

Abstract
The ophiobolin sesterterpenes are notable plant pathogens, which have recently elicited significant chemical and biological attention due to their intriguing carbogenic frameworks, reactive functionality, and emerging anticancer profiles. Herein we report a total synthesis of (+)-6-epi-ophiobolin A in 14 steps, a task which addresses construction of the synthetically challenging spirocyclic tetrahydrofuran motif as well as several other key stereochemical problems. This work demonstrates a streamlined synthetic platform to complex ophiobolins leveraging disparate termination modes of a radical polycyclization cascade for divergent elaboration and functionalization.
Keywords: total synthesis, terpene, natural product, radical reaction, cascade
Graphical Abstract
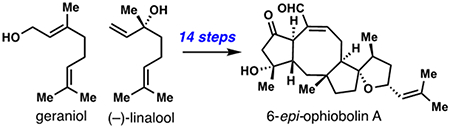
The ophiobolins are a class of biologically active 5,8,5-fused sesterterpene natural products isolated from pathogenic fungi of the Bipolaris and Aspergillus genus (see 1-6, Figure 1A).[1] Initially investigated as a result of their phytotoxic effects and calmodulin A inhibitory properties, most ophiobolins are now known to inhibit the growth of human cancer cells.[1b] In particular, ophiobolin A (1), the most abundant and well-studied member, shows strong cytotoxic properties against glioblastoma multiforme (GBM), breast cancer (MCF-7), multi-drug resistant leukemia (MDR HL-60) and over 30 other cell lines.[1b] The epimeric congener 6-epi-ophiobolin A (2) is also cytotoxic, and amongst evaluated ophiobolins, the relationship between C-6 stereochemistry and cytotoxicity appears to be cell line specific.[1b] Phytotoxic evaluation of 2 also shows unique selectivity patterns relative to 1,[2] implying that these molecules may have different, and context-dependent, biological targets. Despite interest from the synthetic community,[3] only three ophiobolin syntheses have been developed in the past three decades.[4–6] We recently completed a short synthesis of ent-6, a lower oxidation state congener, in nine steps from abundant chiral pool building block (–)-linalool (7) (Figure 1B).[6,7] Our lab has a longstanding interest in being able to prepare all known ophiobolin structural types; herein we describe a route to complex ophiobolins in their correct enantiomeric series also using (–)-7, work which has resulted in a 14-step route to (+)-2. This chemistry greatly expands the ophiobolins that can be prepared using a radical cascade strategy.
