heretofore unexplored area of research that has strong implications for FtH1 uptake and Sema4A-mediated axonal guidance.
Acknowledgements:
The authors would like to thank Jean Copper and the Microscopy Imaging Core at Penn State College of Medicine for invaluable assistance with microscopy imaging. Drs. Amanda Snyder and Acuthamangalan Madhankumar provided technical assistance. Funding for this study was provided by NIH R01 NS077678 (JRC/IAS).
Footnotes
Disclosure: JRC is the founder and chairman of the board of Sidero Biosciences LLC, a company with a product involving oral consumption of ferritin for management of iron deficiency. JRC is an editor for Journal of Neurochemistry.
Open Science Badges
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. The complete Open Science Disclosure form for this article can be found at the end of the article. More information about the Open Practices badges can be found at https://cos.io/our-services/open-science-badges/.
References:
- Algarin C, Karunakaran KD, Reyes S, Morales C, Lozoff B, Peirano P, Biswal B (2017) Differences on Brain Connectivity in Adulthood Are Present in Subjects with Iron Deficiency Anemia in Infancy. Front. Aging Neurosci 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algarín C, Peirano P, Garrido M, Pizarro F, Lozoff B (2003) Iron Deficiency Anemia in Infancy: Long-Lasting Effects on Auditory and Visual System Functioning. Pediatr. Res 53, 217–223. [DOI] [PubMed] [Google Scholar]
- Arosio P, Elia L, Poli M (2017) Ferritin, cellular iron storage and regulation. IUBMB Life 69, 414–422. [DOI] [PubMed] [Google Scholar]
- Arosio P, Ingrassia R, Cavadini P (2009) Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta - Gen. Subj 1790, 589–599. [DOI] [PubMed] [Google Scholar]
- Beard JL, Felt B, Schallert T, Burhans M, Connor JR, Georgieff MK (2006) Moderate iron deficiency in infancy: Biology and behavior in young rats. Behav. Brain Res 170, 224–232. [DOI] [PubMed] [Google Scholar]
- Benkovic SA, Connor JR (1993) Ferritin, transferrin, and iron in selected regions of the adult and aged rat brain. J. Comp. Neurol 338, 97–113. [DOI] [PubMed] [Google Scholar]
- Billings JL, Hare DJ, Nurjono M, Volitakis I, Cherny RA, Bush AI, Adlard PA, Finkelstein DI (2016) Effects of Neonatal Iron Feeding and Chronic Clioquinol Administration on the Parkinsonian Human A53T Transgenic Mouse. ACS Chem. Neurosci 7, 360–366. [DOI] [PubMed] [Google Scholar]
- Black MM (2003) Micronutrient Deficiencies and Cognitive Functioning. J. Nutr 133, 3927S–3931S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight GD, Morgan EH (1983) Ferritin and iron uptake by reticulocytes. Br. J. Haematol 55, 59–71. [DOI] [PubMed] [Google Scholar]
- Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, Subramaniam VN, Powell LW, Anderson GJ, Ramm GA (2003) Disrupted hepcidin regulation in HFE -associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 361, 669–673. [DOI] [PubMed] [Google Scholar]
- Chen TT, Li L, Chung D-H, Allen CDC, Torti SV, Torti FM, Cyster JG, et al. (2005) TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J. Exp. Med 202, 955–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou B, Connor J (2018) Emerging and Dynamic Biomedical Uses of Ferritin. Pharmaceuticals 11, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou B, Lucassen E, Sather M, Kallianpur A, Connor J (2018a) Semaphorin4A and H-ferritin utilize Tim-1 on human oligodendrocytes: A novel neuro-immune axis. Glia 66, 1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou B, Neal EH, Bowman AB, Lippmann ES, Simpson IA, Connor JR (2018b) Pharmaceutical iron formulations do not cross a model of the human blood-brain barrier. PLoS One 13, e0198775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou B, Neal EH, Bowman AB, Lippmann ES, Simpson IA, Connor JR (2018c) Endothelial cells are critical regulators of iron transport in a model of the human blood–brain barrier. J. Cereb. Blood Flow Metab, 0271678X1878337. [DOI] [PMC free article] [PubMed]