Figure 1.
A) Representative ophiobolin sesterterpenes. B) Overarching synthetic strategy leveraging divergent termination modes of a radical cyclization cascade.
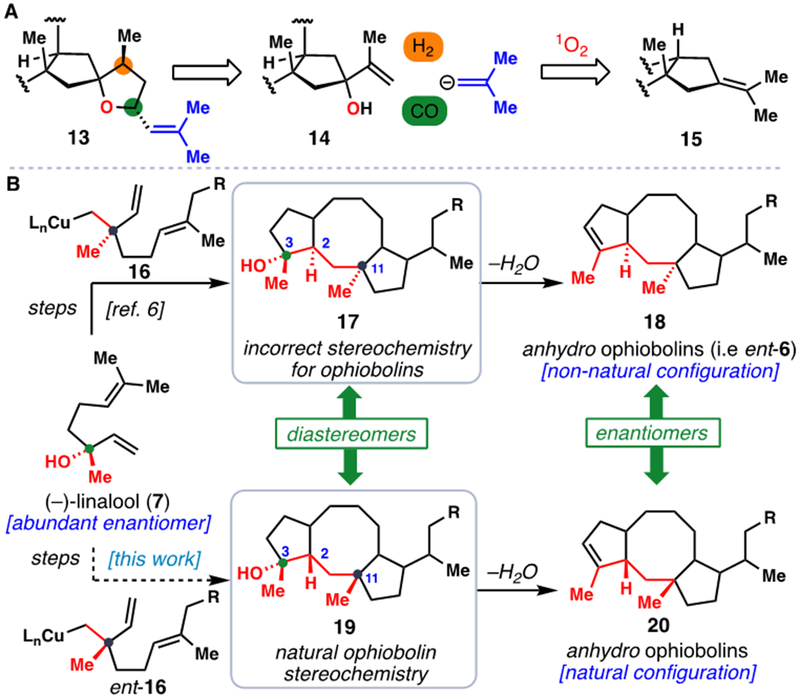
Our prior work toward the ophiobolins leveraged an 8-endo/5-exo radical cascade cyclization of trichloroketone-derived radical 8, itself prepared from (–)-linalool (7), to rapidly assemble the hallmark 5,8,5-fused ring system of these natural products (Figure 1B).[6] During these investigations, several termination modes were available to the radical formed after cyclization (see 9) depending on the conditions employed. In path A (shown in green), an overall reductive process could be achieved leading to saturated motif 10 which was advanced to (–)-6-epi-ophiobolin N (ent-6).[6] Non-reductive pathways leading to 1,1-disubstituted alkene 11 (path B in orange) or internal alkene 12 (path C in blue) were also noted and in fact proved challenging to avoid in our pursuit of path A. While the formation of 10 offered the most expedient entry to the ophiobolins, the minimally oxidized side chain formed presents significant synthetic hurdles when completing the pursuit of more complex members, especially those containing the challenging spirocyclic THF-ring motif found in 1 and 2. On the other hand, tricyclic alkene 15 was viewed as containing an ideal handle for such chemistry in that it could be potentially converted into tertiary alcohol 14 via a stereoselective singlet oxygen (1O2) ene reaction (Figure 2A). From 14, the spirocyclic ether (13) was envisioned to arise through a hydroformylation reaction generating a lactol, which could be merged with a suitable vinyl organometallic nucleophile (shown in blue). If successful, this challenging sector of the most complex ophiobolins could potentially be assembled in only three steps from the products of the radical cyclization cascade.
Figure 2.
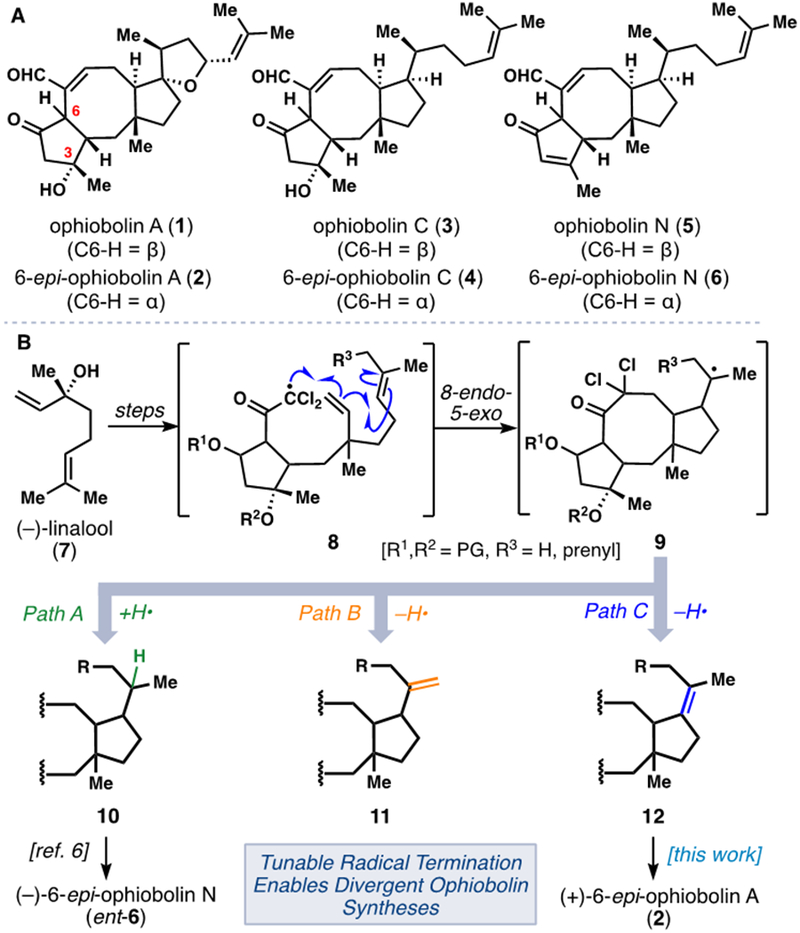
A) Retrosynthetic analysis of the ophiobolin A spirocyclic THF ring motif. B) Stereochemical considerations in past and present routes to ophiobolin members.
In addition to the unique THF ring, several key stereochemical hurdles needed to be surmounted in order to achieve a total synthesis of 2, and in fact most members of the ophiobolin family (Figure 2B). In our previous work toward ent-6, (–)-linalool (7) and a chiral neopentyl copper reagent (16) served as initial sources of chirality ultimately leading to a 5,8,5-fused ring system (see 17) with the indicated stereochemistry. Notably, the relative stereochemistry between the C-3, C-2, and C-11 centers in 17 does not match that found in any ophiobolin member, thereby necessitating a dehydration of C-3 hydroxyl group to produce enone-containing members (see 18). Thus prior to the work reported herein, only enone-containing ophiobolins in their non-natural enantiomeric series could be prepared from the abundant, levorotary enantiomer of linalool.[7,8] Interestingly, (–)-linalool (7) does possess the correct stereochemistry for the C-3 position of the ophiobolins (Figure 2B). Thus, by switching the enantiomer of the copper reagent to ent-16 and inverting the C-2 configuration, it was anticipated that isomer 19 could be accessed which corresponds to the natural ophiobolin stereochemistry. Nevertheless, the success of our previously investigated radical cyclization chemistry was far from certain with this new stereochemical series.
We began by targeting cylopentanone 24 and thus developing a solution to invert the C-2 configuration relative to prior studies (Scheme 1). A two-step procedure was developed to construct this ketone, a product we were unable to form in one-step via direct, diastereoselective conjugate addition.[9] First, geraniol-derived cyclopropyl iodide 21, prepared in two-steps using Charette’s protocol,[10] was converted to its ring-opened lithiate (t-BuLi) and coupled with (–)-linalool-derived enone 22 under the mediation of CuI. In a divergence from prior work, the resulting enolate was quenched with Mukaiyama’s reagent (N-tert-Butylbenzenesulfinimidoyl chloride), leading directly to enone 23 upon warming.[11] Notably, the addition of HMPA to the presumed copper enolate prior to addition of Mukaiyama’s reagent was critical to the success of this process. With 23 in hand we examined 1,4-reduction conditions to set the C-2 stereocenter. We suspected that this conjugate reduction would be extremely challenging due to the neo-pentyl and quaternary centers adjacent to the enone system. Not surprisingly, well- precedented hydride reagents capable of conjugate reduction led to either exclusive carbonyl 1,2-reduction or no reaction at all. After extensive experimentation, we identified that stoichiometric quantities of PhMe2SiCu–H (generated in situ from DIBAL and PhMe2SiCu), reported by Daniewski and co-workers,[12] led to a highly diastereoselective 1,4-reduction – only trace quantities of carbonyl reduction were observed. Following conjugate reduction, methyl lithium was added to increase the reactivity of the in-situ generated aluminum enolate formed, thus allowing for a productive C-acylation event with trichloroacetyl chloride.
Scheme 1.