- Chiou B, Neely E, Kallianpur A, Connor JR (2019) Semaphorin4A causes loss of mature oligodendrocytes and demyelination in vivo. J. Neuroinflammation 16, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Boyer PJ, Menzies SL, Dellinger B, Allen RP, Ondo WG, Earley CJ (2003) Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 61, 304–309. [DOI] [PubMed] [Google Scholar]
- Connor JR, Menzies SL (1996) Relationship of iron to oligodendrocytes and myelination. Glia 17, 83–93. [DOI] [PubMed] [Google Scholar]
- Connor JR, Menzies SL, Martin SM St., Mufson EJ (1992) A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J. Neurosci. Res 31, 75–83. [DOI] [PubMed] [Google Scholar]
- Connor JR, Pavlick G, Karli D, Menzies SL (1995) A Histochemical Study of Iron-Positive Cells in the Developing Rat Brain. J. Comp. Neurol 335, 111–123. [DOI] [PubMed] [Google Scholar]
- Connor JR, Ponnuru P, Wang X-S, Patton SM, Allen RP, Earley CJ, Leader GM (2011) Profile of altered brain iron acquisition in restless legs syndrome. Brain 134, 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Woo S-R, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, et al. (2013) Stability and function of regulatory T cells is maintained by a neuropilin-1–semaphorin-4a axis. Nature 501, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duck KA, Connor JR (2016) Iron uptake and transport across physiological barriers. BioMetals 29, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duck KA, Neely EB, Simpson IA, Connor JR (2018) A role for sex and a common HFE gene variant in brain iron uptake. J. Cereb. Blood Flow Metab 38, 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duck KA, Simpson IA, Connor JR (2017) Regulatory mechanisms for iron transport across the blood-brain barrier. Biochem. Biophys. Res. Commun 494, 70–75. [DOI] [PubMed] [Google Scholar]
- Dunham J, Bauer J, Campbell GR, Mahad DJ, Driel N. van, Pol S. M. A. van der, T Hart BA, et al. (2017) Oxidative Injury and Iron Redistribution Are Pathological Hallmarks of Marmoset Experimental Autoimmune Encephalomyelitis. J. Neuropathol. Exp. Neurol 76, 467–478. [DOI] [PubMed] [Google Scholar]
- Elalfy MS, Aly RH, Azzam H, Aboelftouh K, Shatla RH, Tarif M, Abdatty M, Elsayed RM (2017) Neurocognitive dysfunction in children with β thalassemia major: psychometric, neurophysiologic and radiologic evaluation. Hematology 22, 617–622. [DOI] [PubMed] [Google Scholar]
- Felt BT, Beard JL, Schallert T, Shao J, Aldridge JW, Connor JR, Georgieff MK, Lozoff B (2006) Persistent neurochemical and behavioral abnormalities in adulthood despite early iron supplementation for perinatal iron deficiency anemia in rats. Behav. Brain Res 171, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J, Devraj K, Ingram J, Slagle-Webb B, Madhankumar AB, Liu X, Klinger M, Simpson IA, Connor JR (2007) Ferritin: a novel mechanism for delivery of iron to the brain and other organs. Am J Physiol Cell Physiol 293, C641–9. [DOI] [PubMed] [Google Scholar]
- Fishman JB, Rubin JB, Handrahan JV, Connor JR, Fine RE (1987) Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J. Neurosci. Res 18, 299–304. [DOI] [PubMed] [Google Scholar]
- Girouard J, Giguère Y, Delage R, Rousseau F (2002) Prevalence of HFE gene C282Y and H63D mutations in a French-Canadian population of neonates and in referred patients. Hum. Mol. Genet 11, 185–9. [DOI] [PubMed] [Google Scholar]
- Graham RM, Chua ACG, Carter KW, Delima RD, Johnstone D, Herbison CE, Firth MJ, et al. (2010) Hepatic iron loading in mice increases cholesterol biosynthesis. Hepatology 52, 462–471. [DOI] [PubMed] [Google Scholar]
- Hahn P, Song Y, Ying G-S, He X, Beard J, Dunaief JL (2009) Age-dependent and gender-specific changes in mouse tissue iron by strain. Exp. Gerontol 44, 594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Seaman WE, Di X, Wang W, Willingham M, Torti FM, Torti SV (2011) Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS One 6, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulet S ., Hess E ., Debinski W, Arosio P, Bruce K, Powers S, Connor JR (1999a) Characterization and Distribution of Ferritin Binding Sites in the Adult Mouse Brain. J. Neurochem 72, 868–874. [DOI] [PubMed] [Google Scholar]