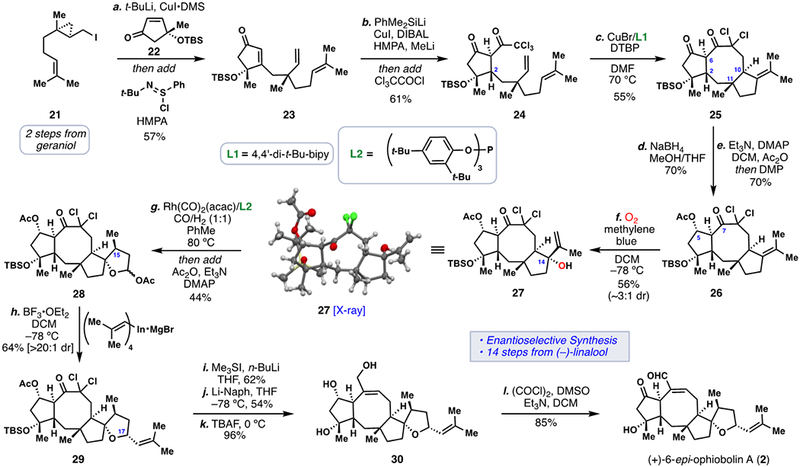
Enantioselective total synthesis of 6-epi-ophiobolin A (2). (a) 21 (1.3 equiv), t-BuLi (1.9 equiv), pentane/Et2O, –78 °C; then add CuI (0.6 equiv), DMS (2.1 equiv), 35 min; then add 22 (1.0 equiv), –78 °C to –40 °C, 4 h; then add HMPA (10.0 equiv), 10 min, –78 °C; then add N-tert-Butylbenzenesulfinimidoyl chloride (1.5 equiv), –78 °C to 25 °C, 2 h (57%); b) PhMe2SiLi, (1.25 equiv), CuI (1.0 equiv), THF, –10 °C, 10 min; then add HMPA (36.0 equiv), DIBAL (3.0 equiv), –50 °C, 30 min; then add 23 (1.0 equiv), –78 °C to –30 °C, 3 h; then add MeLi (3.8 equiv), –78 °C, 30 min; then add trichloroacetyl chloride (6.0 equiv), –78 °C to 25 °C, 3 h (61%); c) CuBr (0.7 equiv), 4,4’-di-tert-butyl-bipyridine (0.9 equiv), 2,6-di-tert-butyl-pyridine (1.0 equiv), DMF, 70 °C, 1 h (55%); d) NaBH4 (15.0 equiv), MeOH/THF (v:v = 4:1), –40 °C, 1 h (70%); e) DMAP (0.5 equiv), Et3N (2.0 equiv), Ac2O (1.0 equiv), DCM, 0 °C, 1 h; then add DMP (10.0 equiv), 0 °C to 25 °C, 1 h (70%); f) Methylene blue (0.01 equiv), O2, hν, DCM, –78 °C, 20 min; then add PPh3 (3.0 equiv), –78 °C to 25 °C, 2 h (56%, 3:1 dr); g) Rh(CO)2(acac) (0.3 equiv), tris(2,4-di-tert-butylphenyl)phosphite (0.6 equiv), 1:1 CO/H2 (100 psi), PhMe, 80 °C, 24 h; then add DMAP (0.6 equiv), Et3N (10.0 equiv), Ac2O (5.0 equiv), 0 °C to 25 °C, 1 h (44%); h) BF3•OEt2 (3.0 equiv) DCM, –78 °C, 5 min; then add In(C4H7)4•MgBr (3.0 equiv), –78 °C, 8 h (64%); i) Me3SI (20 equiv), n-BuLi (7.0 equiv), THF, –20 °C, 10 min (62%); j) Li-naphthalenide (35 equiv), THF, –78 °C, 15 min (54%); k) TBAF (5.0 equiv), THF, 0 °C, 29 h (96%); l) DMSO (15.0 equiv), (COCl)2 (10.0 equiv), DCM, –78 °C, 30 min; then add Et3N (20 equiv), –78 °C to 0 °C, 3 h (85%); DMS = dimethyl sulfide, HMPA = hexamethylphosphoramide, DMAP = 4-dimethylaminopyridine, DMP = Dess-Martin Periodinane, acac = acetylacetone, DIBAL = Diisobutylaluminium hydride, TBAF = Tetrabutylammonium fluoride, DTBP = 2,6-Di-tert-butylpyridine.
With access to trichloroketone 24, we were poised to examine the key radical cyclization cascade in this new diastereomeric series. Pleasingly, we discovered that intramo-lecular atom-transfer radical cyclization using di-tert-butylbipyridine-ligated Cu(I) proceeded efficiently, generating polycycle 25 in 55% isolated yield. Notably, trans stereo-chemistry was observed at the newly formed 5,8-fused ring junction (C-10 and C-11) despite the C-2 and C-6 centers having opposite configurations as compared to our prior work.[6] With 25 secured, we shifted our attention to formation of the formidable THF-ring system. Preliminary studies, however, indicated that various oxidative and late-stage operations would be challenging in the presence of the free 1,3-diketone moiety, thus necessitating selective protection of this section of the molecule. A two-step pro-tocol involving double reduction of the diketone followed by one-pot, C-5 hydroxyl acylation/C-7 hydroxyl oxidation (DMP) generated 26 in 49% over two steps.
Forays into construction of the THF-ring motif could com-mence with 26 in hand (Scheme 1). The singlet oxygen ene reaction of 26 (O2, methylene blue, DCM, –78 °C) proceeded to generate 27 as a separable mixture of C-14 tertiary alcohol diastereomers (60% combined yield, dr ~ 3:1). Single crystal X-ray diffraction of the major isomer confirmed this structural assignment as well as previously generated stereocenters.[13] Next, hydroformylation of 27 was pursued.[14] This transformation proved challenging due to the presence of the dichloroketone motif which was easily dehalogenated, thus limiting the catalyst systems and conditions that could be successfully employed. Ultimately, we found that exposure of 27 to catalytic quantities of Rh(CO)2(acac) and the bulky phosphite ligand tris(2,4-di-tert-butylphenyl)phosphite under a 1:1 atmosphere of CO/H2 (100 psi), produced the lactol-acetate motif in near quantitative yield after in-situ acetylation with acetic anhydride. Under these conditions, the desired C-15-configured diastereomer (28) was generated in 44% yield.[15]
All that remained to construct the THF ring was generation of the C-17 stereocenter. At the outset, we anticipated that the addition of a 2-methyl-propenyl nucleophile to an in-situ generated oxocarbenium ion should favor a trans C-15–C-17 stereochemical relationship due to the steric influence of the C-15 methyl group; in practice this proved correct under several conditions (Table 1). Initial experiments adding 2-methyl-propenyl magnesium bromide to 28 in the presence of BF3•OEt2 led to 36% of the addition product as a 3:1 mixture of C-17 diastereomers favoring the trans-addition product (29) (entry 1). Zinc, cerium, and copper-based organometallics performed poorly in this reaction with respect to both yield and selectivity (entries 2–4). Cognizant of the relatively mild nature of organoindium nucleophiles in various substitution reactions, we began to examine this class of organometallic nucleophiles.[16] These reagents were prepared in-situ from InCl3 and various quantities of 2-methyl-propenyl magnesium bromide (entries 5–7). While InCl2(C4H7) gave poor results (entry 5), In(C4H7)3 provided 54% yield of product (entry 6). Surprisingly, though, no facial selectivity was observed under these conditions. Much to our delight, however, the tetraorganoindate complex (In(C4H7)4•MgBr) coupled smoothly (64%) and with high selectivity for the correct C-17 diastereomer.[17,18] We presume that the anionic nature of this species and increased steric bulk surrounding the tetra-coordinate indium center accounts for the increased reactivity and selectivity of this reagent in the addition reaction.
Table 1.
Synthesis of tetracycle 29: Selected optimization.
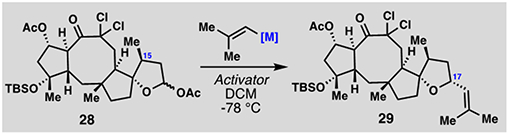 | |||
|---|---|---|---|
| Entry[a] | Activator | Nucleophile | Yield (29: 17-epi-29)[b] |
| 1 | BF3•OEt2 | MgBr(C4H7) | 36% (3:1) |
| 2 | BF3•OEt2 | ZnBr(C4H7) | 11% (2:1) |
| 3 | BF3•OEt2 | CeCl2(C4H7) | 10% (1:1.5) |
| 4 | TMSBr | CuTC(CN)(C4H7) | 17% (1:1) |
| 5 | BF3•OEt2 | InCl2(C4H7) | <5% |
| 6 | BF3•OEt2 | In(C4H7)3 | 54% (1:1) |
| 7 | BF3•OEt2 | In(C4H7)4•MgBr | 64% (>20:1) |
yields and selectivities determined by 1H NMR analysis.
yields based on amount of correct C15 diastereomer in starting 28.