- Hulet S ., Powers S, Connor J. (1999b) Distribution of transferrin and ferritin binding in normal and multiple sclerotic human brains. J. Neurol. Sci 165, 48–55. [DOI] [PubMed] [Google Scholar]
- Hulet SW, Menzies S, Connor JR (2002) Ferritin binding in the developing mouse brain follows a pattern similar to myelination and is unaffected by the jimpy mutation. Dev. Neurosci 24, 208–13. [DOI] [PubMed] [Google Scholar]
- Ito D, Nojima S, Nishide M, Okuno T, Takamatsu H, Kang S, Kimura T, et al. (2015) mTOR Complex Signaling through the SEMA4A–Plexin B2 Axis Is Required for Optimal Activation and Differentiation of CD8 + T Cells. J. Immunol 195, 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur D, Peng J, Chinta SJ, Rajagopalan S, Monte D. A. Di, Cherny RA, Andersen JK (2007) Increased murine neonatal iron intake results in Parkinson-like neurodegeneration with age. Neurobiol. Aging 28, 907–913. [DOI] [PubMed] [Google Scholar]
- Kim BJ, Lee SH, Koh JM, Kim GS (2013) The association between higher serum ferritin level and lower bone mineral density is prominent in women ≥45 years of age (KNHANES 2008–2010). Osteoporos. Int 24, 2627–2637. [DOI] [PubMed] [Google Scholar]
- Kumanogoh A, Marukawa S, Suzuki K, Takegahara N, Watanabe C, Ch’ng E, Ishida I, et al. (2002) Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature 419, 629–633. [DOI] [PubMed] [Google Scholar]
- Leitner DF, Connor JR (2012) Functional roles of transferrin in the brain. Biochim. Biophys. Acta - Gen. Subj 1820, 393–402. [DOI] [PubMed] [Google Scholar]
- Li R, Luo C, Mines M, Zhang J, Fan G-H (2006) Chemokine CXCL12 Induces Binding of Ferritin Heavy Chain to the Chemokine Receptor CXCR4, Alters CXCR4 Signaling, and Induces Phosphorylation and Nuclear Translocation of Ferritin Heavy Chain. J. Biol. Chem 281, 37616–37627. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lee SY, Neely E, Nandar W, Moyo M, Simmons Z, Connor JR (2011) Mutant HFE H63D protein is associated with prolonged endoplasmic reticulum stress and increased neuronal vulnerability. J. Biol. Chem 286, 13161–13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopin KV, Gray IP, Obejero-Paz CA, Thevenod F, Jones SW (2012) Fe2+ Block and Permeation of CaV3.1 (α1G) T-Type Calcium Channels: Candidate Mechanism for Non-Transferrin-Mediated Fe2+ Influx. Mol. Pharmacol 82, 1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie EL, Iwasaki K, Tsuji Y (2008) Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid. Redox Signal 10, 997–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moos T, Morgan EH (2002) A morphological study of the developmentally regulated transport of iron into the brain. Dev. Neurosci 24, 99–105. [DOI] [PubMed] [Google Scholar]
- Nandar W, Connor JR (2011) HFE gene variants affect iron in the brain. J. Nutr 141, 729S–739S. [DOI] [PubMed] [Google Scholar]
- Nandar W, Neely EB, Simmons Z, Connor JR (2014) H63D HFE genotype accelerates disease progression in animal models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta - Mol. Basis Dis 1842, 2413–2426. [DOI] [PubMed] [Google Scholar]
- Nandar W, Neely EB, Unger E, Connor JR (2013) A mutation in the HFE gene is associated with altered brain iron profiles and increased oxidative stress in mice. Biochim. Biophys. Acta 1832, 729–41. [DOI] [PubMed] [Google Scholar]
- Olynyk JK, Trinder D, Ramm GA, Britton RS, Bacon BR (2008) Hereditary hemochromatosis in the post- HFE era. Hepatology 48, 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz E, Pasquini JM, Thompson K, Felt B, Butkus G, Beard J, Connor JR (2004) Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J. Neurosci. Res 77, 681–689. [DOI] [PubMed] [Google Scholar]
- Pantopoulos K (2008) Function of the hemochromatosis protein HFE: Lessons from animal models. World J. Gastroenterol 14, 6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian ZM, To Y, Tang PL, Feng YM (1999) Transferrin receptors on the plasma membrane of cultured rat astrocytes. Exp. Brain Res 129, 473–6. [DOI] [PubMed] [Google Scholar]