To complete the synthesis of 6-epi-ophiobolin A from 29, a four-step endgame was employed in analogy to previous work on ent-6 (Scheme 1). Corey-Chaykovsky epoxidation of the C-7 carbonyl proceeded with concomitant deprotection of the C-5 acetate in 62% yield. Next, tandem reductive epoxide opening and dehalogenation (lithium naphthalenide) followed by silyl ether deprotection (TBAF) generated triol intermediate 30 in 52% yield over two steps. Finally, Swern oxidation of the triol intermediate generated the target in 85% yield, thus completing a 14-step total synthesis of (+)-2 from (–)-linalool and geraniol.
In conclusion, we have leveraged the divergent termination modes of a radical polycyclization cascade to rapidly construct 5,8,5-fused tricyclic ring systems with handles for elaboration into the most complex ophiobolin chemotypes. A number of stereochemical challenges were solved along the way, although solutions to the C-15 stereogenic center remain elusive in this context. Nevertheless, in combination with our prior studies, this work again highlights the unique advantages and flexibility that radicals can offer relative to biomimetic, cationic chemistry in the efficient construction of complex terpene natural products.[19]
Supplementary Material
Acknowledgements
Financial support is acknowledged from the NIH (R01GM116952). Novartis, Amgen, Eli Lilly, and Bristol-Myers Squibb are also thanked for unrestricted research support. T. J. M. is an Arthur C. Cope Scholar. Z. G. B. and D. Q. T. acknowledge the NSF (DGE-1106400) for pre-doctoral fellowships. Z. G. B. also acknowledges UC-Berkeley for a Berkeley Fellowship. K. V. E. in appreciative to the NSERC for a postdoctoral fellowship. J. K. T. thanks the ACS Division of Organic Chemistry for a Summer Undergraduate Research Fellowship (2019 DOC SURF). We are grateful to Dr. Hasan Celik for NMR spectroscopic assistance and NIH grant S10OD024998. Jeffrey S. Derrick is acknowledged for X-ray crystallographic analysis wherein support from NIH Shared Instrument Grant (S10-RR027172) is also acknowledged. We thank the Hartwig group and the LBNL Catalysis Center for their assistance with our hydroformylation studies.
References
- [1].For reviews on ophiobolins, see:; a) Au TK, Chick WSH, Leung PC. Life Sci. 2000, 67, 733; [DOI] [PubMed] [Google Scholar]; b) Tian W, Deng Z, Hong K, Mar. Drugs 2017, 15, 229; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Masi M, Dasari R, Evidente A, Mathieu V, Kornienko A, Bioorg. Med. Chem. Lett 2019, 29, 859. [DOI] [PubMed] [Google Scholar]
- [2].Sugawara F, Strobel G, Strange RN, Siedow JN, Van Duyne GD, Clardy J, Proc. Natl. Acad. Sci 1987, 84, 3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].For approaches toward these natural products, see:; a) Salvati AE, Law JA, Liriano J, Frederich JH, Chem. Sci 2018, 9, 5389; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li K, Wang C, Yin G, Gao S, Org. Biomol. Chem 2013, 11, 7550; [DOI] [PubMed] [Google Scholar]; c) Michalak M, Michalak K, Urbanczyk-Lipkowska Z, Wicha J, J. Org. Chem 2011, 76, 7497; [DOI] [PubMed] [Google Scholar]; d) Ruprah PK, Cros J-P, Pease JE, Whittingham WG, Williams JMJ, Eur. J. Org. Chem 2002, 3145; [Google Scholar]; e) Blake AJ, Highton AJ, Majid TN, Simpkins NS, Org. Lett 1999, 1, 1787; [Google Scholar]; f) Wender PA, Nuss JM, Smith DB, J. Org. Chem 1997, 62, 4908; [Google Scholar]; g) Rigby JH, Senanayake CJ, J. Org. Chem 1987, 52, 4634; [Google Scholar]; h) Paquette LA, Colapret JA, Andrews DR, J. Org. Chem 1985, 50, 201; [Google Scholar]; i) Paquette LA, Andrews DR, Springer JP, J. Org. Chem 1983, 48, 1147; [Google Scholar]; j) Dauben WG, Hart DJ, J. Org. Chem 1977, 42, 922; [Google Scholar]; k) Boeckman RK, Bershas JP, Clardy J, Solheim B, J. Org. Chem 1977, 42, 3630. [Google Scholar]
- [4].Rowley M, Tsukamoto M, Kishi Y, J. Am. Chem. Soc 1989, 111, 2735. [Google Scholar]
- [5].a) Tsuna K, Noguchi N, Nakada M, Angew. Chem. Int. Ed 2011, 50, 9452; [DOI] [PubMed] [Google Scholar]; b) Tsuna K, Noguchi N, Nakada M, Chem. Eur. J 2013, 19, 5476. [DOI] [PubMed] [Google Scholar]
- [6].Brill ZG, Grover H, Maimone TJ, Science 2016, 352, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brill ZG, Condakes ML, Ting CP, Maimone TJ, Chem. Rev 2017, 117, 11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].While (–)-linalool is abundant, either enantiomer can be pre-pared from geraniol in two steps. For details, see: Wu Y-K, Liu H-J, Zhu J-L, Synlett 2008, 621.
- [9].All attempts to use the C-3 hydroxy group, or protected variants, to direct the facial selectivity of the conjugate addition were unsuccessful.
- [10].a) Charette AB,Juteau H, Lebel H, Molinaro C, J. Am. Chem. Soc 1998, 120, 11943; [Google Scholar]; b) Charette AB, Naud J, Tetrahedron Lett. 1998, 39, 7259. [Google Scholar]
- [11].Mukaiyama T, Matsuo J-I, Kitagawa H, Chem. Lett 2000, 1250. [Google Scholar]
- [12].Daniewski AR, Liu W, J. Org. Chem 2001, 66, 626. [DOI] [PubMed] [Google Scholar]
- [13].Structure 27 has been deposited in The Cambridge Crystallographic Data Centre (CCDC# 1963634)
- [14].For related examples, see:; a) Eilbracht P, P. et al. , Chem. Rev 1999, 3329; [DOI] [PubMed] [Google Scholar]; b) Airiau E et al. , Synthesis 2010, 2901. [Google Scholar]
- [15].The purified isomeric mixture could not be further separated on silica gel at this point and the mixture was used in the next step.
- [16].Shen Z-L, Wang S-Y, Chok Y-K, Xu Y-H, Loh T-P, Chem. Rev 2013, 113, 271. [DOI] [PubMed] [Google Scholar]
- [17].For applications of tetraorganoindates, see:; a) Araki S, Shimizu T, Jin S-J, Butsugan Y, J. Chem. Soc. Chem. Commun 1991, 824; [Google Scholar]; b) Araki S, Jin S-J, Butsugan Y. J. Chem. Soc. Perkin Trans 1. 1995, 549; [Google Scholar]; c) Lee PH, Lee SW, Seomoon D, Org. Lett 2003, 5, 4963; [DOI] [PubMed] [Google Scholar]; d) Hirashita T, Hayashi Y, Mitsui K, Araki S, Tetrahedron Lett. 2004, 45, 3225. [Google Scholar]
- [18].Compound 29 could only be separated from its C-15 diastereomer using multiple rounds of preparative TLC. In practical terms, C-15 diastereomers were removed at the stage of triol 30.
- [19].a) Hung K, Hu X, Maimone TJ, Nat. Prod. Rep 2018, 35, 174; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu G, Elkin M, Tantillo DJ, Newhouse TR, Maimone TJ, Angew. Chem. Int. Ed 2017, 56, 12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.