- Raha-Chowdhury R, Raha AA, Forostyak S, Zhao J-W, Stott SRW, Bomford A (2015) Expression and cellular localization of hepcidin mRNA and protein in normal rat brain. BMC Neurosci 16, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncagliolo M, Garrido M, Walter T, Peirano P, Lozoff B (1998) Evidence of altered central nervous system development in infants with iron deficiency anemia at 6 mo: delayed maturation of auditory brainstem responses. Am. J. Clin. Nutr 68, 683–690. [DOI] [PubMed] [Google Scholar]
- Santiago C, Ballesteros A, Tami C, Martínez-Muñoz L, Kaplan GG, Casasnovas JM (2007) Structures of T Cell Immunoglobulin Mucin Receptors 1 and 2 Reveal Mechanisms for Regulation of Immune Responses by the TIM Receptor Family. Immunity 26, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonberg DL, Goldstein EZ, Sahinkaya FR, Wei P, Popovich PG, McTigue DM (2012) Ferritin Stimulates Oligodendrocyte Genesis in the Adult Spinal Cord and Can Be Transferred from Macrophages to NG2 Cells In Vivo. J. Neurosci 32, 5374–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibille JC, Ciriolo M, Kondo H, Crichton RR, Aisen P (1989) Subcellular localization of ferritin and iron taken up by rat hepatocytes. Biochem. J 262, 685–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Ponnuru P, Klinger ME, Myers RL, Devraj K, Coe CL, Lubach GR, Carruthers A, Connor JR (2015) A Novel Model for Brain Iron Uptake: Introducing the Concept of Regulation. J. Cereb. Blood Flow Metab 35, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder AM, Wang X, Patton SM, Arosio P, Levi S, Earley CJ, Allen RP, Connor JR (2009) Mitochondrial Ferritin in the Substantia Nigra in Restless Legs Syndrome. J. Neuropathol. Exp. Neurol 68, 1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EM, Morgan EH (1990) Developmental changes in transferrin and iron uptake by the brain in the rat. Dev. brain Res 55, 35–42. [DOI] [PubMed] [Google Scholar]
- Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR (2009) Oligodendrocytes and myelination: The role of iron. Glia 57, 467–478. [DOI] [PubMed] [Google Scholar]
- Todorich B, Zhang X, Connor JR (2011) H-ferritin is the major source of iron for oligodendrocytes. Glia 59, 927–935. [DOI] [PubMed] [Google Scholar]
- Todorich B, Zhang X, Slagle-Webb B, Seaman WE, Connor JR (2008) Tim-2 is the receptor for H-ferritin on oligodendrocytes. J. Neurochem 107, 1495–1505. [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Yabuki M, Kamei J, Kamei M, Makino N, Kumanogoh A, Hori M (2007) Semaphorin-4A, an activator for T-cell-mediated immunity, suppresses angiogenesis via Plexin-D1. EMBO J 26, 1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger EL, Hurst AR, Georgieff MK, Schallert T, Rao R, Connor JR, Kaciroti N, Lozoff B, Felt B (2012) Behavior and Monoamine Deficits in Prenatal and Perinatal Iron Deficiency Are Not Corrected by Early Postnatal Moderate-Iron or High-Iron Diets in Rats. J. Nutr 142, 2040–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varatharaj A, Galea I (2017) The blood-brain barrier in systemic inflammation. Brain. Behav. Immun 60, 1–12. [DOI] [PubMed] [Google Scholar]
- Wade QW, Chiou B, Connor JR (2019) Iron uptake at the blood-brain barrier is influenced by sex and genotype, in Adv. Pharmacol, pp. 123–145. [DOI] [PubMed]
- Yukawa K, Tanaka T, Bai T, Ueyama T, Owada-Makabe K, Tsubota Y, Maeda M, Suzuki K, Kikutani H, Kumanogoh A (2005) Semaphorin 4A induces growth cone collapse of hippocampal neurons in a Rho/Rho-kinase-dependent manner. Int. J. Mol. Med 16, 115–8. [PubMed] [Google Scholar]
- Zechel S, Huber-Wittmer K, Bohlen und Halbach O. von (2006) Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J. Neurosci. Res 84, 790–800. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gunput R-AF, Pasterkamp RJ (2008) Semaphorin signaling: progress made and promises ahead. Trends Biochem. Sci 33, 161–170. [DOI] [PubMed] [Google Scholar]
- Zimmermann MB, Hurrell RF (2007) Nutritional iron deficiency [DOI] [PubMed]